

Abstract
Protein kinase D is a family of evolutionarily conserved serine/threonine kinases that belongs to the Ca++/Calmodulin-dependent kinase superfamily. Signal transduction pathways mediated by PKD can be triggered by a variety of stimuli including G protein-coupled receptor agonists, growth factors, hormones, and cellular stresses. The regulatory mechanisms and physiological roles of PKD have been well documented including cell proliferation, survival, migration, angiogenesis, regulation of gene expression, and protein/membrane trafficking. However, its precise roles in disease progression, especially in cancer, remain elusive. A plethora of studies documented the cell- and tissue-specific expressions and functions of PKD in various cancer-associated biological processes, while the causes of the differential effects of PKD have not been thoroughly investigated. In this review, we have discussed the structural-functional properties, activation mechanisms, signaling pathways and physiological functions of PKD in the context of human cancer. Additionally, we have provided a comprehensive review of the reported tumor promoting or tumor suppressive functions of PKD in several major cancers types and discussed the discrepancies that have been raised on PKD as a major regulator of malignant transformation.
Keywords: PKD, cancer, structure, regulation, signaling mechanisms, function
1. Introduction
Protein kinases are of utmost importance in maintaining a battery of cellular activities and the human genome encodes over 500 protein kinase genes which constitute about 2% of all human genes, collectively named as the human kinome [1]. The protein kinase D (PKD) family of serine/threonine kinases falls in to the Ca++/Calmodulin-dependent protein kinases (CaMKs) superfamily and consists of three isoforms in mammals, notably, PKD1, PKD2 and PKD3. PKD1 was the first member identified in human and mouse in 1994 [2, 3], although initially it was categorized as a member of the protein kinase C (PKC) family and named PKCμ [2, 4]. It was later reclassified in to CaMK family based on sequence homology in the catalytic domain. PKD3 and PKD2, two additional PKD isoforms, were discovered thereafter [5, 6].
PKDs are evolutionarily highly conserved and homologs are found in several organisms including mice (Mus musculus), rats (Rattus norvegicus), flies (Drosophila melanogaster) and yeast (Saccharomyces cerevisiae) [7]. There is also high sequence homology among PKD isoforms, although structural and functional differences have been noted. For example, PKD3 lacks PDZ (PSD-95/Discs large/ZO-1) binding motif [8] and a Src family kinase phosphorylation motif [9]. Among other organisms, D. melanogaster possesses only one PKD gene [10], whereas, two orthologs termed dfk-1 and dfk-2 are present in C. elegans [11–13]. In a canonical pathway, various stimuli including hormones, phorbol esters, growth factors, cellular stress converge to the activation of PKDs through diacylglycerol (DAG) and classical or novel protein kinase C (c/nPKC) via active phospholipase C (PLC) β and γ [14, 15]. Activated PKD resides in diverse subcellular locations such as cytosol, Golgi apparatus, nucleus, mitochondria to regulate a plethora of cellular functions, especially those related to malignant transformation including cell proliferation, growth, migration/invasion, apoptosis, epidermal-to-mesenchymal transition (EMT) [14, 16, 17].
In this review, we discuss the status quo of PKD isoforms in terms of their modulation of different physiological activities and mechanistic role in development and progression of human diseases focusing on cancer. In light of accumulating scientific evidence, we aim to provide an updated and comprehensive review of each of the PKD isoforms, their differential expression patterns, and how they communicate with the cellular machineries in a wide variety of cell and tissue types to coordinate its biological role in oncogenesis. Furthermore, we discuss potential tumor promoting as well as suppressive properties of PKD in different cancer types and aim to resolve the prevailing functional discrepancies it poses as a regulator of cancer.
2. PKD structural and functional relationships, phosphorylation, activation
All 3 members of the PKD family share discrete structural and functional similarities (Fig. 1). They possess an N-terminal regulatory domain which is subdivided into 2 tandem cysteine-rich Zn-finger like domains (CRD, C1a and C1b), a plekstrin homology (PH) domain and a C-terminal catalytic domain [14, 16]. The regulatory domain plays a critical role by auto-inhibiting the kinase domain and maintaining the protein in an inactivated state and thus deletion or mutation of critical residues of the C1 domains results in constitutively active PKD [14]. The C1 domains bind DAG and phorbol esters, anchor PKD to membranes and modulate the localization of the protein to Golgi, nucleus as well as plasma membrane [14, 18, 19]. Although the C1 domains are active in PKD, there are intrinsic differences in their activity and selectivity for ligands. Our study has demonstrated isoform-specific differences in the ligand binding activities of PKD isoforms with PKD3 being most sensitivity of DAG/phorbol esters [20]. As for individual C1 domains, we have shown that C1a is a high affinity receptor for DAG, while C1b is low affinity for DAG but high affinity for phorbol esters. In the context of full-length protein, C1a in several studies appears to play a more crucial role in DAG binding and membrane targeting of PKD [19–22]. Furthermore, the reduced DAG binding activity of C1b is attributed to a conserved lysine residue within this domain that impairs DAG binding [20, 23]. Overall, C1 domain is central to the spatial and temporal regulation of PKD localization at different subcellular locations. In addition to the intrinsic differences of the twin C1 domains [20, 24, 25], their ligand binding activity, selectivity, and accessibility to ligand in a holoenyme are intricately regulated by PKD phosphorylation [22], kinase activity [19, 21, 22] and interactions with protein binding partners [26], and this regulation becomes more complex with embedded nuclear localization and export signals within the structure of C1 domain [27]. Whether, how, and how much they contribute to the differential biological functions of PKD isoforms remain to be determined.
Figure 1.
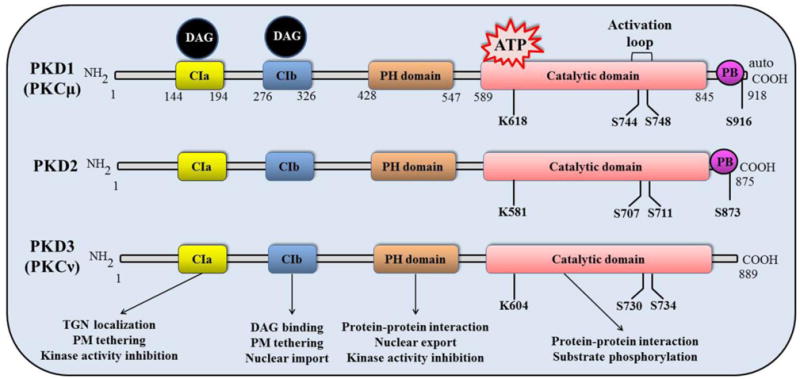
Diagram of protein kinase D structure. The structure of PKD contains an N-terminal regulatory domain which consists of cysteine-rich Zn-finger like motifs (CIa and CIb), a plekstrin homology (PH) domain and a Cterminal catalytic domain, which are shared by all three isoforms. However, PKD3 lacks C-terminal PDZ binding (PB) domain as it is present in both PKD1 and PKD2. The serine residues shown in the catalytic domain represent the conserved activation loop amino acids phosphorylated by the members of the c/nPKCs, which leads to PKD activation. Note: Phosphorylation sites are numbered based on the murine PKD isoforms.
The PH domain has been shown to interact with other proteins and play a role in subcellular localization as well as nuclear export of PKD [14]. The type 1 PDZ (PSD-95/Discs large/ZO-1) binding motif is found at the C-terminus of PKD1 and PKD2 which is responsible for interaction with protein substrates such as NHERF-1 and Kidins220 [8, 28]. The substrate recognition motif of PKD1 (L.X.R. (Q/K/E/M).S.X.X.X.X) displays a unique preference of leucine at the -5 position [16, 29, 30]. There are minor structural differences in PKD2 and PKD3. For example, PKD2 contains a serine-rich region between C1a and C1b motifs and PKD3 lacks any C-terminal PDZ domain [14]. PKD can be activated by a variety of physiological factors, such as bioactive peptides [31], lipids [32], growth factors [33], tumor necrosis factor (TNF) [34], chemokines, and many of which act through binding and activating the G-protein-coupled receptors (GPCR) or receptor tyrosine kinase (RTK) and further activating phospholipase Cs (PLCs) and c/nPKCs. c/nPKCs phosphorylate the conserved serine residues in the activation loop of PKDs (for example, Ser744 and Ser748 of murine PKD1, equivalent to Ser738 and Ser742 of human PKD1), leading to PKD activation [14, 15]. Mutation of active site serine residues in PKD (PKD1 S744A/S748A) abolishes PKD activation [16, 17]. Replacement of both serine residues with glutamic acid (PKD1 S744E/S748E) results in a constitutively active PKD implying that activation loop phosphorylation is an essential mechanism for PKD activation [14]. In further analysis of the canonical PKC-dependent activation of PKD pathway governing the functional facets of PKD in cellular physiology, emerging evidence suggests that different regulatory mechanisms control the phosphorylation at the two sites in the activation loop. For instance, Gq-coupled receptor agonists such as bombesin induce biphasic PKD activation, notably, a first rapid PKC-dependent activation through phosphorylation of PKD1 at Ser744, followed by a second PKC-independent autophosphorylation at Ser748 to sustain PKD activity [35, 36]. Several lines of evidence emerged for supporting the fact that PKD1 autophosphorylation at Ser748 is the major mechanism for late sustained PKD activation in cells treated with GPCR agonists [35]. Beyond the activation loop phosphorylation, there are considerable discrepancies in the understanding of an autophosphorylation site, PKD1 Ser916 (equivalent to human PKD1 Ser910), which has been used as a marker of PKD1 activity status in many studies [37]. The lines of evidence supporting its use as a marker for PKD activation are: 1) PKD1 Ser916 phosphorylation increases when PKD1 is activated by growth factor receptors or phorbol esters, 2) constitutively active PKD1S744E, S748E exhibits high levels of endogenous Ser916 phosphorylation [37, 38]. It was conceived that activation of PKD through phosphorylation of Ser744 and Ser748 residues is often followed by autophosphorylation of Ser916 [14, 37, 39, 40]. However, other evidences support the contrary: 1) PKD1 activation loop phosphorylation and increase in catalytic activity by agonist stimulation do not augment Ser910 phosphorylation [41–43], 2) A catalytically inactive PKD1 mutant (PKD1K612W) displays Ser910 phosphorylation by endogenous PKD1 and other enzymes [8, 44], 3) Ser910 autophosphorylation can be achieved at exceedingly low concentration of ATP that does not require PKD1 phosphorylation of Ser738, 742. Additionally, the phosphorylation status on Ser910 does not correlate well with the inhibition of PKD by certain inhibitors, for example, Ser910 phosphorylation is resistant to Gö6976, an ATP competitive inhibitor [44] and another ATP competitive inhibitor, BPKDi that inhibits HDAC5 phosphorylation, does not inhibit PKD1 Ser910 autophosphorylation [45]. Further studies are necessary to define the precise role of PKD1 Ser910 autophosphorylation and its implication in the regulation of PKD1 catalytic activity.
3. Protein Kinase D and somatic mutations in cancer
Benign cells acquire somatic mutations which cause dysregulation of cell proliferation, migration and invasion, a key phenomenon for oncogenesis. Approaches through comprehensive genomics analysis have provided valuable cues to somatic aberrations that define individual cancers [46–48]. PRKD is generally thought to exhibit low frequency of somatic mutations in pan-cancer analysis. However, in several recent reports, high frequency somatic mutations in PRKD genes have been reported in at least two rare tumors, Polymorphus low-grade adenocarcinoma (PLGA) and angiolipomas [49–51].
