

Abstract
Newborn screening (NBS) for Krabbe disease, a rare neurodegenerative disorder caused by deficient galactocerebrosidase (GALC) enzyme activity, has recently been implemented in a number of US states. However, the spectrum of phenotypic manifestations associated with deficient GALC activity complicates the management of screen-positive newborns and underscores the need to identify clinically relevant biomarkers. Earlier studies with a small number of patients identified psychosine, a substrate of the GALC enzyme, as a potential biomarker for Krabbe disease. In this study, we provide, for the first time, longitudinal data on dried blood spot (DBS) psychosine concentrations in different Krabbe disease phenotypes for both untreated patients and those treated with hematopoietic stem cell transplantation (HSCT). Our cohort included patients previously identified by NBS to be at high risk to develop Krabbe disease. Substantially elevated DBS psychosine concentration during the newborn period was found to be a highly specific marker for infantile Krabbe disease. This finding supports the use of DBS psychosine concentration as a second-tier NBS test to aid in the identification of patients who require urgent evaluation for HSCT. In addition, longitudinal assessments showed that both natural disease progression and treatment with HSCT were associated with decreases in DBS psychosine concentrations. Based on these findings we provide recommendations for the interpretation of psychosine concentrations in DBS specimens collected during the first year of life. Future studies should aim to better delineate the relationship between DBS psychosine concentration and disease onset in patients with later-onset forms of Krabbe disease.
Keywords: Krabbe disease, globoid cell leukodystrophy, psychosine, galactosylsphingosine, newborn screening, tandem mass spectrometry
1. Introduction
Krabbe disease (KD), also known as globoid cell leukodystrophy, is an autosomal recessive neurodegenerative disorder caused by deficient activity of the galactocerebrosidase (GALC) enzyme. This disease has traditionally been divided into four subtypes based on the age at which symptoms appear. Most patients develop the classic form, early-infantile Krabbe disease (EIKD), which is characterized by irritability, stiffness, and developmental delay during the first 6 months of life. Neurodegeneration occurs rapidly in these patients, and untreated infants typically die during early childhood. Children with late-infantile Krabbe disease (LIKD) present with developmental delay and regression, vision loss, and gait abnormalities. The clinical course is highly variable in the juvenile- and adult-onset forms of this disease [1, 2].
Other than supportive measures, hematopoietic stem cell transplantation (HSCT) is currently the only treatment for KD. When performed prior to the onset of symptoms, HSCT has been shown to preserve cognitive and receptive language development of patients with EIKD [3]. However, HSCT is generally not effective at reversing pre-existing neurological damage in infants that are already symptomatic at the time of transplantation; in these patients, survival may be extended, but most continue to deteriorate. The detection of pre-symptomatic or minimally symptomatic patients without a known family history of KD is challenging because of the nonspecific nature of initial symptoms and a lack of familiarity with this rare disorder among primary care providers. Thus, population-based newborn screening (NBS) for KD is likely to be the most effective way to ensure timely access to treatment for infants with this disease.
In 2006, New York State (NYS) became the first US state to initiate NBS for KD. Under the NYS protocol, mass spectrometry is used to measure GALC activity in dried blood spots (DBS) from newborns. Specimens that are found to have GALC activity levels of less than 20% of the daily mean are retested, and if repeat testing reveals an average GALC activity of less than 12% of the daily mean, molecular genetic testing is also performed. Patients are considered screen-positive if at least one potentially pathogenic GALC mutation is identified. Screen-positive patients then undergo confirmatory testing of GALC activity in leukocytes, which is used to designate their level of risk of developing KD (high, moderate, or low/no risk) [4].
Although KD has recently been added to NBS panels in other states, concerns about the ethical and practical implications of screening for KD and the efficacy of treatment have limited its implementation on a national scale. Much of the controversy surrounding NBS for KD has centered on the challenges associated with predicting phenotype and long-term prognosis in patients that are determined to be at risk for KD [5, 6, 7]. Homozygosity or compound heterozygosity for a 30-kb deletion and a number of well-characterized point mutations are known to be associated with EIKD. However, the prediction of phenotype based on genotype is more difficult in patients with mutations of unknown significance or mutations associated with late-onset disease. Moreover, residual GALC enzyme activity has not been shown to reliably predict the disease course [2, 8].
Although a new high-resolution mass spectrometry assay to measure GALC activity in leukocytes has been shown in a research lab to better discriminate between KD phenotypes, the limited prognostic value of current enzyme activity assays and molecular genetic tests complicates clinical decision-making [9]. In providing clinical care to infants with positive screening and confirmatory testing results, the most pressing question that must be addressed by clinicians is whether the patient is likely to develop infantile-onset KD, which requires urgent evaluation for HSCT. Current guidelines recommend that candidacy for HSCT be evaluated using a rating scale that takes into consideration genotype, physical examination findings, and the results of neurodiagnostic testing. If a screen-positive infant is determined not to require immediate transplantation, clinicians are faced with the additional challenge of determining when—if ever—the child is likely to develop a later-onset form of KD. During the first 10 years of screening for KD in NYS, just five of the 52 infants that were classified as moderate- or high-risk developed EIKD, and it is not currently known what proportion of the remainder will go on to develop later-onset KD phenotypes [10, 11]. For these patients and their families, uncertainty about disease status and prognosis may cause undue distress. Moreover, the battery of follow-up neurodiagnostic testing that has been recommended to longitudinally monitor moderate- to high-risk infants for disease onset is burdensome and costly. These considerations underscore the need to identify biomarkers to aid in the prediction of prognosis and monitoring of disease progression.
