

Summary
Cancer cells must evade immune responses at distant sites to establish metastases. The lung is a frequent site for metastasis. We hypothesized that lung-specific immunoregulatory mechanisms create an immunologically permissive environment for tumor colonization. We found that T cell-intrinsic expression of the oxygen-sensing prolyl-hydroxylase (PHD) proteins is required to maintain local tolerance against innocuous antigens in the lung, but powerfully licenses colonization by circulating tumor cells. PHD proteins limit pulmonary type helper (Th)-1 responses, promote CD4+-regulatory T (Treg) cell induction, and restrain CD8+ T cell effector function. Tumor colonization is accompanied by PHD protein-dependent induction of pulmonary Treg cells and suppression of IFN-γ-dependent tumor clearance. T cell-intrinsic deletion or pharmacological inhibition of PHD proteins limits tumor colonization of the lung and improves the efficacy of adoptive cell transfer immunotherapy. Collectively, PHD proteins function in T cells to coordinate distinct immunoregulatory programs within the lung that are permissive to cancer metastasis.
Graphical abstract
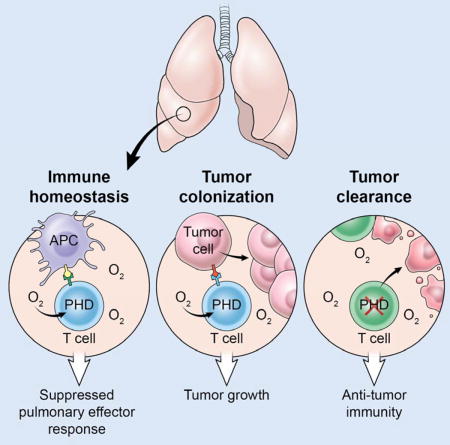
Introduction
Metastasis is the cause of more than 90% of cancer deaths and is a major obstacle for curative therapy (Valastyan and Weinberg, 2011). Metastatic dissemination involves a complex interaction between circulating tumor cells and the site of secondary colonization. Local immunity is an important feature of metastatic sites, and circulating tumor cells must evade local immune responses for successful metastasis (Massague and Obenauf, 2016). The lung is a common metastatic site for numerous cancer types including malignant melanoma (Minn et al., 2005). The extensive capillary network perfusing the lung parenchyma provides an anatomical mechanism for dissemination to this site. We hypothesized that the lungs also form an immunologically favorable site for cancer metastasis.
T cells play a critical role in coordinating immune function. Whereas effector T (Teff) cells promote immune activation and can drive clearance of infections and cancer, regulatory T (Treg) cells, dependent upon the transcription factor Foxp3, suppress their function, preventing excessive autoimmune and allergic reactions (Gavin et al., 2007; Sakaguchi et al., 1985). Inflammatory Teff cell responses are restrained in the lung despite continuous exposure to innocuous foreign antigens (Holt et al., 2008). While many specialized cell types are important regulators of pulmonary tolerance (de Heer et al., 2004; Stumbles et al., 1998), T cell-intrinsic molecular programs may also influence site-specific immunity in the lung.
The prolyl hydroxylase domain containing family of proteins, comprised of PHD1, PHD2, and PHD3, function as intracellular sensors of oxygen (Bruick and McKnight, 2001; Epstein et al., 2001). PHD enzymes are Fe2+-dependent dioxygenases that use a conserved two-histidine, one-carboxylate motif to coordinate Fe2+, 2-oxoglutarate, and free oxygen at the active site (Kaelin and Ratcliffe, 2008). In well-oxygenated environments the PHD enzymes catalyze post-translational hydroxylation of substrate proteins, including hypoxia inducible factors HIF1α and HIF2α (collectively HIFα), which are then degraded (Jaakkola et al., 2001). In T cells HIFα can drive effector responses by promoting differentiation of CD4+ Th17 cells (Dang et al., 2011; Shi et al., 2011), CD8+ T cell effector function (Doedens et al., 2013), and IFN-γ production within T regulatory cells (Lee et al., 2015). This led us to hypothesize that the oxygen-sensing PHD proteins might influence T cell differentiation and function, particularly in the oxygen-rich environment of the lung.
Here, we show that T cell-intrinsic expression of the PHD proteins suppresses pulmonary inflammation against innocuous foreign antigens, but powerfully licenses tumor colonization of the lung. Upon tumor colonization PHD proteins promote Treg cell expansion and restrain IFN-γ-dependent clearance of tumors. Importantly, genetic and pharmacological disruption of PHD proteins limits tumor colonization of the lung and improves the efficacy of T cell-based adoptive cell therapy (ACT). These results indicate that T cell-intrinsic expression of the oxygen-sensing PHD proteins coordinates a tissue-specific immunoregulatory program that suppresses mild inflammatory pathology but readily permits cancer metastasis in the lung.
Results
PHD proteins function within T cells to limit pulmonary effector responses
Pulmonary tolerance requires suppression of inflammatory T cell responses under physiologic conditions. We asked whether T cell-intrinsic expression of the oxygen-sensing PHD proteins contributes to site-specific tolerance in the lung. We generated mice harboring a T cell-specific deletion of all three PHD proteins (henceforth PHD-tKO). Cd4-driven Cre recombinase expression resulted in significant reduction of Egln1, Egln2, and Egln3 mRNA transcripts, which encode PHD2, PHD1, and PHD3 proteins respectively, in CD4+, CD8+, and NKT T cells, but not in other lymphoid cell subsets (Figure S1A). Upon gross evaluation we observed a patchy hemorrhagic appearance within the lungs of PHD-tKO mice that was not present amongst WT littermates (Figure 1A). Histologic examination revealed the presence of diffuse alveolar hemorrhage (DAH) of variable severity in PHD-tKO mice (Figure 1B–C). PHD-tKO mice had increased serum autoantibodies, which can be elevated upon immune-mediated tissue damage (Figure S1B). Pathology in PHD-tKO mice was only observed in the lung, and we did not detect abnormal liver and pancreas enzymes in the blood (Figure 1D).
Figure 1. PHD proteins function within T cells to suppress spontaneous pulmonary inflammation.
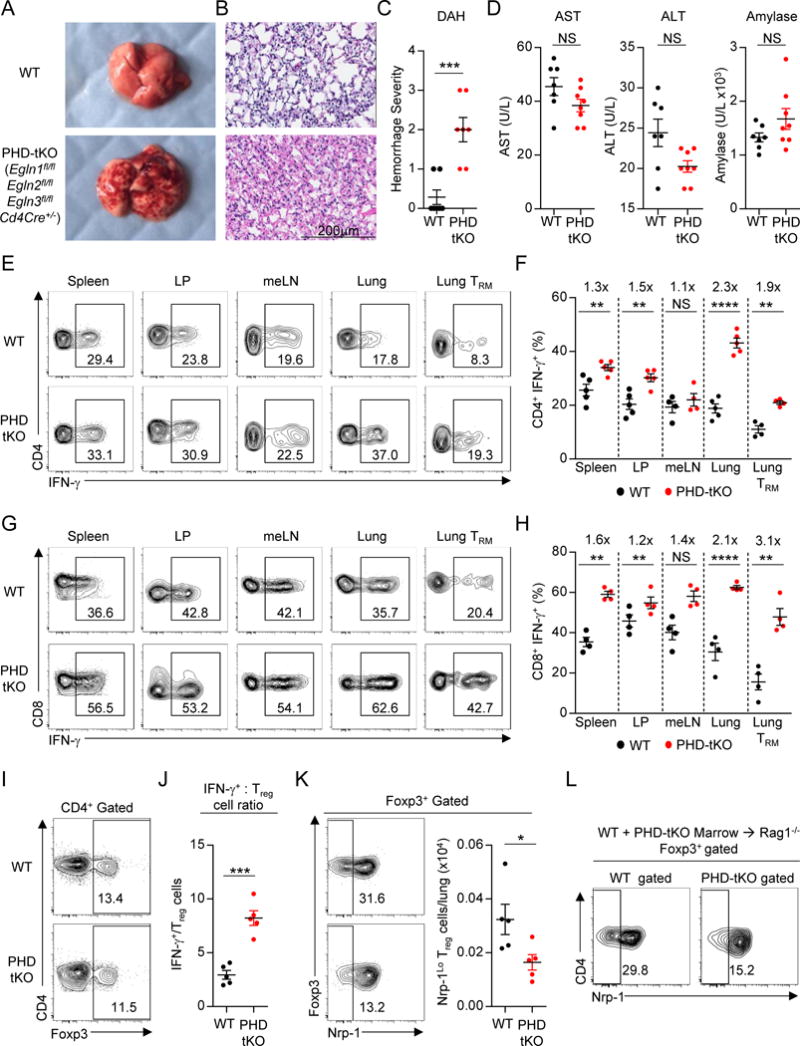
(A) Gross morphology of lungs from Egln1fl/fl Egln2fl/fl Egln3fl/fl CD4Cre+/− triple knockout (PHD-tKO) and Cre-negative wild-type (WT) littermate mice.
(B) Hematoxylin and eosin (H&E) stains of WT and PHD-tKO lungs demonstrating diffuse alveolar hemorrhage (n=7 mice per group).
(C) Histopathology scoring of lung tissue from WT and PHD-tKO mice (n = 7 mice per group).
(D) Titers of hepatic AST and ALT and pancreatic amylase in the sera of WT and PHD-tKO mice
(E–F) IFN-γ production by CD4+ T cells from spleen, small intestine lamina propria (LP), mediastinal lymph node (meLN), lung, and pulmonary tissue resident memory (TRM) cells in WT and PHD-tKO mice. Representative flow cytometry (E) and replicate values (F) are shown. Fold increase in frequency of CD4+ IFN-γ+ T cells in PHD-tKO mice normalized to WT is provided for each organ. (G–H) IFN-γ production by CD8+ T cells in the indicated organs in WT and PHD-tKO mice. Representative flow cytometry (G) and replicate values (H) are shown. Fold increase in frequency of CD8+ IFN-γ+ T cells in PHD-tKO mice normalized to WT is provided for each organ.
(I) Frequency of pulmonary Foxp3+ Treg cells in WT and PHD-tKO mice.
(J) Ratio of total numbers of CD4+ and CD8+ IFN-γ+ Teff cells to Foxp3+ Treg cells in the lungs of WT and PHD-tKO mice.
(K) Distribution of Nrp-1 -Hi and -Lo populations amongst pulmonary Foxp3+ Treg cells in WT and PHD-tKO mice. Representative flow cytometry and absolute numbers of Nrp-1Lo Treg cells are shown.
(L) Representative flow cytometry of Nrp-1 expression by WT and PHD-tKO pulmonary Foxp3+ Treg cells from WT and PHD-tKO mixed bone marrow chimeric mice.
Data are representative of ≥ 2 independent experiments with ≥ 3 mice per genotype. Mice were analyzed at 3 months of age unless otherwise specified. Bars and error represent mean ±SEM of replicate measurements. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (Student’s t-test).
See also Figure S1.
PHD-tKO animals showed no defects in thymocyte number or phenotypic distribution (Figure S1C). Similar numbers of CD4+ and CD8+ T lymphocytes were detected in peripheral blood and secondary lymphoid organs (Figure S1D). In the bone marrow of PHD-tKO mice there was a slight reduction in CD8+ T cells but similar numbers of CD4+ T cells (Figure S1D). As the bone marrow is a reservoir of memory T cells, we evaluated the differentiation state of T cells in WT and PHD-tKO mice. There was a similar distribution of naïve and effector CD4+ T cells in WT and PHD-tKO mice (Figure S1E). However, increased frequencies of terminally differentiated CD8+ T cells were observed in the peripheral blood, spleen, and bone marrow of PHD-tKO mice (Figure S1E–F). This might explain the reduction of CD8+ T cells in the bone marrow of PHD-tKO mice and suggests a role for the PHD proteins in restraining terminal CD8+ T cell differentiation.
T cells can drive immunopathology in the lung. We observed increased numbers of pulmonary CD4+ and CD8+ T cells in PHD-tKO mice (Figure S1G). Additionally, pulmonary T cells in PHD-tKO mice had increased expression of the activation markers CD44 (Figure S1H), CD25, CTLA-4, and GITR (Figure S1I). Importantly, in PHD-tKO mice we measured increased expression of IFN-γ by CD4+ and CD8+ T cells isolated from multiple lymphoid and non-lymphoid organs, yet this was most pronounced in the lung (Figure 1E–H and Figure S1G). Elevated IFN-γ production was more significant in the lung, including among pulmonary tissue resident memory (TRM) T cells, than in T cells residing in mediastinal lymph nodes (meLN) (Figure 1F, H). These data suggest that the PHD proteins predominantly restrain T cell effector function in the lung parenchyma and airway-associated lymphoid tissue. We also measured increased expression of cytolytic effector molecules in pulmonary CD4+ and CD8+ T cells in PHD-tKO mice (Figure S1J). Additionally, we observed increased expression of IFN-γ, but not other effector cytokines, in pulmonary NKT cells from PHD-tKO mice (Figure S1K). Thus, T cell-intrinsic expression of PHD proteins limits effector T cell function and prevents mild spontaneous immune pathology in the lung.
PHD proteins maintain pulmonary Nrp-1Lo Treg cells
Treg cells suppress deleterious effector T cell responses directed against self and innocuous foreign antigens (Strickland et al., 2006). We asked whether increased effector function observed in pulmonary T cells from PHD-tKO mice is secondary to a local deficit in Treg cells. Pulmonary Treg cell frequency and absolute number was similar in PHD-tKO mice compared to WT controls (Figure 1I and Figure S1G). The overall phenotypic profile thus revealed a significantly elevated IFN-γ+ Teff to Treg cell ratio in lungs of PHD-tKO mice (Figure 1J).