Polymorphus low-grade adenocarcinoma (PLGA) is an intra-oral salivary gland malignancy which preferentially affects the minor salivary glands. Weinreb et al. subjected three consecutive PLGAs to massive parallel RNA sequencing and whole exome sequencing (WES) and identified two somatic heterozygous single-nucleotide variations (SNV), which are c.2130A>T and c.2130A>C, affecting highly conserved E710 amino acid at the catalytic loop, resulting a mutant PKD1 (PKD1 E710D) [51]. To further validate the results, the authors analyzed 53 PLGAs by Sanger sequencing and targeted amplicon sequencing of PRKD1 exon 15 and confirmed the presence of somatic c.2130A>T and c.2130A>C mutations in 41.5% and 30.2% of PLGAs, respectively and the total mutation frequency of 72.9%, representing a single high-frequency hotspot mutation that is indicative of a driver oncogene. Homology modelling of PKD1 suggested that this E710D mutation could alter coordination with Mg2+ ion, affect enzyme kinetics as well as interfere with ADP-binding [51]. Cell-free in vitro kinase assay showed significantly increased transphosphorylation of the substrate CREBtide by PKD1E710D mutant protein and its elevated catalytic activity as compared to the wild type PKD1 [51]. Further analysis indicated that the expression of PKD1E710D mutant protein in embryonic kidney epithelial and non-malignant breast epithelial cells caused increased phosphorylation of Ser738/Ser742 and Ser910 of PKD1 as compared to the wild type PKD1. It was also demonstrated that forced expression of PKD1E710D mutant protein in MCF10A and MCF12A breast cells changed the hollow spheroid, acinar-like structures into large, coalescent structures filled with lumens and irregular contours, an increased invasive phenotype typically associated with the overexpression of other oncogenes in this model system [52, 53]. Taken together, these results demonstrate that the somatic mutation in PKD1 encoding PKD1E710D is likely activating driver of PLGA and confers a neoplastic advantage to the epithelial cells [51]. There was another report which aimed to identify somatic mutations in PLGA that affect the kinase domains of PRKD2 or PRKD3 gene and act as a driver of neoplasia [50]. This study found PLGAs that lack PRKD1 somatic mutations or PKD gene family rearrangement; do not harbor somatic mutations in the kinase domains of PRKD2 or PRKD3 genes. These findings appear to bring up an interesting concept that PKD1 is not functionally equivalent to PKD2 and PKD3 in tumorigenesis. There is a lack of evidence for the somatic mutations and their effect on the biology of adipocytic tumors, including angiolipoma as no genetic aberrations or chromosomal rearrangements/deregulations have been reported [54]. In a recent report, Hofvander et al. analyzed a cohort of benign adipocytic tumors including conventional lipoma, hibernoma and angiolipoma by WES and ultra-deep sequencing and demonstrated the presence of somatic mutations (18 out of 21) in the catalytic domain of PKD2 [49]. qRT-PCR confirmed that the level of PRKD2 but not PRKD1 or PRKD3 was higher in angiolipoma than in lipomas. The authors argued that the finding of PRKD2 mutations in 80% of the tumors strongly correlate with the neoplastic origin of angiolipoma. Further studies are necessary to evaluate the significance of the mutations in the catalytic domain of PRKD2 gene. Collectively, these studies not only highlight the significance of PKD family in oncogenesis, but also reveal its potential utility as molecular diagnostic markers or therapeutic targets for certain tumors.
4. PKD regulates major biological processes that contribute to development and progression of cancer
PKD contributes to a broad spectrum of cellular processes including cell survival, proliferation, EMT, angiogenesis, gene transcription, secretion and vesicle transport through TGN and innate immunity (Fig.2). Once activated, PKD rapidly localizes to different subcellular locations including plasma membrane, nucleus and mitochondria. In this section we discuss several major biological events and pathways regulated by PKD that directly contribute to development and progression of cancer.
Figure 2.
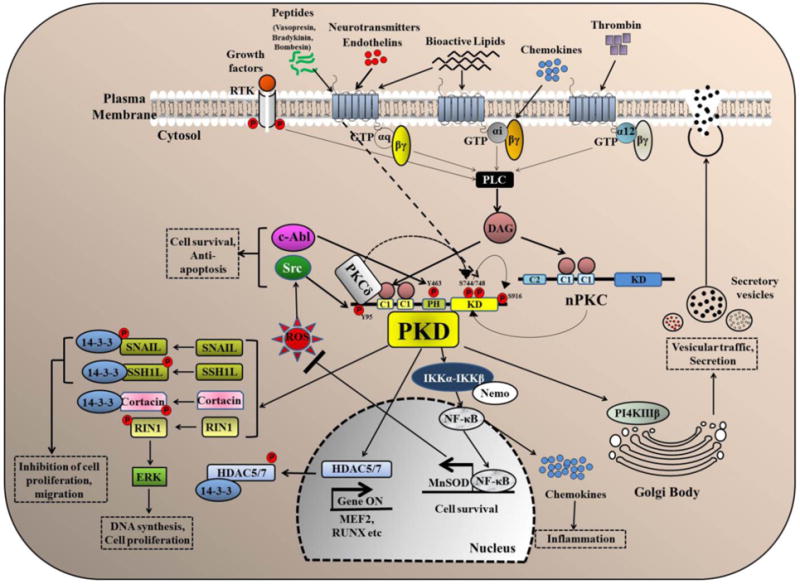
Major signaling pathways and biological functions of PKD. Several extracellular stimuli activate phospholipase C (PLC) which catalyzes the formation of diacylglycerol (DAG). DAG recruits PKD and PKC to the plasma membrane inducing the activation of PKC which then further phosphorylates PKD at two serine (Ser744,748) residues resulting in the activation of PKD. PKD can also be activated by PKC-independent pathways and the activity of PKD can be sustained through autophosphorylation at Ser748 residue (dashed line). Activated PKD regulates an array of cancer-associated functions including cell proliferation, migration, survival, regulation of gene transcription, protein/vesicle trafficking and secretion through several major signaling pathways.
4.1. Cell growth and proliferation
A major characteristic of cancer cells is uncontrolled cellular growth and proliferation. In this section, we will review the roles of PKD in modulating the biological pathways that control cell growth, proliferation and survival in context of neoplasia.
PKD is activated by many mitogenic GPCR agonists that mediate their response through Gq, Gi and G12 implying a role of PKD in cell proliferation [55–58]. Gq-coupled receptor agonists including bombesin and vasopressin-induced activation of PKD1 and PKD2 and subsequent increased DNA replication and cell proliferation has been shown in Swiss 3T3 fibroblast [36, 59–61]. Extracellular-regulated protein kinase (ERK) pathway is a major mediator of GPCR agonists-induced mitogenic effect [62]. PKD contributes to the duration and intensity of MEK/ERK/RSK activation in GPCR agonist-stimulated cells leading to the induction of c-Fos that stimulates cell cycle progression [36, 59].
4.2 Cell survival
Oxidative stress can be accounted as an imbalance between the systemic production of reactive oxygen species and a cell’s ability to readily detoxify the reactive intermediates or to repair the resulting damage. Abnormalities in the normal redox state of cells can cause toxic effects through the production of reactive oxygen species (ROS), such as O2− (superoxide radical), OH (hydroxyl radical) and H2O2 (hydrogen peroxide). Oxidative stress can activate PKD via nonreceptor tyrosine kinase c-Abl and Src along with PKCδ. Reactive oxygen species (ROS) produces DAG via PLD1 and phosphatidic acid phosphatase (PAP)-mediated catalysis and further recruitment of PKD and PKCδ at the outer mitochondrial membrane [63]. A colocalized c-Abl phosphorylates Tyr463 in the PH domain of PKD1 which causes a conformational change, allowing the YGLY domain to be released from PH domain-mediated intramolecular autoinhibition [9]. Src-dependent phosphorylation of Tyr95 creates a priming site for the C1b domain of PKCδ. PKCδ efficiently activates PKD1 by phosphorylating Ser746/Ser748. Activated PKD in turn activates IKKα-IKKβ-Nemo complex and nuclear import of NF-κB which results in induction of antiapoptotic and, or antioxidant genes such as manganese superoxide dismutase (MnSOD) and promotes cell survival [64]. Hence, a complete understanding of the molecular mechanism underlying ROS-induced PKD-mediated cell survival and identification of PKD substrate that activate NF-κB signaling pathway will help us better decipher the mechanism behind cancer cell survival.
4.3 Epithelial-to-mesenchymal transition (EMT)
Epithelial-to-mesenchymal transition (EMT) is a prominent neoplastic characteristic, where cell-cell adhesion is disrupted leading to enhanced cell motility and invasiveness [65]. Epithelial cells undergoing EMT process show mesenchymal cell properties including expression of mesenchymal markers and ability to migrate and invade [66]. E-cadherin is a master regulator of EMT process where it binds to β-catenin to form a protein complex and maintains cell-cell adhesion by interacting with actin and microtubule cytoskeleton owing to its antiproliferative, antimetastatic and anti-invasion properties [67, 68]. Different mechanisms of E-cadherin repression in malignant tumor have been shown including mutation, transcriptional repression, epigenetic silencing [68] and many transcription factors are involved in repression of E-cadherin and induction of EMT such as Snail and Twist [4, 69–71]. Regulation of Snail protein is achieved by multiple pathways. The NF-κB signaling pathway positively modulates and stabilizes Snail protein promoting cell migration [72], whereas, phosphorylation of Snail by p21-activated kinase 1 (PAK1) [73] and GSK3β [74, 75] increase Snail repression activity. Snail represses E-cadherin expression in prostate and breast cancer. PKD1 phosphorylates Snail at Ser11 residue triggering nuclear exclusion of Snail by 14-3-3 adaptor protein. As a result, Snail-repressed genes are de-repressed and cell migration is suppressed via production of E-cadherin and other proteins that mediate cell-cell adhesion. Hence, PKD negatively regulates the function of Snail and inhibits EMT.
4.4 Cellular motility, migration and invasion
Cellular motility and invasion are coupled to remodeling of actin cytoskeleton [76] and degradation of extracellular matrix (ECM) [77, 78]. Cellular movement is achieved when Cofilin slices actin filaments at the leading edge of motile cells, generating a supply of actin monomers and orchestrating the formation of WAVE-2-cortactin-ARP2/3 complex which ultimately creates an expanded, branched network of F-actin [76]. LIM kinase suppresses cellular migration by phosphorylating Ser3 residue of cofilin [79] and motility is restored when a protein phosphatase slingshot 1 like (SSH1L) dephosphorylates cofilin [80]. The original work by the Storz group showed that PKD phosphorylates SSH1L by complexing with SSH1L and F-actin at lamellipodium [81, 82]. Phosphorylated SSH1L is sequestered to cytosol by 14-3-3 adaptor protein [83], consequently, pSer3-cofilin concentration rises and cell migration is inhibited. A follow-up study by the Storz group also reported PKD2/PKD3-mediated regulation of SSH1L and p21-activated kinase 4 (PAK4) resulting in phosphorylation of cofilin and decrease in cell migration [84]. PKD inhibits cytoskeleton remodeling by phosphorylating the Ras effector RIN1 [85]. Phosphorylated RIN1 activates tyrosine kinase c-Abl and the RIN1-c-Abl complex phosphorylates and brings conformational change in a scaffold protein CRK that recruits F-actin remodeling proteins [85, 86], resulting in suppression of cell motility. Matrix metalloproteinases (MMPs) are a class of proteases (collagenases, e.g. MMP-1; gelatinases, e.g. MMP-2) that mediate cell migration through ECM degradation. It has been reported that PKD inhibits breast cancer cell invasion by negatively regulating the transcription of several MMPs including MMP-2, MMP-7, MMP-9, MMP-10, MMP-11, MMP-13, MMP-14 and MMP-15 [81]. Histone deacetylases (HDACs) have been shown to regulate MMP expression [87, 88] and PKD1 is shown to be a negative regulator of HDACs [89]. Therefore, it is conceivable that PKD may negatively regulate MMPs via HDAC regulation. PKD has also been reported to be a positive regulator of cell migration. PKD2 and PKD3 have been shown to increase prostate cancer cell invasion and migration by promoting NF-κB and urokinase-type plasminogen activator (uPA) expression/activation [90]. Yamamoto et al. has shown that Wnt5a-JNK-PKD1 axis positively regulates cell proliferation and migration of prostate cancer [91]. It has also been shown that vascular endothelial growth factors (VEGF) promote cell proliferation and migration via PKD-mediated phosphorylation of class IIa HDACs and subsequent expression of VEGF-responsive genes [92, 93]. In summary, several PKD-regulated pathways converge on the promotion as well as inhibition of cell proliferation, invasion and migration.
4.5 Angiogenesis
Angiogenesis is a process where new blood capillaries are formed and it is essential for many physiological processes such as embryonic development, wound healing and many pathological processes including tumorigenesis [94]. VEGFs are prominent in angiogenesis [95, 96]. There are two related receptor tyrosine kinases that bind VEGF, VEGFR-1 and VEGFR-2 [97, 98] and induce downstream signal that activates a variety of proteins such as PLC-γ, PI3-Kinase and the Src family [99, 100].
As one of the best characterized angiogenic factors, VEGF exerts different biological functions in endothelial cells, such as: 1) stimulation of endothelial cell proliferation and migration [96], 2) promotion of endothelial cell survival by inducing expression of anti-apoptotic proteins such as Bcl-2 and death antagonist A1[101–105]. PKD1 has been shown to be activated downstream of VEGFR2-PLCγ-PKC to activate ERK1/2 pathway and stimulate endothelial cell proliferation [33]. Hao et al. found that PKD2 was a major PKD isoform that mediates endothelial cell proliferation and migration [106]. In mouse embryonic stem cells, PKD2 activity is required for angiogenesis [107]. Mechanistically, PKD-phosphorylation of class IIa HDACs enable them to be sequestered to the cytoplasm by 14-3-3 proteins leading to derepression of target genes. VEGF-stimulated, PKD-mediated phosphorylation at Ser259/498 and concomitant nuclear export of HDAC5 induces MEF2-dependent genes and endothelial cell migration [89]. HDAC7 is involved in regulating endothelial cell morphology and migration [92]. VEGF induces PKD-mediated phosphorylation of HDAC7 at Ser178/344/479 and its subsequent nuclear exclusion by 14-3-3 proteins. This results in the expression of VEGF-responsive genes and promotion of endothelial cell proliferation and migration, MMP expression and EMT [89, 92]. Therefore, it can be concluded that PKD plays a key role in signaling pathways that regulate angiogenesis in endothelial cells.