Galactosylsphingosine, also known as psychosine, is a substrate of the GALC enzyme that shows promise to aid in the diagnosis and follow-up of at-risk infants identified through NBS. To date, two small studies have reported elevated psychosine concentrations in newborn DBS specimens from infants that developed EIKD [12, 13]. Although these preliminary findings are promising, a number of unanswered questions must be addressed before psychosine can be used for clinical follow-up and monitoring purposes. The aims of the present study were threefold: to determine whether DBS psychosine concentration at birth is predictive of disease onset and clinical phenotype; to assess the relationship between disease progression and longitudinal changes in DBS psychosine concentration in patients with KD; and to evaluate the effects of treatment with HSCT on DBS psychosine concentration. This is the first study to report longitudinal measurements of DBS psychosine concentrations in a cohort of treated and untreated patients with the early-infantile, late-infantile, and juvenile-onset forms of KD.
2. Methods
2.1 DBS collection
From 2010 to 2016, DBS samples were prospectively collected from 69 patients with a confirmed diagnosis of KD, four sibling carriers, and two asymptomatic NBS-positive patients during clinical evaluations at the Program for the Study of Neurodevelopment in Rare Disorders (NDRD). Based on the age at which symptoms appeared, patients with KD were classified as having EIKD (0–6 months at onset), LIKD (6–48 months at onset), or juvenile-onset KD (4–18 years at onset). The specimens were stored in sealed bags at room temperature with desiccant. This study received institutional review board approval from the University of North Carolina at Chapel Hill (08-0237) and the University of Pittsburgh (PRO11050036). Informed consent was obtained in all cases.
In addition to the samples collected at the time of clinical evaluation at the NDRD, DBS cards from birth were obtained directly from state NBS programs for 8 patients with KD, 2 sibling carriers, and 9 NBS-positive infants who have not developed KD but were classified as moderate- or high-risk at the time of screening. Psychosine measurements from 75 newborn DBS samples were used to establish the normal range of psychosine concentrations in non-KD infants.
2.2 Determination of DBS psychosine concentration
Blood psychosine concentration was measured by extraction of DBS 3-mm punches with methanol followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using a modification of the published method that has been described separately [9, 13]. A Waters TQS-MS/MS instrument was used, which provided a limit of quantification of approximately 0.1 nM, below that reported in a previous study [13]. The chemically identical but isotopically substituted d3-psychosine (Avanti Polar Lipids) was used to convert the observed mass spectrometry signals to nanomolar concentrations of psychosine in blood, assuming a 3-mm DBS punch contains 3.1 μL blood. The laboratory was blinded with regards to the clinical identity associated with the specimen being tested.
2.3 Analysis of psychosine concentration
To assess intra-specimen variability, 114 replicate samples were tested. The variance of the within-specimen measures was calculated and graphed. It has been noted that the coefficient of variation (CV) is not the ideal measure of variability when the number of replicates varies between samples. However, because of its familiarity we report both the CV and the standard error percent (SE% = (SE/mean) × 100). The SE% was proposed by Eisenberg to account for the greater precision in samples with more replicates [14].
DBS psychosine concentrations were plotted on a log scale and graphed against age to allow for longitudinal assessment. Samples were grouped according to disease status (EIKD, LIKD, juvenile-onset KD, asymptomatic NBS-positive, carrier, unaffected newborn) and HSCT status (treated or untreated) at the time of specimen collection.
3. Results
3.1 Variability
To evaluate intra-specimen variability, 114 punches from 39 DBS specimens were analyzed. The number of repeat punches per individual blood draw ranged from 2 to 20. The variability was found to be correlated with the mean level of psychosine (Figure 1), with a mean CV of 12.1% and SE% of 8.6%. Greater absolute variability was observed in the EIKD samples, which tended to have the highest psychosine concentrations, compared to samples from patients with later-onset forms of KD.
Fig. 1.
