

Abstract
Background
Atherosclerotic plaque formation results from chronic inflammation and fibroproliferative remodeling in the vascular wall. We previously demonstrated that both human and mouse atherosclerotic plaques show elevated expression of EphA2, a guidance molecule involved in cell-cell interactions and tumorigenesis.
Methods
Here, we assessed EphA2's role in atherosclerosis by deleting EphA2 in a mouse model of atherosclerosis (Apoe-/-) and by assessing EphA2 function in multiple vascular cell culture models. Following 8-16 weeks Western diet, male and female mice were assessed for atherosclerotic burden in the large vessels, and plasma lipid levels were analyzed.
Results
Despite enhanced weight gain and plasma lipid levels compared to Apoe-/- controls, EphA2-/-Apoe-/- knockout mice show diminished atherosclerotic plaque formation, characterized by reduced proinflammatory gene expression and plaque macrophage content. While plaque macrophages express EphA2, EphA2 deletion does not affect macrophage phenotype, inflammatory responses, and lipid uptake, and bone marrow chimeras suggest hematopoietic EphA2 deletion does not affect plaque formation. In contrast, endothelial EphA2 knockdown significantly reduces monocyte firm adhesion under flow. In addition, EphA2-/-Apoe-/- mice show reduced progression to advanced atherosclerotic plaques with diminished smooth muscle and collagen content. Consistent with this phenotype, EphA2 shows enhanced expression following smooth muscle transition to a synthetic phenotype, and EphA2 depletion reduces smooth muscle proliferation, mitogenic signaling, and extracellular matrix deposition both in atherosclerotic plaques and in vascular smooth muscle cells in culture.
Conclusions
Together these data identify a novel role for EphA2 in atherosclerosis, regulating both plaque inflammation and progression to advanced atherosclerotic lesions. Cell culture studies suggest that endothelial EphA2 contributes to atherosclerotic inflammation by promoting monocyte firm adhesion, whereas smooth muscle EphA2 expression may regulate the progression to advanced atherosclerosis by regulating smooth muscle proliferation and extracellular matrix deposition.
Keywords: atherosclerosis, extracellular matrix, inflammation, smooth muscle cell, proliferation, fibrosis
Introduction
Atherosclerosis, a chronic inflammatory disease of the large arteries, involves the focal accumulation of low density lipoproteins (LDL) in the vessel intima, stimulating endothelial cell activation and macrophage recruitment. As lipid accumulates, macrophage clearance of LDL becomes dysfunctional resulting in foam cell formation visible histologically as fatty streaks. Smooth muscle fibroproliferative remodeling critically regulates the progression of atherosclerosis from early fatty streaks to advanced atheromas1. As fatty streaks form, underlying medial smooth muscle cells undergo a phenotypic transition from their contractile, quiescent phenotype to a synthetic phenotype characterized by reduced contractile gene expression, enhanced cell proliferation and migration, and heightened deposition of extracellular matrix proteins2. While this fibroproliferative remodeling can be deleterious by enhancing plaque size to promote stenotic lesions, formation of a protective fibrous cap over the plaque's necrotic core provides stability to the plaque by preventing rupture and thrombosis1.
The Eph family of receptor tyrosine kinases, the largest family in the mammalian genome, interact with ephrin ligands on adjacent cells to mediate cell adhesion/repulsion signaling during axonal guidance, tissue patterning, leukocyte homing, and metastasis3. The 14 mammalian Eph receptors are divided between nine EphA receptors (EphA1-8, A10) and five EphB (EphB1-4, B6) receptors based on sequence homology and ligand binding preferences3. With some exceptions, EphA receptors interact predominantly with GPI-linked ephrinA ligands (ephrinA1-5), whereas EphB receptors interact predominantly with transmembrane ephrinB ligands (ephrinB1-3).
Accumulating evidence suggests that Eph/ephrin interactions regulate various aspects of inflammation4. Multiple Ephs and ephrins show enhanced expression during endothelial cell activation, including ephrinB2, ephrinA1, and EphA25, 6. Activation of EphA2 promotes endothelial ICAM-1 and VCAM-1 expression in response to several stimuli6, 7, whereas activation of EphA4 enhances endothelial stiffness to promote monocyte binding8. In addition to endothelial activation, Eph/ephrin interactions also affect leukocyte adhesion to the activated endothelium. Monocyte EphB2 receptors bind ephrinB2 on activated endothelial cells, resulting in enhanced monocyte adhesion5. Similarly, ligation of ephrinA ligands by endothelial EphA receptors promotes lymphocyte integrin activation and firm adhesion4. In addition to leukocyte homing, activation of macrophage EphB receptors enhances interleukin-8 and monocyte chemotactic protein-1 expression, suggesting that Eph/ephrin signaling affects leukocyte function5.
We previously demonstrated enhanced endothelial and macrophage EphA2 expression in both human and murine atherosclerotic plaques6, and Jiang et al. showed that injecting EphA2 shRNA into atherosclerosis-prone Apoe-/- mice reduced plaque formation9. Intriguingly, the EphA2 gene is located at the 1p36 locus in humans10, which is linked to premature myocardial infarction11, and the athsq1 locus in mice, which is linked to susceptibility to atherosclerosis12. However, the contributions of EphA2 to vascular disease remains poorly characterized. The data herein define novel, multifactorial effects of EphA2 deletion on atherosclerotic plaque formation.
Materials and Methods
An expanded material and methods section can be found in the Supplemental Materials.
Animal models and tissue harvest
Animal protocols were approved by the LSU Health Sciences Center Animal Care and Use Committee, and all animals were cared for according to the National Institute of Health Guidelines for the Care and Use of Laboratory Animals. Apoe-/- mice were crossed with EphA2+/+ or EphA2-/- mice and fed Western diet for 8, 12, 16, or 24 weeks. Weight and blood glucose were monitored weekly. Following diet regimens, mice were euthanized, perfused with PBS, and tissues were fixed in 3.7% neutral-buffered formaldehyde prior to paraffin embedding or staining with Oil Red O. Some vessels were utilized for mRNA extraction prior to fixation. For detailed information, see Supplemental Methods.
Plaque analysis and immunohistochemistry
The aortic root, aorta, innominate artery, and carotid arteries were harvested for analysis of atherosclerotic plaque burden. All paraffin-embedded tissues were sectioned at 5 μm thickness. Stains were visualized on a Nikon Eclipse Ti inverted epifluorescence microscope equipped with a photometrics CoolSNAP120 ES2 camera and NIS Elements 3.00, SP5 imaging software. Analysis was performed using NIS Elements software. Scale bars provided in figure legends. For detailed information, see Supplemental Methods.
Cell culture and treatments
Human peripheral blood monocytes were isolated using Percoll and Ficoll density gradients from blood drawn from human donors following a LSU Health Sciences Center Institutional Review Board approved protocol. Peritoneal macrophages were isolated following thioglycollate injection. HAEC (Lonza) and RAW264.7 macrophages (ATCC) were cultured as previously described6, 13. LDL (intracel) was oxidized as described previously14. For detailed information, see Supplemental Methods.
Lipid uptake
Peritoneal macrophages isolated from mice fed a high fat Western diet for 8 weeks were stained with Nile Red to assess lipid content. Uptake of Dil-oxLDL was also assessed using peritoneal macrophages and quantified using fluorescence microscopy. For detailed information, see Supplemental Methods.
Quantitative real-time PCR
qRT-PCR was performed as described previously6. mRNA was extracted with TRIzol reagent and cDNA was synthesized using the iScript cDNA synthesis kit (BioRad). qRT-PCR primer sequences can be found in Supplemental Tables 1 and 2. For detailed information, see Supplemental Methods.
Immunoblotting and immunocytochemistry
Cell lysis, Western blotting and TNFα ELISA assays (eBioscience) were performed as described previously13, 15. For detailed information, see Supplemental Methods.
Matrix Deposition Analysis
Deoxycholate extraction was performed as described previously16. For detailed information, see Supplemental Methods.
Statistical analyses
All statistical analyses were performed with GraphPad Prism Software. The number of experiments performed and statistical test employed are provided in figure legends. Values are expressed as mean ± SEM. For detailed information, see Supplemental Methods.
Results
EphA2 deletion reduces plaque size in multiple vascular beds
To determine whether EphA2 contributes to atherosclerosis, we crossed EphA2-/- mice with Apoe-/- mice prone to hypercholesterolemia and atherosclerosis. Apoe-/- (EphA2 WT) and EphA2-/-Apoe-/- (EphA2 KO) mice were fed a high fat, Western diet for up to 12 weeks. Mouse weight and blood glucose were monitored weekly, and plasma lipid profile and atherosclerotic plaque formation were assessed at the 8- and 12-week time points. For the initial analysis, both male and female mice were used to assess gender-specific differences in plaque formation following EphA2 deletion. However, both male and female EphA2 KO mice showed reduced atherosclerotic plaque formation at the 8- and 12-week time points as assessed by Oil Red O staining of the mouse aorta (Figure 1A-D), so subsequent analysis focused on plaque characterization in male mice. While effects on plaque formation may differ by vascular site17, plaque area in cross-sections of the innominate artery and carotid sinus were similarly reduced in male EphA2 KO mice, suggesting that EphA2 deletion reduces atherosclerosis across multiple vascular beds (Figure 1E/F). While EphA2 KO mice showed predilection for smaller plaques (Supplemental Figure 1A), the stage of plaque progression (classic Stary scoring system18, 19) remained similar between EphA2 KO and EphA2 WT mice after 8 weeks Western diet (Supplemental Figure 1B). Blood glucose levels remained similar between groups (Supplemental Figure 2A), but EphA2 KO mice demonstrated enhanced weight gain (Supplemental Figure 2B) compared to EphA2 WT controls. Furthermore, despite reductions in atherosclerotic plaque formation, analysis of plasma lipids levels showed elevated total cholesterol, HDL cholesterol, and plasma triglycerides in male EphA2 KO mice compared to EphA2 WT mice (Supplemental Figure 2C), whereas female mice demonstrated a similar lipid profile between the two groups. Similarly exacerbated plasma lipid profiles were observed in male EphA2 KO mice, but not female EphA2 KO mice, fed a standard chow diet (Supplemental Figure 2D). The similar reduction in plaque size between male and female EphA2 KO mice suggests that the slight elevation in plasma HDL observed in male EphA2 KO mice does not likely contribute to the atheroprotective effect.