4.6 Bone development and innate immunity
Of note, it is noteworthy to mention that PKD is also involved in bone formation and innate immunity. Bone morphogenic proteins (BMPs), a family of multifunctional growth factors belonging to the transforming growth factor β (TGFβ) superfamily maintain skeletal integrity by modulating signaling pathways that converge on Runt-related transcription factors (RUNX), regulator of osteoblast gene transcription. It has been shown that BMP-2 induces PKD activation via PKC-independent pathway during osteoblast lineage progression [108] and PKD activation is required for the BMP-2 mediated osteoblast differentiation [42]. Our recent report using conditional PKD1-knockout mice model has shown that PKD1 positively regulates bone development and osteoblast differentiation which could be linked to the activity of the STAT3/p38 MAPK signaling pathway [109]. Cancer mortality and morbidity are mainly caused by metastasis and bone is the 3rd most common site of metastasis. Given the important role of PKD in bone homeostasis, it is conceivable that PKD may play an important role in bone metastasis of malignant tumors [110]. PKD has been shown to be involved in innate immunity in many different ways, such as: 1) functional regulator of T and B lymphocyte [14], 2) regulator of class IIa HDACs in lymphocytes [111–113], 3) modulator of β1 integrin activity in T lymphocytes [114], 4) regulator of IL-2 via TCR stimulation [115], 5) downstream target in Toll-like receptor 9 (TLR9) signaling in macrophages [116] and TLR2 in mouse bone marrow-derived mast cells [117], 7) promoter of cell proliferation in chronic myelogenous leukemia (CML) [118] and 8) regulator of neutrophil chemotaxis [119].
5. PKD: A friend or foe in cancer development and progression?
Among other hallmarks such as sustaining proliferative signaling, enabling replicative immortality and evading cell death by apoptosis are of utmost significance in cancer development [120]. Accumulating evidence shows prominent link between tumor development and diverse signal transduction pathways that are modulated by PKD. The precise role of PKD in tumor progression remains elusive as evidence suggests that PKD plays a critical role as both the potent tumor promoter and suppressor of tumor development. In this section, we discuss the role of PKD isoforms in cancer development and progression focusing on several major cancer types and attempt to discuss the prevailing discrepancies that are associated with these tumor-specific studies (Fig 3).
Figure 3.
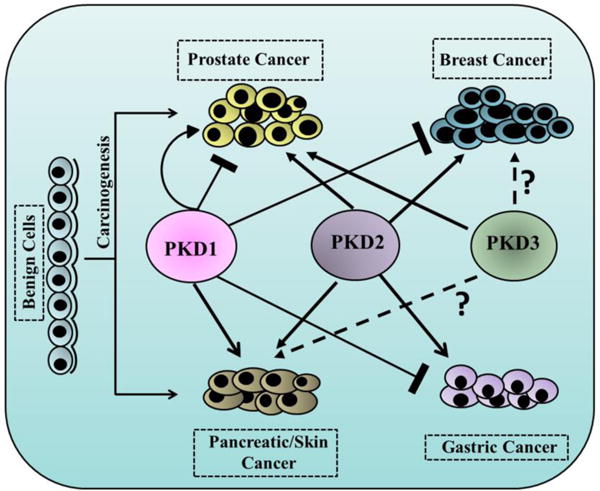
Tumor-specific roles of PKD isoforms in several major cancer types. Benign cells transform into neoplasm such as carcinoma of prostate, breast, pancreas, skin and gastric. Three isoforms of PKD, namely PKD1, PKD2 and PKD3 either promote (solid arrow) or inhibit (bar-headed solid line) cancer progression in highly tumor-specific manner.
5.1 PKD in prostate cancer
Prostate cancer is one of the most common malignancies in male and accounts for 13% of cancer-related deaths in the USA [121]. Although early diagnosis and screening methods have advanced, effective treatments of late-stage metastasized tumors are scarce [122]. Although elevated levels of PKD is observed in human prostate carcinoma tissue compared to normal prostate epithelium, differential expression and distribution of PKD isoforms have been reported and functional analysis of the PKD isoforms has revealed their different roles in prostate cancer progression.
PKD1 is highly expressed in cultured androgen-sensitive, less metastatic LNCaP cell line; whereas, in contrast, PC3 and DU145, two androgen-insensitive, highly metastatic prostate cancer cell lines fail to express PKD1 but found to have high levels of PKD2 and significant PKD3 expression [123]. PKD1 has been reported to be a negative regulator of cell proliferation of prostate cancer cells [124–126]. It has been shown that PKD1 once overexpressed blocks migration through E-cadherin phosphorylation and regulation of β-catenin activity [125–127]. PKD1 has also been shown to downregulate androgen receptor (AR) function in prostate cancer cells [124]. This study demonstrated that PKD1 physically associates with AR through its kinase domain and makes a transcriptional complex. The binding of PKD1 with AR initiates transcription of target genes and this PKD1-AR interaction might be a novel signaling mechanism responsible for prostate cancer progression. In contrast to these studies showing a tumor-suppressive function of PKD1, several lines of independent studies have shown that PKD in fact can serve as an oncogenic factor in prostate cancer [91]. Its role as an enhancer of cell migration and invasion has been linked to Wnt5a overexpression. This study has demonstrated that Wnt5a activates Jun-N-terminal kinase via PKD and PKD is required for Wnt5a-mediated induction of MMP-1 expression, cell migration and invasion.
PKD3, on the other hand, has been shown to promote cell proliferation and survival of prostate cancer by increasing prolonged activation of Akt and ERK1/2 [123, 128]. It is know that hyperactive Akt in PTEN-null prostate cancer has been linked to angiogenesis, invasion and metastasis [129, 130]. Moreover, our study has reported that PKD2 and PKD3 positively regulate prostate cancer cell invasion by upregulating NF-κB signaling and HDAC1-mediated urokinase-type plasminogen activator (uPA) expression [90]. and PKD2 and PKD3 were responsible for increased matrix metalloprotease-9 (MMP-9) expression, a key player for EMT [128] Additionally, our previous study has demonstrated that PKD3 contributes to the growth and survival of prostate cancer cells through PKCε/PKD3 pathway that is downstream of Akt and ERK-1/2 [123]. Activated PKD1 and PKD2 were shown to protect LNCaP prostate cancer cells from phorbol ester PMA-induced apoptosis by promoting downstream ERK-1/2 and NF-κB activities [131]. Using stable inducible PKD3 knockdown prostate cancer cell lines we have demonstrated that knockdown of PKD3 inhibits secretion of multiple key tumor-promoting factors including MMP-9, IL-6, IL-8, and GROα and inducible depletion of PKD3 in a subcutaneous xenograft model suppressed tumor growth and decreased levels of intratumoral GROα in mice. Furthermore, we have shown that androgen represses PKD expression in androgen-sensitive prostate cancer cells in an androgen receptor (AR)-dependent manner and the response is mediated by fibroblast growth factor receptor substrate 2 (FRS2) [132] and thus, we envision that upregulation of PKD as a result of loss or inhibition of AR may promote prostate cancer tumor cell survival.
In support of our view, using several classes of structurally distinct small molecule inhibitors of PKD discovered by our group, including CID755673 and its analogs SD-208, kb-NB142-70, 1-naphthyl PP1 (1-NA-PP1), Compound 139 [128, 133–136], which are all nanomolar cell-active pan-PKD inhibitors, we demonstrated that targeted inhibition of PKD by these inhibitors led to reduced proliferation, migration and invasion of prostate cancer cells. Altogether, these studies have validated PKD as a potential therapeutic target for prostate cancer. In the future, detailed investigation should be carried out to decipher the precise role of each PKD isoforms at different stages of prostate cancer development, and a PKD genetically engineered mouse model of prostate cancer will further validate the functional role of PKD in prostate carcinogenesis and tumor progression.
5.2 PKD in breast cancer
Breast cancer is the leading cause of cancer death in women and it is expected to account for 29% all new cancer diagnoses in the USA by 2017 [121]. PKD was first linked to breast cancer in a study by Bowden et al. demonstrating the association of PKD1 in a complex with cortactin and paxillin in invadopodia at sites of extracellular matrix degradation [137]. Later studies have shown that PKD1 expression, but not PKD2 or PKD3, is reduced in over 95% of invasive breast cancer samples compared to benign breast tissue [81] and thus established a tumor-suppressive role of PKD1 in breast cancer. Eiseler et al. have shown that the loss of PKD1 in breast cancer is associated with higher degree of tumor invasiveness [81]. Highly aggressive breast cancer cell lines such as MDA-MB-231 and BT-20 do not express PKD1, whereas; less invasive MCF-7 and normal mammary epithelial cells such as MCF-10A show significant PKD1 expression. It has been demonstrated that hypermethylation of the PRKD1 promoter causes loss of PKD1 expression in invasive breast cancer [138]. In MCF-7 cell, silencing of PKD1 enhanced its migration, whereas; overexpression of constitutively active form of PKD1 in MDA-MB-231 cells led to decreased cell invasion [81]. Multicellular spheroid/3D cell culture assay using MDA-MB-231 cells demonstrated that expression of active PKD1 inhibited cell invasion compared to normal breast epithelial cells [81]. At molecular level, PKD confers its role as tumor-suppressor in breast cancer by 1) suppressing the expression of many matrix metalloproteases (MMP) such as MMP-2, MMP-7, MMP-9, MMP-10, MMP-11, MMP-13 and MMP-14 [81], 2) inhibiting EMT via inactivation of Snail function by phosphorylating Ser11 residue [139, 140], 3) inducing the expression of epithelial-to-mesenchymal markers (vimentin, E-cadherin) [139] and 4) phosphorylating SSH1L at Ser937 and Ser978 residues and negatively regulating cofilin phosphorylation and interacting with cortactin and paxillin at lamellipodia, thereby inhibits cell motility and invasion [81, 83, 137, 141]. In contrast, opposing effects of PKD has also been demonstrated in breast cancer. Kennett et al. and Palmantier et al. have demonstrated a tumor-promoting function of PKD1 in breast cancer cells [142, 143].
Although accumulating evidence indicate PKD1 as a gross tumor-suppressor and a major contributor to the maintenance of epithelial phenotype in breast cancer [81, 139–141], PKD2 and PKD3 have been assigned roles of tumor-promoters where they induce cell proliferation, invasiveness and chemoresistance [144–146]. Huck et al. have demonstrated that PKD3 is a tumor-promoter in triple-negative breast cancer (TNBC) [147]. PKD3 triggered the activation of S6 kinase 1 (S6K1) which is the main downstream target of the mammalian target of rapamycin complex 1 (mTORC1). The authors have also shown that PKD3 depletion inhibited cell proliferation of TNBC and hence, identifying PKD3 as a potent chemotherapeutic target. Borges et al. have demonstrated that PKD3 is highly upregulated in estrogen receptor (ER)-negative (ER−) invasive ductal carcinoma (IDC) which is associated with triple-negative phenotype [148]. This study showed that ER directly binds to the PRKD3 gene promoter and inhibits PKD3 expression. Hence, loss of ER leads to upregulation of PKD3 which eventually induce increased cell proliferation, migration and invasion. Hao et al. have shown that silencing PKD2 or PKD3 significantly inhibited proliferation of HCC1806 triple-negative breast cancer cell line and PKD3 knockdown inhibited Hsp27 and HDAC4/5/7 phosphorylation [144]. Further investigation into different PKD isoforms at cellular levels as well as using mouse genetic models will help us to better understand the biology of this family of protein kinases in the development of breast neoplasia.
5.3 PKD in pancreatic cancer
Ductal adenocarcinoma of pancreas is a very aggressive, chemotherapy-resistant type of cancer [149] and according to the American Cancer Society, Pancreatic cancer will account for about 3% of all cancers in the US and about 7% of all cancer deaths by 2017. Almost 95% of all pancreatic ductal adenocarcinoma (PDAC) display somatic activating mutations of Kras [150] and increased epidermal growth factor (EGFR) signaling [151, 152]. As compared to the normal pancreas, human ductal adenocarcinoma of the pancreas shows increased PKD1 expression and kinase activity [39, 153]. An important study from the Storz group has demonstrated a positive role of PKD1 in the malignant transformation of PDAC. The authors have demonstrated that in a 3D explant model, using PKD inhibitors or PKD knockdown approach that PKD1 is necessary for TGFα- and Kras-mediated formation of duct-like structures originating from acinar cells, a process called acinar-to-ductal metaplasia (ADM), which converts to pancreatic intraepithelial neoplasia (PanIN), the premalignant neoplastic precursor of PDAC. Using in vivo mouse model (p48cre, KrasG12D mice), the authors have knocked down PKD1 in acinar cells and demonstrated decreased progression of acinar-to-ductal metaplasia (ADM) to PanIN [154]. This study provided strong support for a role of PKD1 in the pathogenesis of PDAC. Additionally, other studies at cellular level support pro-proliferative and pro-survival effects of PKD1 in pancreatic cancer cells. Specifically, Trauzold et al. has shown that overexpression of PKD1 in pancreatic cancer cell decreased CD95-mediated apoptosis, increased cell proliferation rate and upregulated surviving [153]. PKD1 mediates the mitogenic effects of neurotensin in pancreatic cancer cells [155, 156]. A study by Yuan et al. has shown that neurotensin increases Hsp27 phosphorylation of Ser82 via p38 MAPK-PKD2 signaling axis in pancreatic cancer cells [157]. Hsp27 level is markedly increased in many cancers and its elevated expression contributes to increased tumorigenicity and chemoresistance [158–161]. Another recent study has demonstrated a pro-oncogenic role of PKD2 in pancreatic cancer where PKD2 functions upstream of MMP-7 and -9 in pancreatic cancer cells and induces invasion and angiogenesis in vivo and in vitro [162]. PKD2 does so by stimulating expression and secretion of MMP-7 and -9 and induces invasion in 3D extracellular matrix (ECM) culture, and furthermore, PKD2-activated MMP9 induces tumor angiogenesis by releasing ECM-bound VEGF-A [162]. Although PKD1 and PKD2 have emerged as tumor-promoters in pancreatic cancer respectively, the role of PKD3 remains obscure and further research is needed to address the function of individual PKD isoform in pancreatic cancer.