The stability of Foxp3+ Treg cells is an important parameter in their immunoregulatory function. Treg cell stability is proportional to the level of demethylation within the Treg specific demethylated region (TSDR) of the Foxp3 locus (Floess et al., 2007). TSDR CpG methylation was similar in WT and PHD-tKO Treg cells (Figure S1L). Consistently, the stability of Foxp3 expression was similar in WT and PHD-tKO Treg cells following restimulation in vitro or upon in vivo transfer into Rag1−/− hosts (Figure S1M–O).
Treg cell populations are comprised of thymically-derived Treg (tTreg) cells and peripherally induced iTreg cells (Bluestone and Abbas, 2003). iTreg cells are particularly important in suppressing effector T cell responses at mucosal sites (Josefowicz et al., 2012). Neuropilin-1 (Nrp-1) has been proposed as a marker to distinguish tTreg cells from iTreg cells (Weiss et al., 2012; Yadav et al., 2012). We detected a reduction in the frequency and number of Nrp-1Lo Treg cells in the lungs of PHD-tKO mice (Figure 1K). Nrp-1 expression can be induced on Treg cells in inflammatory environments (Weiss et al., 2012). This raised the possibility that the observed reduction in pulmonary Nrp-1Lo Treg cells in PHD-tKO mice is the result of induced Nrp-1 expression in the inflamed lung environment. To exclude this possibility, we tested whether differences in Nrp-1 expression between WT and PHD-tKO Treg cells exist when cells are exposed to an identical pulmonary environment. We reconstituted lethally ablated Rag1−/− mice with bone marrow isolated from congenically distinguishable WT or PHD-tKO mice in a 1:1 mixture and evaluated pulmonary Treg cells following reconstitution. Importantly, we observed a reduced frequency of Nrp-1Lo cells within PHD-tKO pulmonary Treg cells (Figure 1L). Thus, the reduced frequency of Nrp-1Lo Treg cells in lungs of PHD-tKO mice is not secondary to the inflamed environment, but instead is a cell-intrinsic phenomenon.
PHD proteins restrain Th1 inflammation against innocuous foreign antigens
Respiratory surfaces are chronically exposed to immunogenic and normally non-pathogenic environmental antigens. These stimuli predominantly induce non-inflammatory immunosuppressive Treg cell responses or Th2 responses, which in certain instances can lead to the development of asthma (Strickland et al., 2006; Stumbles et al., 1998). We hypothesized that the mild steady-state pathology observed in lungs of PHD-tKO mice was caused by excessive inflammatory responses against innocuous environmental antigens. To test this, we sensitized and challenged the airway of WT and PHD-tKO mice with house dust mite (HDM) extract.
HDM challenge promoted similar expansion of pulmonary CD4+ T cells in WT and PHD-tKO mice (Figure S2A). HDM challenge induced a robust expansion of pulmonary Th2 cells in WT mice (Figure 2A, C), but failed to do so in PHD-tKO mice. Remarkably, we instead observed a dramatic increase in IFN-γ+ Th1 cells in lungs of PHD-tKO mice upon HDM challenge (Figure 2A, C). Additionally, HDM challenge induced accumulation of pulmonary Treg cells that was reduced in PHD-tKO mice compared to WT controls (Figure 2B–C). PHD-tKO mice also exhibited greater expansion of CD8+ T cells and induction of IFN-γ expression amongst these cells upon HDM challenge (Figure 2D–E, Figure S2B). Consequently, PHD-tKO mice suffered from more severe inflammation following chronic HDM exposure that was consistent with Th1 rather than Th2 driven immunopathology including vasculitis and diffuse tissue damage leading to alveolar hemorrhage (Figure 2F–H). Thus, the PHD proteins coordinate a T cell-intrinsic program that restrains inflammatory T cell effector responses against innocuous foreign antigens in the lung.
Figure 2. T cell-intrinsic PHD proteins restrain Th1 inflammation against innocuous antigens.
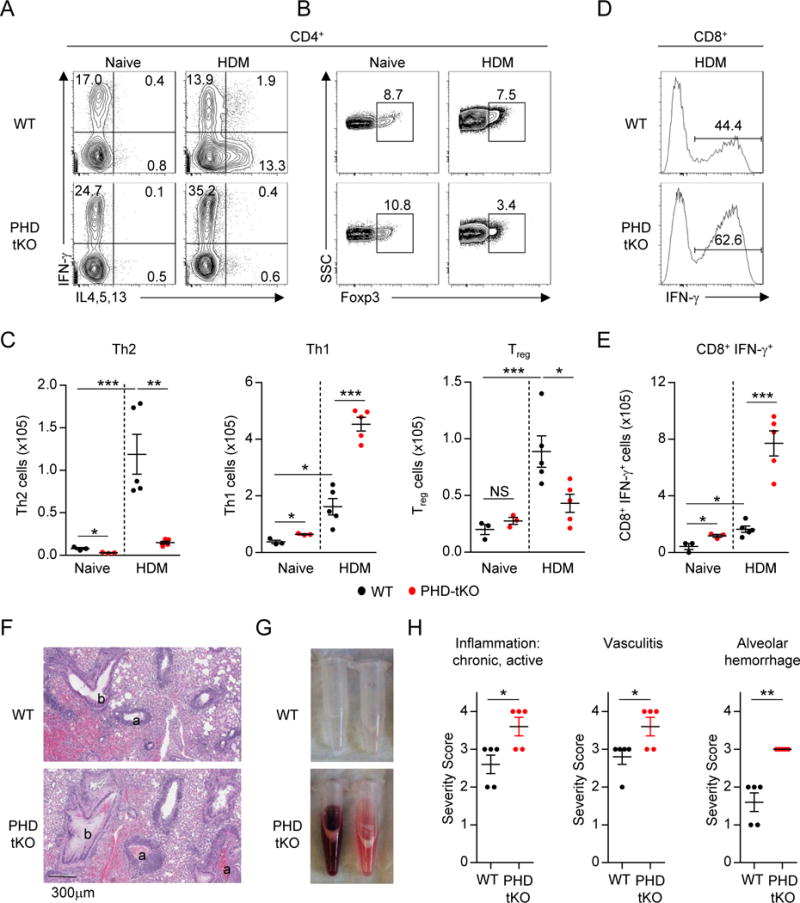
(A–B) Flow cytometry analysis of Th1 and Th2 cytokine production (A) and Foxp3 expression (B) by pulmonary CD4+ T cells from naïve and house dust mite (HDM) challenged WT and PHD-tKO mice.
(C) Total numbers of pulmonary Th2, Th1, and Treg cells in naïve and HDM challenged WT and PHD-tKO mice.
(D–E) IFN-γ production by pulmonary CD8+ T cells from HDM challenged WT and PHD-tKO mice. Representative flow cytometry (D) and total numbers of CD8+ IFN-γ+ T cells (E) are shown.
(F) Representative hematoxylin and eosin (H&E) stains of WT and PHD-tKO lungs following airway sensitization and challenge with house dust mite extract. a, arteriole; b, bronchiole.
(G) Representative bronchoalveolar lavage fluid appearance from WT and PHD-tKO mice.
(H) Histopathology scoring of lung tissue from HDM challenged WT and PHD-tKO mice
Data are representative of ≥ 2 independent experiments with ≥ 3 mice per group. Bars and error represent mean ±SEM of replicate measurements. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (Student’s t-test).
See also Figure S2.
PHD proteins regulate reciprocal iTreg and Th1 differentiation programs
The decrease in steady state pulmonary Nrp-1Lo Treg cells and reduction in HDM-induced Treg cell accumulation suggested that the PHD proteins might promote iTreg cell differentiation. Stimulation of naïve CD4+ T cells in the presence of IL-2 and TGF-β promotes the development of iTreg cells in vitro and in vivo (Chen et al., 2003; Li et al., 2006). To determine if PHD proteins are required for iTreg cell differentiation, we evaluated the gene expression profile of PHD-tKO and WT CD4+ T cells primed in vitro in conditions favoring iTreg cell generation. RNA sequencing revealed widespread gene expression differences between PHD-tKO and WT cells (545 upregulated and 344 downregulated genes in PHD-tKO). Importantly, a significant reduction in Foxp3 expression and induction of genes encoding the Th1 lineage specifying transcription factor T-bet (Tbx21) and effector cytokine IFN-γ (Ifng) was observed in PHD-tKO cells (Figure 3A). By contrast, genes encoding Th2 and Th17 lineage specifying transcription factors and effector cytokines were expressed at similar levels in WT and PHD-tKO cells (Figure 3A). Competitive antagonism of PHD protein enzymatic activity using a pan-PHD inhibitor, dimethyloxalylglycine (DMOG), caused similar changes in global gene expression as those observed in PHD-tKO T cells (Figure S3A–B).
Figure 3. PHD proteins regulate reciprocal iTreg and Th1 differentiation programs.
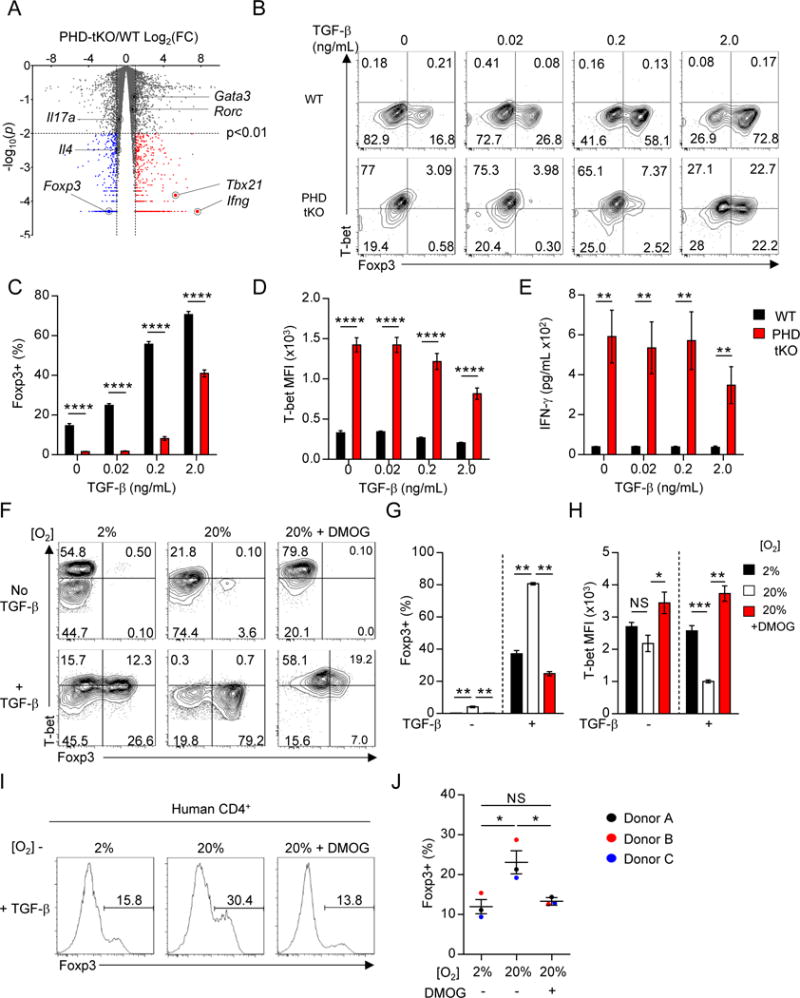
(A) Volcano plot of expressed transcripts (RPKM≥1) in PHD-tKO compared with WT CD4+ T cells stimulated in vitro. Transcripts significantly (p<0.01) over-expressed (FC≥2, red) and under-expressed (FC≤0.5, blue) in PHD-tKO cells are indicated.
(B–D) Foxp3 and T-bet expression in WT and PHD-tKO CD4+ T cells stimulated in vitro in the presence of indicated amounts of TGF-β. Representative flow cytometry (B) and replicate values for Foxp3 (C) and T-bet (D) are shown.
(E) ELISA quantification of IFN-γ in culture supernatants from WT and PHD-tKO CD4+ T cells stimulated as described in (B).
(F–H) Foxp3 and T-bet expression in WT CD4+ T cells stimulated in vitro under the indicated environmental oxygen concentrations ± DMOG. Representative flow cytometry (F) and replicate values (G–H) shown.
(I–J) Foxp3+ iTreg fate specification of human CD4+ T cells stimulated in vitro under the indicated environmental oxygen concentrations ± DMOG. Representative flow cytometry histograms (I) and replicate values are shown (J).
Data are representative of ≥ 2 independent experiments. Bars and error represent mean ±SEM of replicate measurements. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (Student’s t-test).
See also Figure S3.
To further evaluate the requirement for PHD proteins in iTreg cell differentiation, we stimulated naïve CD4+ T cells isolated from WT and PHD-tKO mice in the presence of titrated concentrations of TGF-β. PHD proteins were required for TGF-β-driven induction of Foxp3 and repression of T-bet (Figure 3B–D). Consistently, higher amounts of IFN-γ were detected in supernatants collected from PHD-tKO cell cultures (Figure 3E). Naïve PHD-tKO CD4+ T cells also demonstrated impaired iTreg conversion in vivo following transfer into Rag1−/− recipient animals (Figure S3C). These results indicate that PHD proteins regulate reciprocal iTreg and Th1 differentiation programs.