Figure 1. EphA2 deletion reduces plaque size in multiple vascular beds.
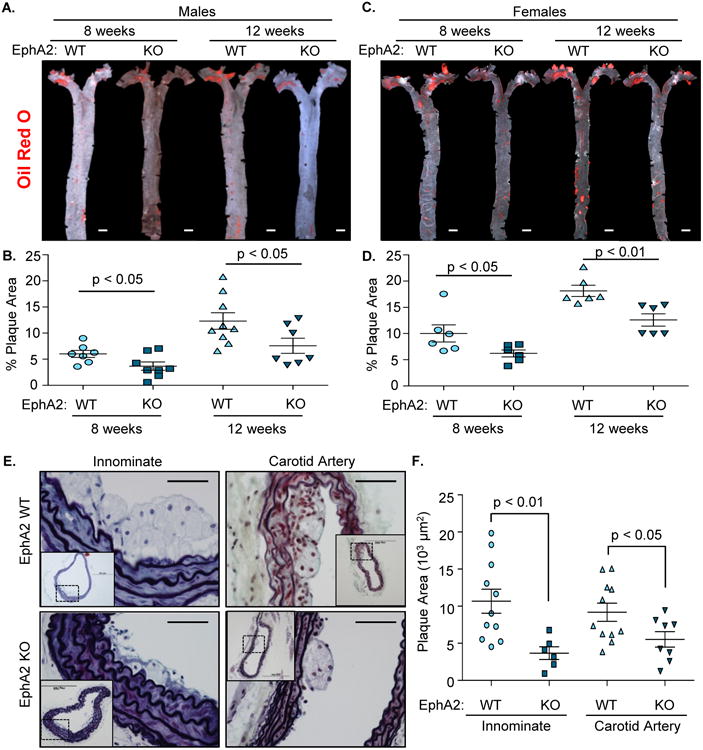
A-D) Plaque formation in mice fed Western diet for 8 or 12 weeks was determined following Oil Red O staining. Representative micrographs are shown (A/C), and plaque area was calculated as a percent of the aorta staining positive for Oil Red O (B/D). n = 7-9 male, 6 female. Scale bar = 1 mm E, F) Male Apoe-/- controls (EphA2 WT) and EphA2-/-Apoe-/- (EphA2 KO) mice were fed Western diet for 8 weeks, and the innominate and carotid arteries were stained with Movat pentachrome stain. Plaque area was quantified as the neointimal area within the internal elastic lamina. n = 6-11 innominate artery, 8-11 carotid sinus. Scale bar = 100 μm. Data are expressed as mean ± SEM. Student's T-tests were used for statistical comparisons.
EphA2 deletion reduced macrophage accumulation and inflammatory gene expression in vivo
Since EphA2 has been implicated in inflammation in a variety of systems6, we compared leukocyte accumulation and proinflammatory gene expression during early atherogenesis between EphA2 WT and EphA2 KO mice. Following 8 weeks Western diet, macrophage area (Mac2-positive) was reduced in atherosclerotic lesions from EphA2 KO mice in both the innominate and carotid arteries compared to EphA2 WT mice (Figure 2A/B). Similar reductions in plaque area and macrophage area were observed in the aortic root of EphA2 KO mice, consistent with the reduction in plaque formation observed by Jiang et al. following EphA2 siRNA injection (Supplemental Figure 1C-G) 9. mRNA isolated from the atherosclerosis-prone aortic arch of EphA2 WT mice showed elevated CD68 expression (macrophage marker) and CD3 expression (T cells) compared to the atherosclerosis-resistant thoracic aorta (Figure 2C); however, this induction was significantly reduced in EphA2 KO mice indicative of reduced leukocyte content. In addition, markers of endothelial activation (ICAM-1, VCAM-1) showed a similar reduction in EphA2 KO mice compared to EphA2 WT mice (Figure 2D). Together, these data suggest EphA2 deletion reduces endothelial activation and leukocyte accumulation during early atherosclerosis.
Figure 2. Reduced inflammation in early atherosclerotic plaque formation.
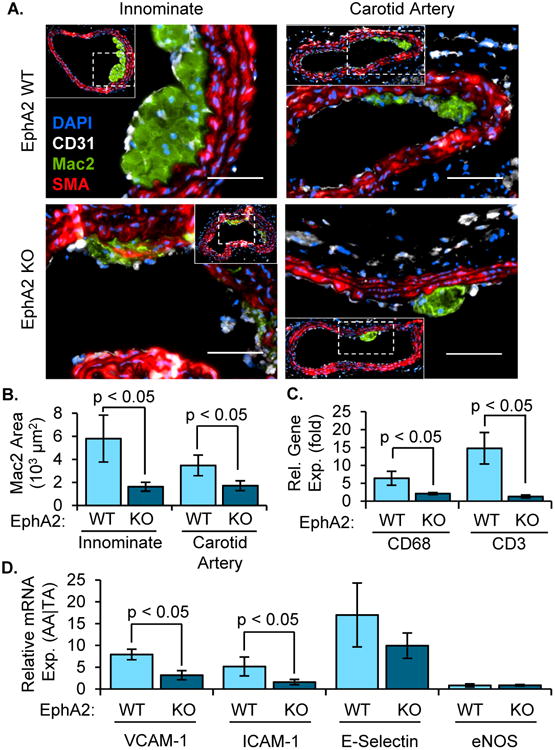
Male EphA2 WT) and EphA2 KO mice were fed Western diet for 8 weeks. A,B) Macrophage area (Mac2 positive, green) was determined by immunohistochemistry in both the innominate (n = 6-10) and carotid arteries (n = 14-21). Smooth muscle (SMA-positive, red) and endothelial (CD31-positive, white) cell staining are also shown. Quantification of average macrophage area (B) is provided. Scale bar = 100 μm. C/D) Relative gene expression between atherosclerosis-prone (aortic arch (AA)) and protected (thoracic aorta (TA)) regions was determined by mRNA isolation and qRT-PCR and compared between EphA2 WT and EphA2 KO mice. Expression of (C) macrophage (CD68) and T cell (CD3) marker genes or (D) markers of endothelial activation (VCAM-1, ICAM-1, E-selectin) were normalized to the housekeeping genes PPIA and Rpl13a. n = 5-8. Data are expressed as mean ± SEM. Student's T-tests were used for statistical comparisons.
Deletion of EphA2 has no effect on macrophage gene expression or lipid uptake in vitro
Since plaque macrophages express EphA26, we next sought to determine if EphA2 contributes to macrophage function and gene expression. We assessed the Eph-ephrin gene expression profile and observed similar expression profiles in human peripheral blood monocytes, murine peritoneal macrophages, and the RAW264.7 human macrophage cell line, with prominent monocyte/macrophage expression of EphA2, EphA4, and ephrinA1 (Supplemental Figure 3A). However, bone marrow-derived macrophages and the THP-1 monocyte-like cell line showed a disparate Eph/ephrin expression profile limiting their usefulness for assessing EphA2 function in macrophages (Supplemental Figure 3A). Thioglycollate-elicited peritoneal macrophages isolated from EphA2 KO mice showed ablated EphA2 expression without compensatory upregulation of EphA4 (Figure 3A). In contrast, ephrinA1 expression was enhanced in EphA2 KO mice consistent with previous studies suggesting antagonistic expression patterns between EphA2 and ephrinA120.
Figure 3. EphA2 does not contribute to macrophage inflammatory response or lipid uptake.
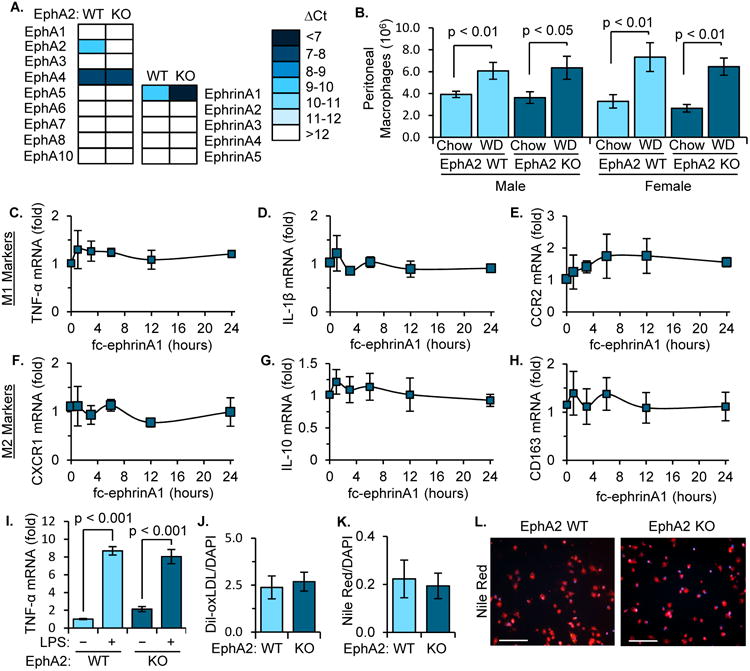
A/B) EphA2 WT and EphA2 KO mice were fed either standard chow (Chow) or Western diet (WD) for 8 weeks and peritoneal macrophages were isolated following thioglycollate injection. A) mRNA isolated from WT and KO peritoneal macrophages was assessed for EphA and ephrinA family gene expression normalized to GAPDH. n = 6. B) Quantification of peritoneal macrophages content. n = 8-18 male, n = 6-9 female. C-H) RAW264.7 macrophages were treated with Fc-ephrinA1 (1 μg/mL) for the indicated times, and changes in classic M1 markers (TNFα, IL-1β, CCR2) and M2 markers (CXCR1, IL-10, CD163) were assessed by qRT-PCR. Results are normalized to Rpl13a and expressed as a fold change compared to unstimulated conditions. n = 4. I) Peritoneal macrophages were treated with LPS (100 ng/ml, 24 hours) and TNF-α gene expression was quantified by qRT-PCR and normalized to GAPDH. n = 6. J) Uptake of dil-oxLDL (20 μg/mL, 4 hours) by WT and KO peritoneal macrophages was assessed. n = 4. K, L) WT and KO mice were fed WD for 8 weeks, and the lipid levels in peritoneal macrophages was assessed by Nile Red staining, n = 5-6. Scale bar = 100 μm. Data are expressed as mean ± SEM. Statistical comparisons were made using Two-way (B, I) or One-way ANOVA (C-H) with Bonferroni post-test or Student's T-test (J, K).