5.4 PKD in skin cancer
Basal cell carcinoma (BCC) is the most common form of malignant skin cancer in the world. An estimated 83,000 new cases of skin cancer (6% of all cancer types) have been documented in the USA by 2016 [121]. In normal epidermis, PKD1 is primarily expressed in stratum basilis, the proliferative compartment of the skin which supports a notion that PKD1 may promote hyperproliferative disorders in skin [163] and PKD1 expression was shown to be upregulated in mouse carcinomas and human hyperplastic disorders including BCC [163–165]. PKD1 has been shown to repress keratinocyte differentiation and promote cellular proliferation through modulation of MEK/ERK1/2 pathway [166]. UVB radiation is a key risk factor for developing BCC and it has been reported that activation of PKD1 by Src family of tyrosine kinases in primary mouse keratinocytes exposed to UVB reduced UVB-induced apoptosis and this activation of PKD1 was PKC-independent [167]. Rashel et al. have demonstrated a pro-proliferative role of PKD1 in epidermal adaptive response, wound healing and skin carcinogenesis [168]. Using PKD1- conditional knockout (cKO) mouse model, the authors have presented evidence that: 1) keratinocyte cells in PKD1-cKO mice showed delayed wound healing and reduced proliferative response, 2) PKD1 is a positive regulator of epidermal hyperplasia and inflammation in response to phorbol esters and 3) PKD1-cKO mice are resistant to tumor formation when subjected to two-stage chemically-induced skin carcinogenesis [168]. In a recent study, Ryvkin et al. have demonstrated an opposing role of PKD2 and PKD3 isoforms in human keratinocyte proliferation and differentiation [169]. The authors have shown that loss of PKD2 resulted in enhanced keratinocyte proliferation suggesting an anti-tumorigenic role of PKD2. Whereas, silencing of PKD3 showed proliferation defect, loss of clonogenicity and diminished tissue regenerative ability, implying a pro-oncogenic role of PKD3. It is to be noted that PKD1 is not expressed in human keratinocytes and thus, PKD2 and PKD3 play a key role in maintaining human epidermal homeostasis, loss of which results in BCC. Further studies are needed to decipher precise roles of PKD isoforms in skin cancer.
5.5 PKD in gastric cancer
PKD1 has been shown to be a negative regulator of gastric cancer [170]. Gastric carcinoma cells as well as patient tissue samples showed decreased PKD1 expression [170]. Gene silencing of PKD1 using siRNA increased cell invasion of gastric cancer and it was found that PKD1 was epigenetically silenced in this tissue type [170]. In an independent study, Shabelnik et al. have demonstrated opposing roles of PKD1 and PKD2 in gastric cancer where PKD1 acts as tumor-suppressor and PKD2 as tumor-promoter [171]. Using tumor samples of different histological variants of primary gastric cancer and gastric adenocarcinoma cell line AGS, the authors have shown that PKD1 and PKD2 are differentially expressed, i.e., lower and higher expression of PKD1 and PKD2 respectively, in both mRNA and protein levels. pcDNA3.1-mediated overexpression of PKD1 resulted in the inhibition of cell proliferation, migration and colony formation, whereas, that of PKD2 enhanced cell proliferation, migration and colony formation abilities in AGS cells [171]. The role of PKD3 isoform in gastric cancer development and progression remains elusive.
5.6 PKD and other cancer types
The role of PKD in other cancer types is poorly defined, although some reports link PKD to certain cancers. For example, activation of PKD by phorbol esters and bombesin via PKC has been shown in small cell lung cancer (SCLC) cell lines h69, H345 and H510 [172]. Using PKC and PKD inhibitors, Brenner et al. has shown that PKD1 is involved in renal cell carcinoma and it promotes tumor progression by positively regulating the adhesion of renal carcinoma cells to endothelial cells [173]. Studies on human malignant lymphoma cells did not conclude distinct roles of PKD as PKD1 levels were undetected and there was no change in PKD2 expression in the malignant cells as compared to benign tissues [174]. In a recent study, PKD2 has been demonstrated to be a potent mediator of glioblastoma where it promotes tumor progression by upregulating integrin α-2 and -4 (ITGA2 and -4), plasminogen activator urokinase (PLAU), plasminogen activator urokinase receptor (PLAUR), and matrix metallopeptidase 1 (MMP1) [175]. When overexpressed in SW480 colon cancer cells, PKD1 suppresses nuclear β-catenin accumulation and inhibits colon cancer [176]. On the contrary, in another study using RKO human colon cancer cell line, it has been demonstrated that targeted inhibition of PKD by small molecule inhibitor suppressed AKT/ERK signaling and NF-κB activity [177]. Hence, PKD might be a potent chemotherapeutic target for the treatment of colorectal cancer.
6. Therapeutic targeting of PKD in cancer
Accumulating evidence indicate that PKD expression is deregulated in many cancers and PKD plays a crucial role in a wide range of cancer-associated cellular processes such as cell proliferation, migration, apoptosis, EMT, and angiogenesis. This makes PKD an attractive therapeutic target for cancer and has since embarked efforts in the discovery and development of novel PKD inhibitors. Starting with the discovery of a non-ATP-competitive PKD inhibitor CID755673 [128], we have subsequently reported several first-in-class structurally distinct PKD small molecule inhibitors, including the non-ATP-competitive CID755673 and its derivatives kb-NB142-70 and KMG-NB4-23 [128, 178–181], three dual PKD inhibitors (compound 139, 1-NA-PP1, and SD208), and three cell-active PKD inhibitors (CID2011756, CID5389142 and CID1893668) [134–136, 181], which are all nanomolar PKD small molecule inhibitors that potently block prostate cancer cell proliferation, migration, and invasion [128, 180]. CID755673, in particular, have shown in vivo efficacy in other disease models [182].
In addition to our efforts, several highly potent and selective PKD inhibitors have also emerged from the pharmaceutical industry [45, 183–185] including an aminopyridine arene CRT0066101 (CRT101) [186] and an aminopyrimidine phenol CRT0066051 (CRT051) [187] from Cancer Research Technology Ltd. Additionally, in a recent study, Golkowski et al. have identified novel PKD inhibitors, namely compounds 1553, 1561, 1649 and 1369 using kinobead-based proteomic assay [188]. In this study, compound 1369 was found to be highly selective and potent pan-PKD inhibitor and an important tool compound to identify the roles of PKD isoforms in cellular as well as in vivo models. Among all available PKD SMIs, CRT101 is by far the most potent, selective, and cell-permeable PKD small molecule inhibitor with demonstrated in vivo antitumor activity in multiple cancer models [148, 177, 189]. Specifically, CRT0066101 was specifically used in pancreatic cancer tumor xenografts [189]. CRT0066101 was also found to cause significant inhibition of tumor growth in HCT116 xenograft nude mice, supporting the therapeutic potential of this inhibitor in colon cancer [177]. CRT0066101 has been shown to be a potent anticancer agent against highly aggressive ER negative breast cancer [148]. This study showed that similar to PKD3 knockdown effect, CRT0066101 significantly reduced breast cancer cell proliferation, migration and invasion both in vitro and in vivo.
It is interesting to note that despite the differential roles of PKD isoforms in different cancer types as implicated in our discussion above PKD inhibitors have unequivocally exhibited antitumor activities in various in vitro and in vivo cancer models. It is possible the overall selectivity profiles of these inhibitors favor anticancer activity with PKD being the primary target. Nonetheless, with the growing reports of differential functions of PKD isoforms in different cancers, the development of isoform-selective PKD inhibitors is a well-justified direction for future studies.
7. Perspectives and concluding remarks
Growing evidence supports PKD as a key signaling molecule that orchestrates various cancer-associated biological functions such as cell proliferation, survival, EMT, migration, invasion, secretion and angiogenesis upon activation by a battery of stimuli. Despite recent advances of our knowledge about the role of PKD in various pathological conditions including cancer, cardiac hypertrophy and inflammation, the similar or opposing roles of the same PKD isoform in different cancers or of the same cancer for different PKD isoforms has raised more unanswered questions.
To summarize briefly, emerging evidence suggests that PKD1 can function as a tumor-suppressive protein in breast and gastric cancers, where PKD1 inhibits cell survival, proliferation, migration by negatively regulating several key target proteins including SSH1L, Snail and MMPs. However, in other cancers, such as pancreatic and skin cancers, PKD1 emerged as a driver of neoplasia. It does so by many mechanisms such as activating MEK1/2 pathway that increases DNA replication, inhibits apoptosis and promotes proliferation by positively regulating ERK/MAPK pathway. Compared to the opposing roles of PKD1 in cancer development and progression, majority of studies have shown that PKD2 is a tumor-promoting protein in a wide range of cancers. It activates different biological functions such as NF-κB signaling, MMP expression, induction of angiogenesis and inhibition of apoptosis and promotes some common cancers including carcinomas of prostate, breast, pancreas, stomach and also some other types of cancers such as glioblastoma. The actual role of PKD3 in cancer is elusive. It shows pro-oncogenic properties in case of prostate, breast and skin cancer but what it does in pancreatic and gastric cancer, is unknown.
In this review, we have discussed the major signaling pathways involved in development and progression of neoplasm that are modulated by PKD, the molecular mechanisms of regulation of cellular phenomenon orchestrated by PKD and manifestation of each type of PKD isotypes in the context of major carcinomas. Although PKD emerged as a potential target for chemotherapeutic intervention and pan-PKD inhibitors have shown potent anti-cancer activity in multiple cancer models both in vivo and in vitro. Many cellular studies have demonstrated differential effects of PKD isoforms on different biological processes. These differences seem to be isoform- and tumor type-dependent, which raise questions on whether it is appropriate and how to target PKD for cancer treatment. As developed in this review and going forward, there remain many unanswered questions. What are the molecular cues that direct precise and selective role of PKD in disease progression? Is it the isoform-specific function that enables PKD to selectively choose the cell type to exert its biological effect? What would be the best strategy to develop effective small molecule inhibitors of PKD that will preserve the tumor-suppressing capacity of PKD while eradicating those that are tumor-promoting? Further knowledge defining the precise role of PKD isoforms in different tumor models will provide a much clearer picture for targeting this family of protein kinases for cancer therapy.
Table-1.