Foxp3+ iTreg cells derived from PHD-tKO cells or WT cells stimulated in the presence of DMOG expressed high levels of T-bet (Figure 3B, F). T-bet expression can be induced in Treg cells in response to IFN-γ (Koch et al., 2012). Indeed, blockade of IFN-γ limited T-bet expression in PHD-tKO Foxp3+ iTreg cells (Figure S3D). Foxp3+ T-bet+ Treg cells exert suppressive function, especially in inflammatory contexts (Koch et al., 2009). Consistently, FACS-purified Foxp3-GFP+ iTreg cells generated in the presence of DMOG, which express high levels of T-bet, were as suppressive in vitro as T-betLo iTreg cells (Figure S3E–F).
Downstream substrates of PHD enzymatic activity including HIF1α have been implicated in Th17 differentiation and CD8+ T cell effector function (Dang et al., 2011; Doedens et al., 2013). We evaluated whether PHD proteins restrain differentiation and function of these lineages. IL-17A production was similar in CD4+ T cells isolated from the lung and small intestine LP of WT and PHD-tKO mice (Figure S3G). Moreover, no differences in RORγt expression or IL-17A production were detected between WT and PHD-tKO T cells stimulated under Th17 polarizing conditions in vitro (Figure S3H). Consistent with the increased frequency of effector CD8+ T cells in PHD-tKO mice, we observed increased terminal differentiation and production of cytolytic molecules amongst PHD-tKO CD8+ T cells stimulated in vitro (Figure S3I–J).
Collectively, these results demonstrate that PHD proteins function within the CD4+ T cell lineage to promote iTreg cell differentiation while restraining the differentiation of Th1 cells. Within the CD8+ T cell lineage, PHD proteins restrain the acquisition of effector cytokine and cytolytic functions.
Extracellular oxygen promotes iTreg cell differentiation in a PHD-dependent manner
Local environmental factors influence immune responses in a site-specific manner. Orally ingested food antigens, vitamins, and commensal microbes and their metabolite byproducts promote local iTreg cell differentiation in the small intestine (Arpaia et al., 2013; Atarashi et al., 2011; Mucida et al., 2005; Sun et al., 2007). T cells in the lung reside in an oxygen rich environment (≥13% O2) (Semenza, 2010). We asked whether extracellular oxygen availability influences the efficiency of iTreg and Th1 cell differentiation. WT CD4+ T cells were stimulated in vitro under fixed extracellular oxygen concentrations. Cells stimulated under higher oxygen tensions demonstrated increased iTreg and reduced Th1 cell differentiation, and this phenomenon was dependent on the oxygen-sensing PHD proteins (Figure 3F–H). High extracellular oxygen also promoted human iTreg cell differentiation in a PHD dependent manner (Figure 3I–J). Thus, PHD proteins enable T cell-intrinsic detection of environmental oxygen, which influences reciprocal iTreg and Th1 cell differentiation efficiency.
PHD proteins are functionally redundant in T lymphocytes
Many biologic systems exhibit functional redundancy. We asked whether a single PHD protein is functionally predominant in T cells, or if they function in a redundant manner. We generated mice with T cell-specific deletions of one, two, or all three PHD proteins. We hypothesized that if any one PHD protein was functionally predominant, then removal of this factor should phenocopy the effect of deletion of all three. Deletion of one or two PHD proteins did not significantly affect IFN-γ production or Nrp-1Lo Treg cell frequency amongst pulmonary CD4+ T cells (Figure 4A–B). Only loss of all three PHD proteins resulted in a dramatic increase in the pulmonary IFN-γ+ Teff to Nrp-1Lo Treg cell ratio (Figure 4E). Elevated IFN-γ production in pulmonary CD8+ T cells was only detected when all three PHD proteins were ablated (Figure 4C–D). Deletion of one PHD protein or the combined removal of PHD1/PHD2 or PHD1/PHD3 did not affect iTreg cell differentiation in vitro. PHD2/3 dKO T cells demonstrated a modest reduction in iTreg cell differentiation and reciprocal increase in Th1 cell differentiation. However, this defect was most pronounced in PHD-tKO cells (Figure S4A–C). Collectively, these results provide evidence that PHD proteins function redundantly in T cells.
Figure 4. PHD proteins are functionally redundant in T lymphocytes.
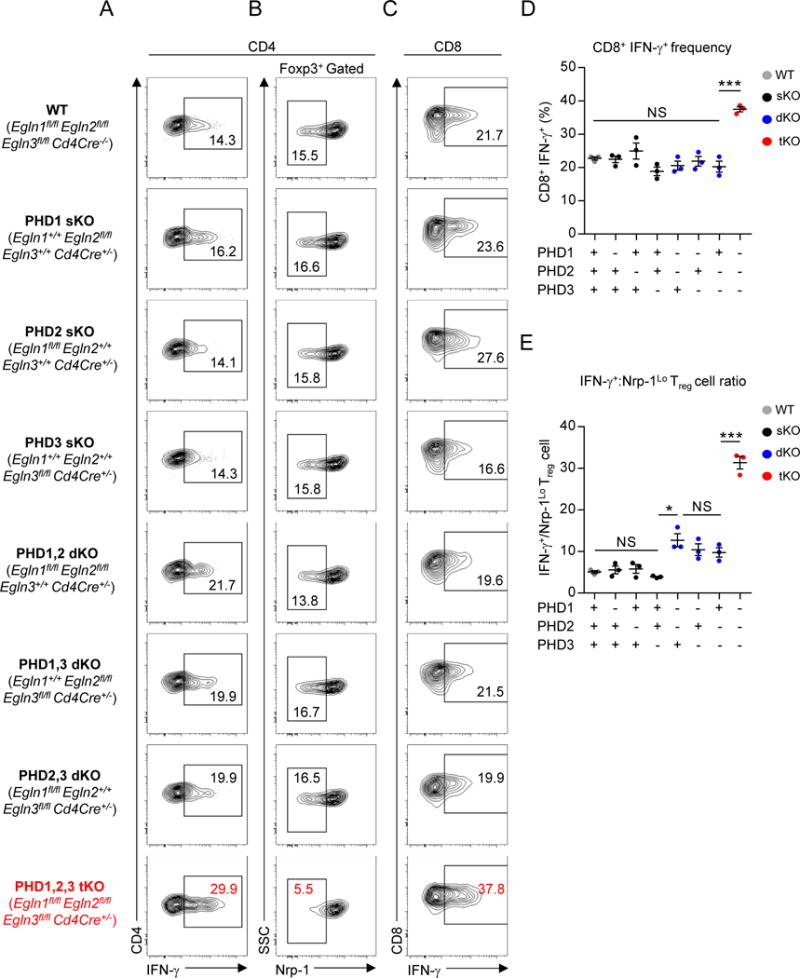
(A–B) Phenotype of pulmonary CD4+ T cells in mice with indicated PHD protein deficiency. Representative flow cytometry of IFN-γ expression (A) and Nrp-1 expression by Foxp3+ Treg cells (B).
(C–D) IFN-γ expression by pulmonary CD8+ T cells in mice with indicated PHD protein deficiency. Representative flow cytometry (C) and replicate values (E) are shown.
(E) Ratio of CD4+ IFN-γ+ Teff cells to Nrp-1Lo Foxp3+ Treg cells of total pulmonary CD4+ T cells in mice with the indicated PHD protein deficiency.
Data are representative of ≥ 2 independent experiments with ≥ 3 mice per genotype. Bars and error represent mean ±SEM of replicate measurements. *P<0.05, ***P<0.001 (Student’s t-test).
See also Figure S4.
PHD proteins repress HIF-driven glycolytic metabolism to mediate T cell fate specification
We next asked how PHD proteins mediate reciprocal iTreg and Th1 cellular differentiation programs. Gene set enrichment analysis (GSEA) of the global transcriptional differences between PHD-tKO and WT CD4+ T cells activated in vitro revealed significant enrichment of genes involved in hypoxic responses and transcriptional targets of hypoxia inducible factors in PHD-tKO cells (Figure S5A). Nuclear accumulation of HIFα proteins correlates inversely with PHD hydroxylase activity (Kaelin and Ratcliffe, 2008). We therefore measured the dynamics of HIF1α mRNA and protein expression in WT and PHD-tKO T cells. Naïve and activated WT and PHD-tKO CD4+ T cells had similar expression of Hif1a mRNA (Figure S5B). HIF1α protein expression was similar in naïve cells, but significantly elevated in PHD-tKO T cells upon activation (Figure S5C). Consistent with their functional redundancy, each PHD protein limited HIF1α accumulation in T cells (Figure 5A). Furthermore, HIF1α accumulation was suppressed in CD4+ T cells stimulated in high oxygen environments in a PHD dependent manner (Figure 5B).
Figure 5. PHD proteins control T cell differentiation through repression of HIF-driven glycolytic metabolism.
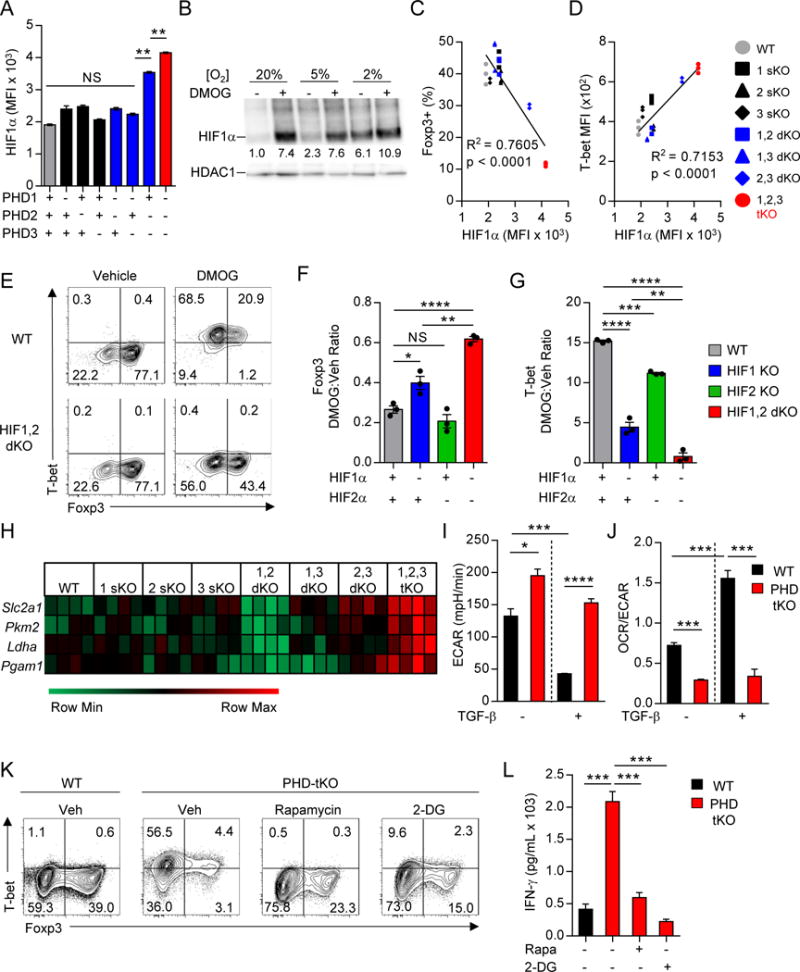
(A) Flow cytometry detection of HIF1α expression in CD4+ T cells isolated from mice with the indicated PHD KO genotype and stimulated in vitro. Average HIF1α MFI is shown.
(B) Immunoblot of HIF1α and HDAC1 from nuclear lysate of WT CD4+ T cells stimulated in vitro in the indicated environmental oxygen concentrations ± DMOG. Numbers indicate densitometry quantification of HIF1α band intensity relative to HDAC1 loading control. All values are normalized to HIF1α/HDAC1 density in cells stimulated in 20% oxygen in the absence of DMOG (Lane 1).
(C–D) Correlation between HIF1α and Foxp3 (C) and T-bet (D) expression in CD4+ T cells isolated from the indicated PHD KO genotype and stimulated as described in (A).
(E) Representative flow cytometry of Foxp3 and T-bet expression in WT and HIF1,2α double knockout (HIF1,2 dKO) CD4+ T cells stimulated in vitro in the presence of TGF-β ± DMOG.
(F–G) Foxp3 (F) and T-bet (G) expression in CD4+ T cells isolated from HIF1α sKO, HIF2α sKO, HIF1,2α dKO, or WT mice and stimulated in vitro in the presence of TGF-β ± DMOG. Protein expression levels in DMOG treated cells are normalized to vehicle. Black dots represent biologic replicates.
(H) mRNA expression of glycolytic genes in CD4+ T cells isolated from the indicated PHD KO genotype and stimulated in vitro. Heatmap normalized to row minimum and maximum for each gene.
(I–J) ECAR (I) and OCR:ECAR ratio (J) as determined by Seahorse bioassay in CD4+ T cells isolated from WT and PHD-tKO mice and stimulated in vitro.
(K) Foxp3 and T-bet expression in WT and PHD-tKO CD4+ T cells stimulated in vitro in the presence of TGF-β with or without the addition of compounds which inhibit glycolysis directly (2-DG) or indirectly (Rapamycin).
(L) IFN-γ ELISA from supernatants of WT and PHD-tKO CD4+ T cells stimulated in vitro as described in (K).
Data are representative of ≥ 2 independent experiments. Bars and error represent mean ±SEM of replicate measurements. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (Student’s t-test).
See also Figure S5.