Since EphA2 deletion has shown both positive and negative effects on leukocyte homing in multiple model systems4, we analyzed macrophage infiltration following thioglycollate-induced sterile inflammation in the peritoneal cavity. Although Western diet-fed mice exhibited enhanced peritoneal macrophage recruitment in both models, there was no difference in macrophage recruitment between EphA2 KO mice and EphA2 WT mice regardless of diet (Figure 3B). Analysis of M1 and M2 marker gene expression showed no consistent trends between EphA2 KO and EphA2 WT peritoneal macrophages (Supplemental Figure 3B), and ligation of EphA2 in RAW264.7 macrophages using soluble ephrinA1 did not affect M1 or M2 marker expression (Figure 3C-H). Furthermore, polarization of RAW264.7 macrophages toward M1 or M2 phenotypes did not affect EphA2 expression (Supplemental Figure 3C-F). While peritoneal macrophages from EphA2 KO mice showed enhanced mRNA expression of the M1 marker gene TNFα under chow-fed conditions (Supplemental Figure 3B), this association was lost following Western diet feeding. Furthermore, EphA2 KO peritoneal macrophages showed no difference in LPS-induced TNFα expression (Figure 3I), and siRNA-mediated EphA2 knockdown in RAW264.7 macrophages did not affect TNFα induction by oxLDL (Supplemental Figure 4A/B). Similarly, EphA2 KO peritoneal macrophages and EphA2-depleted RAW264.7 macrophages showed no deficiency in Dil-oxLDL uptake (Figure 3J; Supplemental Figure 4C), and peritoneal macrophages from EphA2 KO mice and EphA2 WT mice showed no difference in lipid uptake, identified by Nile Red staining, following Western diet feeding (Figure 3K/L). Taken together, these data suggest that EphA2 signaling does not significantly affect macrophage phenotype, proinflammatory gene expression, or lipid uptake.
Deletion of EphA2 confers protection against atherosclerosis in resident lineages compared with hematopoietic lineage
To determine the specific contributions of EphA2 in hematopoietic or resident cell lineages in vivo, bone marrow chimeras were generated and atherosclerotic plaque formation was assessed following 12 weeks on Western diet. Alterations in hematopoietic EphA2 expression did not affect plasma lipid levels in this model, suggesting that the disparate lipid profile likely derives from changes in hepatic lipid handling and not altered reverse cholesterol transport (Supplemental Figure 5). Consistent with our in vitro data suggesting EphA2 does not significantly affect macrophage function, EphA2 KO mice still showed reduced plaque formation when reconstituted with EphA2 WT bone marrow (Figure 4A/B), whereas EphA2 WT mice reconstituted with EphA2 KO bone marrow showed only minor, insignificant reductions compared to reconstitution with EphA2 WT marrow. Cross-sectional analysis of atherosclerotic plaques in the innominate artery showed significant reductions in plaque size only when EphA2 was deleted in resident lineages and not with EphA2-deleted bone marrow (Figure 4C/D). Similarly, plaque macrophage area was significantly reduced in EphA2 KO mice even when reconstituted with EphA2 WT bone marrow (Figure 4C/E), whereas plaque macrophage content was unaltered in EphA2 WT mice whether reconstituting with EphA2 WT or EphA2 KO marrow. While these data suggest that vascular EphA2 expression plays a primary role in regulating atherosclerotic inflammation, we cannot rule out a role for EphA2 expression in macrophages since no statistically significant differences were observed between EphA2 deletion in the recipient mice and EphA2 deletion in the marrow-derived cells. Supporting a primary role for endothelial EphA2 in atherogenic inflammation, EphA2 knockdown in human aortic endothelial cells reduced TNFα-induced firm adhesion of human peripheral blood monocytes (Figure 4F/G) and THP-1 monocytes (Supplemental Figure 6A/B) under flow conditions but had no effect on monocyte rolling. Indeed, monocytes exhibit enhanced spreading and adhesion when plated onto recombinant EphA2 in vitro, but showed no affect when plated onto recombinant ephrinA1 ligand (Supplemental Figure 7A-C). EphA2/ephrinA1 interactions induce EphA2 phosphorylation, and prominent EphA2 phosphorylation (Y772) was observed in plaque endothelial cells bound to monocytes (Supplemental Figure 7D). However, EphA2 does not appear to regulate monocyte targeting to all vascular beds, as monocyte recruitment to the peritoneal cavity following thioglycollate injection was similar between EphA2 WT and EphA2 KO mice (Figure 3B). Taken together, these data suggest that endothelial EphA2 contributes to monocyte recruitment into atherosclerotic lesions.
Figure 4. EphA2 deletion from resident cells but not hematopoietic cells confers atheroprotection.
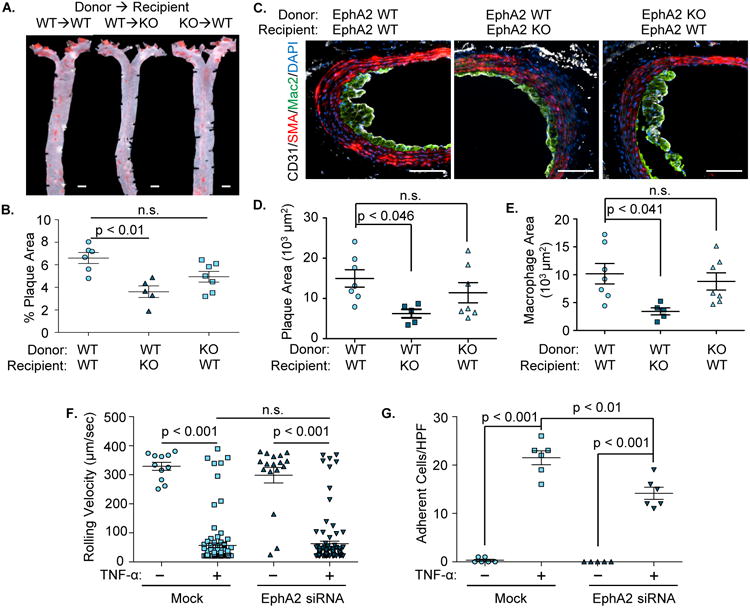
A-E). EphA2 WT and EphA2 KO mice were irradiated and received either EphA2 WT or KO bone marrow cells. Four weeks post-irradiation, mice were fed Western diet for 12 weeks. Atherosclerotic plaque formation was assessed in (A/B) the aorta by Oil Red O staining and (C/D/E) the innominate artery by immunohistochemistry for macrophages (Mac2, green), smooth muscle (SMA, red), and endothelial cells (CD31, white). Scale bar = 1 mm (A) or 100 μm (C). n = 5-7. D) Plaque area was quantified as the neointimal area within the internal elastic lamina. E) Macrophage area was quantified as the total area of positive Mac2 staining. F/G) Human aortic endothelial cells with or without EphA2 knockdown (siRNA) were treated with TNF-α (10 ng/mL, 4 hours) and labeled primary human monocytes were perfused over the endothelial monolayer under physiological flow. Rolling velocity and firmly adherent monocytes were analyzed per high powered field averaged over six fields. n = 3 in duplicate. Data are expressed as mean ± SEM, Statistical comparisons were performed using One-way (B-E) or Two-way ANOVA (F, G) with Bonferroni post-tests.
EphA2 deletion attenuates progression to advanced atherosclerotic plaques
While EphA2 deletion did not affect the stage of plaque progression at early time points following Western diet feeding (8 weeks), we next investigated whether EphA2 contributes to plaque progression to advanced atherosclerosis following 16 weeks of Western diet feeding. Consistent with earlier time points, deletion of EphA2 reduced plaque area as demonstrated by Oil Red O staining of the aorta (Figure 5A/B). Analysis of plaque stage with the Stary scoring system revealed a reduced incidence of advanced atheromas (type III and IV plaques) in EphA2 KO mice compared to EphA2 WT controls (Figure 5C). The reduction in plaque progression was associated with attenuated smooth muscle actin (SMA)-positive smooth muscle area in the atherosclerotic lesions from the innominate artery, carotid artery (Figure 5D-F), and aortic root (Supplemental Figure 8A-D). Consistent with this staining pattern, EphA2 KO mice also show reduced staining for the smooth muscle marker gene smooth muscle myosin heavy chain (SM-MHC) in the atherosclerotic plaques (Supplemental Figure 8E/F), suggesting that EphA2 expression affects smooth muscle incorporation into the atherosclerotic lesion. While plaque angiogenesis is associated with advanced atherosclerotic plaques21 and endothelial EphA2 contributes to angiogenesis22, we did not detect any differences in capillary density in either the plaque or vessel adventitia in EphA2 KO mice compared to EphA2 WT mice (Supplemental Figure 9A-D).