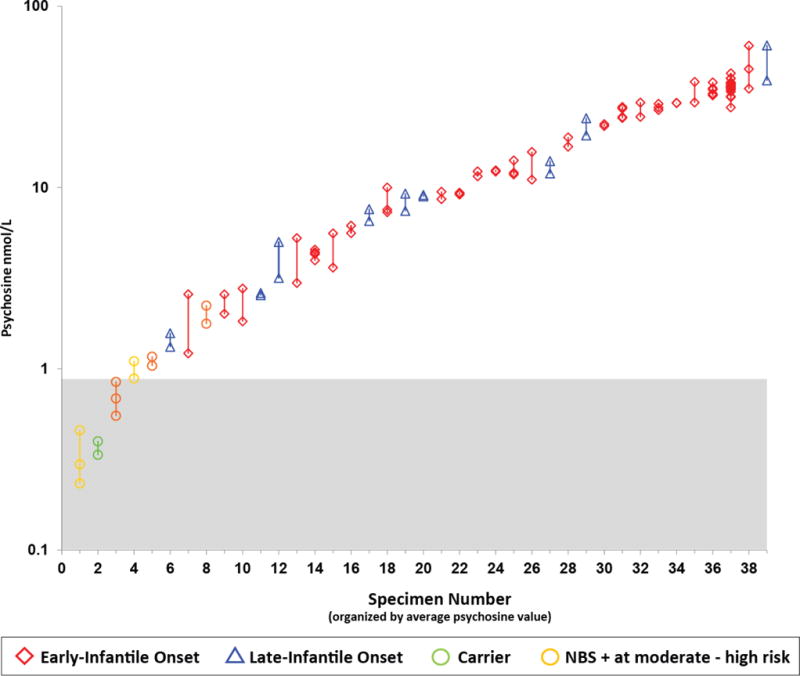
Within-specimen variability in DBS psychosine concentrations. Psychosine concentrations were plotted on a log scale for each DBS specimen, each of which was tested 2–20 times. Vertical lines connect replicates from the same specimen. The red diamonds represent EIKD patients; blue triangles represent LIKD patients; green circles represent sibling carriers; gold circles represent newborn screen-positive (NBS+) patients classified as moderate to high risk of developing KD. Each test was carried out using a separate 3-mm punch from the same DBS or from multiple DBS from the same bleed.
3.2 Controls
Analysis of DBS samples from 75 normal newborns showed that psychosine concentrations ranged from 0.14 to 0.53 nmol/L, with a mean of 0.31 nmol/L and a standard deviation of 0.10. The normal range of blood psychosine concentration was defined to include four standard deviations above or below the mean (0–0.71 nmol/L), which would be expected to encompass more than 99.9% of the unaffected population.
3.3 NBS samples
Results from newborn DBS were available for 8 patients (6 with EIKD, 1 with LIKD, 1 with juvenile-onset KD), 9 moderate- or high-risk NBS-positive infants, and 2 sibling carriers (Figure 2). Psychosine was elevated in all newborn DBS samples obtained from patients with KD. The highest values were observed in the samples from the six patients who developed EIKD (range: 5.2–44 nmol/L). The single available LIKD NBS sample tested at 5.0 nmol/L, and the juvenile-onset NBS sample tested at 2.3 nmol/L. Of the nine NBS-positive infants with available newborn DBS, the median psychosine concentration was 0.98 nmol/L (range: 0.21–2.7). In the NBS samples obtained from carriers, DBS psychosine concentration was just outside the normal range in one patient (0.76 nmol/L) but was normal in the other (0.35 nmol/L).
Fig. 2.
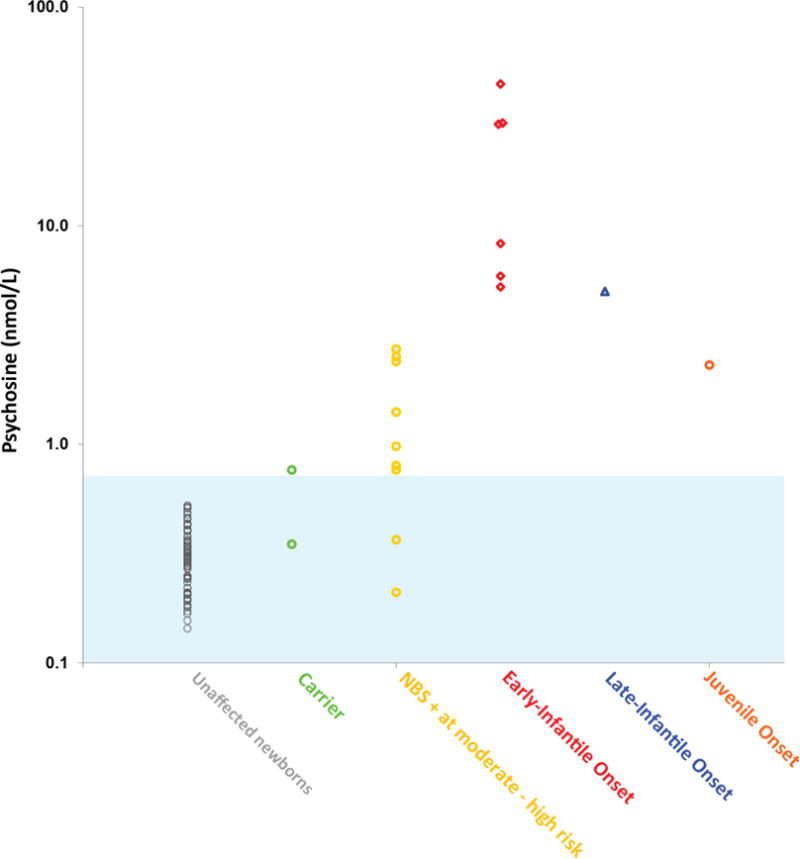
Psychosine concentrations in newborn DBS specimens. The blue shaded area is the estimated 8 standard deviation range (mean ± 4 SDs), which should include >99.9% of the unaffected population. This estimate is based on the distribution of the 75 unaffected newborns (gray). The mean psychosine concentration of one of the carrier DBS specimens (green) was similar to that of the unaffected newborns but was elevated in the other specimen. The newborn screen-positive (NBS+) infants considered to be at moderate to high risk based on GALC enzyme activity levels and mutational analysis are shown in gold. Two of these infants had psychosine levels within the range of the unaffected newborns, whereas seven infants showed elevated levels. Psychosine levels were elevated in all specimens from patients with EIKD (red), LIKD (blue), and juvenile-onset (orange) KD. The patient with juvenile-onset KD had a psychosine concentration lower than that of three samples in the NBS+ group.