We asked whether PHD-mediated suppression of HIF1α accumulation is required for appropriate control of iTreg and Th1 cell differentiation. We analyzed HIF1α, Foxp3, and T-bet protein expression in CD4+ T cells isolated from each PHD KO genotype. HIF1α accumulation inversely correlated with iTreg cell differentiation (Figure 5C) and positively correlated with Th1 effector cell differentiation (Figure 5D). Moreover, loss of HIF1α and HIF2α partially rescued iTreg cell differentiation and reversed excessive Th1 differentiation when the enzymatic activity of PHD proteins was inhibited using DMOG (Figure 5E–G). Loss of HIF1α alone only partially reversed the phenotype induced by DMOG, while loss of HIF2α had little effect on the phenotype (Figure 5F–G, Figure S5D–E). Thus, PHD proteins regulate CD4+ T cell differentiation in part by controlling the expression of HIFα proteins.
HIF transcriptional activity drives expression of genes involved in multiple cellular programs including glycolysis. Expression of components of the glycolytic machinery were elevated in PHD-tKO and DMOG treated WT T cells in a HIFα dependent manner (Figure 5H, Figure S5F–G). T cells stimulated in the presence of TGF-β demonstrated a PHD dependent reduction in extracellular acidification rate (ECAR), a measure of glycolytic activity (Figure 5I). PHD-tKO CD4+ T cells demonstrated increased glucose uptake (Figure S5H) and adopted an anaerobic metabolic signature (Figure 5J). PHD proteins also restrained glycolysis in CD8+ T cells (Figure S5I).
Metabolic programs direct T cell fate specification and effector function (Chang et al., 2013; Cui et al., 2015; Gerriets et al., 2015). We asked whether PHD-mediated repression of glycolysis is required for appropriate iTreg and Th1 cell specification. Strikingly, pharmacological blockade of mTOR-driven glycolytic programs or glycolysis with rapamycin and 2-deoxyglucose, respectively, completely abrogated spontaneous Th1 differentiation and IFN-γ production and partially restored iTreg cell differentiation in PHD-tKO T cells (Figure 5K–L, Figure S5J). Collectively, these findings suggest that PHD proteins coordinate a transcriptional and metabolic program that regulates the reciprocal differentiation of Th1 and iTreg cells.
T cell-intrinsic expression of PHD proteins licenses tumor colonization of the lung
We found that PHD proteins coordinate a T cell-intrinsic immunoregulatory program to sustain tolerance against harmless environmental antigens in the lung. We reasoned that infiltrating pre-metastatic cancer cells might resemble innocuous foreign antigens through their expression of mutated neoantigenic epitopes. We therefore hypothesized that PHD proteins limit effector responses against infiltrating tumor cells in the lung. We evaluated the ability of circulating tumor cells to colonize lungs of WT and PHD-tKO mice. As a control, we also evaluated subcutaneous tumor growth. WT and PHD-tKO mice were injected with B16 melanoma tumors subcutaneously in the flank and intravenously (IV) through the tail vein to introduce tumor cells at each site within the same animal (Figure 6A). Strikingly, while subcutaneous tumor growth was similar in WT and PHD-tKO mice (Figure 6B), PHD-tKO mice were significantly protected from tumor colonization in the lung. PHD-tKO animals had fewer detectable lung tumors upon gross and microscopic evaluation (Figure 6C–D), resulting in a significant reduction in total pulmonary tumor burden (Figure 6E).
Figure 6. T cell-intrinsic expression of PHD proteins licenses tumor colonization of the lung.
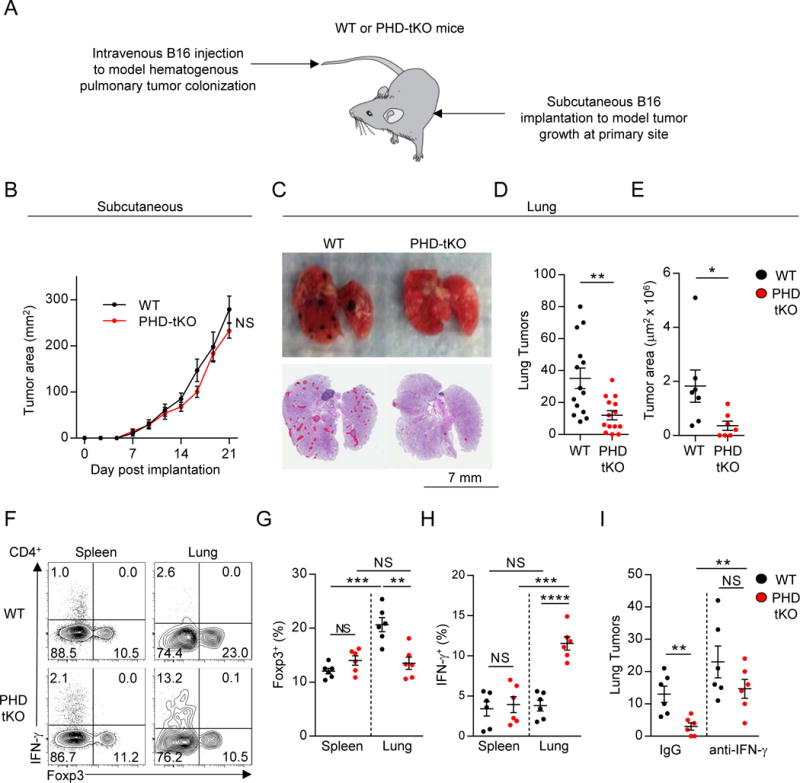
(A) Experimental schema of primary subcutaneous and secondary pulmonary tumor colonization in WT and PHD-tKO mice.
(B) Growth kinetics of subcutaneous B16 melanoma in PHD-tKO or WT mice.
(C) Gross morphology and H&E analysis of lungs from PHD-tKO or WT mice following intravenous injection of B16 melanoma. Micrometastatic lesions are outlined in red.
(D–E) Quantification of total metastatic nodules (D) and total area of tumor burden (E) in lung H&E sections from mice described in (C).
(F–H) Foxp3 and IFN-γ expression in CD4+ T lymphocytes isolated from the spleen and lungs of PHD-tKO and WT mice following IV B16 melanoma injection. Representative flow cytometry (D) and replicate values for Foxp3 (F) and IFN-γ (G) are shown.
(I) Quantification of pulmonary tumor nodules in PHD-tKO or WT mice injected intravenously with B16 melanoma and administered IFN-γ neutralizing or IgG control antibodies.
Data are representative of ≥ 2 independent experiments with > 5 mice per group. Bars and error represent mean ±SEM of replicate measurements. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (Student’s t-test).
We next analyzed the immune phenotype of CD4+ T cells in WT and PHD-tKO mice that were injected IV with B16 melanoma. The frequency of splenic Treg cells was similar in WT and PHD-tKO mice (Figure 6F). However, we observed an increase in the frequency of pulmonary Treg cells following IV tumor administration that was absent in PHD-tKO mice (Figure 6F–G). Additionally, we detected increased frequencies of IFN-γ expressing pulmonary CD4+ T cells in PHD-tKO mice compared to WT controls upon tumor colonization (Figure 6F, H).
Th1 cells play an important role in orchestrating anti-tumor immunity (Pardoll and Topalian, 1998). We asked whether PHD proteins support pulmonary tumor colonization through suppression of IFN-γ mediated anti-tumor immunity. PHD-tKO mice were injected intravenously with B16 melanoma and received IFN-γ neutralizing or control antibodies at serial time points following tumor implantation. Strikingly, the protection from lung tumor colonization observed in PHD-tKO mice was abrogated by IFN-γ neutralization (Figure 6I). Thus, PHD proteins limit IFN-γ-mediated tumor clearance in the lung.
These results indicate that T cell-intrinsic expression of PHD proteins restrains anti-tumor immunity in the lung, thus creating favorable conditions for metastatic tumor colonization.
Inhibition of PHD proteins improves adoptive cell transfer immunotherapy
Our findings suggested that inhibition of PHD proteins in T cells may improve the efficacy of cancer immunotherapy. To explore this hypothesis we utilized the TRP-1 TCR-transgenic system to model adoptive cell transfer immunotherapy (ACT) (Muranski et al., 2008). In this model, antigen specific TRP-1 CD4+ T cells are expanded ex vivo and transferred into mice bearing established subcutaneous or pulmonary B16 melanoma tumors (Figure 7A).
Figure 7. Inhibition of PHD proteins improves adoptive cell transfer immunotherapy.
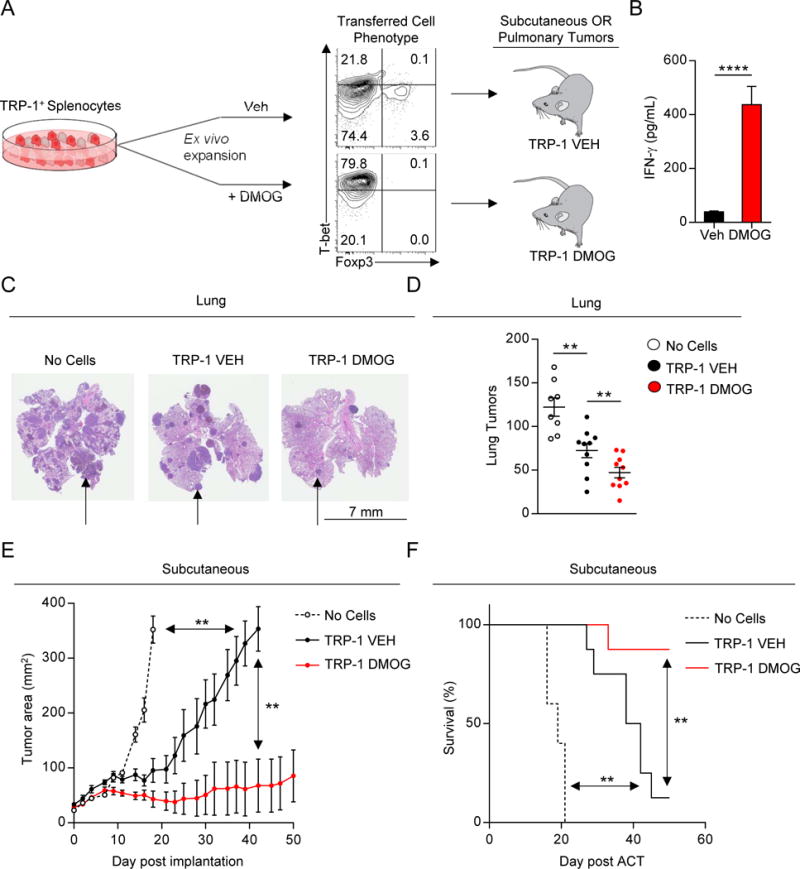
(A) Experimental schema of adoptive cell transfer immunotherapy (ACT). Pre-transfer levels of T-bet and Foxp3 expression is shown.
(B) IFN-γ ELISA from culture supernatants of TRP-1 splenocytes stimulated in vitro ± DMOG.
(C–D) Pulmonary tumor burden in mice receiving ACT of indicated cells. Representative H&E (C) and total lung tumor metastatic nodules (D) are shown. Black arrows indicate representative area of tumor burden.
(E–F) Subcutaneous tumor growth in mice receiving ACT of indicated cells. Tumor area (E) and overall survival (F) are shown. Survival significance was assessed by a Log-rank Mantel-Cox test.
Data are representative of ≥ 2 independent experiments with > 5 mice per group. Bars and error represent mean ±SEM of replicate measurements. **P<0.01, ****P<0.0001 (Student’s t-test).
See also Figure S6.
We expanded TRP-1 CD4+ T cells in the presence or absence of DMOG. DMOG treated cultures (TRP-1 DMOG) demonstrated increased Th1 differentiation and IFN-γ production compared to vehicle treated cultures (TRP-1 VEH) (Figure 7A–B). Cells were adoptively transferred into WT mice that had established B16 melanoma metastases in the lung. Consistent with their enhanced effector phenotype at the time of infusion, TRP-1 DMOG cells mediated superior clearance of lung metastases (Figure 7C–D). Additionally, TRP-1 DMOG cells mediated superior regression of established subcutaneous tumors (Figure 7E). This led to an increase in overall survival of tumor bearing mice (Figure 7F). We have previously found that Th1 cells are more effective than Th0 cells at inducing tumor rejection (Muranski et al., 2008). Augmented anti-tumor function in mice administered TRP-1 DMOG cells is consistent with the induction of a Th1 phenotype in these cells. Indeed, TRP-1 DMOG cells were as effective in mediating tumor regression as cells polarized under conventional Th1 cell specifying conditions (Figure S6A–B). Thus, inhibition of PHD proteins provides a novel strategy to enforce Th1 cell differentiation under conditions conventionally used for ACT, thereby providing a potential means to improve the efficacy of ACT.
Discussion
In this study we have demonstrated that three proteins comprising the PHD protein family function redundantly within T cells to promote pulmonary immune homeostasis in the face of innocuous foreign antigens. However, this immunoregulatory mechanism drives deleterious immunosuppression upon invasion of the lung by circulating tumor cells. Over a century ago Stephen Paget developed the “seed and soil” hypothesis, according to which properties of pre-metastatic cancer cells (“seed”) and secondary sites (“soil”) are required for the development and distribution of cancer metastases. While many studies have focused on cancer cell-intrinsic programs that contribute to metastatic potential, our investigations indicate that PHD proteins function within T cells to establish the lung as an immunologically permissive metastatic site. These results provide an immunological basis for the predisposition of many cancers to metastasize to the lung.