Summary of differential expression and physiological roles of PKD isoforms in major cancers
| Cancer type | PKD isoform | Function in tumorigenesis | Mode of action | References |
|---|---|---|---|---|
| Prostate | PKD1 | anti-tumorigenic |
|
[124] [126] [125] |
| pro-oncogenic | Wnt5 signaling pathway activation of Jun-N-terminal kinase. | [91] | ||
| PKD2, PKD3 | pro-oncogenic |
|
[128, 190] [90] |
|
| Breast | PKD1 | anti-tumorigenic |
|
[81, 83, 137, 141] [81] [139] |
| pro-oncogenic | Promotes cell adhesion. | [142, 143] | ||
| PKD2/PKD3 | Pro-oncogenic |
|
[144] [145] |
|
| Pancreas | PKD1 | pro-oncogenic |
|
[155, 156] [153] [154] |
| PKD2 | pro-oncogenic |
|
[157] [162] |
|
| Skin | PKD1 | pro-oncogenic |
|
[166] [167] |
| PKD2 | anti-tumorigenic | Correlated with upregulation of CDK4/6 inhibitor p15INK4B and induction of p53-independent G1 cell cycle arrest. | [169] | |
| PKD3 | Pro-oncogenic | [169] | ||
| Gastric | PKD1 | anti-tumorigenic |
|
[170] |
| PKD2 | pro-oncogenic |
|
[177] [177] [177] |
Acknowledgments
This study was supported in part by the National Institutes of Health grant R01CA142580 (Wang), Department of Defense award PC150190, and Oversea Hong Kong & Macao Scholars Collaborative Research Fund of NSFC in China (Grant#813228020, Deng and Wang)
Abbreviations
- PKC
protein kinase C
- PKD
protein kinase D
- PLC
phospholipase C
- DAG
diacylglycerol
- CRD
cysteine-rich domain
- GPCR
G-protein-coupled receptor
- TNF
tumor necrosis factor
- TGN
Trans-Golgi Network
- EMT
epithelial-to-mesenchymal transition
- MMP
matrix metalloprotease
- HDAC
histone deacetylase
- BMP
bone morphogenic protein
- AR
androgen receptor
- RUNX
Runt-related transcription factors
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interests
The authors declare no conflict of interest
References
- 1.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 2.Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc Natl Acad Sci U S A. 1994;91:8572–8576. doi: 10.1073/pnas.91.18.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johannes FJ, Prestle J, Eis S, Oberhagemann P, Pfizenmaier K. PKCu is a novel, atypical member of the protein kinase C family. J Biol Chem. 1994;269:6140–6148. [PubMed] [Google Scholar]
- 4.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 5.Hayashi A, Seki N, Hattori A, Kozuma S, Saito T. PKCnu, a new member of the protein kinase C family, composes a fourth subfamily with PKCmu. Biochim Biophys Acta. 1999;1450:99–106. doi: 10.1016/s0167-4889(99)00040-3. [DOI] [PubMed] [Google Scholar]
- 6.Sturany S, Van Lint J, Muller F, Wilda M, Hameister H, Hocker M, Brey A, Gern U, Vandenheede J, Gress T, Adler G, Seufferlein T. Molecular cloning and characterization of the human protein kinase D2. A novel member of the protein kinase D family of serine threonine kinases. J Biol Chem. 2001;276:3310–3318. doi: 10.1074/jbc.M008719200. [DOI] [PubMed] [Google Scholar]
- 7.Ellwanger K, Hausser A. Physiological functions of protein kinase D in vivo. IUBMB Life. 2013;65:98–107. doi: 10.1002/iub.1116. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez-Ruiloba L, Cabrera-Poch N, Rodriguez-Martinez M, Lopez-Menendez C, Jean-Mairet RM, Higuero AM, Iglesias T. Protein kinase D intracellular localization and activity control kinase D-interacting substrate of 220-kDa traffic through a postsynaptic density-95/discs large/zonula occludens-1-binding motif. J Biol Chem. 2006;281:18888–18900. doi: 10.1074/jbc.M603044200. [DOI] [PubMed] [Google Scholar]
- 9.Doppler H, Storz P. A novel tyrosine phosphorylation site in protein kinase D contributes to oxidative stress-mediated activation. J Biol Chem. 2007;282:31873–31881. doi: 10.1074/jbc.M703584200. [DOI] [PubMed] [Google Scholar]
- 10.Maier D, Hausser A, Nagel AC, Link G, Kugler SJ, Wech I, Pfizenmaier K, Preiss A. Drosophila protein kinase D is broadly expressed and a fraction localizes to the Golgi compartment. Gene Expr Patterns. 2006;6:849–856. doi: 10.1016/j.modgep.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 11.Feng H, Ren M, Chen L, Rubin CS. Properties, regulation, and in vivo functions of a novel protein kinase D: Caenorhabditis elegans DKF-2 links diacylglycerol second messenger to the regulation of stress responses and life span. J Biol Chem. 2007;282:31273–31288. doi: 10.1074/jbc.M701532200. [DOI] [PubMed] [Google Scholar]
- 12.Feng H, Ren M, Rubin CS. Conserved domains subserve novel mechanisms and functions in DKF-1, a Caenorhabditis elegans protein kinase D. J Biol Chem. 2006;281:17815–17826. doi: 10.1074/jbc.M511898200. [DOI] [PubMed] [Google Scholar]
- 13.Feng H, Ren M, Wu SL, Hall DH, Rubin CS. Characterization of a novel protein kinase D: Caenorhabditis elegans DKF-1 is activated by translocation-phosphorylation and regulates movement and growth in vivo. J Biol Chem. 2006;281:17801–17814. doi: 10.1074/jbc.M511899200. [DOI] [PubMed] [Google Scholar]
- 14.Rozengurt E, Rey O, Waldron RT. Protein kinase D signaling. J Biol Chem. 2005;280:13205–13208. doi: 10.1074/jbc.R500002200. [DOI] [PubMed] [Google Scholar]
- 15.Wang QJ. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol Sci. 2006;27:317–323. doi: 10.1016/j.tips.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Rykx A, De Kimpe L, Mikhalap S, Vantus T, Seufferlein T, Vandenheede JR, Van Lint J. Protein kinase D: a family affair. FEBS Letters. 2003;546:81–86. doi: 10.1016/s0014-5793(03)00487-3. [DOI] [PubMed] [Google Scholar]
- 17.Van Lint J, Rykx A, Maeda Y, Vantus T, Sturany S, Malhotra V, Vandenheede JR, Seufferlein T. Protein kinase D: an intracellular traffic regulator on the move. Trends Cell Biol. 2002;12:193–200. doi: 10.1016/s0962-8924(02)02262-6. [DOI] [PubMed] [Google Scholar]
- 18.Wang QJ, Fang TW, Yang D, Lewin NE, Van Lint J, Marquez VE, Blumberg PM. Ligand structure-activity requirements and phospholipid dependence for the binding of phorbol esters to protein kinase D. Mol Pharmacol. 2003;64:1342–1348. doi: 10.1124/mol.64.6.1342. [DOI] [PubMed] [Google Scholar]
- 19.Anderson G, Chen J, Wang QJ. Individual C1 domains of PKD3 in phorbol ester-induced plasma membrane translocation of PKD3 in intact cells. Cell Signal. 2005;17:1397–1411. doi: 10.1016/j.cellsig.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 20.Chen J, Deng F, Li J, Wang QJ. Selective binding of phorbol esters and diacylglycerol by individual C1 domains of the PKD family. Biochem J. 2008;411:333–342. doi: 10.1042/BJ20071334. [DOI] [PubMed] [Google Scholar]
- 21.Spitaler M, Emslie E, Wood CD, Cantrell D. Diacylglycerol and protein kinase D localization during T lymphocyte activation. Immunity. 2006;24:535–546. doi: 10.1016/j.immuni.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 22.Maeda Y, Beznoussenko GV, Van Lint J, Mironov AA, Malhotra V. Recruitment of protein kinase D to the trans-Golgi network via the first cysteine-rich domain. EMBO J. 2001;20:5982–5990. doi: 10.1093/emboj/20.21.5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang QJ, Fang TW, Nacro K, Marquez VE, Wang S, Blumberg PM. Role of hydrophobic residues in the C1b domain of protein kinase C delta on ligand and phospholipid interactions. J Biol Chem. 2001;276:19580–19587. doi: 10.1074/jbc.M010089200. [DOI] [PubMed] [Google Scholar]
- 24.Irie K, Nakahara A, Ohigashi H, Fukuda H, Wender PA, Konishi H, Kikkawa U. Synthesis and phorbol ester-binding studies of the individual cysteine-rich motifs of protein kinase D. Bioorg Med Chem Lett. 1999;9:2487–2490. doi: 10.1016/s0960-894x(99)00413-8. [DOI] [PubMed] [Google Scholar]
- 25.Iglesias T, Waldron RT, Rozengurt E. Identification of in vivo phosphorylation sites required for protein kinase D activation. J Biol Chem. 1998;273:27662–27667. doi: 10.1074/jbc.273.42.27662. [DOI] [PubMed] [Google Scholar]
- 26.Pusapati GV, Krndija D, Armacki M, von Wichert G, von Blume J, Malhotra V, Adler G, Seufferlein T. Role of the second cysteine-rich domain and Pro275 in protein kinase D2 interaction with ADP-ribosylation factor 1, trans-Golgi network recruitment, and protein transport. Mol Biol Cell. 2010;21:1011–1022. doi: 10.1091/mbc.E09-09-0814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Auer A, von Blume J, Sturany S, von Wichert G, Van Lint J, Vandenheede J, Adler G, Seufferlein T. Role of the regulatory domain of protein kinase D2 in phorbol ester binding, catalytic activity, and nucleocytoplasmic shuttling. Mol Biol Cell. 2005;16:4375–4385. doi: 10.1091/mbc.E05-03-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kunkel MT, Garcia EL, Kajimoto T, Hall RA, Newton AC. The protein scaffold NHERF-1 controls the amplitude and duration of localized protein kinase D activity. J Biol Chem. 2009;284:24653–24661. doi: 10.1074/jbc.M109.024547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doppler H, Storz P, Li J, Comb MJ, Toker A. A phosphorylation state-specific antibody recognizes Hsp27, a novel substrate of protein kinase D. J Biol Chem. 2005;280:15013–15019. doi: 10.1074/jbc.C400575200. [DOI] [PubMed] [Google Scholar]
- 30.Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, Turk BE. A rapid method for determining protein kinase phosphorylation specificity. Nat Methods. 2004;1:27–29. doi: 10.1038/nmeth708. [DOI] [PubMed] [Google Scholar]
- 31.Guo J, Gertsberg Z, Ozgen N, Sabri A, Steinberg SF. Protein kinase D isoforms are activated in an agonist-specific manner in cardiomyocytes. J Biol Chem. 2011;286:6500–6509. doi: 10.1074/jbc.M110.208058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durand N, Borges S, Storz P. Protein Kinase D Enzymes as Regulators of EMT and Cancer Cell Invasion. J Clin Med. 2016;5 doi: 10.3390/jcm5020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong C, Jin ZG. Protein kinase C-dependent protein kinase D activation modulates ERK signal pathway and endothelial cell proliferation by vascular endothelial growth factor. J Biol Chem. 2005;280:33262–33269. doi: 10.1074/jbc.M503198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoo J, Chung C, Slice L, Sinnett-Smith J, Rozengurt E. Protein kinase D mediates synergistic expression of COX-2 induced by TNF-{alpha} and bradykinin in human colonic myofibroblasts. Am J Physiol Cell Physiol. 2009;297:C1576–1587. doi: 10.1152/ajpcell.00184.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jacamo R, Sinnett-Smith J, Rey O, Waldron RT, Rozengurt E. Sequential protein kinase C (PKC)-dependent and PKC-independent protein kinase D catalytic activation via Gq-coupled receptors: differential regulation of activation loop Ser(744) and Ser(748) phosphorylation. J Biol Chem. 2008;283:12877–12887. doi: 10.1074/jbc.M800442200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinnett-Smith J, Jacamo R, Kui R, Wang YM, Young SH, Rey O, Waldron RT, Rozengurt E. Protein kinase D mediates mitogenic signaling by Gq-coupled receptors through protein kinase C-independent regulation of activation loop Ser744 and Ser748 phosphorylation. J Biol Chem. 2009;284:13434–13445. doi: 10.1074/jbc.M806554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matthews SA, Rozengurt E, Cantrell D. Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/Protein kinase Cmu. J Biol Chem. 1999;274:26543–26549. doi: 10.1074/jbc.274.37.26543. [DOI] [PubMed] [Google Scholar]
- 38.Steinberg SF. Regulation of protein kinase D1 activity. Mol Pharmacol. 2012;81:284–291. doi: 10.1124/mol.111.075986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guha S, Tanasanvimon S, Sinnett-Smith J, Rozengurt E. Role of protein kinase D signaling in pancreatic cancer. Biochem Pharmacol. 2010;80:1946–1954. doi: 10.1016/j.bcp.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaggi M, Du C, Zhang W, Balaji KC. Protein kinase D1: a protein of emerging translational interest. Front Biosci. 2007;12:3757–3767. doi: 10.2741/2349. [DOI] [PubMed] [Google Scholar]
- 41.Brandlin I, Hubner S, Eiseler T, Martinez-Moya M, Horschinek A, Hausser A, Link G, Rupp S, Storz P, Pfizenmaier K, Johannes FJ. Protein kinase C (PKC)eta-mediated PKC mu activation modulates ERK and JNK signal pathways. J Biol Chem. 2002;277:6490–6496. doi: 10.1074/jbc.M106083200. [DOI] [PubMed] [Google Scholar]