3.4 Samples from affected patients not treated with HSCT
We obtained DBS from 69 affected patients (44 with EIKD, 15 with LIKD, 6 with juvenile-onset KD) at various stages of disease who had not undergone HSCT at the time of sample collection. The median DBS psychosine concentration was 24 nmol/L (range: 1.7–52) in the EIKD samples, 4.7 nmol/L (range: 1.3–50) in the LIKD samples, 1.5 nmol/L (range: 0.64–2.3) in the juvenile-onset samples, and 0.98 nmol/L (range: 0.21–2.7) in the NBS-positive samples (Figures 3 and 4). These ranges include results from NBS DBS samples in addition to DBS samples that were collected during clinical evaluations. Longitudinal data prior to treatment were available for twelve EIKD and six LIKD patients (Figure 3).
Fig. 3.
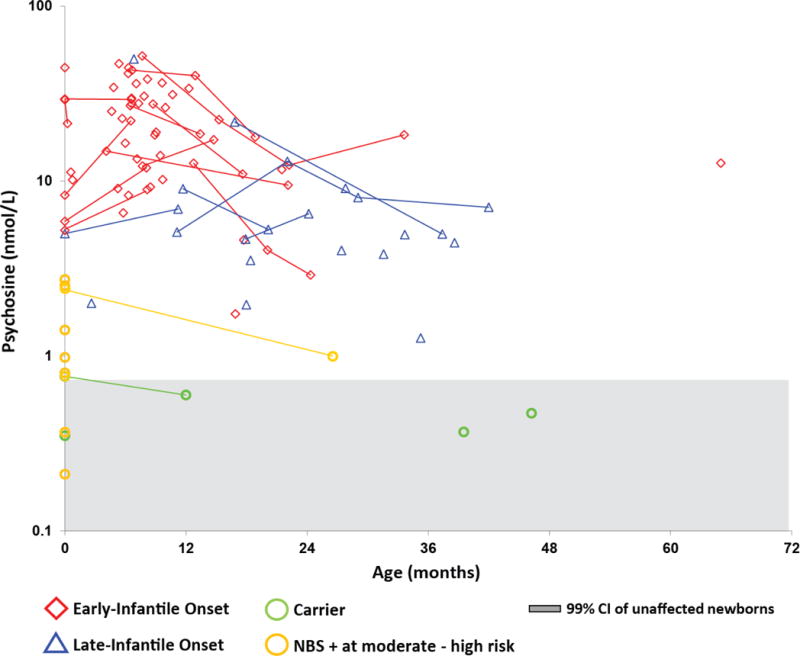
Psychosine concentrations in untreated infantile KD patients. Average DBS psychosine concentrations from EIKD and LIKD patients who had not undergone HSCT at the time of sample collection were plotted on a log scale against age. Lines indicate longitudinal data. The red diamonds represent EIKD patients; blue triangles represent LIKD patients; green circles represent sibling carriers; and gold circles represent newborn screen-positive (NBS+) infants classified as moderate to high risk of developing KD. Psychosine concentrations from newborn dried blood spots are shown in bold and plotted on the y-axis (age = 0). The gray shaded area indicates the 99.9% CI (mean ± 4 standard deviations) for unaffected newborns.
Fig. 4.