It is reasonable to hypothesize that cancers preferentially metastasize to sites where local immune responses are suppressed. From an evolutionary perspective the selective pressure to restrain pathologic inflammation likely outweighed the pressure to protect against tumor cell metastasis. T cells in the lung experience continuous exposure to innocuous foreign antigens. The default immune response against these stimuli is non-inflammatory and immunosuppressive. Our findings reveal a T cell-intrinsic program whereby the PHD proteins contribute to pulmonary tolerance by restraining inflammatory CD4+ and CD8+ T cell responses and permitting immunosuppressive iTreg cell differentiation. Infectious respiratory pathogens can bypass the “default state” as pathogen-associated molecular pattern antigen stimulate pulmonary dendritic cells to promote inflammatory T cell responses (Wikstrom and Stumbles, 2007). Elucidating whether PHD proteins influence T cell responses against respiratory pathogens will be an important area of investigation.
We observed a local PHD-dependent increase in Treg cell frequency and number following pulmonary challenge with HDM or B16 melanoma. Whether this is the result of de novo induction of antigen specific iTreg cells, expansion of existing pulmonary Treg cells, or recruitment of Treg cells from distant sites is incompletely resolved. The antigen driven increase in pulmonary Treg cells was absent in PHD-tKO animals, which have normal baseline tTreg cells but reduced iTreg cells and differentiation potential. These findings raise the possibility that de novo specification of iTreg cells following exposure to innocuous antigens or colonizing cancer cells is required to maintain pulmonary tolerance and support metastasis to the lung.
PHD proteins restrain CD4+ and CD8+ T cell effector function in many tissues, yet this was most pronounced in the lung – even when compared to mediastinal lymph nodes. However, these data do not exclude the possibility that PHD proteins also play a role in effector cell specification in secondary lymphoid organs. Moreover, we only detected increased numbers of CD4+ and CD8+ T cells in the lungs of PHD-tKO mice, but not other organs, suggesting that PHD proteins are involved in lymphocyte trafficking or site-specific survival and proliferation. This may be related to increased T-bet expression, which drives T cell effector differentiation and localization to peripheral non-lymphoid tissues (Joshi et al., 2007; Lord et al., 2005). The development of lung pathology in PHD-tKO mice is likely caused by inappropriate inflammatory responses against innocuous antigens in a well-oxygenated tissue. T cells in the gut also experience continuous antigen exposure. However, in the less well oxygenated gut microenvironment, PHD-independent mechanisms are the predominant mediators of tolerance. PHD proteins also appear dispensable for steady-state immune tolerance in sterile tissues. However, PHD proteins might restrain inflammation in these organs upon the introduction of antigen, such as an infectious pathogen or a metastasizing cancer cell.
PHD proteins limit HIF-driven glycolytic programs to inhibit spontaneous Th1 differentiation in conditions that promote iTreg cell commitment. PHD proteins also suppress NFκB signaling through the hydroxylation and inactivation of IKKβ (Cummins et al., 2006). Thus, in addition to constitutive HIF activity, PHD-tKO or DMOG-treated WT cells may experience increased NFκB signaling, which in excess can prevent iTreg cell differentiation (Molinero et al., 2011). Elucidating new and existing PHD substrates and dissecting the contributions of each in T cell differentiation will be an important area of future study.
Genetic and pharmacological inhibition of T cell-intrinsic PHD proteins limits tumor dissemination into the lung and improves the efficacy of ACT. Inhibition of PHD proteins could offer a viable clinical strategy to limit lung metastasis. However, systemic administration of a PHD inhibitor may not be effective. HIF has been shown to increase proliferation in some cancer types (Keith et al., 2012). HIF also promotes the development of immunosuppressive macrophages (Colegio et al., 2014; Doedens et al., 2010). Instead, a clinical strategy that permits PHD inhibition selectively in T cells, such as ACT, should be considered. In models of ACT that depend on therapeutic vaccination, inhibition of glycolysis in vitro improves expansion and anti-tumor efficacy in vivo (Sukumar et al., 2013). However, in the non-vaccine setting, which is more similar to ACT protocols in humans, enforcing effector capacity ex vivo might improve the anti-tumor efficacy of tumor specific lymphocytes. Promoting glycolytic metabolism might also improve their ability to compete for nutrients in the tumor microenvironment (Chang et al., 2015; Ho et al., 2015). Consistently, T cells expanded in the presence of DMOG were highly glycolytic, spontaneously acquired Th1 effector properties, and ultimately mediated superior tumor regression in mouse models of ACT. Addition of a PHD inhibitor to established clinical expansion protocols for human ACT using tumor infiltrating lymphocytes (TIL) or chimeric antigen receptor (CAR) transduced T cells is a feasible and potentially effective therapeutic strategy to improve the functional quality of tumor-specific T cells.
In summary, our findings provide an understanding of how site-specific immunoregulatory mechanisms contribute to the permissivity of distinct anatomical locations to tumor colonization in a manner amenable to therapeutic intervention.
METHODS AND RESOURCES
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Nicholas P Restifo (restifo@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Experiments were approved by the Institutional Animal Care and Use Committees of the NCI and performed in accordance with NIH guidelines. C57BL/6J, Rag1−/− (B6.129S7-Rag1tm1Mom/J), Ly5.1+/+ (B6.SJL-PtprcaPepcb/BoyJ), TRP-1 (B6.Cg-Rag1tm1MomTyrp1B−wTg(Tcra,Tcrb)9Rest/J), and Cd4-Cre (Tg(CD4-Cre)1Cwi/BfluJ) were purchased from The Jackson Laboratory. The following mice have been described: Egln1fl/fl, Egln2fl/fl, and Egln3fl/fl mice (Takeda et al., 2006), Hif1afl/fl mice (Ryan et al., 2000), Epas1fl/fl mice (Gruber et al., 2007). Deletion of loxP-flanked genes in T cells was achieved by crossing to Cd4-Cre mice to obtain animals with homozygous loxP-flanked alleles without Cre or hemizygous for Cre. All mice were previously backcrossed over ten generations to the C57BL/6 background. For all in vivo studies (experimental metastasis, house dust mite challenge) age and sex matched WT and PHD-tKO mice were used with at least 5 mice per genotype.
METHOD DETAILS
In vitro T cell differentiation
CD4+ T cells from spleens and lymph nodes of 6–12-week-old mice were purified by negative magnetic selection (Miltenyi) followed by sorting of naive CD4+ CD62L+ CD44− CD25− cells using a FACSAria II sorter (BD). Naive CD4+ T cells were activated with plate-bound anti-CD3 and soluble anti-CD28 (5 μg ml−1 each; eBioscience) in media for 72h either under: Th0 conditions (media alone); Th1 conditions (IL-12 (10 ng ml−1, R&D Systems), anti-IL-4 neutralizing antibodies (10 μg ml−1); Th17 conditions (IL-6 (20 ng ml−1, R&D Systems), human TGF-β1 (0.2 ng ml−1, R&D Systems), anti-IFN-γ neutralizing antibodies (10 μg ml−1) and anti-IL-4 neutralizing antibodies (10 μg ml−1); or iTreg conditions (human TGF-β1 (0–2 ng ml−1, as indicated)). For human in vitro iTreg induction assays, naïve CD4+CD45RA+CD45RO−CD62L+CCR7+ cells were FACS purified from three biologically independent healthy donor buffy coats and activated with plate-bound human anti-CD3 and soluble anti-CD28 (5 μg ml−1 each; eBioscience) with human TGF-β1 (5 ng mL−1, R&D Systems) and recombinant human IL-2 (20 CU). Mouse and human cells were cultured under standard incubator conditions or 5% or 2% oxygen as indicated (Thermo Scientific-Hera Cell incubator equipped to replace oxygen with nitrogen) through the entire duration of culture. Where indicated, the PHD protein inhibitor DMOG (0.5 – 1.0 mM, Sigma-Aldrich), Rapamycin (50 nM, Sigma-Aldrich), or 2-deoxyglucose (1 μM, Sigma-Aldrich) was added for the duration of in vitro culture.
Histopathology
Lungs were isolated from WT and PHD-tKO mice and fixed in 10% formalin, embedded in methylacrylate, sectioned, and H&E stained. Sections were blindly evaluated by a veterinary pathologist. The presence of chronic, active inflammation, vasculitis, and hemorrhage in the alveoli was quantified on a standardized severity score system (0 least severe, 3 most severe).
House Dust Mite Airway Challenge
WT and PHD-tKO mice were sensitized with 25μg of house dust mite (HDM, D. Pteronyssinus; low-endotoxin, Greer) delivered to the retropharyngeal space in sterile PBS on days 0, 1 and 2. Animals were challenged with 5μg of HDM on days 15, 16, 17 and 18, and euthanized on day 19.
Antibodies and flow cytometry
The following fluorescent dye-conjugated antibodies against surface and intracellular antigens were used: anti-FOXP3 (FJK-16s), anti-IL-17A (eBio17B7), anti-IFN-γ (XMG1.2), anti-Tbet (eBio4B10), anti-Ror gamma (t) (B2D), anti-CD304 (Nrp-1, 3DS304M), anti-mouse TCR-beta (H57-597), anti-IL-13 (eBio13A), anti-Granzyme B (NGZB), and anti-Perforin (eBioOMAK-D) (eBioscience); anti-CD45.1 (A20), anti-CD4 (RM4-5), anti-CD25 (PC61), anti-CD62L (MEL-14), anti-CD44 (IM7), anti-CD8a (53-6.7), anti-CD152 (CTLA4, UC10-4F10-11), anti-GITR (DTA-1), anti-IL-4 (11B11), anti-IL-5 (TRFK5) (BD Biosciences); Anti-mouse IgG Fab2 (Cell Signaling); Anti-mouse HIF1α (Abcam). For the identification of NKT cells, α-galactosyl ceramide loaded, PE-conjugated recombinant CD1d tetramers were used (Proimmune). Cells were incubated with specific antibodies for 30 min on ice in the presence of 2.4G2 monoclonal antibody to block FcγR binding. All samples were acquired with a Fortessa or LSR flow cytometer (Becton Dickinson) and analyzed using FlowJo software (TreeStar). Intranuclear staining was carried out using the FOXP3 staining kit (eBioscience). To determine cytokine expression, cellular suspensions containing T cells were stimulated in media containing phorbol 12-myristate 13-acetate, ionomycin and brefeldin-A (Leukocyte activation cocktail with Golgiplug; BD biosciences) for 4 h. After stimulation, cells were stained an amine-reactive exclusion-based viability dye (Invitrogen) and with antibodies against cell-surface antigens, fixed and permeabilized followed by intracellular staining with specific anti-cytokine antibodies. Single-cell suspensions from lung tissues were prepared by mechanical disruption (GentleMACS, Miltenyi). Countbright beads were spiked-in for the flow cytometric quantification of absolute cell number (Invitrogen).
RNA Sequencing and analysis
All RNA-Seq analyses were performed using ≥2 biological replicates. Raw data from replicate measurements are publically available from the GEO repository. Total RNA was prepared from cells using the RNeasy Plus Mini kit (Qiagen). 200 ng of total RNA was subsequently used to prepare RNA-Seq library by using TruSeq RNA sample prep kit (Illumina) according to manufacturer’s instructions. Paired-end RNA sequencing was performed on a HiSeq 2000 (Illumina). Sequenced reads were aligned to the mouse genome (NCBI37/mm9) with Tophat 2.0.11(Kim and Salzberg, 2011) and uniquely mapped reads were used to calculate gene expression. RefSeq gene database (mm9) was downloaded from the UCSC genome browser for RNA-Seq analysis. RNA-Seq reads were mapped to mm9 (UCSC) using Bowtie. Gene expression of annotated transcripts was calculated from mapped RNA-Seq reads using Cuffdiff to obtain RPKM-normalized gene expression values (Trapnell et al., 2012). Two-tailed t tests were performed to identify differentially expressed genes after applying the Benjamini-Hochberg correction for multiple testing. Gene set enrichment analysis was performed as previously described (Subramanian et al., 2005). RNA-sequencing data have been deposited under the Gene Expression Omnibus (GEO) accession code GSExxxxx.
Autoantibody enzyme-linked immunosorbent assay (ELISA)
For measurement of antinuclear (ANA) and anti-dsDNA autoantibodies, ELISA assays were performed on mouse serum according to manufacturer’s instructions (Alpha Diagnostic International). ELISA quantification of IFN-γ in cell supernatants collected after 72h or stimulation was performed according to manufacturer’s instructions (Ebioscience).
Quantitative reverse-transcription polymerase chain reaction (qRT–PCR)
Cells were sorted or transferred into RNALater solution (Ambion) and stored at −80 °C. Total RNA from pelleted cells was isolated using the RNeasy Plus mini kit (Qiagen). First-strand cDNA synthesis was performed using random priming with the high-capacity cDNA synthesis kit (Applied Biosystems) in the presence of SuperaseIn RNase inhibitor (Ambion). cDNA was used as a template for quantitative PCR reactions using Taqman primer-probes against specified mRNA transcripts (Applied Biosystems). Reactions were performed using Universal PCR Mastermix (Applied Biosystems). FAM channel intensity were normalized to ROX intensity, and Ct values were calculated using automatically determined threshold values using SDS software (Applied Biosystems).
Immunoblot analysis
Nuclear extracts were isolated (NE-PER kit; Pierce) and ~15 μg protein was loaded in each lane of a mini-protean precast TGX gel (Biorad). Protein was transferred onto activated PVDF membrane (Biorad), blocked with 5% BSA in TBST at RT for 1h, and incubated in 1° antibody at 4° C overnight. HIF1 α (NB100-449, Novus) and loading control HDAC1 (10E2, Cell Signaling Technology) antibodies were used at 1:1000 dilution in TBST. Samples were washed 5× with TBST, incubated with 2° antibody at 1:5000 dilution in TBST for 2h, and washed 5× with TBST. Protein was visualized with enhanced chemiluminescence (ThermoScientific) and autoradiography film. Densitometry was calculated using ImageJ software.