- 42.Celil AB, Campbell PG. BMP-2 and insulin-like growth factor-I mediate Osterix (Osx) expression in human mesenchymal stem cells via the MAPK and protein kinase D signaling pathways. J Biol Chem. 2005;280:31353–31359. doi: 10.1074/jbc.M503845200. [DOI] [PubMed] [Google Scholar]
- 43.Storz P, Doppler H, Toker A. Activation loop phosphorylation controls protein kinase D-dependent activation of nuclear factor kappaB. Mol Pharmacol. 2004;66:870–879. doi: 10.1124/mol.104.000687. [DOI] [PubMed] [Google Scholar]
- 44.Rybin VO, Guo J, Steinberg SF. Protein kinase D1 autophosphorylation via distinct mechanisms at Ser744/Ser748 and Ser916. J Biol Chem. 2009;284:2332–2343. doi: 10.1074/jbc.M806381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meredith EL, Beattie K, Burgis R, Capparelli M, Chapo J, Dipietro L, Gamber G, Enyedy I, Hood DB, Hosagrahara V, Jewell C, Koch KA, Lee W, Lemon DD, McKinsey TA, Miranda K, Pagratis N, Phan D, Plato C, Rao C, Rozhitskaya O, Soldermann N, Springer C, van Eis M, Vega RB, Yan W, Zhu Q, Monovich LG. Identification of potent and selective amidobipyridyl inhibitors of protein kinase D. J Med Chem. 2010;53:5422–5438. doi: 10.1021/jm100076w. [DOI] [PubMed] [Google Scholar]
- 46.Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, Gao J, Socci ND, Solit DB, Olshen AB, Schultz N, Taylor BS. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34:155–163. doi: 10.1038/nbt.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 48.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hofvander J, Arbajian E, Stenkula KG, Lindkvist-Petersson K, Larsson M, Nilsson J, Magnusson L, von Steyern FV, Rissler P, Hornick JL, Mertens F. Frequent low-level mutations of protein kinase D2 in angiolipoma. J Pathol. 2017 doi: 10.1002/path.4865. [DOI] [PubMed] [Google Scholar]
- 50.Piscuoglio S, Fusco N, Ng CK, Martelotto LG, da Cruz Paula A, Katabi N, Rubin BP, Skalova A, Weinreb I, Weigelt B, Reis-Filho JS. Lack of PRKD2 and PRKD3 kinase domain somatic mutations in PRKD1 wild-type classic polymorphous low-grade adenocarcinomas of the salivary gland. Histopathology. 2016;68:1055–1062. doi: 10.1111/his.12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weinreb I, Piscuoglio S, Martelotto LG, Waggott D, Ng CK, Perez-Ordonez B, Harding NJ, Alfaro J, Chu KC, Viale A, Fusco N, da Cruz Paula A, Marchio C, Sakr RA, Lim R, Thompson LD, Chiosea SI, Seethala RR, Skalova A, Stelow EB, Fonseca I, Assaad A, How C, Wang J, de Borja R, Chan-Seng-Yue M, Howlett CJ, Nichols AC, Wen YH, Katabi N, Buchner N, Mullen L, Kislinger T, Wouters BG, Liu FF, Norton L, McPherson JD, Rubin BP, Clarke BA, Weigelt B, Boutros PC, Reis-Filho JS. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet. 2014;46:1166–1169. doi: 10.1038/ng.3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 53.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 54.Bartuma H, Nord KH, Macchia G, Isaksson M, Nilsson J, Domanski HA, Mandahl N, Mertens F. Gene expression and single nucleotide polymorphism array analyses of spindle cell lipomas and conventional lipomas with 13q14 deletion. Genes Chromosomes Cancer. 2011;50:619–632. doi: 10.1002/gcc.20884. [DOI] [PubMed] [Google Scholar]
- 55.Yuan J, Slice L, Walsh JH, Rozengurt E. Activation of protein kinase D by signaling through the alpha subunit of the heterotrimeric G protein G(q) J Biol Chem. 2000;275:2157–2164. doi: 10.1074/jbc.275.3.2157. [DOI] [PubMed] [Google Scholar]
- 56.Yuan J, Slice LW, Gu J, Rozengurt E. Cooperation of Gq, Gi, and G12/13 in protein kinase D activation and phosphorylation induced by lysophosphatidic acid. J Biol Chem. 2003;278:4882–4891. doi: 10.1074/jbc.M211175200. [DOI] [PubMed] [Google Scholar]
- 57.Yuan J, Slice LW, Rozengurt E. Activation of protein kinase D by signaling through Rho and the alpha subunit of the heterotrimeric G protein G13. J Biol Chem. 2001;276:38619–38627. doi: 10.1074/jbc.M105530200. [DOI] [PubMed] [Google Scholar]
- 58.Zugaza JL, Waldron RT, Sinnett-Smith J, Rozengurt E. Bombesin, vasopressin, endothelin, bradykinin, and platelet-derived growth factor rapidly activate protein kinase D through a protein kinase C-dependent signal transduction pathway. J Biol Chem. 1997;272:23952–23960. doi: 10.1074/jbc.272.38.23952. [DOI] [PubMed] [Google Scholar]
- 59.Sinnett-Smith J, Zhukova E, Hsieh N, Jiang X, Rozengurt E. Protein kinase D potentiates DNA synthesis induced by Gq-coupled receptors by increasing the duration of ERK signaling in swiss 3T3 cells. J Biol Chem. 2004;279:16883–16893. doi: 10.1074/jbc.M313225200. [DOI] [PubMed] [Google Scholar]
- 60.Sinnett-Smith J, Zhukova E, Rey O, Rozengurt E. Protein kinase D2 potentiates MEK/ERK/RSK signaling, c-Fos accumulation and DNA synthesis induced by bombesin in Swiss 3T3 cells. J Cell Physiol. 2007;211:781–790. doi: 10.1002/jcp.20984. [DOI] [PubMed] [Google Scholar]
- 61.Zhukova E, Sinnett-Smith J, Rozengurt E. Protein kinase D potentiates DNA synthesis and cell proliferation induced by bombesin, vasopressin, or phorbol esters in Swiss 3T3 cells. J Biol Chem. 2001;276:40298–40305. doi: 10.1074/jbc.M106512200. [DOI] [PubMed] [Google Scholar]
- 62.Rozengurt E. Mitogenic signaling pathways induced by G protein-coupled receptors. J Cell Physiol. 2007;213:589–602. doi: 10.1002/jcp.21246. [DOI] [PubMed] [Google Scholar]
- 63.Cowell CF, Doppler H, Yan IK, Hausser A, Umezawa Y, Storz P. Mitochondrial diacylglycerol initiates protein-kinase D1-mediated ROS signaling. J Cell Sci. 2009;122:919–928. doi: 10.1242/jcs.041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Storz P, Doppler H, Toker A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol Cell Biol. 2005;25:8520–8530. doi: 10.1128/MCB.25.19.8520-8530.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 67.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 68.van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008;65:3756–3788. doi: 10.1007/s00018-008-8281-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 70.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 71.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 72.Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15:416–428. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang Z, Rayala S, Nguyen D, Vadlamudi RK, Chen S, Kumar R. Pak1 phosphorylation of snail, a master regulator of epithelial-to-mesenchyme transition, modulates snail’s subcellular localization and functions. Cancer Res. 2005;65:3179–3184. doi: 10.1158/0008-5472.CAN-04-3480. [DOI] [PubMed] [Google Scholar]
- 74.Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, Fearon ER, Weiss SJ. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398–1406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- 75.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 76.Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773:642–652. doi: 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 78.Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scott RW, Olson MF. LIM kinases: function, regulation and association with human disease. J Mol Med (Berl) 2007;85:555–568. doi: 10.1007/s00109-007-0165-6. [DOI] [PubMed] [Google Scholar]
- 80.Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 2002;108:233–246. doi: 10.1016/s0092-8674(01)00638-9. [DOI] [PubMed] [Google Scholar]
- 81.Eiseler T, Doppler H, Yan IK, Goodison S, Storz P. Protein kinase D1 regulates matrix metalloproteinase expression and inhibits breast cancer cell invasion. Breast Cancer Res. 2009;11:R13. doi: 10.1186/bcr2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eiseler T, Doppler H, Yan IK, Kitatani K, Mizuno K, Storz P. Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat Cell Biol. 2009;11:545–556. doi: 10.1038/ncb1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peterburs P, Heering J, Link G, Pfizenmaier K, Olayioye MA, Hausser A. Protein kinase D regulates cell migration by direct phosphorylation of the cofilin phosphatase slingshot 1 like. Cancer Res. 2009;69:5634–5638. doi: 10.1158/0008-5472.CAN-09-0718. [DOI] [PubMed] [Google Scholar]
- 84.Doppler H, Bastea LI, Borges S, Spratley SJ, Pearce SE, Storz P. Protein kinase d isoforms differentially modulate cofilin-driven directed cell migration. PLoS One. 2014;9:e98090. doi: 10.1371/journal.pone.0098090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ziegler S, Eiseler T, Scholz RP, Beck A, Link G, Hausser A. A novel protein kinase D phosphorylation site in the tumor suppressor Rab interactor 1 is critical for coordination of cell migration. Mol Biol Cell. 2011;22:570–580. doi: 10.1091/mbc.E10-05-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hu H, Bliss JM, Wang Y, Colicelli J. RIN1 is an ABL tyrosine kinase activator and a regulator of epithelial-cell adhesion and migration. Curr Biol. 2005;15:815–823. doi: 10.1016/j.cub.2005.03.049. [DOI] [PubMed] [Google Scholar]
- 87.Klampfer L, Huang J, Shirasawa S, Sasazuki T, Augenlicht L. Histone deacetylase inhibitors induce cell death selectively in cells that harbor activated kRasV12: The role of signal transducers and activators of transcription 1 and p21. Cancer Res. 2007;67:8477–8485. doi: 10.1158/0008-5472.CAN-07-0210. [DOI] [PubMed] [Google Scholar]
- 88.Liu LT, Chang HC, Chiang LC, Hung WC. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer Res. 2003;63:3069–3072. [PubMed] [Google Scholar]
- 89.Ha CH, Wang W, Jhun BS, Wong C, Hausser A, Pfizenmaier K, McKinsey TA, Olson EN, Jin ZG. Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J Biol Chem. 2008;283:14590–14599. doi: 10.1074/jbc.M800264200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zou Z, Zeng F, Xu W, Wang C, Ke Z, Wang QJ, Deng F. PKD2 and PKD3 promote prostate cancer cell invasion by modulating NF-kappaB- and HDAC1-mediated expression and activation of uPA. J Cell Sci. 2012;125:4800–4811. doi: 10.1242/jcs.106542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yamamoto H, Oue N, Sato A, Hasegawa Y, Yamamoto H, Matsubara A, Yasui W, Kikuchi A. Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene. 2010;29:2036–2046. doi: 10.1038/onc.2009.496. [DOI] [PubMed] [Google Scholar]
- 92.Mottet D, Bellahcene A, Pirotte S, Waltregny D, Deroanne C, Lamour V, Lidereau R, Castronovo V. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ Res. 2007;101:1237–1246. doi: 10.1161/CIRCRESAHA.107.149377. [DOI] [PubMed] [Google Scholar]
- 93.Ha CH, Jhun BS, Kao HY, Jin ZG. VEGF stimulates HDAC7 phosphorylation and cytoplasmic accumulation modulating matrix metalloproteinase expression and angiogenesis. Arterioscler Thromb Vasc Biol. 2008;28:1782–1788. doi: 10.1161/ATVBAHA.108.172528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- 95.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 96.Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2:795–803. doi: 10.1038/nrc909. [DOI] [PubMed] [Google Scholar]
- 97.Chen H, Chedotal A, He Z, Goodman CS, Tessier-Lavigne M. Neuropilin-2, a novel member of the neuropilin family, is a high affinity receptor for the semaphorins Sema E and Sema IV but not Sema III. Neuron. 1997;19:547–559. doi: 10.1016/s0896-6273(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 98.Kolodkin AL, Levengood DV, Rowe EG, Tai YT, Giger RJ, Ginty DD. Neuropilin is a semaphorin III receptor. Cell. 1997;90:753–762. doi: 10.1016/s0092-8674(00)80535-8. [DOI] [PubMed] [Google Scholar]
- 99.Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. 1999;4:915–924. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- 100.Guo D, Jia Q, Song HY, Warren RS, Donner DB. Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. Association with endothelial cell proliferation. J Biol Chem. 1995;270:6729–6733. doi: 10.1074/jbc.270.12.6729. [DOI] [PubMed] [Google Scholar]