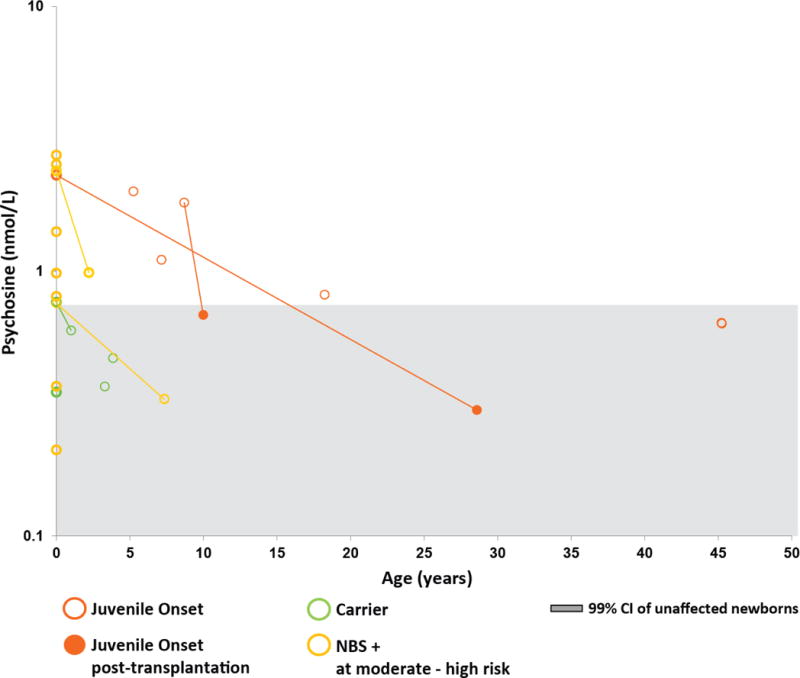
Psychosine concentrations in patients with juvenile-onset KD. Average DBS psychosine concentrations were plotted on a log scale against age. Lines indicate longitudinal data. The orange circles represent patients with juvenile-onset KD; green circles represent sibling carriers; and gold circles represent newborn screen-positive (NBS+) infants classified as moderate to high risk of developing KD. The solid circles show values after hematopoietic stem cell transplantation. Psychosine concentrations from newborn DBS are shown in bold and plotted on the y-axis (age = 0). The gray shaded area indicates the 99.9% CI (mean ± 4 standard deviations) for unaffected newborns.
Samples from transplanted patients
We collected DBS from 17 patients (9 with EIKD, 6 with LIKD, and 2 with juvenile-onset KD) following treatment with HSCT. In all patients for whom pre- and post-transplant DBS specimens were available, DBS psychosine was lower in the most recent post-transplant sample than in the first sample prior to HSCT (Figures 4 and 5). DBS psychosine concentrations at the most recent post-HSCT follow-up ranged from 1.1–8.1 nmol/L in patients with EIKD and from 0.60–7.2 nmol/L in patients with LIKD (Figure 5). Two patients with juvenile-onset KD were assessed post-HSCT (Figure 4). In one patient, who was transplanted at 9 years of age, DBS psychosine was elevated four months prior to HSCT (1.8 nmol/L) and was within the normal range (0.68 nmol/L) in a specimen collected one year after treatment. In the other juvenile-onset KD patient, who underwent HSCT at 16 years of age, DBS specimens were not available from within 10 years of transplantation; however, DBS psychosine concentrations in that patient were elevated at birth (2.3 nmol/L) and within the normal range (0.30 nmol/L) in a specimen collected twelve years after transplantation.
Fig. 5.
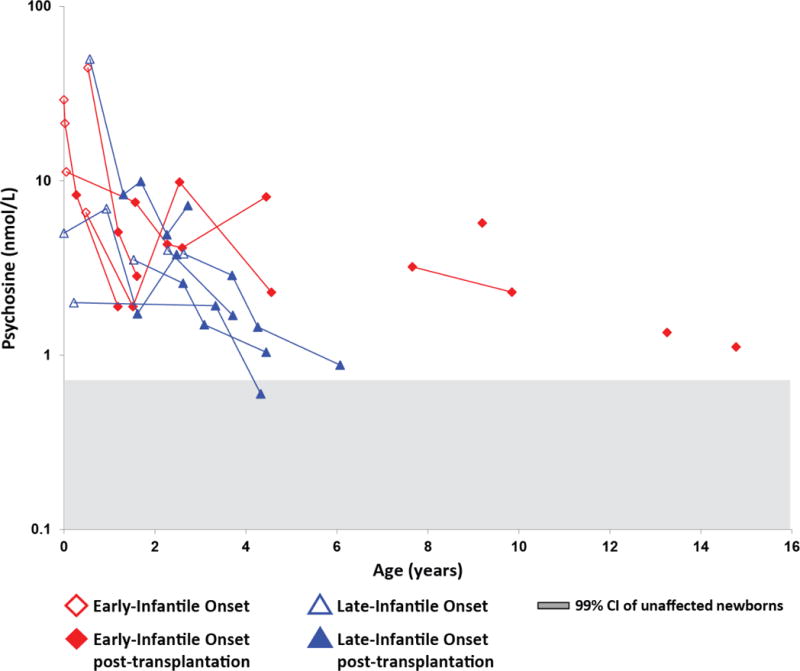
Psychosine concentrations in KD patients who underwent HSCT. Average DBS psychosine concentrations from early-infantile KD (EIKD) and late-infantile KD (LIKD) patients that underwent HSCT were plotted on a log scale against age. Lines indicate longitudinal data. The red diamonds represent EIKD patients; blue triangles represent LIKD patients. Open shapes represent samples obtained before HSCT; shaded shapes represent samples obtained after HSCT. All patients underwent HSCT within 6 weeks of the collection of the most recent pre-HSCT sample. The gray shaded area indicates the 99% CI (mean ± 4 standard deviations) for unaffected newborns.