Bone marrow chimeras
For bone marrow reconstitution experiments, Rag1−/− mice were administered 1,000 Gy total-bodγ-radiation from a 137Cs source before intravenous injection of BM cells depleted of mature lineages from single-cell bone-marrow preparations using antibody-coupled magnetic beads (Miltenyi). Bone marrow from 6–10-week-old donor mice were used.
In vivo iTreg induction
Rag1−/− mice were injected intravenously with 4 × 105 CD4+ CD25− CD45RBhigh cells from wild-type or PHD-tKO mice. On day 21 to 23, transferred cells were isolated and analyzed for FoxP3 expression by flow cytometry.
In vivo Treg stability
Rag1−/− mice were injected intravenously with 4 × 105 CD4+ CD25+ cells from wild-type or PHD-tKO mice. On day 7, transferred cells were isolated and analyzed for FoxP3 expression by flow cytometry.
In vitro Treg stability
CD4+ CD25+ Treg cells were FACS purified from wild-type or PHD-tKO mice and stimulated in vitro with anti-CD3 (1 μg mL−1) in the presence of IL-2 (100 IU). Foxp3 expression was analyzed by flow cytometry 72h post-stimulation.
In vitro suppression assay
Naïve CD4+ T cells were FACS purified from Foxp3-GFP mice and stimulated in vitro in the presence of TGF-β ± DMOG for 72h. Foxp3-GFP+ iTreg cells were then FACS purified from VEH and DMOG treated cultures. VEH and DMOG iTreg cells were cultured in 96-well round-bottom plates with 5×104 CFSE-labelled naïve CD45.1 CD4+ CD25− (Tresp) cells along with 1×104 CD11c+ dendritic cells used as antigen-presenting cells, isolated by immunomagnetic selection (Miltenyi). Cells were stimulated with anti-CD3 antibody (1 μg mL−1, BD Biosciences) for 72h at 37 and 5% CO2. Tresp cell proliferation was measured by CFSE dilution by flow cytometry.
Extracellular Acidification Rate and Glucose Uptake
Extracellular acidification rates (ECAR) and oxygen consumption rates (OCR) were measured at 37°C using an XF24 extracellular analyzer (Seaho rse Bioscience). ECAR was measured in XF media (nonbuffered RPMI 1640 containing 25 mM glucose, 2 mM L-glutamine, and 1 mM sodium pyruvate) under basal conditions. To determine glucose uptake, T cells were incubated with 100 μM 2-NBDG (Invitrogen) for 2 hours before measuring fluorescence by flow cytometry.
Foxp3 TSDR Methylation
500 ng of sample DNA from WT and PHD-tKO FACS purified CD4+ CD25+ Treg and CD4+ CD25− non-Treg cells was bisulfite treated (Epigendx) and column purified (Zymogen). PCR amplification of the Foxp3 TSDR locus region (nucleotides −2369 to −2207 relative to Foxp3 start codon) was performed using biotinylated PCR primer (Epigendx). PCR products were bound to Streptavidin Sepharose HP (GE Healthcare Life Sciences), washed, and denatured with NaOH. Pyrosequencing primer was annealed to the purified single-stranded PCR product and sequencing was performed using the Pyrosequencing PSQ96 HS System according to manufacturer’s instructions (Pyrosequencing, Qiagen). The methylation status of each locus was analyzed individually as a T/C SNP using QCpG software (Pyrosequencing, Qiagen).
Experimental Metastasis
B16F10 (B16) melanoma cell line was obtained from the NCI tumor repository and passaged in Dulbecco’s Modified Eagle Medium (Invitrogen) supplemented with 10% fetal calf serum. 2.5×105 B16 cells were injected subcutaneously in flanks and intravenously through the tail vein. Tumor implantation experiments were performed in littermate mice of 8–12 weeks of age. Subcutaneous tumors were measured at serial time points following implantation using digital calipers and the tumor area was calculated as the product of perpendicular diameter. Lung tumor nodules were enumerated by gross count of visible sites of disease. Micrometastatic lesions were quantified following H&E stain of lung sections, and sections consistent with melanoma histology were circumscribed by a certified veterinarian. All subcutaneous and pulmonary tumor measurements were performed in a blinded manner. IFN-γ depletion was performed using intraperitoneal injection of 250 μg anti-IFN-γ (XMG1.2, BioXcell) at indicated time points.
Adoptive Cell Transfer
Mice 6 to 12 weeks of age (n = 6–10 for all groups) were injected intravenously or subcutaneously with 2.5×105 B16 melanoma cells. 5–10 days later mice were treated with adoptively transferred TRP-1 specific CD4+ T cells derived from TCR transgenic splenocytes and stimulated in vitro in the presence or absence of the PHD inhibitor DMOG. At the time of ACT mice received 500 Gy total body irradiation.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the exact value of n, the definition of center, dispersion and precision measures (mean ± SEM) and statistical significance are reported in the Figures and Figure Legends. Data is judged to be statistically significant when p < 0.05 by two-tailed Student’s T-test. In figures, asterisks denote statistical significance as calculated by Student’s T-test (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001). Survival significance in adoptive cell transfer studies was determined by a Log-rank Mantel-Cox test. Statistical analysis was performed in GraphPad PRISM 6.
DATA AND SOFTWARE AVAILABILITY
Data Resources
Raw data files for the RNA sequencing analysis have been deposited in the NCBI Gene Expression Omnibus under accession number XXXXXX.
TABLE FOR AUTHOR TO COMPLETE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse/Rat Foxp3 (clone FJK-16s) | Ebioscience | Cat# 17-5773 |
| Anti-Mouse IFN-γ (clone XMG1.2) | Ebioscience | Cat# 17-7311 |
| Anti-Mouse/Rat IL-17A (clone eBio17B7) | Ebioscience | Cat# 25-7177 |
| Anti-Human/Mouse T-bet (clone eBio4B10) | Ebioscience | Cat# 12-5825 |
| Anti-Mouse ROR gamma (t) (clone B2D) | Ebioscience | Cat# 12-6981 |
| Anti-Mouse CD304 (Neuropilin-1) (clone 3DS304M) | Ebioscience | Cat# 25-3041 |
| Anti-Mouse TCR beta (clone H57-597) | Ebioscience | Cat# 47-5961 |
| Anti-Mouse IL-13 (clone eBio13A) | Ebioscience | Cat# 12-7133 |
| Anti-Mouse Granzyme B (clone NGZB) | Ebioscience | Cat# 12-8898 |
| Anti-Mouse Perforin (clone eBioOMAK-D) | Ebioscience | Cat# 12-9392 |
| Anti-Human Foxp3 (PCH101) | Ebioscience | Cat# 17-4776 |
| Anti-Mouse CD45.1 (clone A20) | BD Biosciences | Cat# 558701 |
| Anti-Mouse CD4 (clone RM4-5) | BD Biosciences | Cat# 563727 |
| Anti-Mouse CD25 (clone PC61) | BD Biosciences | Cat# 553866 |
| Anti-Mouse CD62L (clone MEL-14) | BD Biosciences | Cat# 553150 |
| Anti-Mouse CD44 (clone IM7) | BD Biosciences | Cat# 559250 |
| Anti-Mouse CD8a (clone 53-6.7) | BD Biosciences | Cat# 553035 |
| Anti-Mouse CD152 (CTLA4) (clone UC10-4F10-11) | BD Biosciences | Cat# 553720 |
| Anti-Mouse GITR (clone DTA-1) | BD Biosciences | Cat# 558119 |
| Anti-Mouse IL-4 (clone 11B11) | BD Biosciences | Cat# 562044 |
| Anti-Mouse IL-5 (clone TRFK5) | BD Biosciences | Cat# 554395 |
| Anti-Human CD4 (clone RPA-T4) | BD Biosciences | Cat# 560158 |
| Anti-Human CD45RO (clone UCHL1) | BD Biosciences | Cat# 562791 |
| Anti-Human CD45RA (clone HI100) | BD Biosciences | Cat# 555489 |
| Anti-Human CD62L (clone DREG-56) | BD Biosciences | Cat# 555544 |
| Anti-Human CD197 (CCR7) (clone 3D12) | BD Biosciences | Cat# 560548 |
| Anti-Mouse IgG Fab2 | Cell Signaling Technology | Cat# 4410S |
| Mouse monoclonal HDAC1 (clone 10E2) | Cell Signaling Technology | Cat# 5356S |
| Mouse polyclonal HIF-1 alpha | Novus Biologicals | Cat# NB100-479 |
| InVivoMAb anti-mouse IFN-γ neutralizing antibody (clone XMG1.2) | BioXCell | Cat# BE0055 |
| InVivoMAb anti-mouse IL-4 neutralizing antibody (clone 11B11) | BioXcell | Cat# BE0045 |
| Anti-Mouse CD3 Functional Grade Purified (clone 17A2) | Ebioscience | Cat# 14-0032 |
| Anti-Mouse CD28 Functional Grade Purified (clone 37.51) | Ebioscience | Cat# 16-0281 |
| Anti-Human CD3 Functional Grade Purified (clone OKT3) | Ebioscience | Cat# 16-0037 |
| Anti-Human CD28 Functional Grade Purified (clone CD28.2) | Ebioscience | Cat# 16-0289 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| α-GalCer Loaded Recombinant CD1d Tetramer | Proimmune | Cat# E001-2A |
| Dimethyloxalylglycine (DMOG) | Sigma-Aldrich | Cat# D3695 |
| Rapamycin | Sigma-Aldrich | Cat# R8781 |
| 2-Deoxy-D-glucose (2-DG) | Sigma-Aldrich | Cat# D6134 |
| 2-Deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-D-glucose (2-NBDG) | Sigma-Aldrich | Cat# 72987 |
| Recombinant Human IL-2 Protein | R&D Systems | Cat# 202-IL |
| Recombinant Human IL-12 Protein | R&D Systems | Cat# 219-IL |
| Recombinant Human IL-6 Protein | R&D Systems | Cat# 206-IL |
| Recombinant Human TGF-β Protein | R&D Systems | Cat# 240-B |
| House Dust Mite extract (D. Pteronyssinus; low-endotoxin) | Stallergenes Greer | Cat# B70 |
| Taqman PCR Master Mix, no AmpErase UNG | Applied Biosystems | Cat# 4324018 |
| Critical Commercial Assays | ||
| RNeasy Plus Mini Kit | Qiagen | Cat# 74134 |
| TruSeq RNA Library Preparation Kit v2 | Illumina | Cat# RS-122-2001 |
| Mouse anti-dsDNA Ig’s ELISA kit | Alpha Diagnostic | Cat# 5110 |
| Mouse Anti-Nuclear Antigens (ANA/ENA) Ig’s | Alpha Diagnostic | Cat# 5210 |
| High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor | Applied Biosystems | Cat# 4374966 |
| Mouse IFN gamma ELISA Ready-SET-Go | Ebioscience | Cat# |
| NE-PER Nuclear and Cytoplasmic Extraction Reagents | Thermo Scientific | Cat# 78833 |
| Pierce BCA Protein Assay Kit | Thermo Scientific | Cat# 23225 |
| Seahorse Bioassay | Agilent Technologies | Cat# 102416 |
| Mouse Foxp3 Methylation Panel | EpigenDx | http://epigendx.com/d/products/methylation-assay-panels/foxp3-panel/ |
| Deposited Data | ||
| Raw data files for RNA sequencing | NCBI Gene Expression Omnibus | GEOxxxx |
| Experimental Models: Cell Lines | ||
| Mouse: B16-F10 melanoma | ATCC | Cat# CRL-6475 |
| Mouse: primary T lymphocytes | This paper | N/A |
| Human: primary healthy donor T lymphocytes | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Stock No: 000664 |
| Mouse: B6.129S7-Rag1tm1Mom/J | The Jackson Laboratory | Stock No: 002216 |
| Mouse: B6.Cg-Rag1 tm1Mom Tyrp1B−w Tg(Tcra,Tcrb)9Rest/J | The Jackson Laboratory | Stock No: 008684 |
| Mouse: Tg(CD4-cre)1Cwi/BfluJ | The Jackson Laboratory | Stock No: 017336 |
| Mouse: Egln1fl/fl | Takeda et al., 2006 | N/A |
| Mouse: Egln2fl/fl | Takeda et al., 2006 | N/A |
| Mouse: Egln3fl/fl | Takeda et al., 2006 | N/A |
| Mouse: Hif1afl/fl | Ryan et al., 2000 | N/A |
| Mouse: Epas1fl/fl | Gruber et al., 2007 | N/A |
| Recombinant DNA | ||
| Sequence-Based Reagents | ||
| Software and Algorithms | ||
| TopHat (v2.0.11) | Kim and Salzberg et al., 2011 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks (v2.12.0) | Trapnell et al., 2012 | http://cole-trapnelllab.github.io/cufflinks/ |
| GSEA (v2.2.2) | Broad Institute | http://software.broadinstitute.org/gsea/index.jsp |
| Other | ||
Supplementary Material
(A) Expression of Egln1 (PHD2), Egln2 (PHD1), and Egln3 (PHD3) mRNA transcripts in CD4+ and CD8+ T cells, NKT cells, γδ-T cells and B cells FACS purified from the spleens of littermate matched Egln1fl/fl Egln2fl/fl Egln3fl/fl CD4Cre−/− (WT) or Egln1fl/fl Egln2fl/fl Egln3fl/fl CD4Cre+/− (PHD-tKO) mice. Expression of each transcript is normalized to Actb. Heatmap is row normalized to minimum and maximum value for each Egln transcript across indicated cell type and mouse genotype.
