

Abstract
Currently, approximately 30% of patients with epilepsy do not have adequate seizure control. A greater understanding of the underlying mechanisms by which seizures start or propagate could lead to new therapeutic strategies. The recent development of optogenetics, because of its unprecedented precision for controlling activity within distinct neuronal populations, has revolutionized neuroscience, including epilepsy research. This Review discusses recent breakthroughs made with optogenetics in epilepsy research. These breakthroughs include new insights into the key roles that different cell types play in mediating seizures as well as in the development of epilepsy. Subsequently, we discuss how targeting different brain regions and cell populations has opened up the possibility of highly specific therapies that can stop seizures on demand. Finally, we illustrate how combining newly available neuroscience tools with whole‐brain imaging techniques will allow researchers to understand better the spread of seizures on a network level. © 2016 The Authors. Journal of Neuroscience Research Published by Wiley Periodicals, Inc.
Keywords: optogenetics, epilepsy, seizures
Epileptic seizures are characterized by transient bursts of pathological activity permeating brain networks. Since the middle of the 20th century, epilepsy researchers have used electrical and pharmacological tools to probe epileptic networks, leading to considerable improvements in treatments for epilepsy (Putnam and Merritt, 1937; Spiegel, 1937; Goddard et al., 1969; for excellent historical reviews of epilepsy drug development, see Shorvon, 2009a, 2009b; Brodie, 2010). However, approximately 30% of patients with epilepsy do not respond to antiepileptic medications, and at this time we do not fully understand the conditions required for seizures to start, propagate, or stop. The recent development of optogenetics, because of its unprecedented precision, has revolutionized neuroscience, including epilepsy research; it has allowed us to target specific cell types and to switch them on or off with exquisite temporal control, revealing the details of seizure circuits like never before. Although the use of optogenetics in epilepsy research is still in its infancy, exciting work has already emerged, including new insights into the key roles of certain cell types in mediating seizures and epileptogenesis. Optogenetics has also revealed the potential of very specific targeted interventions that could allow on‐demand seizure interventions. Combining these new research tools with imaging technologies such as functional magnetic resonance imaging (fMRI) will allow us to have a better understanding of seizure circuits over the whole brain. This Review discusses some of the most recent breakthroughs in the field of optogenetics and epilepsy research. First, we discuss how optogenetics has been used to further our understanding of seizure circuits, and then we consider the future potential of this technique to aid both our understanding and the treatment of epilepsy.
UNDERSTANDING SEIZURE CIRCUITS
Epilepsy research has a long history of using electrical stimulation of brain regions to drive seizure‐like afterdischarges, and this technique is widely used in animal models (Goddard et al., 1969; Racine, 1972; Pinel and Rovner, 1978; Leung, 1987; Nissinen et al., 2000; Englot et al., 2008). However, electrical stimulation is nonspecific, exciting all types of neurons as well as nonneuronal cells, thus making it difficult to elucidate the role played by individual cell types during the generation of seizures (Pitkänen et al., 2006). The optogenetics toolbox has helped to disentangle these roles and, in this way, has aided our understanding of the conditions that allow seizures to emerge.
Optogenetics uses light at different wavelengths to excite or inhibit the activity of opsin‐expressing cells with millisecond control (Boyden et al., 2005; Deisseroth, 2015). The nonselective cation channel channelrhodopsin‐2 (Chr2) and its variants are used for depolarizing cells, and halorhodopsin‐3, a chloride pump, archaeorhodopsin‐3, a proton pump, or their variants are commonly used for inhibition or hyperpolarization. By tying the expression of these genes to specific promoters and delivering light onto targeted regions, the activity of particular cell populations can be rapidly modulated in a given circuit.