4. Discussion
Psychosine has been proposed as a biomarker to assess and monitor infants who are diagnosed with or determined to be at risk for KD. However, prior studies of DBS psychosine concentrations have been limited to cross-sectional measurements in small numbers of patients. This is the first study to assess the clinical utility of this biomarker based on longitudinal measurements of DBS psychosine in a cohort of untreated and treated patients with KD.
The breadth of the phenotypic spectrum associated with deficient GALC enzyme activity complicates the clinical management of infants with positive NBS and confirmatory testing results. Given that the effectiveness of HSCT is greatest when performed prior to the onset of neurological deterioration, the most urgent question that must be addressed for an NBS-positive infant is whether the patient is likely to develop infantile disease requiring immediate treatment. Thus, a major aim of this study was to determine whether DBS psychosine concentration at birth can predict disease onset and clinical phenotype. Overall, the findings of this study most clearly support the prognostic value of DBS psychosine in newborns with substantially elevated concentrations. In this cohort, DBS psychosine concentration above 3 nmol/L showed perfect specificity as a marker for infantile KD; all specimens with DBS psychosine concentrations above this threshold were associated with infantile KD phenotypes (EIKD or LIKD). Among the infantile KD patients with available NBS specimens, the degree of DBS psychosine elevation at birth did not appear to correlate with disease severity or clinical outcome. These findings suggest that prompt evaluation for HSCT candidacy is warranted in any infant with substantially elevated DBS psychosine concentration during the newborn period.
Interpreting the prognostic significance of DBS psychosine in newborns is likely to be more challenging in patients with mildly elevated or borderline psychosine concentrations. Although all of those who tested above 3 nmol/L have developed EIKD or LIKD, the range between this value and the upper limit of the control range (0.71 nmol/L) included a mix of KD patients, carriers, and NBS-positive infants who have not developed signs of disease. The two first-year KD specimens in this range included an NBS sample from a patient who developed juvenile-onset KD (2.3 nmol/L) and a specimen collected at 2 months of age from an LIKD patient (2.0 nmol/L) who was evaluated pre-symptomatically due to a family history of disease. Three of the NBS samples from asymptomatic NBS-positive patients had higher DBS psychosine concentrations (2.4 – 2.7 nmol/L) than these KD patients. Since it is currently not known which, if any, of the NBS-positive patients will ultimately develop KD, it is unclear whether the range of newborn DBS psychosine concentrations found in pre-symptomatic KD patients overlaps with the range observed in NBS-positive individuals who never develop symptoms of KD despite having abnormal GALC activity. Notably, DBS psychosine was slightly elevated, at 0.76 nmol/L, in an NBS specimen from a carrier, suggesting that mild elevations in DBS psychosine may occur at birth even in phenotypically normal individuals. Overall, our results suggest that, although substantially elevated DBS psychosine concentration (>3 nmol/L) during the newborn period is associated with a high probability of developing infantile KD, mildly elevated (0.7 – 3 nmol/L) DBS psychosine in a newborn neither indicates that the patient is certain to develop KD nor excludes the possibility that they will develop an infantile form of KD requiring urgent treatment. Thus, NBS-positive infants who test within this range will need to be followed closely. Longitudinal, prospective studies are needed to better delineate the relationship between mildly elevated DBS psychosine at birth and long-term clinical outcomes in NBS-positive patients.
Long-term clinical follow-up beyond the newborn period is vital to ensure optimal outcomes for all patients with KD. Thus, in addition to assessing the predictive value of a single DBS psychosine measurement obtained shortly after birth, this study sought to determine whether longitudinal changes in DBS psychosine concentration correlate with disease onset and progression. A crucial question is whether the onset of symptoms is associated with changes in DBS psychosine concentrations in patients with KD. For four EIKD patients in this cohort, psychosine was measured both in newborn DBS and in DBS collected at 6–9 months of age, by which time neurological disease was apparent. Between these two samplings, psychosine increased by more than 10% in three patients and remained stable at 29 nmol/L in the other patient. In the one LIKD patient for whom DBS were available before and after the onset of symptoms, which occurred at 7 months, DBS psychosine increased from 5.0 nmol/L at birth to 6.9 nmol/L at 11 months. The relationship between disease onset and changes in DBS psychosine in patients with later-onset forms of KD is unclear, since longitudinal pre-HSCT data were not available for any of the juvenile-onset KD patients in this cohort. Given that the latency period between birth and the appearance of overt manifestations of neurological disease may last for years or even decades in later-onset KD phenotypes, the identification of a biomarker for the onset of neurological disease in this group could aid in the monitoring of NBS-positive patients who do not develop infantile KD. Prospectively measuring DBS psychosine over time in patients who are at risk for juvenile-onset KD may help to clarify the utility of psychosine for this purpose.