(B) Titer of anti-dsDNA and anti-nuclear antibodies (ANA) in the sera of WT and PHD-tKO animals. Relative titer normalized to average WT levels.
(C) Phenotypic distribution and total thymocyte count in the thymus of WT and PHD-tKO mice as assessed by flow cytometry.
(D) Total number of CD4+ and CD8+ T lymphocytes in the spleen, mesenteric lymph nodes (mLN), blood, and bone marrow (BM) from WT or PHD-tKO mice.
(E) Representative flow cytometry demonstrating steady state expression of markers of T cell differentiation CD44 and CD62L in CD4+ and CD8+ T cells in the peripheral blood of WT and PHD-tKO mice.
(F) Differentiation state (Naïve, CM, EM) of CD8+ T cells in the peripheral blood, spleen, and bone marrow of WT and PHD-tKO mice as determined by flow cytometry expression of CD44 and CD62L.
(G) Total numbers of the indicated pulmonary T cell populations in the lungs of WT and PHD-tKO mice.
(H) CD44 expression on pulmonary CD4+ and CD8+ T cells from WT and PHD-tKO mice. Representative flow cytometry histograms and replicate values are shown.
(I) Mean fluorescence intensity (MFI) of T cell activation markers CD25, CTLA-4, and GITR, as determined by flow cytometry, expressed on pulmonary CD4+ Foxp3−, CD4+ Foxp3+, and CD8+ T cells from WT and PHD-tKO mice. Average MFI ± SEM are reported in the table. * indicates significant increase in expression in PHD-tKO mice compared to WT for each indicated marker and cell type.
(J) Expression of cytolytic effector molecules Granzyme B (Gzmb) and Perforin (Prf1) in pulmonary CD4+ and CD8+ T cells purified from WT and PHD-tKO mice. Gene expression is normalized to Actb.
(K) Pulmonary NKT cell expression of effector cytokines IFN-γ and IL-17A in WT and PHD-tKO mice. Representative flow cytometry and replicate values are shown.
(L) Methylation status of CpG motifs of the TSDR region of the Foxp3 locus (CpG sites between −2369 to −2207 from Foxp3 start codon) in Foxp3+ and Foxp3− CD4+ T cells isolated from the spleen of WT and PHD-tKO mice.
(M) Foxp3 expression in CD4+ CD25+ T cells FACS purified from WT and PHD-tKO mice.
(N) Foxp3 expression in CD4+ CD25+ T cells FACS purified from WT and PHD-tKO mice and stimulated in vitro for 72h.
(O) Foxp3 expression in CD4+ CD25+ T cells FACS purified from WT and PHD-tKO mice one week following injection into Rag1−/− host mice. Rag1−/− splenocytes were used to determine Foxp3-negative gate. Representative flow cytometry and replicate values are shown.
Dots represent individual mice. Data are representative of ≥ 2 independent experiments with ≥ 3 mice per group. Bars and error represent mean +/− SEM of replicate measurements. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (Student’s t-test).
(A-B) Total number of CD4+ (A) and CD8+ (B) T cells in the lungs of WT and PHD-tKO mice following sensitization and challenge with HDM extract.
(A) Volcano plot of all expressed transcripts (RPKM≥1) determined by RNA-seq analysis of CD4+ T cells stimulated in vitro (anti-CD3, anti-CD28 5 μg mL−1 each, TGF-β 2.0 ng mL−1) in the presence or absence of DMOG (1mM). Transcripts significantly (p<0.01) over-expressed (FC≥2, red) and under-expressed (FC≤0.5, blue) in DMOG treated cells are highlighted.
(B) Geneset enrichment analysis with a gene-set composed of all significantly over-expressed transcripts in PHD-tKO vs WT CD4+ T cells in T cells stimulated in the presence of DMOG compared to Vehicle. Transcripts over-expressed in the presence of DMOG significantly enrich with transcripts over-expressed in PHD-tKO cells compared to WT. GSEA significance was tested using a weighted Kolmogorov-Smirnov test.
(C) In vivo iTreg cell conversion of naïve WT and PHD-tKO CD4+ T cells. Foxp3 was assessed by flow cytometry, replicate values are shown.
(D) T-bet expression in Foxp3+ iTreg cells generated from WT and PHD-tKO CD4+ T cells stimulated in the presence of TGF-β (0.2 ng mL−1) ± the addition of IFN-γ blocking antibodies (XMG1.2). T-bet MFI determined by flow cytometry, replicate values are shown.
(E) Gating strategy to isolate Foxp3-GFP+ iTreg cells derived from naïve CD4+ T cells purified from Foxp3-GFP mice and stimulated in vitro in the presence of TGF-β ± DMOG (0.5mM).
(F) Suppressive capacity of Foxp3-GFP+ iTreg cells derived in the absence (VEH Treg) or presence of DMOG (DMOG Treg) (as described in E). VEH Treg and DMOG Treg were mixed with CFSE labeled naïve CD4+ CD45.1+ responder cells, soluble CD3 (1μg/mL), and CD11c+ cells purified by magnetic bead isolation. Proliferation of responder cells was determined by CFSE dilution. Representative flow cytometry and replicate values are shown.
(G) IL-17A production from small intestine lamina propria and lung CD4+ T cells isolated from WT and PHD-tKO mice and stimulated briefly ex vivo. Representative flow cytometry and replicate values are shown.
(H) IL-17A and RORγt expression in CD4+ T cells isolated from WT and PHD-tKO mice and stimulated in vitro under established Th17 polarizing conditions (anti-CD3, anti-CD28 5 μg mL−1 each, TGF-β 0.2ng mL−1, IL-6 20ng mL−1, anti-IFN-γ 10μg mL−1, anti-IL4 10μg mL−1).
Representative flow cytometry and replicate values for IL-17A+ percentage and RORγt MFI are shown.
(I) CD44 and CD62L expression in CD8+ T cells isolated from WT and PHD-tKO mice and stimulated in vitro (anti-CD3, anti-CD28 1 μg mL−1 each, IL-2 100 IU) for 48 h. Cells were removed from stimulation and expanded in the presence of IL-2 for an additional 72h. Representative flow cytometry and replicate values are shown. SCM: CD44− CD62L+, CM: CD44+ CD62L+, EM: CD44+ CD62L−
(J) Intracellular expression of cytolytic molecules Granzyme B and Perforin in WT and PHD-tKO CD8+ T cells stimulated as described in (I).
Data are representative of ≥ 2 independent experiments with ≥ 3 mice per group. Bars and error represent mean +/− SEM of replicate measurements. *P<0.05, **P<0.01, ****P<0.0001 (Student’s t-test).
(A-C) Flow cytometry of Foxp3 and T-bet expression in CD4+ T cells isolated from the indicated PHD knockout genotype and stimulated in vitro in the presence of TGF-β (0.2 ng mL−1) for 72h. Representative flow cytometry (A) and replicate values for Foxp3 (B) and T-bet (C) are shown.
Data are representative of ≥ 2 independent experiments with ≥ 3 mice per group. Bars and error represent mean +/− SEM of replicate measurements. *P<0.05, **P<0.01, ***P<0.001 (Student’s t-test).
(A) Gene set enrichment analysis with indicated gene sets in PHD-tKO compared with WT CD4+ T cells stimulated in vitro (anti-CD3, anti-CD28 5 μg ml−1, TGF-β 0.2 ng ml−1). Non-random distribution of genes comprising Qi_Hypoxia and Elvidge_HIF1A_AND_HIF2A_TARGETS in PHD-tKO compared to WT cells. NES = normalized enrichment score. Significance was tested using a weighted Kolmogorov-Smirnov test.
(B) Hif1a mRNA expression in WT and PHD-tKO naïve CD4+ T cells freshly isolated ex vivo (Pre-stim) or 24h following in vitro stimulation (Post-stim).
(C) HIF1α protein expression as determined by flow cytometry in WT and PHD-tKO naïve CD4+ T cells freshly isolated ex vivo (Pre-stim) or 24h following in vitro stimulation (Post-stim).
(D) Foxp3 and T-bet expression in CD4+ T cells isolated from WT and HIF1α KO mice and stimulated in vitro in the presence of TGF-β (0.2 ng mL−1) ± DMOG (1mM) for 72h. Representative flow cytometry and replicate values are shown. Dots represent individual mice.
(E) Foxp3 and T-bet expression in CD4+ T cells isolated from WT and HIF2α KO mice and stimulated in vitro in the presence of TGF-β (0.2 ng mL−1) ± DMOG (1mM) for 72h.
Representative flow cytometry and replicate values are shown. Dots represent individual mice.
(F-G) Expression of genes involved in glycolysis Slc2a1 (F) and Ldha (G) in WT and HIF1,2 dKO CD4+ T cells stimulated in vitro in the presence of TGF-β (0.2 ng mL−1) ± DMOG (1mM) for 72h.
(H) 2-NBDG uptake in WT and PHD-tKO CD4+ T cells stimulated as described in (A). Representative flow cytometry and replicate values are shown.
(I) Extracellular acidification rate (ECAR) as determined by Seahorse bioassay in CD8+ T cells isolated from WT and PHD-tKO mice. Cells were stimulated in vitro for 48h and were removed from stimulation and rested for 24h prior to Seahorse analysis.
(J) Replicate measurements of Foxp3 and T-bet expression in WT and PHD-tKO CD4+ T cells stimulated in vitro in the presence of TGF-β (0.2 ng mL−1) with or without the addition of inhibitors of glycolytic metabolism rapamycin (Rapa) and 2-deoxyglucse (2DG).
Data are representative of ≥ 2 independent experiments with ≥ 3 mice per group. Bars and error represent mean +/− SEM of replicate measurements. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (Student’s t-test).
(A) Subcutaneous tumor growth in mice receiving ACT of indicated cells. Adoptively transferred cells were expanded ex vivo under non-polarizing conditions ±DMOG (1mM) or Th1 specifying conditions (IL-12 10ng mL−1, anti-IL-4 10μg mL−1)
(B) Overall survival of tumor bearing mice treated as described in (A). Survival significance was assessed by a Log-rank Mantel-Cox test.
Data are representative of ≥ 2 independent experiments with > 5 mice per group. Bars and error represent mean ±SEM of replicate measurements. **P<0.01.
Acknowledgments
This research was supported by the Intramural Research Programs of the NCI and NIAID and by charitable gifts from Li Jinyuan and the Tiens Charitable Foundation and the Milstein Family Foundation. R.R. was supported by the Wellcome Trust/Royal Society and the UK Biotechnology and Biological Sciences Research Council. M.G.C. is a Cancer Research Institute Irvington Fellow supported by the Cancer Research Institute. We thank G.H. Fong for providing Egln1-3fl/fl mice, D. Haines for pathologic analysis of H&E stained tissues, R. Johnson, A. Palazon, and P. Tyrakis for technical advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, D.C. and N.P.R. Methodology, D.C. and R.R. Investigation, D.C., R.R., M.G.C., M.H.A., M.S., R.L.E., H.H., Z.Y, and J.H.P. Resources, A.W.G, A.T.P., and J.G. Review and Editing, N.P.R., Y.B., R.R., A.W.G., C.A.K., D.C.P., and L.G. Supervision, N.P.R. and Y.B.