Figure 5. EphA2 deletion reduces advanced atherosclerotic progression.
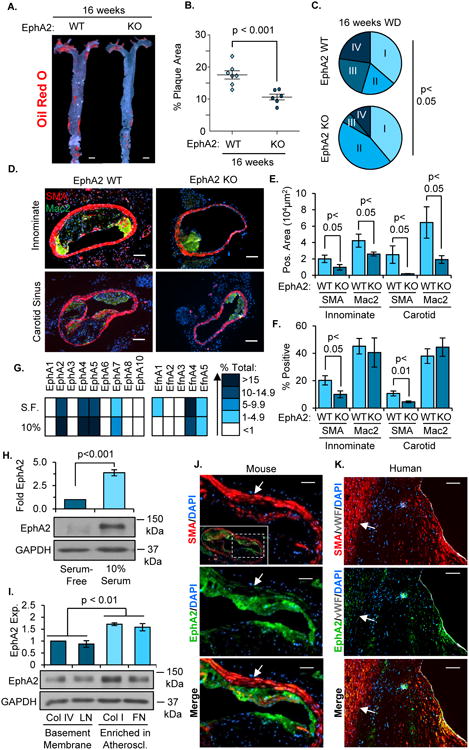
A, B) EphA2 WT and EphA2 KO mice were fed Western diet for 16 weeks, and atherosclerosis in the aortas was visualized by Oil Red O staining. n = 6-7. Scale bar = 1 mm. C) Atherosclerotic plaques from innominate arteries and carotid sinuses were graded for plaque scores based on a modified Stary scoring system. n = 22 plaques from 8 WT mice, 18 plaques from 6 KO mice. D-F) Atherosclerotic plaque composition was assessed by immunohistochemistry for macrophages (Mac2, green) and smooth muscle cells (SMA, red). Total macrophage and smooth muscle area were analyzed, and percent of the plaque area staining positive for SMA or Mac2 was calculated. Scale bar = 100 μm, n = 6-8. G/H) Human coronary artery smooth muscle cells (hCoASMCs) from 4 separate donors were either serum-starved or treated with 10% complete media for 3 days. G) mRNA was isolated and the expression of EphA and ephrinA family genes was quantified by qRT-PCR and normalized to GAPDH. Representative expression of each gene was determined and a heat map generated. H) EphA2 protein expression was determined by Western blot and normalized to GAPDH, n = 4. I) hCoASMCs were plated onto tissue culture dishes coated with collagen I (40 μg/mL), collagen IV (20 μg/mL), fibronectin (10 μg/mL), or laminin (20 μg/mL) and cultured for 3 days in serum-free conditions. EphA2 protein expression was assessed by Western blotting and normalized to GAPDH. n = 5. J/K) Smooth muscle EphA2 expression in atherosclerotic plaques was assessed by immunohistochemistry. Plaques from (J) the carotid artery of Apoe-/- mice fed Western diet for 16 weeks or (K) grade V human atherosclerotic lesions from coronary arteries were stained for EphA2 (green) and smooth muscle cells (SMA, red). White arrows indicate medial smooth muscle cells. Scale bar = 100 μm, n = 4-5. Data are expressed as mean ± SEM, Statistical comparisons were made using Student's T-test (B, E, F, H), One-way ANOVA (I) with Bonferroni post-test, or Chi Squared test (C) comparing early stage (I and II) and advanced stage (III and IV) plaques.
EphA2 shows differential expression during vascular smooth muscle phenotypic modulation
Since vascular smooth muscle phenotypic modulation critically regulates atherosclerotic plaque progression2, we next sought to define whether EphA2 expression is altered during smooth muscle phenotypic modulation in vitro and in vivo. In cell culture, vascular smooth muscle cells maintain the contractile, quiescent phenotype in serum-depleted conditions but transition to a synthetic phenotype in response to serum (Supplemental Figure 10A-D)23. Human coronary artery vascular smooth muscle cells (hCoASMCs) expressed several EphA receptors and ephrinA ligands, including EphA2, EphA4, EphA5, EphA7, ephrinA1, ephrinA4, and ephrinA5. Interestingly, serum-induced transition to the synthetic phenotype promoted the mRNA expression of EphA receptors while reducing expression of ephrinA ligands (Figure 5G) and promoted EphA2 protein expression in a dose-dependent and time-dependent manner (Figure 5H, Supplemental Figure 10F/G). We observed a similar phenotypic transition in human aortic smooth muscle cells (hAoSMCs, Supplemental Figure 11A-E) associated with enhanced EphA2 expression (Supplemental Figure 11F). In addition to serum, smooth muscle cells undergo phenotypic transition in response to a variety of extracellular matrix proteins, with basement membrane proteins (collagen IV, laminin) promoting the quiescent, contractile phenotype and plaque-associated interstitial matrix proteins (unpolymerized collagen I, fibronectin) promoting a synthetic phenotype24, 25. Like the smooth muscle response to serum, culturing either hCoASMCs or hAoSMCs on collagen I and fibronectin promoted EphA2 expression compared to cells on basement membrane proteins (Figure 5I, Supplemental Figure 11G). Consistent with in vitro models, plaque-associated smooth muscle cells (SMA-positive) showed enhanced EphA2 expression compared to medial smooth muscle cells in both murine carotid and human coronary atherosclerotic lesions (Figure 5J/K). Together, these data suggest that smooth muscle cells in the atherosclerotic plaque express EphA2.
EphA2 depletion reduces proliferation and matrix remodeling in atherosclerotic plaques and vascular smooth muscle cells
During atherosclerotic progression, migration and proliferation of smooth muscle cells into the neointima contributes to plaque size2 . Several known genetic loci for cardiovascular disease involve genes that regulate cell proliferation and carcinogenesis26, 27, and EphA2 upregulation contributes to proliferation in a variety of cancer models28, 29. Therefore, we sought to determine if EphA2 expression affects proliferation within atherosclerotic lesions. Using Ki67 as a marker for proliferation, immunohistochemistry showed a significant reduction in proliferation within the atherosclerotic lesions of EphA2 KO mice compared to EphA2 WT mice both in the innominate (Figure 6A/B) and carotid arteries (Figure 6C/D), and this reduction was apparent in both the plaque itself as well as in vessel adventitia. Consistent with reduced proliferation in the absence of EphA2, siRNA-mediated EphA2 knockdown in hCoASMCs reduced both Ki67-staining (Figure 6C/D) and BrdU incorporation (Figure 6E/F) in response to serum. Similar reductions in smooth muscle Ki67 staining were observed in hAoSMCs and using a second siRNA construct (Supplemental Figure 12A-D), and EphA2 knockdown was verified at both at mRNA and protein level for both siRNAs (Supplemental Figure 13A-D). In addition, time-lapse videomicroscopy of hCoASMC wound healing in the presence of 1% serum shows a significant reduction in scratch wound closure in EphA2 siRNA-treated cells that was apparent at 11 hours post-scratch and maintained for at least 18 hours (Figure 6G/H), suggesting that maximal serum-induced proliferation and scratch wound closure requires EphA2 expression.
Figure 6. EphA2 deficiency attenuates proliferation in atherosclerotic lesions and hCoASMCs in vitro.
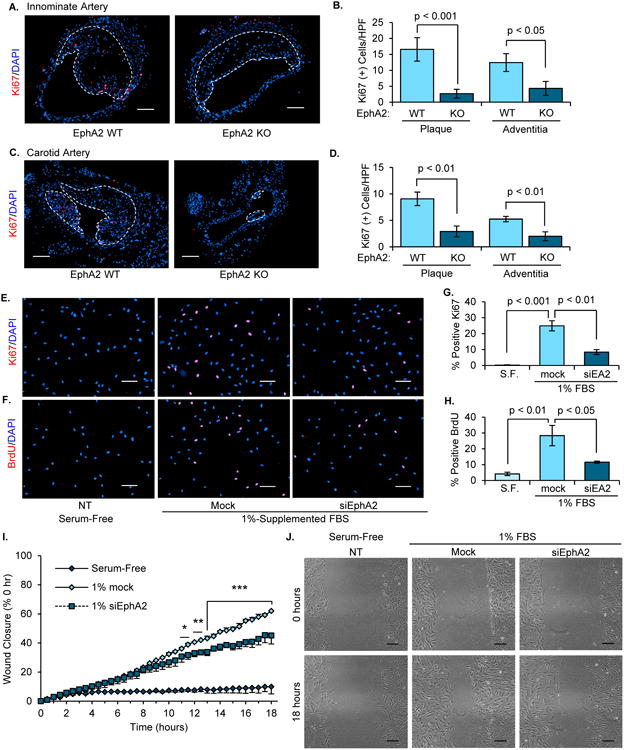
A-D) EphA2 WT and EphA2 KO mice were fed Western diet for 16 weeks, and proliferation in the (A/B) innominate and (C/D) carotid arteries were assessed by staining for Ki67 (red). The number of Ki67-positive cells in the plaque or adventitia was assessed, and plaque borders are indicated with a white dotted line. Scale bar = 100 μm. n = 6-7. E-H) hCoASMCs treated with or without EphA2 siRNA were maintained in either serum-free or 1% serum-containing conditions overnight, and proliferation was assessed by (E/G) staining for Ki67 (red) or (F/H) treatment with BrdU (10 μM) for 2 hours and staining for BrdU (red). The percentage of proliferating cells was determined for each condition. n = 4. Scale bar = 100 μm. I/J) hCoASMCs were plated onto collagen I (40 μg/mL) and a scratch was created. Wound closure was monitored by time-lapse imaging (3 regions per wound) for 18 hours. Wound closure was calculated as percent closure from t=0. J) Representative images from scratch wound assay. Scale bar = 100 μm. n = 4. Data are expressed as mean ± SEM. Statistical comparisons were made using Student's T-test (B, D, G, H) or One-way repeated measures ANOVA (I) with Bonferroni post-test. * p < 0.05, ** p < 0.01, *** p < 0.001 comparing EphA2 siRNA to Mock control.