- 101.Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103:159–165. doi: 10.1172/JCI5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Benjamin LE, Keshet E. Conditional switching of vascular endothelial growth factor (VEGF) expression in tumors: induction of endothelial cell shedding and regression of hemangioblastoma-like vessels by VEGF withdrawal. Proc Natl Acad Sci U S A. 1997;94:8761–8766. doi: 10.1073/pnas.94.16.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J Biol Chem. 1998;273:13313–13316. doi: 10.1074/jbc.273.21.13313. [DOI] [PubMed] [Google Scholar]
- 104.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 105.Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK. Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc Natl Acad Sci U S A. 1996;93:14765–14770. doi: 10.1073/pnas.93.25.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hao Q, Wang L, Zhao ZJ, Tang H. Identification of protein kinase D2 as a pivotal regulator of endothelial cell proliferation, migration, and angiogenesis. J Biol Chem. 2009;284:799–806. doi: 10.1074/jbc.M807546200. [DOI] [PubMed] [Google Scholar]
- 107.Muller M, Schroer J, Azoitei N, Eiseler T, Bergmann W, Kohntop R, Lin Q, Costa IG, Zenke M, Genze F, Weidgang C, Seufferlein T, Liebau S, Kleger A. A time frame permissive for Protein Kinase D2 activity to direct angiogenesis in mouse embryonic stem cells. Sci Rep. 2015;5:11742. doi: 10.1038/srep11742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lemonnier J, Ghayor C, Guicheux J, Caverzasio J. Protein kinase C-independent activation of protein kinase D is involved in BMP-2-induced activation of stress mitogen-activated protein kinases JNK and p38 and osteoblastic cell differentiation. J Biol Chem. 2004;279:259–264. doi: 10.1074/jbc.M308665200. [DOI] [PubMed] [Google Scholar]
- 109.Li S, Xu W, Xing Z, Qian J, Chen L, Gu R, Guo W, Lai X, Zhao W, Li S, Wang Y, Wang QJ, Deng F. A Conditional Knockout Mouse Model Reveals a Critical Role of PKD1 in Osteoblast Differentiation and Bone Development. Sci Rep. 2017;7:40505. doi: 10.1038/srep40505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nandana S, Tripathi M, Duan P, Chu CY, Mishra R, Liu C, Jin R, Yamashita H, Zayzafoon M, Bhowmick NA, Zhau HE, Matusik RJ, Chung LW. Bone metastasis of prostate cancer can be therapeutically targeted at the TBX2-WNT signaling axis. Cancer Res. 2017 doi: 10.1158/0008-5472.CAN-16-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dequiedt F, Van Lint J, Lecomte E, Van Duppen V, Seufferlein T, Vandenheede JR, Wattiez R, Kettmann R. Phosphorylation of histone deacetylase 7 by protein kinase D mediates T cell receptor-induced Nur77 expression and apoptosis. J Exp Med. 2005;201:793–804. doi: 10.1084/jem.20042034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Matthews SA, Liu P, Spitaler M, Olson EN, McKinsey TA, Cantrell DA, Scharenberg AM. Essential role for protein kinase D family kinases in the regulation of class II histone deacetylases in B lymphocytes. Mol Cell Biol. 2006;26:1569–1577. doi: 10.1128/MCB.26.4.1569-1577.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Parra M, Kasler H, McKinsey TA, Olson EN, Verdin E. Protein kinase D1 phosphorylates HDAC7 and induces its nuclear export after T-cell receptor activation. J Biol Chem. 2005;280:13762–13770. doi: 10.1074/jbc.M413396200. [DOI] [PubMed] [Google Scholar]
- 114.Medeiros RB, Dickey DM, Chung H, Quale AC, Nagarajan LR, Billadeau DD, Shimizu Y. Protein kinase D1 and the beta 1 integrin cytoplasmic domain control beta 1 integrin function via regulation of Rap1 activation. Immunity. 2005;23:213–226. doi: 10.1016/j.immuni.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 115.Irie A, Harada K, Tsukamoto H, Kim JR, Araki N, Nishimura Y. Protein kinase D2 contributes to either IL-2 promoter regulation or induction of cell death upon TCR stimulation depending on its activity in Jurkat cells. Int Immunol. 2006;18:1737–1747. doi: 10.1093/intimm/dxl108. [DOI] [PubMed] [Google Scholar]
- 116.Park JE, Kim YI, Yi AK. Protein kinase D1: a new component in TLR9 signaling. J Immunol. 2008;181:2044–2055. doi: 10.4049/jimmunol.181.3.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Murphy TR, Legere HJ, 3rd, Katz HR. Activation of protein kinase D1 in mast cells in response to innate, adaptive, and growth factor signals. J Immunol. 2007;179:7876–7882. doi: 10.4049/jimmunol.179.11.7876. [DOI] [PubMed] [Google Scholar]
- 118.Yoo N, Lee HR, Son JM, Kang HB, Lee HG, Yoon SR, Yoon SY, Kim JW. Genkwadaphnin promotes leukocyte migration by increasing CD44 expression via PKD1/NF-kappaB signaling pathway. Immunol Lett. 2016;173:69–76. doi: 10.1016/j.imlet.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 119.Xu X, Gera N, Li H, Yun M, Zhang L, Wang Y, Wang QJ, Jin T. GPCR-mediated PLCbetagamma/PKCbeta/PKD signaling pathway regulates the cofilin phosphatase slingshot 2 in neutrophil chemotaxis. Mol Biol Cell. 2015;26:874–886. doi: 10.1091/mbc.E14-05-0982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 121.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 122.Shirai T. Significance of chemoprevention for prostate cancer development: experimental in vivo approaches to chemoprevention. Pathol Int. 2008;58:1–16. doi: 10.1111/j.1440-1827.2007.02182.x. [DOI] [PubMed] [Google Scholar]
- 123.Chen J, Deng F, Singh SV, Wang QJ. Protein kinase D3 (PKD3) contributes to prostate cancer cell growth and survival through a PKCepsilon/PKD3 pathway downstream of Akt and ERK 1/2. Cancer Res. 2008;68:3844–3853. doi: 10.1158/0008-5472.CAN-07-5156. [DOI] [PubMed] [Google Scholar]
- 124.Mak P, Jaggi M, Syed V, Chauhan SC, Hassan S, Biswas H, Balaji KC. Protein kinase D1 (PKD1) influences androgen receptor (AR) function in prostate cancer cells. Biochem Biophys Res Commun. 2008;373:618–623. doi: 10.1016/j.bbrc.2008.06.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jaggi M, Johansson SL, Baker JJ, Smith LM, Galich A, Balaji KC. Aberrant expression of E-cadherin and beta-catenin in human prostate cancer. Urol Oncol. 2005;23:402–406. doi: 10.1016/j.urolonc.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 126.Hassan S, Biswas MH, Zhang C, Du C, Balaji KC. Heat shock protein 27 mediates repression of androgen receptor function by protein kinase D1 in prostate cancer cells. Oncogene. 2009;28:4386–4396. doi: 10.1038/onc.2009.291. [DOI] [PubMed] [Google Scholar]
- 127.Jaggi M, Rao PS, Smith DJ, Wheelock MJ, Johnson KR, Hemstreet GP, Balaji KC. E-cadherin phosphorylation by protein kinase D1/protein kinase C{mu} is associated with altered cellular aggregation and motility in prostate cancer. Cancer Res. 2005;65:483–492. [PubMed] [Google Scholar]
- 128.Sharlow ER, Giridhar KV, LaValle CR, Chen J, Leimgruber S, Barrett R, Bravo-Altamirano K, Wipf P, Lazo JS, Wang QJ. Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J Biol Chem. 2008;283:33516–33526. doi: 10.1074/jbc.M805358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nelson EC, Evans CP, Mack PC, Devere-White RW, Lara PN., Jr Inhibition of Akt pathways in the treatment of prostate cancer. Prostate Cancer Prostatic Dis. 2007;10:331–339. doi: 10.1038/sj.pcan.4500974. [DOI] [PubMed] [Google Scholar]
- 130.Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res. 2009;15:4799–4805. doi: 10.1158/1078-0432.CCR-08-0125. [DOI] [PubMed] [Google Scholar]
- 131.Chen J, Giridhar KV, Zhang L, Xu S, Wang QJ. A protein kinase C/protein kinase D pathway protects LNCaP prostate cancer cells from phorbol ester-induced apoptosis by promoting ERK1/2 and NF-{kappa}B activities. Carcinogenesis. 2011;32:1198–1206. doi: 10.1093/carcin/bgr113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang L, Zhao Z, Xu S, Tandon M, LaValle CR, Deng F, Wang QJ. Androgen suppresses protein kinase D1 expression through fibroblast growth factor receptor substrate 2 in prostate cancer cells. Oncotarget. 2017;8:12800–12811. doi: 10.18632/oncotarget.14536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Guo J, Clausen DM, Beumer JH, Parise RA, Egorin MJ, Bravo-Altamirano K, Wipf P, Sharlow ER, Wang QJ, Eiseman JL. In vitro cytotoxicity, pharmacokinetics, tissue distribution, and metabolism of small-molecule protein kinase D inhibitors, kb-NB142–70 and kb-NB165–09, in mice bearing human cancer xenografts. Cancer Chemother Pharmacol. 2013;71:331–344. doi: 10.1007/s00280-012-2010-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tandon M, Johnson J, Li Z, Xu S, Wipf P, Wang QJ. New pyrazolopyrimidine inhibitors of protein kinase d as potent anticancer agents for prostate cancer cells. PLoS One. 2013;8:e75601. doi: 10.1371/journal.pone.0075601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tandon M, Salamoun JM, Carder EJ, Farber E, Xu S, Deng F, Tang H, Wipf P, Wang QJ. SD-208, a novel protein kinase D inhibitor, blocks prostate cancer cell proliferation and tumor growth in vivo by inducing G2/M cell cycle arrest. PLoS One. 2015;10:e0119346. doi: 10.1371/journal.pone.0119346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tandon M, Wang L, Xu Q, Xie X, Wipf P, Wang QJ. A targeted library screen reveals a new inhibitor scaffold for protein kinase D. PLoS One. 2012;7:e44653. doi: 10.1371/journal.pone.0044653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bowden ET, Barth M, Thomas D, Glazer RI, Mueller SC. An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene. 1999;18:4440–4449. doi: 10.1038/sj.onc.1202827. [DOI] [PubMed] [Google Scholar]
- 138.Borges S, Doppler H, Perez EA, Andorfer CA, Sun Z, Anastasiadis PZ, Thompson E, Geiger XJ, Storz P. Pharmacologic reversion of epigenetic silencing of the PRKD1 promoter blocks breast tumor cell invasion and metastasis. Breast Cancer Res. 2013;15:R66. doi: 10.1186/bcr3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Du C, Zhang C, Hassan S, Biswas MH, Balaji KC. Protein kinase D1 suppresses epithelial-to-mesenchymal transition through phosphorylation of snail. Cancer Res. 2010;70:7810–7819. doi: 10.1158/0008-5472.CAN-09-4481. [DOI] [PubMed] [Google Scholar]
- 140.Bastea LI, Doppler H, Balogun B, Storz P. Protein kinase D1 maintains the epithelial phenotype by inducing a DNA-bound, inactive SNAI1 transcriptional repressor complex. PLoS One. 2012;7:e30459. doi: 10.1371/journal.pone.0030459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Eiseler T, Schmid MA, Topbas F, Pfizenmaier K, Hausser A. PKD is recruited to sites of actin remodelling at the leading edge and negatively regulates cell migration. FEBS Lett. 2007;581:4279–4287. doi: 10.1016/j.febslet.2007.07.079. [DOI] [PubMed] [Google Scholar]
- 142.Palmantier R, George MD, Akiyama SK, Wolber FM, Olden K, Roberts JD. Cis-polyunsaturated fatty acids stimulate beta1 integrin-mediated adhesion of human breast carcinoma cells to type IV collagen by activating protein kinases C-epsilon and -mu. Cancer Res. 2001;61:2445–2452. [PubMed] [Google Scholar]
- 143.Kennett SB, Roberts JD, Olden K. Requirement of protein kinase C micro activation and calpain-mediated proteolysis for arachidonic acid-stimulated adhesion of MDA-MB-435 human mammary carcinoma cells to collagen type IV. J Biol Chem. 2004;279:3300–3307. doi: 10.1074/jbc.M305734200. [DOI] [PubMed] [Google Scholar]
- 144.Hao Q, McKenzie R, Gan H, Tang H. Protein kinases D2 and D3 are novel growth regulators in HCC1806 triple-negative breast cancer cells. Anticancer Res. 2013;33:393–399. [PubMed] [Google Scholar]
- 145.Chen J, Lu L, Feng Y, Wang H, Dai L, Li Y, Zhang P. PKD2 mediates multi-drug resistance in breast cancer cells through modulation of P-glycoprotein expression. Cancer Lett. 2011;300:48–56. doi: 10.1016/j.canlet.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 146.Borges S, Storz P. Protein kinase D isoforms: new targets for therapy in invasive breast cancers? Expert Rev Anticancer Ther. 2013;13:895–898. doi: 10.1586/14737140.2013.816460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Huck B, Duss S, Hausser A, Olayioye MA. Elevated protein kinase D3 (PKD3) expression supports proliferation of triple-negative breast cancer cells and contributes to mTORC1-S6K1 pathway activation. J Biol Chem. 2014;289:3138–3147. doi: 10.1074/jbc.M113.502633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Borges S, Perez EA, Thompson EA, Radisky DC, Geiger XJ, Storz P. Effective Targeting of Estrogen Receptor-Negative Breast Cancers with the Protein Kinase D Inhibitor CRT0066101. Mol Cancer Ther. 2015;14:1306–1316. doi: 10.1158/1535-7163.MCT-14-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Philip PA. Targeted therapies for pancreatic cancer. Gastrointest Cancer Res. 2008;2:S16–19. [PMC free article] [PubMed] [Google Scholar]