References
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nature reviews Immunology. 2003;3:253–257. doi: 10.1038/nri1032. [DOI] [PubMed] [Google Scholar]
- Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. The Journal of experimental medicine. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513:559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, Kaech SM. IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8+ T Cell Longevity. Cell. 2015;161:750–761. doi: 10.1016/j.cell.2015.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:18154–18159. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Heer HJ, Hammad H, Soullie T, Hijdra D, Vos N, Willart MA, Hoogsteden HC, Lambrecht BN. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. The Journal of experimental medicine. 2004;200:89–98. doi: 10.1084/jem.20040035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, Johnson RS, Goldrath AW. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nature immunology. 2013;14:1173–1182. doi: 10.1038/ni.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, Coussens LM, Karin M, Goldrath AW, Johnson RS. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer research. 2010;70:7465–7475. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal ablation of Hif-2alpha results in anemia. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2301–2306. doi: 10.1073/pnas.0608382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162:1217–1228. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt PG, Strickland DH, Wikstrom ME, Jahnsen FL. Regulation of immunological homeostasis in the respiratory tract. Nature reviews Immunology. 2008;8:142–152. doi: 10.1038/nri2236. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, Umetsu DT, Rudensky AY. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature. 2012;482:395–399. doi: 10.1038/nature10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Molecular cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nature reviews Cancer. 2012;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome biology. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Thomas KR, Perdue NR, Smigiel KS, Srivastava S, Campbell DJ. T-bet(+) Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor beta2. Immunity. 2012;37:501–510. doi: 10.1016/j.immuni.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nature immunology. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Elly C, Park Y, Liu YC. E3 Ubiquitin Ligase VHL Regulates Hypoxia-Inducible Factor-1alpha to Maintain Regulatory T Cell Stability and Suppressive Capacity. Immunity. 2015;42:1062–1074. doi: 10.1016/j.immuni.2015.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Lord GM, Rao RM, Choe H, Sullivan BM, Lichtman AH, Luscinskas FW, Glimcher LH. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood. 2005;106:3432–3439. doi: 10.1182/blood-2005-04-1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306. doi: 10.1038/nature17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinero LL, Miller ML, Evaristo C, Alegre ML. High TCR stimuli prevent induced regulatory T cell differentiation in a NF-kappaB-dependent manner. J Immunol. 2011;186:4609–4617. doi: 10.4049/jimmunol.1002361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucida D, Kutchukhidze N, Erazo A, Russo M, Lafaille JJ, Curotto de Lafaille MA. Oral tolerance in the absence of naturally occurring Tregs. The Journal of clinical investigation. 2005;115:1923–1933. doi: 10.1172/JCI24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM, Topalian SL. The role of CD4+ T cell responses in antitumor immunity. Current opinion in immunology. 1998;10:588–594. doi: 10.1016/s0952-7915(98)80228-8. [DOI] [PubMed] [Google Scholar]
- Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, Johnson RS. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer research. 2000;60:4010–4015. [PubMed] [Google Scholar]
- Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. The Journal of experimental medicine. 1985;161:72–87. doi: 10.1084/jem.161.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Oxygen homeostasis. Wiley interdisciplinary reviews Systems biology and medicine. 2010;2:336–361. doi: 10.1002/wsbm.69. [DOI] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. The Journal of experimental medicine. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland DH, Stumbles PA, Zosky GR, Subrata LS, Thomas JA, Turner DJ, Sly PD, Holt PG. Reversal of airway hyperresponsiveness by induction of airway mucosal CD4+CD25+ regulatory T cells. The Journal of experimental medicine. 2006;203:2649–2660. doi: 10.1084/jem.20060155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumbles PA, Thomas JA, Pimm CL, Lee PT, Venaille TJ, Proksch S, Holt PG. Resting respiratory tract dendritic cells preferentially stimulate T helper cell type 2 (Th2) responses and require obligatory cytokine signals for induction of Th1 immunity. The Journal of experimental medicine. 1998;188:2019–2031. doi: 10.1084/jem.188.11.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. The Journal of clinical investigation. 2013;123:4479–4488. doi: 10.1172/JCI69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. The Journal of experimental medicine. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Molecular and cellular biology. 2006;26:8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, Xiong H, Dolpady J, Frey AB, Ruocco MG, et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. The Journal of experimental medicine. 2012;209:1723–1742. S1721. doi: 10.1084/jem.20120914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrom ME, Stumbles PA. Mouse respiratory tract dendritic cell subsets and the immunological fate of inhaled antigens. Immunol Cell Biol. 2007;85:182–188. doi: 10.1038/sj.icb.7100039. [DOI] [PubMed] [Google Scholar]
- Yadav M, Louvet C, Davini D, Gardner JM, Martinez-Llordella M, Bailey-Bucktrout S, Anthony BA, Sverdrup FM, Head R, Kuster DJ, et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. The Journal of experimental medicine. 2012;209:1713–1722. S1711–1719. doi: 10.1084/jem.20120822. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Expression of Egln1 (PHD2), Egln2 (PHD1), and Egln3 (PHD3) mRNA transcripts in CD4+ and CD8+ T cells, NKT cells, γδ-T cells and B cells FACS purified from the spleens of littermate matched Egln1fl/fl Egln2fl/fl Egln3fl/fl CD4Cre−/− (WT) or Egln1fl/fl Egln2fl/fl Egln3fl/fl CD4Cre+/− (PHD-tKO) mice. Expression of each transcript is normalized to Actb. Heatmap is row normalized to minimum and maximum value for each Egln transcript across indicated cell type and mouse genotype.
(B) Titer of anti-dsDNA and anti-nuclear antibodies (ANA) in the sera of WT and PHD-tKO animals. Relative titer normalized to average WT levels.
(C) Phenotypic distribution and total thymocyte count in the thymus of WT and PHD-tKO mice as assessed by flow cytometry.
(D) Total number of CD4+ and CD8+ T lymphocytes in the spleen, mesenteric lymph nodes (mLN), blood, and bone marrow (BM) from WT or PHD-tKO mice.
(E) Representative flow cytometry demonstrating steady state expression of markers of T cell differentiation CD44 and CD62L in CD4+ and CD8+ T cells in the peripheral blood of WT and PHD-tKO mice.
(F) Differentiation state (Naïve, CM, EM) of CD8+ T cells in the peripheral blood, spleen, and bone marrow of WT and PHD-tKO mice as determined by flow cytometry expression of CD44 and CD62L.
(G) Total numbers of the indicated pulmonary T cell populations in the lungs of WT and PHD-tKO mice.
(H) CD44 expression on pulmonary CD4+ and CD8+ T cells from WT and PHD-tKO mice. Representative flow cytometry histograms and replicate values are shown.
(I) Mean fluorescence intensity (MFI) of T cell activation markers CD25, CTLA-4, and GITR, as determined by flow cytometry, expressed on pulmonary CD4+ Foxp3−, CD4+ Foxp3+, and CD8+ T cells from WT and PHD-tKO mice. Average MFI ± SEM are reported in the table. * indicates significant increase in expression in PHD-tKO mice compared to WT for each indicated marker and cell type.
(J) Expression of cytolytic effector molecules Granzyme B (Gzmb) and Perforin (Prf1) in pulmonary CD4+ and CD8+ T cells purified from WT and PHD-tKO mice. Gene expression is normalized to Actb.
(K) Pulmonary NKT cell expression of effector cytokines IFN-γ and IL-17A in WT and PHD-tKO mice. Representative flow cytometry and replicate values are shown.
(L) Methylation status of CpG motifs of the TSDR region of the Foxp3 locus (CpG sites between −2369 to −2207 from Foxp3 start codon) in Foxp3+ and Foxp3− CD4+ T cells isolated from the spleen of WT and PHD-tKO mice.
(M) Foxp3 expression in CD4+ CD25+ T cells FACS purified from WT and PHD-tKO mice.
(N) Foxp3 expression in CD4+ CD25+ T cells FACS purified from WT and PHD-tKO mice and stimulated in vitro for 72h.
(O) Foxp3 expression in CD4+ CD25+ T cells FACS purified from WT and PHD-tKO mice one week following injection into Rag1−/− host mice. Rag1−/− splenocytes were used to determine Foxp3-negative gate. Representative flow cytometry and replicate values are shown.
Dots represent individual mice. Data are representative of ≥ 2 independent experiments with ≥ 3 mice per group. Bars and error represent mean +/− SEM of replicate measurements. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (Student’s t-test).
(A-B) Total number of CD4+ (A) and CD8+ (B) T cells in the lungs of WT and PHD-tKO mice following sensitization and challenge with HDM extract.
(A) Volcano plot of all expressed transcripts (RPKM≥1) determined by RNA-seq analysis of CD4+ T cells stimulated in vitro (anti-CD3, anti-CD28 5 μg mL−1 each, TGF-β 2.0 ng mL−1) in the presence or absence of DMOG (1mM). Transcripts significantly (p<0.01) over-expressed (FC≥2, red) and under-expressed (FC≤0.5, blue) in DMOG treated cells are highlighted.
(B) Geneset enrichment analysis with a gene-set composed of all significantly over-expressed transcripts in PHD-tKO vs WT CD4+ T cells in T cells stimulated in the presence of DMOG compared to Vehicle. Transcripts over-expressed in the presence of DMOG significantly enrich with transcripts over-expressed in PHD-tKO cells compared to WT. GSEA significance was tested using a weighted Kolmogorov-Smirnov test.
(C) In vivo iTreg cell conversion of naïve WT and PHD-tKO CD4+ T cells. Foxp3 was assessed by flow cytometry, replicate values are shown.
(D) T-bet expression in Foxp3+ iTreg cells generated from WT and PHD-tKO CD4+ T cells stimulated in the presence of TGF-β (0.2 ng mL−1) ± the addition of IFN-γ blocking antibodies (XMG1.2). T-bet MFI determined by flow cytometry, replicate values are shown.
(E) Gating strategy to isolate Foxp3-GFP+ iTreg cells derived from naïve CD4+ T cells purified from Foxp3-GFP mice and stimulated in vitro in the presence of TGF-β ± DMOG (0.5mM).
(F) Suppressive capacity of Foxp3-GFP+ iTreg cells derived in the absence (VEH Treg) or presence of DMOG (DMOG Treg) (as described in E). VEH Treg and DMOG Treg were mixed with CFSE labeled naïve CD4+ CD45.1+ responder cells, soluble CD3 (1μg/mL), and CD11c+ cells purified by magnetic bead isolation. Proliferation of responder cells was determined by CFSE dilution. Representative flow cytometry and replicate values are shown.
(G) IL-17A production from small intestine lamina propria and lung CD4+ T cells isolated from WT and PHD-tKO mice and stimulated briefly ex vivo. Representative flow cytometry and replicate values are shown.
(H) IL-17A and RORγt expression in CD4+ T cells isolated from WT and PHD-tKO mice and stimulated in vitro under established Th17 polarizing conditions (anti-CD3, anti-CD28 5 μg mL−1 each, TGF-β 0.2ng mL−1, IL-6 20ng mL−1, anti-IFN-γ 10μg mL−1, anti-IL4 10μg mL−1).
Representative flow cytometry and replicate values for IL-17A+ percentage and RORγt MFI are shown.
(I) CD44 and CD62L expression in CD8+ T cells isolated from WT and PHD-tKO mice and stimulated in vitro (anti-CD3, anti-CD28 1 μg mL−1 each, IL-2 100 IU) for 48 h. Cells were removed from stimulation and expanded in the presence of IL-2 for an additional 72h. Representative flow cytometry and replicate values are shown. SCM: CD44− CD62L+, CM: CD44+ CD62L+, EM: CD44+ CD62L−
(J) Intracellular expression of cytolytic molecules Granzyme B and Perforin in WT and PHD-tKO CD8+ T cells stimulated as described in (I).
Data are representative of ≥ 2 independent experiments with ≥ 3 mice per group. Bars and error represent mean +/− SEM of replicate measurements. *P<0.05, **P<0.01, ****P<0.0001 (Student’s t-test).
(A-C) Flow cytometry of Foxp3 and T-bet expression in CD4+ T cells isolated from the indicated PHD knockout genotype and stimulated in vitro in the presence of TGF-β (0.2 ng mL−1) for 72h. Representative flow cytometry (A) and replicate values for Foxp3 (B) and T-bet (C) are shown.
Data are representative of ≥ 2 independent experiments with ≥ 3 mice per group. Bars and error represent mean +/− SEM of replicate measurements. *P<0.05, **P<0.01, ***P<0.001 (Student’s t-test).
(A) Gene set enrichment analysis with indicated gene sets in PHD-tKO compared with WT CD4+ T cells stimulated in vitro (anti-CD3, anti-CD28 5 μg ml−1, TGF-β 0.2 ng ml−1). Non-random distribution of genes comprising Qi_Hypoxia and Elvidge_HIF1A_AND_HIF2A_TARGETS in PHD-tKO compared to WT cells. NES = normalized enrichment score. Significance was tested using a weighted Kolmogorov-Smirnov test.
(B) Hif1a mRNA expression in WT and PHD-tKO naïve CD4+ T cells freshly isolated ex vivo (Pre-stim) or 24h following in vitro stimulation (Post-stim).
(C) HIF1α protein expression as determined by flow cytometry in WT and PHD-tKO naïve CD4+ T cells freshly isolated ex vivo (Pre-stim) or 24h following in vitro stimulation (Post-stim).
(D) Foxp3 and T-bet expression in CD4+ T cells isolated from WT and HIF1α KO mice and stimulated in vitro in the presence of TGF-β (0.2 ng mL−1) ± DMOG (1mM) for 72h. Representative flow cytometry and replicate values are shown. Dots represent individual mice.
(E) Foxp3 and T-bet expression in CD4+ T cells isolated from WT and HIF2α KO mice and stimulated in vitro in the presence of TGF-β (0.2 ng mL−1) ± DMOG (1mM) for 72h.
Representative flow cytometry and replicate values are shown. Dots represent individual mice.
(F-G) Expression of genes involved in glycolysis Slc2a1 (F) and Ldha (G) in WT and HIF1,2 dKO CD4+ T cells stimulated in vitro in the presence of TGF-β (0.2 ng mL−1) ± DMOG (1mM) for 72h.
(H) 2-NBDG uptake in WT and PHD-tKO CD4+ T cells stimulated as described in (A). Representative flow cytometry and replicate values are shown.
(I) Extracellular acidification rate (ECAR) as determined by Seahorse bioassay in CD8+ T cells isolated from WT and PHD-tKO mice. Cells were stimulated in vitro for 48h and were removed from stimulation and rested for 24h prior to Seahorse analysis.
(J) Replicate measurements of Foxp3 and T-bet expression in WT and PHD-tKO CD4+ T cells stimulated in vitro in the presence of TGF-β (0.2 ng mL−1) with or without the addition of inhibitors of glycolytic metabolism rapamycin (Rapa) and 2-deoxyglucse (2DG).
Data are representative of ≥ 2 independent experiments with ≥ 3 mice per group. Bars and error represent mean +/− SEM of replicate measurements. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (Student’s t-test).
(A) Subcutaneous tumor growth in mice receiving ACT of indicated cells. Adoptively transferred cells were expanded ex vivo under non-polarizing conditions ±DMOG (1mM) or Th1 specifying conditions (IL-12 10ng mL−1, anti-IL-4 10μg mL−1)
(B) Overall survival of tumor bearing mice treated as described in (A). Survival significance was assessed by a Log-rank Mantel-Cox test.
Data are representative of ≥ 2 independent experiments with > 5 mice per group. Bars and error represent mean ±SEM of replicate measurements. **P<0.01.