In a variety of cancer models, enhanced EphA2 expression promotes activation of extracellular regulated kinase (ERK1/2) and Akt1, classic mitogenic signaling pathways, through ligand-independent receptor signaling30. To assess whether EphA2 expression affects mitogenic signaling in smooth muscle cells, we assessed serum-induced signaling responses following EphA2 knockdown with siRNA. hCoASMCs were serum-deprived for 24 hours following EphA2 knockdown, followed by stimulation with serum either acutely (120 minutes) or chronically (24 hours). While EphA2 knockdown did not affect the early induction of either ERK1/2 or Akt1 phosphorylation in response to serum (5-30 minutes), EphA2 depletion greatly diminished the sustained activation of these pathways at the later time points (Figure 7A-C). Similarly, lower levels of ERK1/2 and Akt1 phosphorylation were observed after 24 hours of serum stimulation in both hCoASMCs (Figure 7D/E) and hAoSMCs (Supplemental Figure 14A/B), consistent with a role for EphA2 in sustained activation of these pathways. Similarly, atherosclerotic plaques from EphA2 KO mice show reduced phospho-ERK1/2 (pERK1/2) (Figure 7F/G) and phospho-Akt1 (pAkt1) staining (Figure 7H/I) in the innominate and carotid arteries (Supplemental Figure 14C-F) both in the total plaque area and the SMA-positive plaque area compared to EphA2 WT controls. Taken together, these results suggest that EphA2 deletion reduces smooth muscle mitogenic signaling, consistent with reductions in smooth muscle and plaque-associated proliferation.
Figure 7. EphA2 depletion decreases hCoASMC mitogenic signaling in vitro and in atherosclerotic plaques.
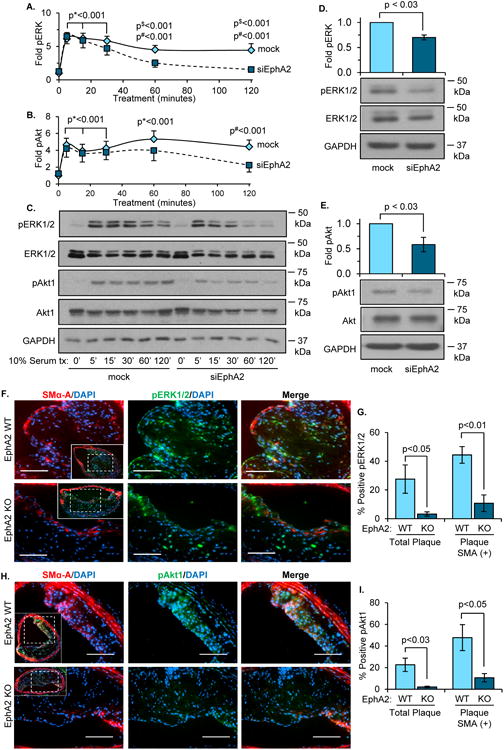
A-D) hCoASMCs were treated with or without EphA2 siRNA and maintained in 1% serum conditions overnight. A-C) For early timepoints (0-120 minutes), cells were serum-starved four hours prior to 10% serum treatments at the indicated timepoints. ERK1/2 (A) or Akt1 (B) phosphorylation was determined by Western blot and normalized to total ERK1/2 or Akt1, respectively. Representative blots shown in (C). Following 24 hours incubation in 1% serum, ERK1/2 (D) and Akt1 (E) phosphorylation was assessed by Western blot. F-I) EphA2 WT or EphA2 KO mice were fed Western diet for 16 weeks, and plaques from the innominate artery were assessed. F) Phospho-ERK1/2 area (pERK1/2-positive, green) or (H) phospho-Akt1 area (pAkt1-positive, green) was measured with smooth muscle cell area (SMA-positive, red). Percent positive area of phospho-ERK1/2 (G) or phospho-Akt1 (I) to total plaque or plaque-associated SMA was determined. Scale bars = 100 μm. n = 4-5. Data are expressed as mean ± SEM. Statistical comparisons were made using Two-way ANOVA with Bonferroni post-test (A/B) or Student's T-test (D, E, G, I). p* indicates values for both mock and siEphA2 timepoints compared to their 0 minute controls. p# indicates values only for mock timepoints compared to 0 minute controls. p$ indicates values for mock compared to siEphA2.
Extracellular matrix deposition and remodeling by intraplaque vascular smooth muscle cells significantly contributes to atherosclerotic plaque progression31. Since EphA2 KO mice show reduced progression to advanced stage plaques at 16 weeks of Western diet feeding (Figure 5C), we next tested whether EphA2 alters fibrosis, a critical feature of advanced atherosclerosis, in the atherosclerotic plaque. Compared to EphA2 WT mice, EphA2 KO mice exhibited a reduction in plaque collagen content, as evidenced by Masson's Trichrome (Figure 8A/B) and polarized Picrosirius Red staining (Figure 8C/D), and a reduction in plaque fibronectin levels (Figure 8E/F). In addition to reduced fibrous area in the plaque, the percent of plaque area staining positive for Masson's Trichrome, polarized Picrosirius Red, and fibronectin were all reduced in EphA2 KO mice (Supplemental Figure 15A-C). Similar reductions in plaque collagen staining were observed in the aortic root of EphA2 KO mice, suggesting this reduction in fibrous content extends across multiple vascular beds (Supplemental Figure 16). However, analysis of hCoASMC mRNA failed to identify significant differences in matrix gene expression following EphA2 knockdown in vitro (Supplemental Figure 17A), although a trend for reduced fibronectin expression was observed upon EphA2 depletion. To assess changes in matrix deposition, we isolated the deoxycholate (DOC)-insoluble matrix from serum-treated smooth muscle cells with or without EphA2 knockdown. Consistent with the in vivo data, EphA2 depletion significantly reduced smooth muscle deposition of DOC-insoluble fibronectin fibrils, including both total fibronectin (Figure 8G, Supplemental Figure 17B/D) and cell-derived fibronectin containing the extradomain A (EDA) alternative splice site (Figure 8H, Supplemental Figure 17C/E) in both hCoASMCs and in hAoSMCs (Supplemental Figure 18A-C). Since collagen deposition requires fibronectin polymerization in models of vascular injury32, our data suggests that EphA2 expression may modulate smooth muscle-driven fibrosis in atherosclerotic plaques by regulating fibronectin deposition.
Figure 8. Deletion of EphA2 reduces plaque fibrosis in vivo and hCoASMC matrix deposition in vitro.
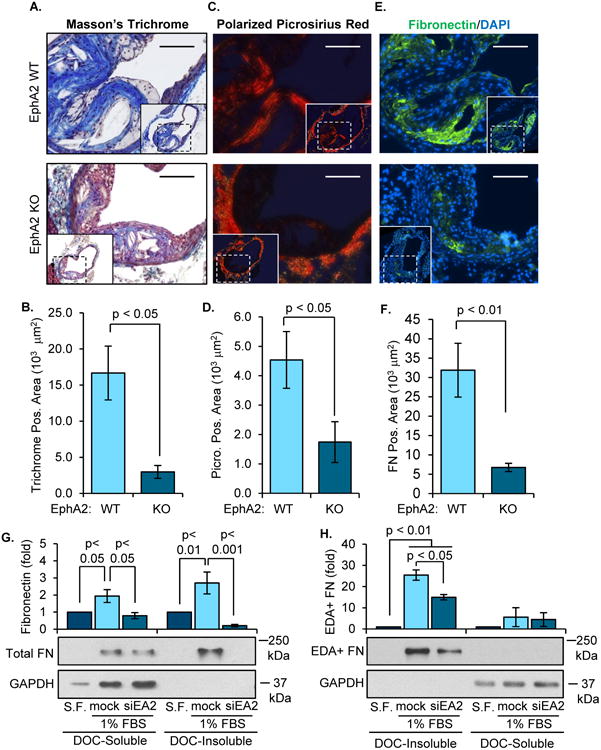
A-F) EphA2 WT or EphA2 KO mice were fed Western diet for 16 weeks, and the matrix remodeling in plaques from the innominate artery and carotid sinus was assessed by histology and immunohistochemistry. Collagen content was assessed using (A/B) Masson's Trichrome stain or (D/E) Picrosirius Red stain, and the area of positive staining in the plaque was quantified. E, F) Fibronectin content was determined by immunohistochemistry for fibronectin (green), and fibronectin-positive area was quantified. n = 6-8. Scale bar = 100 μm. (G, H) hCoASMCs treated with or without EphA2 siRNA were plated onto basement membrane proteins (matrigel, 1:50 dilution) and maintained in either serum-free or 1% serum-containing conditions overnight. Cultures were fractionated into the deposited extracellular matrix (deoxycholate (DOC)-insoluble) fraction or the cell-associated (DOC-soluble) fraction. Total and extradomain A (EDA)-positive fibronectin was assessed in the DOC-soluble and DOC-insoluble fractions by Western blotting. n = 4. Data are expressed as mean ± SEM, Statistical comparisons were made using Student's T-test (B, D, F), or One-way ANOVA (G, H) with Bonferroni post-tests.