Following symptom onset, the untreated EIKD patients who were evaluated beyond the first year of life typically showed longitudinal declines in DBS psychosine concentrations as neurological deterioration progressed. These results are consistent with a study by Turgeon et. al., which found that DBS psychosine concentrations in samples collected from KD patients within 1 year of symptom onset were significantly higher than those of controls, whereas psychosine concentrations in samples collected more than 5 years after symptom onset did not differ significantly from those of controls. Longitudinal decreases in DBS psychosine concentrations in patients with EIKD may be a consequence of end-stage axonopathy and oligodendrocyte loss [13].
The final aim of this study was to assess the effect of HSCT on DBS psychosine concentrations. We observed a large and rapid decline in DBS psychosine following HSCT in the patients with the highest baseline DBS psychosine values. However, even after these declines occurred, DBS psychosine concentrations remained above the normal range in all patients with EIKD and in all but one patient with LIKD. Our finding that treatment with HSCT is associated with a decrease in DBS psychosine supports the benefit of this treatment at the biological level. However, further research is needed to determine whether psychosine levels at baseline or after HSCT correlate with clinical outcomes. Longitudinal changes in psychosine level following treatment should also be investigated in the context of novel therapies as these become available in the future.
Analysis of replicate samples showed that the mean SE% in these DBS samples was 8.6%. Factors that contribute to variability in psychosine measurements from the same DBS specimen are not well understood. Blood clotting and aggregation of white cells due to collection technique have previously been hypothesized to contribute to within-sample variability in GALC activity measurements in DBS [15]. Psychosine concentrations may also be higher in DBS collected for NBS because of the generally higher hematocrit and leukocyte counts in newborns (Dietrich Matern, personal communication). However, the extent to which these and other factors affect measurements of DBS psychosine concentrations is currently unknown and should be the subject of future research. In the context of clinical decision-making, we recommend analyzing DBS psychosine concentrations in multiple punches per patient.
Collectively, the findings of this study suggest that measuring DBS psychosine may aid in the management of patients who are diagnosed with KD or determined to be at risk for KD on the basis of abnormal NBS results. Our results indicate that NBS-positive infants with high DBS psychosine concentrations at birth should be immediately referred for HSCT evaluation. Infants with borderline or slightly elevated psychosine levels should be monitored closely, and DBS psychosine data should be used in conjunction with genotype data to better assess the risk of developing KD. Psychosine may be of most value in those individuals identified through NBS with elevated psychosine concentrations and genotypes that have never been reported for any known KD cases. Prospective, longitudinal studies of NBS-positive infants and patients with confirmed KD are needed to determine the relationship between disease onset and progression and changes in psychosine concentrations. Future research should also aim to identify factors that contribute to the within-sample variability of DBS psychosine measurements. Further characterization of psychosine and other potential biomarkers for KD is vital to ensure that clinicians are able to provide the highest level of care to patients and their families.
Table 1.
Recommendations for interpretation of psychosine concentrations in dried blood spot (DBS) samples collected during the first year of life. In this cohort, all patients with DBS psychosine concentrations above 3 nmol/L developed early-infantile Krabbe disease (EIKD) or late-infantile Krabbe disease (LIKD). None of the KD patients had psychosine concentrations within the normal/carrier range (<0.71 nmol/L). The 0.71–3 nmol/L range included a mix of KD patients, carriers, and asymptomatic, NBS-positive infants. Based on these findings, we provide recommendations for the management of newborns based on DBS psychosine concentration. Only DBS collected during the first year of life were considered for the purposes of this table.
| DBS psychosine range | Number of patients | Patient phenotypes | Interpretation and Recommendation |
|---|---|---|---|
| Elevated (>3 nmol/L) | 42 | All have developed EIKD or LIKD | High risk of developing infantile KD; evaluate immediately for HSCT candidacy |
| Borderline (0.71–3 nmol/L) | 10 | Includes 1 carrier; 2 KD patients (1 LIKD, 1 juvenile-onset KD); 7 NBS-positive infants who have not developed KD | At risk of developing KD (late infantile or juvenile-onset) |
| Normal (<0.71 nmol/L) | 78 | None have developed KD; includes 1 carrier, 75 controls, and 2 NBS-positive infants followed for >30 months | Unlikely to develop infantile KD |
HSCT, hemamiddleoietic stem cell transplantation.
Highlights.
DBS psychosine concentrations were evaluated in a large cohort of KD patients.
Elevated DBS psychosine at birth showed high specificity as a marker for KD.
Disease progression was associated with longitudinal decreases in DBS psychosine.
DBS psychosine typically declined following treatment with HSCT.