- 150.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 151.Korc M. Role of growth factors in pancreatic cancer. Surg Oncol Clin N Am. 1998;7:25–41. [PubMed] [Google Scholar]
- 152.Hruban RH, Wilentz RE, Goggins M, Offerhaus GJ, Yeo CJ, Kern SE. Pathology of incipient pancreatic cancer. Ann Oncol. 1999;10(Suppl 4):9–11. [PubMed] [Google Scholar]
- 153.Trauzold A, Schmiedel S, Sipos B, Wermann H, Westphal S, Roder C, Klapper W, Arlt A, Lehnert L, Ungefroren H, Johannes FJ, Kalthoff H. PKCmu prevents CD95-mediated apoptosis and enhances proliferation in pancreatic tumour cells. Oncogene. 2003;22:8939–8947. doi: 10.1038/sj.onc.1207001. [DOI] [PubMed] [Google Scholar]
- 154.Liou GY, Doppler H, Braun UB, Panayiotou R, Scotti Buzhardt M, Radisky DC, Crawford HC, Fields AP, Murray NR, Wang QJ, Leitges M, Storz P. Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat Commun. 2015;6:6200. doi: 10.1038/ncomms7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Guha S, Lunn JA, Santiskulvong C, Rozengurt E. Neurotensin stimulates protein kinase C-dependent mitogenic signaling in human pancreatic carcinoma cell line PANC-1. Cancer Res. 2003;63:2379–2387. [PubMed] [Google Scholar]
- 156.Guha S, Rey O, Rozengurt E. Neurotensin induces protein kinase C-dependent protein kinase D activation and DNA synthesis in human pancreatic carcinoma cell line PANC-1. Cancer Res. 2002;62:1632–1640. [PubMed] [Google Scholar]
- 157.Yuan J, Rozengurt E. PKD, PKD2, and p38 MAPK mediate Hsp27 serine-82 phosphorylation induced by neurotensin in pancreatic cancer PANC-1 cells. J Cell Biochem. 2008;103:648–662. doi: 10.1002/jcb.21439. [DOI] [PubMed] [Google Scholar]
- 158.Xu L, Bergan RC. Genistein inhibits matrix metalloproteinase type 2 activation and prostate cancer cell invasion by blocking the transforming growth factor beta-mediated activation of mitogen-activated protein kinase-activated protein kinase 2–27-kDa heat shock protein pathway. Mol Pharmacol. 2006;70:869–877. doi: 10.1124/mol.106.023861. [DOI] [PubMed] [Google Scholar]
- 159.Shin KD, Lee MY, Shin DS, Lee S, Son KH, Koh S, Paik YK, Kwon BM, Han DC. Blocking tumor cell migration and invasion with biphenyl isoxazole derivative KRIBB3, a synthetic molecule that inhibits Hsp27 phosphorylation. J Biol Chem. 2005;280:41439–41448. doi: 10.1074/jbc.M507209200. [DOI] [PubMed] [Google Scholar]
- 160.McCollum AK, Teneyck CJ, Sauer BM, Toft DO, Erlichman C. Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res. 2006;66:10967–10975. doi: 10.1158/0008-5472.CAN-06-1629. [DOI] [PubMed] [Google Scholar]
- 161.Chen Y, Arrigo AP, Currie RW. Heat shock treatment suppresses angiotensin II-induced activation of NF-kappaB pathway and heart inflammation: a role for IKK depletion by heat shock? Am J Physiol Heart Circ Physiol. 2004;287:H1104–1114. doi: 10.1152/ajpheart.00102.2004. [DOI] [PubMed] [Google Scholar]
- 162.Wille C, Kohler C, Armacki M, Jamali A, Gossele U, Pfizenmaier K, Seufferlein T, Eiseler T. Protein kinase D2 induces invasion of pancreatic cancer cells by regulating matrix metalloproteinases. Mol Biol Cell. 2014;25:324–336. doi: 10.1091/mbc.E13-06-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rennecke J, Rehberger PA, Furstenberger G, Johannes FJ, Stohr M, Marks F, Richter KH. Protein-kinase-Cmu expression correlates with enhanced keratinocyte proliferation in normal and neoplastic mouse epidermis and in cell culture. Int J Cancer. 1999;80:98–103. doi: 10.1002/(sici)1097-0215(19990105)80:1<98::aid-ijc19>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 164.Ristich VL, Bowman PH, Dodd ME, Bollag WB. Protein kinase D distribution in normal human epidermis, basal cell carcinoma and psoriasis. Br J Dermatol. 2006;154:586–593. doi: 10.1111/j.1365-2133.2005.07073.x. [DOI] [PubMed] [Google Scholar]
- 165.Ivanova P, Atanasova G, Poumay Y, Mitev V. Knockdown of PKD1 in normal human epidermal keratinocytes increases mRNA expression of keratin 10 and involucrin: early markers of keratinocyte differentiation. Arch Dermatol Res. 2008;300:139–145. doi: 10.1007/s00403-008-0832-7. [DOI] [PubMed] [Google Scholar]
- 166.Jadali A, Ghazizadeh S. Protein kinase D is implicated in the reversible commitment to differentiation in primary cultures of mouse keratinocytes. J Biol Chem. 2010;285:23387–23397. doi: 10.1074/jbc.M110.105619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Arun SN, Kaddour-Djebbar I, Shapiro BA, Bollag WB. Ultraviolet B irradiation and activation of protein kinase D in primary mouse epidermal keratinocytes. Oncogene. 2011;30:1586–1596. doi: 10.1038/onc.2010.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rashel M, Alston N, Ghazizadeh S. Protein kinase D1 has a key role in wound healing and skin carcinogenesis. J Invest Dermatol. 2014;134:902–909. doi: 10.1038/jid.2013.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ryvkin V, Rashel M, Gaddapara T, Ghazizadeh S. Opposing growth regulatory roles of protein kinase D isoforms in human keratinocytes. J Biol Chem. 2015;290:11199–11208. doi: 10.1074/jbc.M115.643742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kim M, Jang HR, Kim JH, Noh SM, Song KS, Cho JS, Jeong HY, Norman JC, Caswell PT, Kang GH, Kim SY, Yoo HS, Kim YS. Epigenetic inactivation of protein kinase D1 in gastric cancer and its role in gastric cancer cell migration and invasion. Carcinogenesis. 2008;29:629–637. doi: 10.1093/carcin/bgm291. [DOI] [PubMed] [Google Scholar]
- 171.Shabelnik MY, Kovalevska LM, Yurchenko MY, Shlapatska LM, Rzepetsky Y, Sidorenko SP. Differential expression of PKD1 and PKD2 in gastric cancer and analysis of PKD1 and PKD2 function in the model system. Exp Oncol. 2011;33:206–211. [PubMed] [Google Scholar]
- 172.Paolucci L, Rozengurt E. Protein kinase D in small cell lung cancer cells: rapid activation through protein kinase C. Cancer Res. 1999;59:572–577. [PubMed] [Google Scholar]
- 173.Brenner W, Beitz S, Schneider E, Benzing F, Unger RE, Roos FC, Thuroff JW, Hampel C. Adhesion of renal carcinoma cells to endothelial cells depends on PKCmu. BMC Cancer. 2010;10:183. doi: 10.1186/1471-2407-10-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kovalevska LM, Yurchenko OV, Shlapatska LM, Berdova GG, Mikhalap SV, Van Lint J, Sidorenko SP. Immunohistochemical studies of protein kinase D (PKD) 2 expression in malignant human lymphomas. Exp Oncol. 2006;28:225–230. [PubMed] [Google Scholar]
- 175.Bernhart E, Damm S, Wintersperger A, DeVaney T, Zimmer A, Raynham T, Ireson C, Sattler W. Protein kinase D2 regulates migration and invasion of U87MG glioblastoma cells in vitro. Exp Cell Res. 2013;319:2037–2048. doi: 10.1016/j.yexcr.2013.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sundram V, Ganju A, Hughes JE, Khan S, Chauhan SC, Jaggi M. Protein kinase D1 attenuates tumorigenesis in colon cancer by modulating beta-catenin/T cell factor activity. Oncotarget. 2014;5:6867–6884. doi: 10.18632/oncotarget.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wei N, Chu E, Wipf P, Schmitz JC. Protein kinase d as a potential chemotherapeutic target for colorectal cancer. Mol Cancer Ther. 2014;13:1130–1141. doi: 10.1158/1535-7163.MCT-13-0880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bravo-Altamirano K, George KM, Frantz MC, Lavalle CR, Tandon M, Leimgruber S, Sharlow ER, Lazo JS, Wang QJ, Wipf P. Synthesis and Structure-Activity Relationships of Benzothienothiazepinone Inhibitors of Protein Kinase D. ACS Med Chem Lett. 2011;2:154–159. doi: 10.1021/ml100230n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.George KM, Frantz MC, Bravo-Altamirano K, Lavalle CR, Tandon M, Leimgruber S, Sharlow ER, Lazo JS, Wang QJ, Wipf P. Design, Synthesis, and Biological Evaluation of PKD Inhibitors. Pharmaceutics. 2011;3:186–228. doi: 10.3390/pharmaceutics3020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lavalle CR, Bravo-Altamirano K, Giridhar KV, Chen J, Sharlow E, Lazo JS, Wipf P, Wang QJ. Novel protein kinase D inhibitors cause potent arrest in prostate cancer cell growth and motility. BMC Chem Biol. 2010;10:5. doi: 10.1186/1472-6769-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sharlow ER, Mustata Wilson G, Close D, Leimgruber S, Tandon M, Reed RB, Shun TY, Wang QJ, Wipf P, Lazo JS. Discovery of diverse small molecule chemotypes with cell-based PKD1 inhibitory activity. PLoS One. 2011;6:e25134. doi: 10.1371/journal.pone.0025134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yuan J, Liu Y, Tan T, Guha S, Gukovsky I, Gukovskaya A, Pandol SJ. Protein kinase d regulates cell death pathways in experimental pancreatitis. Front Physiol. 2012;3:60. doi: 10.3389/fphys.2012.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Monovich L, Vega RB, Meredith E, Miranda K, Rao C, Capparelli M, Lemon DD, Phan D, Koch KA, Chapo JA, Hood DB, McKinsey TA. A novel kinase inhibitor establishes a predominant role for protein kinase D as a cardiac class IIa histone deacetylase kinase. FEBS Lett. 2010;584:631–637. doi: 10.1016/j.febslet.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 184.Meredith EL, Ardayfio O, Beattie K, Dobler MR, Enyedy I, Gaul C, Hosagrahara V, Jewell C, Koch K, Lee W, Lehmann H, McKinsey TA, Miranda K, Pagratis N, Pancost M, Patnaik A, Phan D, Plato C, Qian M, Rajaraman V, Rao C, Rozhitskaya O, Ruppen T, Shi J, Siska SJ, Springer C, van Eis M, Vega RB, von Matt A, Yang L, Yoon T, Zhang JH, Zhu N, Monovich LG. Identification of orally available naphthyridine protein kinase D inhibitors. J Med Chem. 2010;53:5400–5421. doi: 10.1021/jm100075z. [DOI] [PubMed] [Google Scholar]
- 185.Gamber GG, Meredith E, Zhu Q, Yan W, Rao C, Capparelli M, Burgis R, Enyedy I, Zhang JH, Soldermann N, Beattie K, Rozhitskaya O, Koch KA, Pagratis N, Hosagrahara V, Vega RB, McKinsey TA, Monovich L. 3,5-diarylazoles as novel and selective inhibitors of protein kinase D. Bioorg Med Chem Lett. 2011;21:1447–1451. doi: 10.1016/j.bmcl.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 186.Harikumar KB, Kunnumakkara AB, Ochi N, Tong Z, Deorukhkar A, Sung B, Kelland L, Jamieson S, Sutherland R, Raynham T, Charles M, Bagherazadeh A, Foxton C, Boakes A, Farooq M, Maru D, Diagaradjane P, Matsuo Y, Sinnett-Smith J, Gelovani J, Krishnan S, Aggarwal BB, Rozengurt E, Ireson CR, Guha S. A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther. 2010;9:1136–1146. doi: 10.1158/1535-7163.MCT-09-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Evans IM, Bagherzadeh A, Charles M, Raynham T, Ireson C, Boakes A, Kelland L, Zachary IC. Characterization of the biological effects of a novel protein kinase D inhibitor in endothelial cells. Biochem J. 2010;429:565–572. doi: 10.1042/BJ20100578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Golkowski M, Vidadala RS, Lombard CK, Suh HW, Maly DJ, Ong SE. Kinobead and Single-Shot LC-MS Profiling Identifies Selective PKD Inhibitors. J Proteome Res. 2017;16:1216–1227. doi: 10.1021/acs.jproteome.6b00817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Harikumar KB, Kunnumakkara AB, Ochi N, Tong Z, Deorukhkar A, Sung B, Kelland L, Jamieson S, Sutherland R, Raynham T, Charles M, Bagherzadeh A, Foxton C, Boakes A, Farooq M, Maru D, Diagaradjane P, Matsuo Y, Sinnett-Smith J, Gelovani J, Krishnan S, Aggarwal BB, Rozengurt E, Ireson CR, Guha S. A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Molecular cancer therapeutics. 2010;9:1136–1146. doi: 10.1158/1535-7163.MCT-09-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chen CD, Sawyers CL. NF-kappa B activates prostate-specific antigen expression and is upregulated in androgen-independent prostate cancer. Mol Cell Biol. 2002;22:2862–2870. doi: 10.1128/MCB.22.8.2862-2870.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]