Discussion
Early atherosclerotic plaques are dominated by inflammatory crosstalk between relatively few key cell types, which elicits endothelial cell activation, leukocyte recruitment, and smooth muscle invasion and fibro-proliferative remodeling within the developing plaque. Many previous studies demonstrate that endothelial EphA2 expression is enhanced and activated during inflammatory contexts, including within the endothelium overlying atherosclerotic plaques6, 9, 33. Using the Apoe-/- model of atherogenesis, we now demonstrate that EphA2 deletion reduced atherosclerotic burden associated with reduced inflammation in the early plaque. While plaque macrophages express EphA2, bone marrow chimeras and in vitro functional studies strongly implicated EphA2 expression in the vessel wall as the pro-inflammatory and pro-atherogenic EphA2 pool. ICAM-1 and VCAM-1 expression are attenuated in early atheromas of EphA2 KO mice, consistent with a role for EphA2 in endothelial activation, and blunting endothelial EphA2 expression reduced monocyte firm adhesion in vitro. While EphA2 deletion does not affect the stage of plaque progression at early time points, EphA2 KO mice show significantly reduced late-stage plaque progression associated with reductions in smooth muscle and fibrous tissue content. Consistent with this finding, EphA2 expression is enhanced in activated, synthetic hCoASMCs and hAoSMCs, and blunting EphA2 expression reduces vascular smooth muscle proliferation, mitogenic signaling (ERK1/2, Akt1), and fibronectin deposition both in vivo and in vitro. Together, these data demonstrate that EphA2 expression critically regulates atherosclerotic plaque formation and progression, potentially through dual effects on both inflammation and fibroproliferative remodeling.
Multiple lines of evidence suggest that EphA2 contributes to inflammatory endothelial activation. EphA2 expression is enhanced in a variety of proinflammatory conditions, including atherosclerosis, acute lung injury, ischemia/reperfusion, and psoriasis6, 33-35, often concomitant with upregulation of its ligand ephrinA1. During early atherogenesis, endothelial activation promotes NF-κB-dependent ICAM-1 and VCAM-1 expression to facilitate leukocyte recruitment and extravasation36, 37. EphA2 deletion blunts NF-κB activation, endothelial permeability, and chemokine expression in LPS-induced lung injury models7, 38, and a study by Jiang et al. showed that infection of Apoe-/- mice with EphA2 shRNA reduced atherosclerotic lesions and diminished both NF-κB activation and proinflammatory gene expression9. Atherosclerotic plaques in EphA2 KO mice similarly show reduced VCAM-1 and ICAM-1 expression compared to EphA2 WT mice (Figure 2D), indicating that EphA2 contributes to the endothelial proinflammatory phenotype in atherosclerosis. Eph-ephrin interactions also regulate leukocyte adhesion/repulsion responses and migration through control of integrin activation and cytoskeletal dynamics39, 40. While EphA2 deletion does not affect TNFα-induced proinflammatory gene expression6, EphA2-depleted endothelial cells show a reduced capacity to support monocyte firm adhesion following TNFα treatment (Figure 4G, Supplemental Figure 6B). In addition, substrate-bound EphA2 significantly enhances both monocyte adhesion to recombinant ICAM-1/VCAM-1 and monocyte spreading on fibronectin (Supplemental Figure 7). Monocyte adhesion and spreading is unaltered when recombinant ephrinA1 is added to the substrates (Supplemental Figure 7A/B), further suggesting monocytic EphA2 does not contribute to monocyte adhesion during leukocyte homing. However, EphA2 does not promote leukocyte recruitment under all contexts, as monocyte recruitment during sterile (thioglycollate-elicited) inflammation remains unchanged in EphA2 deficient mice (Figure 3B). Collectively, these data indicate that endothelial EphA2 expression modulates endothelial-monocyte interactions through multiple mechanisms.
Progression from early fatty streaks to advanced atheromas involves the recruitment of vascular smooth muscle cells into the growing plaque, with fibroproliferative remodeling driving the formation of the protective fibrous cap1. To accomplish this, smooth muscle cells undergo a dynamic shift from a quiescent, contractile phenotype in the medial layer to a pro-migratory, proliferative, and pro-fibrotic phenotype in the atherosclerotic plaque. We now show that smooth muscle transition to this activated phenotype results in the enhanced expression of EphA2 both in cell culture models and in atherosclerotic plaques (Figure 5G-K). Elevated EphA2 expression promotes proliferation and migration in several cancer models28, 29, and deletion of EphA2 reduces plaque progression, plaque smooth muscle content, and plaque-associated proliferation, as well as smooth muscle proliferation, mitogenic signaling (pERK1/2, pAkt1) and scratch wound healing in culture (Figure 5-7). Furthermore, deletion of EphA2 reduces plaque fibronectin and collagen content (Figure 8A-F), and EphA2 knockdown in vascular smooth muscle cells attenuates fibronectin deposition (Figure 8G/H). The reduced collagen content in EphA2-deficient plaques could result from reduced collagen degradation through proteases, such as matrix metalloproteinase 2 (MMP2) and MMP9. While EphA2 depletion does enhance MMP2 and MMP9 mRNA expression, these proteases do not show changes in protein expression or activity following EphA2 knockdown (Supplemental Figure 19). While EphA2 can promote fibronectin expression in cancer models41, EphA2 did not significantly affect smooth muscle matrix gene expression (Supplemental Figure 9), although fibronectin expression was reduced, albeit insignificantly, upon EphA2 silencing. Depletion of EphA2 reduces fibronectin deposition (Figure 8G/H, Supplemental Figure 17/18), and plating smooth muscle cells on a fibronectin matrix abrogates the effect of EphA2 deletion on smooth muscle scratch wound healing (Supplemental Figure 20), suggesting EphA2-dependent fibronectin deposition may contribute to EphA2-dependent regulation of smooth muscle function. Since fibronectin drives vascular smooth muscle proliferation and migration25, EphA2-induced fibronectin deposition may promote a permissible environment for vascular smooth muscle infiltration into atherosclerotic lesions. Furthermore, this relationship between EphA2 and fibronectin may incur an unusual feed-forward response, as fibronectin alone was shown to upregulate EphA2 expression independent of any other stimulus (Figure 5I, Supplemental Figure 11G). Taken together, these data provide the first report of an EphA2-dependent transition to advanced atherosclerosis through the regulation of smooth muscle fibroproliferative remodeling.
EphA2 appears to play a complex role in the pathogenesis of atherosclerotic cardiovascular disease, with EphA2 deletion inducing opposing effects on plasma lipid levels and plaque formation. While both male and female EphA2 KO mice display reduced plaque formation (Figure 1A-D), male EphA2 KO mice demonstrated significantly enhanced weight gain and plasma lipid levels when fed either standard chow or a high fat Western diet (Supplemental Figure 2A-D). Since EphA2 expression does not affect macrophage lipid uptake and hematopoietic EphA2 deletion does not affect plasma lipid levels in bone marrow chimera (Supplemental Figure 4C, 5), the effect of EphA2 deletion on plasma lipid levels may result from a previously unrecognized role for EphA2 in hepatocyte function. While no studies to date have shown a role for EphA2 in metabolic abnormalities, the chromosomal region containing the EphA2 gene (1p36.13) is associated with metabolic syndrome in multiple studies42-44. Furthermore, GWAS analysis links both EphA2 and ephrinA1 to the presence of liver enzymes in plasma45, and both EphA2 and ephrinA1 showed reduced expression in a rat model of nonalcoholic fatty liver disease46. While assessment of EphA2's role in cholesterol transport responses should serve as an interesting direction for future research, the reduction in atherosclerotic plaque formation despite the elevations in plasma lipid levels underscore the important role of EphA2 signaling in vascular remodeling responses during atherogenesis.
The data shown herein highlight two opposing functions of EphA2 in atherosclerosis: the propagation of early atherogenic inflammation and the stabilization of late stage plaques through fibroproliferative remodeling. However, recent publications highlight the often inverse function of EphA2 signaling due to differential ligand-dependent and ligand-independent signaling. Endothelial EphA2 relies on ephrinA1-dependent activation for proinflammatory marker expression6 and likely interacts with ephrinA1 to promote monocyte firm adhesion (Figure 4G, Supplemental Figure 6). Therefore, ligand-dependent signaling of endothelial EphA2 may contribute to early atherosclerosis by propagating atherogenic inflammation. While EphA2 ligation inhibits proliferative signaling cascades47, enhanced expression of unligated EphA2 promotes migration and proliferation20, 48, 49. Unlike endothelial cells, mRNA analysis reveals a downregulation of ephrinA1 in the proliferative vascular smooth muscle phenotype (Figure 5G), suggesting a potential contribution of ligand-independent signaling from smooth muscle EphA2 in late-stage atherosclerosis. Therefore, pharmacological approaches that limit EphA2 ligand-dependent signaling (blocking antibodies, kinase inhibitors) may reduce EphA2's proinflammatory effects without inhibiting EphA2's beneficial effect on smooth muscle proliferation and extracellular matrix deposition.
Supplementary Material
Clinical Perspective.
What is New?
While Eph receptors and their ephrin ligands play well characterized roles in angiogenesis and cancer, we present the first evidence that deletion of EphA2 in mouse models of atherosclerosis diminishes atherosclerotic plaque formation and limits atherosclerotic plaque progression.
We demonstrate that EphA2 deletion reduces proinflammatory gene expression and early monocyte recruitment to the plaque, associated with reductions in monocyte firm adhesion under flow.
We demonstrate the first link between smooth muscle phenotypic modulation and EphA2 expression, and we define a novel role for smooth muscle EphA2 expression in driving smooth muscle proliferation and extracellular matrix deposition.
What Are the Clinical Implications?