Measuring DBS psychosine may serve as a useful second-tier newborn screening test
Acknowledgments
The authors would like to thank Professor Christiane Auray-Blais for her generous gift of 75 newborn dried blood spots from normal infants, The Legacy of Angels Foundation for their continued support of the NDRD Program biorepository, and Kathleen LaPoint for assistance with editing the manuscript. This research was partially funded by the National Institutes of Health Grant R01DK067859 to MHG.
Abbreviations
- EIKD
early-infantile Krabbe disease
- KD
Krabbe disease
- LIKD
late-infantile Krabbe disease
- NBS
newborn screening
- HSCT
hematopoietic stem cell transplantation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors declare no conflict of interest.
Author Contributions
JJO, MLE, MDP, MHG, and DM conceived and designed the experiments; XH and MHG performed the experiments; MDP analyzed the data; BTK, ES, and MLE wrote the paper.
References
- 1.Suzuki K. Globoid cell leukodystrophy (Krabbe’s disease): update. J Child Neurol. 2003;18:595–603. doi: 10.1177/08830738030180090201. [DOI] [PubMed] [Google Scholar]
- 2.Wenger DA. Krabbe Disease. 2000 Jun 19 [Updated 2011 Mar 31] In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet] Seattle WA: University of Washington, Seattle; 1993–2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1238/. Accessed March 6, 2017. [Google Scholar]
- 3.Escolar ML, Poe MD, Provenzale JM, Richards KC, Allison J, Wood S, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med. 2005;352:2069–2081. doi: 10.1056/NEJMoa042604. [DOI] [PubMed] [Google Scholar]
- 4.Orsini JJ, Kay DM, Saavedra-Matiz CA, Wenger DA, Duffner PK, Erbe RW, et al. Newborn screening for Krabbe disease in New York State: the first eight years’ experience. Genet Med. 2016;18:239–248. doi: 10.1038/gim.2015.211. [DOI] [PubMed] [Google Scholar]
- 5.Kemper AR, Knapp AA, Green NS, Comeau AM, Metterville DR, Perrin JM. Weighing the evidence for newborn screening for early-infantile Krabbe disease. Genet Med. 2010;12:539–543. doi: 10.1097/GIM.0b013e3181e85721. [DOI] [PubMed] [Google Scholar]
- 6.Ross LF. Newborn screening for lysosomal storage diseases: an ethical and policy analysis. J Inherit Metab Dis. 2012;35:627–634. doi: 10.1007/s10545-011-9435-0. [DOI] [PubMed] [Google Scholar]
- 7.Dees RH, Kwon JM. The ethics of Krabbe newborn screening. Public Health Ethics. 2013;6:114–128. doi: https://doi.org/10.1093/phe/phs033. [Google Scholar]
- 8.Jalal K, Carter R, Yan L, Barczykowski A, Duffner PK. Does galactocerebrosidase activity predict Krabbe phenotype? Pediatr Neurol. 2012;47:324–329. doi: 10.1016/j.pediatrneurol.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Liao HC, Spacil Z, Ghomashchi F, Escolar ML, Kurtzberg J, Orsini JJ, et al. Lymphocyte galactocerebrosidase activity by liquid chromatography-tandem mass spectrometry for post-newborn screening evaluation of Krabbe disease. Clin Chem. 2017 doi: 10.1373/clinchem.2016.264952. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orsini JJ, Caggana M. Newborn screening for Krabbe disease and other lysosomal storage disorders: Broad lessons learned. Int J Neonatal Screen. 2017;3:3. doi: 10.3390/ijns3010003. [DOI] [Google Scholar]
- 11.Wasserstein MP, Andriola M, Arnold G, Aron A, Duffner P, Erbe RW, et al. Clinical outcomes of children with abnormal newborn screening results for Krabbe disease in New York State. Genet Med. 2016;18:1235–1243. doi: 10.1038/gim.2016.35. [DOI] [PubMed] [Google Scholar]
- 12.Chuang WL, Pacheco J, Zhang XK, Martin MM, Biski CK, Keutzer JM, et al. Determination of psychosine concentration in dried blood spots from newborns that were identified via newborn screening to be at risk for Krabbe disease. Clin Chim Acta. 2013;419:73–76. doi: 10.1016/j.cca.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 13.Turgeon CT, Orsini JJ, Sanders KA, Magera MJ, Langan TJ, Escolar ML, et al. Measurement of psychosine in dried blood spots—a possible improvement to newborn screening programs for Krabbe disease. J Inherit Metab Dis. 2015;38:923–929. doi: 10.1007/s10545-015-9822-z. doi:0.1007/s10545-015-9822-z. [DOI] [PubMed] [Google Scholar]
- 14.Eisenberg DT, Kuzawa CW, Hayes MG. Improving qPCR telomere length assays: Controlling for well position effects increases statistical power. Am J Hum Biol. 2015;27:570–575. doi: 10.1002/ajhb.22690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orsini JJ, Saavedra-Matiz CA, Gelb MH, Caggana M. Newborn screening for Krabbe’s disease. J Neurosc Res. 2016;94:1063–1075. doi: 10.1002/jnr.23781. [DOI] [PMC free article] [PubMed] [Google Scholar]