While EphA2 contributes to monocyte recruitment to the atherosclerotic plaque, EphA2 expression does not affect multiple other monocyte proinflammatory responses, suggesting that blunting EphA2 ligation may selectively reduce plaque-associated inflammation.
While EphA2 expression also contributes to the smooth muscle fibroproliferative remodeling that drives the stable plaque phenotype, EphA2's effect on proliferation appears to be largely ligand independent, unlike inflammation.
Blunting EphA2 ligation may limit inflammation while leaving smooth muscle fibroproliferative remodeling intact, thereby promoting a stable plaque phenotype.
Acknowledgments
The authors thank Randa Eshaq, Dr. Norman Harris (gelatin zymography), Dr. Chowdhury Abdullah, Dr. Shenuarin Bhuiyan (ChemiDoc Touch Imaging System) and Dr. Sushil Jain (THP-1 monocytes) (LSUHSC-Shreveport) for providing reagents and expertise.
Sources of Funding: This work was supported by American Heart Association Grant-In-Aid 13GRNT17050093 to AWO, Predoctoral Fellowship 14PRE18660003 to AYJ and 17PRE33440111 to ACF, NIH R01 AI56077 and P30GM110703 to A.Y., and by a Malcolm Feist Predoctoral Fellowship to ACF and SDF.
Footnotes
Disclosures: The authors have declared that no conflict of interest exists.
References
- 1.Bennett MR, Sinha S, Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res. 2016;118:692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 3.Kania A, Klein R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat Rev Mol Cell Biol. 2016;17:240–256. doi: 10.1038/nrm.2015.16. [DOI] [PubMed] [Google Scholar]
- 4.Funk SD, Orr AW. Ephs and ephrins resurface in inflammation, immunity, and atherosclerosis. Pharmacol Res. 2013;67:42–52. doi: 10.1016/j.phrs.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 5.Braun J, Hoffmann SC, Feldner A, Ludwig T, Henning R, Hecker M, Korff T. Endothelial cell ephrinB2-dependent activation of monocytes in arteriosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:297–305. doi: 10.1161/ATVBAHA.110.217646. [DOI] [PubMed] [Google Scholar]
- 6.Funk SD, Yurdagul A, Jr, Albert P, Traylor JG, Jr, Jin L, Chen J, Orr AW. EphA2 activation promotes the endothelial cell inflammatory response: a potential role in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:686–695. doi: 10.1161/ATVBAHA.111.242792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan B, Sukhatme VP. Receptor tyrosine kinase EphA2 mediates thrombin-induced upregulation of ICAM-1 in endothelial cells in vitro. Thromb Res. 2009;123:745–752. doi: 10.1016/j.thromres.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jellinghaus S, Poitz DM, Ende G, Augstein A, Weinert S, Stutz B, Braun-Dullaeus RC, Pasquale EB, Strasser RH. Ephrin-A1/EphA4-mediated adhesion of monocytes to endothelial cells. Biochim Biophys Acta. 2013;1833:2201–2211. doi: 10.1016/j.bbamcr.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 9.Jiang H, Li X, Zhang X, Liu Y, Huang S, Wang X. EphA2 knockdown attenuates atherosclerotic lesion development in ApoE(-/-) mice. Cardiovasc Pathol. 2014;23:169–174. doi: 10.1016/j.carpath.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Sulman EP, Tang XX, Allen C, Biegel JA, Pleasure DE, Brodeur GM, Ikegaki N. ECK, a human EPH-related gene, maps to 1p36.1, a common region of alteration in human cancers. Genomics. 1997;40:371–374. doi: 10.1006/geno.1996.4569. [DOI] [PubMed] [Google Scholar]
- 11.Wang Q, Rao S, Shen GQ, Li L, Moliterno DJ, Newby LK, Rogers WJ, Cannata R, Zirzow E, Elston RC, Topol EJ. Premature myocardial infarction novel susceptibility locus on chromosome 1P34-36 identified by genomewide linkage analysis. Am J Hum Genet. 2004;74:262–271. doi: 10.1086/381560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Welch CL, Bretschger S, Latib N, Bezouevski M, Guo Y, Pleskac N, Liang CP, Barlow C, Dansky H, Breslow JL, Tall AR. Localization of atherosclerosis susceptibility loci to chromosomes 4 and 6 using the Ldlr knockout mouse model. Proc Natl Acad Sci U S A. 2001;98:7946–7951. doi: 10.1073/pnas.141239098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navratil AR, Vozenilek AE, Cardelli JA, Green JM, Thomas MJ, Sorci-Thomas MG, Orr AW, Woolard MD. Lipin-1 contributes to modified low-density lipoprotein-elicited macrophage pro-inflammatory responses. Atherosclerosis. 2015;242:424–432. doi: 10.1016/j.atherosclerosis.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yurdagul A, Jr, Green J, Albert P, McInnis MC, Mazar AP, Orr AW. alpha5beta1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:1362–1373. doi: 10.1161/ATVBAHA.114.303863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol. 2005;169:191–202. doi: 10.1083/jcb.200410073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green J, Yurdagul A, Jr, McInnis MC, Albert P, Orr AW. Flow patterns regulate hyperglycemia-induced subendothelial matrix remodeling during early atherogenesis. Atherosclerosis. 2014;232:277–284. doi: 10.1016/j.atherosclerosis.2013.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.VanderLaan PA, Reardon CA, Getz GS. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. 2004;24:12–22. doi: 10.1161/01.ATV.0000105054.43931.f0. [DOI] [PubMed] [Google Scholar]
- 18.Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–1374. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- 19.Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W, Jr, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1994;89:2462–2478. doi: 10.1161/01.cir.89.5.2462. [DOI] [PubMed] [Google Scholar]
- 20.Macrae M, Neve RM, Rodriguez-Viciana P, Haqq C, Yeh J, Chen C, Gray JW, McCormick F. A conditional feedback loop regulates Ras activity through EphA2. Cancer Cell. 2005;8:111–118. doi: 10.1016/j.ccr.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 21.Khurana R, Simons M, Martin JF, Zachary IC. Role of angiogenesis in cardiovascular disease: a critical appraisal. Circulation. 2005;112:1813–1824. doi: 10.1161/CIRCULATIONAHA.105.535294. [DOI] [PubMed] [Google Scholar]
- 22.Brantley-Sieders DM, Caughron J, Hicks D, Pozzi A, Ruiz JC, Chen J. EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J Cell Sci. 2004;117:2037–2049. doi: 10.1242/jcs.01061. [DOI] [PubMed] [Google Scholar]
- 23.Bowers CW, Dahm LM. Maintenance of contractility in dissociated smooth muscle: low-density cultures in a defined medium. Am J Physiol. 1993;264:C229–236. doi: 10.1152/ajpcell.1993.264.1.C229. [DOI] [PubMed] [Google Scholar]
- 24.Orr AW, Lee MY, Lemmon JA, Yurdagul A, Jr, Gomez MF, Bortz PD, Wamhoff BR. Molecular mechanisms of collagen isotype-specific modulation of smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. 2009;29:225–231. doi: 10.1161/ATVBAHA.108.178749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hedin U, Bottger BA, Forsberg E, Johansson S, Thyberg J. Diverse effects of fibronectin and laminin on phenotypic properties of cultured arterial smooth muscle cells. J Cell Biol. 1988;107:307–319. doi: 10.1083/jcb.107.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AF, Barbalic M, Gieger C, Absher D, Aherrahrou Z, Allayee H, Altshuler D, Anand SS, Andersen K, Anderson JL, Ardissino D, Ball SG, Balmforth AJ, Barnes TA, Becker DM, Becker LC, Berger K, Bis JC, Boekholdt SM, Boerwinkle E, Braund PS, Brown MJ, Burnett MS, Buysschaert I, Carlquist JF, Chen L, Cichon S, Codd V, Davies RW, Dedoussis G, Dehghan A, Demissie S, Devaney JM, Diemert P, Do R, Doering A, Eifert S, Mokhtari NE, Ellis SG, Elosua R, Engert JC, Epstein SE, de Faire U, Fischer M, Folsom AR, Freyer J, Gigante B, Girelli D, Gretarsdottir S, Gudnason V, Gulcher JR, Halperin E, Hammond N, Hazen SL, Hofman A, Horne BD, Illig T, Iribarren C, Jones GT, Jukema JW, Kaiser MA, Kaplan LM, Kastelein JJ, Khaw KT, Knowles JW, Kolovou G, Kong A, Laaksonen R, Lambrechts D, Leander K, Lettre G, Li M, Lieb W, Loley C, Lotery AJ, Mannucci PM, Maouche S, Martinelli N, McKeown PP, Meisinger C, Meitinger T, Melander O, Merlini PA, Mooser V, Morgan T, Muhleisen TW, Muhlestein JB, Munzel T, Musunuru K, Nahrstaedt J, Nelson CP, Nothen MM, Olivieri O, Patel RS, Patterson CC, Peters A, Peyvandi F, Qu L, Quyyumi AA, Rader DJ, Rallidis LS, Rice C, Rosendaal FR, Rubin D, Salomaa V, Sampietro ML, Sandhu MS, Schadt E, Schafer A, Schillert A, Schreiber S, Schrezenmeir J, Schwartz SM, Siscovick DS, Sivananthan M, Sivapalaratnam S, Smith A, Smith TB, Snoep JD, Soranzo N, Spertus JA, Stark K, Stirrups K, Stoll M, Tang WH, Tennstedt S, Thorgeirsson G, Thorleifsson G, Tomaszewski M, Uitterlinden AG, van Rij AM, Voight BF, Wareham NJ, Wells GA, Wichmann HE, Wild PS, Willenborg C, Witteman JC, Wright BJ, Ye S, Zeller T, Ziegler A, Cambien F, Goodall AH, Cupples LA, Quertermous T, Marz W, Hengstenberg C, Blankenberg S, Ouwehand WH, Hall AS, Deloukas P, Thompson JR, Stefansson K, Roberts R, Thorsteinsdottir U, O'Donnell CJ, McPherson R, Erdmann J, Samani NJ. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Motterle A, Pu X, Wood H, Xiao Q, Gor S, Ng FL, Chan K, Cross F, Shohreh B, Poston RN, Tucker AT, Caulfield MJ, Ye S. Functional analyses of coronary artery disease associated variation on chromosome 9p21 in vascular smooth muscle cells. Hum Mol Genet. 2012;21:4021–4029. doi: 10.1093/hmg/dds224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen P, Huang Y, Zhang B, Wang Q, Bai P. EphA2 enhances the proliferation and invasion ability of LNCaP prostate cancer cells. Oncol Lett. 2014;8:41–46. doi: 10.3892/ol.2014.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X, Wang X, Ruan A, Han W, Zhao Y, Lu X, Xiao P, Shi H, Wang R, Chen L, Chen S, Du Q, Yang H, Zhang X. miR-141 is a key regulator of renal cell carcinoma proliferation and metastasis by controlling EphA2 expression. Clin Cancer Res. 2014;20:2617–2630. doi: 10.1158/1078-0432.CCR-13-3224. [DOI] [PubMed] [Google Scholar]
- 30.Beauchamp A, Debinski W. Ephs and ephrins in cancer: ephrin-A1 signalling. Semin Cell Dev Biol. 2012;23:109–115. doi: 10.1016/j.semcdb.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adiguzel E, Ahmad PJ, Franco C, Bendeck MP. Collagens in the progression and complications of atherosclerosis. Vasc Med. 2009;14:73–89. doi: 10.1177/1358863X08094801. [DOI] [PubMed] [Google Scholar]
- 32.Sottile J, Shi F, Rublyevska I, Chiang HY, Lust J, Chandler J. Fibronectin-dependent collagen I deposition modulates the cell response to fibronectin. Am J Physiol Cell Physiol. 2007;293:C1934–1946. doi: 10.1152/ajpcell.00130.2007. [DOI] [PubMed] [Google Scholar]
- 33.Hong JY, Shin MH, Chung KS, Kim EY, Jung JY, Kang YA, Kim YS, Kim SK, Chang J, Park MS. EphA2 Receptor Signaling Mediates Inflammatory Responses in Lipopolysaccharide-Induced Lung Injury. Tuberc Respir Dis (Seoul) 2015;78:218–226. doi: 10.4046/trd.2015.78.3.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thundyil J, Manzanero S, Pavlovski D, Cully TR, Lok KZ, Widiapradja A, Chunduri P, Jo DG, Naruse C, Asano M, Launikonis BS, Sobey CG, Coulthard MG, Arumugam TV. Evidence that the EphA2 receptor exacerbates ischemic brain injury. PLoS One. 2013;8:e53528. doi: 10.1371/journal.pone.0053528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gordon K, Kochkodan JJ, Blatt H, Lin SY, Kaplan N, Johnston A, Swindell WR, Hoover P, Schlosser BJ, Elder JT, Gudjonsson JE, Getsios S. Alteration of the EphA2/Ephrin-A signaling axis in psoriatic epidermis. J Invest Dermatol. 2013;133:712–722. doi: 10.1038/jid.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;107:1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kitagawa K, Matsumoto M, Sasaki T, Hashimoto H, Kuwabara K, Ohtsuki T, Hori M. Involvement of ICAM-1 in the progression of atherosclerosis in APOE-knockout mice. Atherosclerosis. 2002;160:305–310. doi: 10.1016/s0021-9150(01)00587-1. [DOI] [PubMed] [Google Scholar]
- 38.Carpenter TC, Schroeder W, Stenmark KR, Schmidt EP. Eph-A2 promotes permeability and inflammatory responses to bleomycin-induced lung injury. Am J Respir Cell Mol Biol. 2012;46:40–47. doi: 10.1165/rcmb.2011-0044OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saeki N, Nishino S, Shimizu T, Ogawa K. EphA2 promotes cell adhesion and spreading of monocyte and monocyte/macrophage cell lines on integrin ligand-coated surfaces. Cell Adh Migr. 2015;9:469–482. doi: 10.1080/19336918.2015.1107693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2:62–69. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
- 41.Hu M, Carles-Kinch KL, Zelinski DP, Kinch MS. EphA2 induction of fibronectin creates a permissive microenvironment for malignant cells. Mol Cancer Res. 2004;2:533–540. [PubMed] [Google Scholar]
- 42.Hoffmann K, Mattheisen M, Dahm S, Nurnberg P, Roe C, Johnson J, Cox NJ, Wichmann HE, Wienker TF, Schulze J, Schwarz PE, Lindner TH. A German genome-wide linkage scan for type 2 diabetes supports the existence of a metabolic syndrome locus on chromosome 1p36.13 and a type 2 diabetes locus on chromosome 16p12.2. Diabetologia. 2007;50:1418–1422. doi: 10.1007/s00125-007-0658-4. [DOI] [PubMed] [Google Scholar]
- 43.Hoffmann K, Planitz C, Ruschendorf F, Muller-Myhsok B, Stassen HH, Lucke B, Mattheisen M, Stumvoll M, Bochmann R, Zschornack M, Wienker TF, Nurnberg P, Reis A, Luft FC, Lindner TH. A novel locus for arterial hypertension on chromosome 1p36 maps to a metabolic syndrome trait cluster in the Sorbs, a Slavic population isolate in Germany. J Hypertens. 2009;27:983–990. doi: 10.1097/HJH.0b013e328328123d. [DOI] [PubMed] [Google Scholar]
- 44.Edwards KL, Wan JY, Hutter CM, Fong PY, Santorico SA. Multivariate linkage scan for metabolic syndrome traits in families with type 2 diabetes. Obesity (Silver Spring) 2011;19:1235–1243. doi: 10.1038/oby.2010.299. [DOI] [PubMed] [Google Scholar]
- 45.Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P, Holm H, Sanna S, Kavousi M, Baumeister SE, Coin LJ, Deng G, Gieger C, Heard-Costa NL, Hottenga JJ, Kuhnel B, Kumar V, Lagou V, Liang L, Luan J, Vidal PM, Mateo Leach I, O'Reilly PF, Peden JF, Rahmioglu N, Soininen P, Speliotes EK, Yuan X, Thorleifsson G, Alizadeh BZ, Atwood LD, Borecki IB, Brown MJ, Charoen P, Cucca F, Das D, de Geus EJ, Dixon AL, Doring A, Ehret G, Eyjolfsson GI, Farrall M, Forouhi NG, Friedrich N, Goessling W, Gudbjartsson DF, Harris TB, Hartikainen AL, Heath S, Hirschfield GM, Hofman A, Homuth G, Hypponen E, Janssen HL, Johnson T, Kangas AJ, Kema IP, Kuhn JP, Lai S, Lathrop M, Lerch MM, Li Y, Liang TJ, Lin JP, Loos RJ, Martin NG, Moffatt MF, Montgomery GW, Munroe PB, Musunuru K, Nakamura Y, O'Donnell CJ, Olafsson I, Penninx BW, Pouta A, Prins BP, Prokopenko I, Puls R, Ruokonen A, Savolainen MJ, Schlessinger D, Schouten JN, Seedorf U, Sen-Chowdhry S, Siminovitch KA, Smit JH, Spector TD, Tan W, Teslovich TM, Tukiainen T, Uitterlinden AG, Van der Klauw MM, Vasan RS, Wallace C, Wallaschofski H, Wichmann HE, Willemsen G, Wurtz P, Xu C, Yerges-Armstrong LM, Abecasis GR, Ahmadi KR, Boomsma DI, Caulfield M, Cookson WO, van Duijn CM, Froguel P, Matsuda K, McCarthy MI, Meisinger C, Mooser V, Pietilainen KH, Schumann G, Snieder H, Sternberg MJ, Stolk RP, Thomas HC, Thorsteinsdottir U, Uda M, Waeber G, Wareham NJ, Waterworth DM, Watkins H, Whitfield JB, Witteman JC, Wolffenbuttel BH, Fox CS, Ala-Korpela M, Stefansson K, Vollenweider P, Volzke H, Schadt EE, Scott J, Jarvelin MR, Elliott P, Kooner JS. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 2011;43:1131–1138. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alisi A, Da Sacco L, Bruscalupi G, Piemonte F, Panera N, De Vito R, Leoni S, Bottazzo GF, Masotti A, Nobili V. Mirnome analysis reveals novel molecular determinants in the pathogenesis of diet-induced nonalcoholic fatty liver disease. Lab Invest. 2011;91:283–293. doi: 10.1038/labinvest.2010.166. [DOI] [PubMed] [Google Scholar]
- 47.Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, Sloan AE, Cohen ML, Wang B. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9–20. doi: 10.1016/j.ccr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang J, Hu W, Bottsford-Miller J, Liu T, Han HD, Zand B, Pradeep S, Roh JW, Thanapprapasr D, Dalton HJ, Pecot CV, Rupaimoole R, Lu C, Fellman B, Urbauer D, Kang Y, Jennings NB, Huang L, Deavers MT, Broaddus R, Coleman RL, Sood AK. Cross-talk between EphA2 and BRaf/CRaf is a key determinant of response to Dasatinib. Clin Cancer Res. 2014;20:1846–1855. doi: 10.1158/1078-0432.CCR-13-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subbarayal P, Karunakaran K, Winkler AC, Rother M, Gonzalez E, Meyer TF, Rudel T. EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis. PLoS Pathog. 2015;11:e1004846. doi: 10.1371/journal.ppat.1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.