The emergence of seizures has classically been attributed to a shift in the balance between excitation and inhibition neuronal activity toward excitation. The mechanisms that underlie this transition to seizure are not well understood, but evidence suggests that there is significant interplay between pyramidal cells and interneurons in the emergence of epileptic phenomena. For example, in in vitro hippocampal slices from rats, simultaneous whole‐cell recordings from pyramidal cells and interneurons have revealed that both types of cells are active at different times during the course of a seizure; interneurons are more active at onset of epileptiform discharges but then undergo a period of depolarization block, during which there is an increase in pyramidal cell firing (Ziburkus et al., 2006). Toyoda and colleagues (2015) found a similar response in vivo using unit recordings in the postpilocarpine epileptic rat. Most interneurons recorded in the hippocampus (most notably in the subiculum) increased in firing rate up to 4 min prior to the onset of spontaneous seizures. During this time, a smaller population of interneurons decreased in firing rate. After seizure onset, many of the examined interneurons briefly stopped firing before resuming activity. The mechanism of this pause is not known, but Toyoda and colleagues (2015) speculated that it could be due to depolarization block resulting from elevated extracellular potassium ion concentrations.
The preictal change in interneuronal firing rate could indicate a possible role for them in the initiation of spontaneous seizures. With respect to evoked seizures, remarkably, blocking GABAergic synapses with bicuculline in hippocampal slices enhances the generation of short‐duration interictal bursts but prevents electrically induced afterdischarges altogether (Higashima et al., 1996). Studying epileptic tissue from the human hippocampus, Huberfeld and colleagues (2011) found that the transition to seizure is preceded by pyramidal cell firing and dependent on glutamatergic signaling, whereas interictal epileptiform activity is preceded by interneuron firing and involves both glutamatergic and GABAergic mechanisms. Nevertheless, because of complex interdependencies, it has been difficult to disentangle the precise role of individual cell populations. The development of optogenetics has afforded researchers the possibility of driving specific cell populations, with the aim of investigating their roles in seizure mechanisms.
Osawa and colleagues (2013) found that repetitive optogenetic stimulation of hippocampal neurons at 10 or 20 Hz efficiently evokes seizure‐like afterdischarges in rats anesthetized under ketamine and xylazine. Furthermore, selective stimulation of the excitatory hippocampal pyramidal cell population via the Ca2+/calmodulin‐dependent protein kinase IIα (CamKII) promoter (Weitz et al., 2015) also evokes seizure‐like afterdischarges in awake and anesthetized rats, confirming the hypothesis that excessive tetanic stimulation of excitatory neurons can result in seizures. Alongside the benefits of inducing epileptic activity from specific cell types with temporal precision, another key advantage of optogenetic seizure initiation is that simultaneous electrophysiology remains uncontaminated by electrical stimulation artefacts during the stimulation period so that the dynamics of the stimulation period may be investigated. However, artefacts on electrophysiology can occur if the light delivered for stimulation hits the recording electrode, inducing a photovoltaic effect. These artefacts may be reduced or eliminated by moving the electrode away from light contamination (Cardin et al., 2010). Stimulating rat dorsal hippocampal neurons (expressing ChR2 under the Thy1.2 promoter) in vivo, Osawa and colleagues (2013) noted that, at the start of stimulation, evoked potentials were time locked to the light pulses, but, eventually, abnormal spontaneous activity emerged in addition to the evoked potentials that persisted beyond the end of the stimulation period. A Granger causality analysis on the local field potential (LFP) traces recorded from the septal and temporal hippocampus suggested that the direction of causal influence shifts during the development of afterdischarges from initially being greater in the septotemporal direction at onset to being greater in the temporal‐septal direction toward the end. Using fMRI during selective stimulation of CamKII hippocampal neurons allowed the visualization of distinct frequency‐dependent networks, which depended on the location along the septal‐temporal axis at which the stimulation was applied (Weitz et al., 2015). For example, dorsal stimulations resulted in activity restricted to limbic regions, whereas intermediate stimulations led to widespread brain activity, including recruitment of the cortical and subcortical regions. Awake‐behavioral experiments also suggested a more severe phenotype after stimulation of the intermediate compared with the dorsal hippocampus.
Modulating excitatory neurons is not the only means for driving seizures optogenetically; other research has suggested that activity within the inhibitory system also plays a critical role in this shift toward excitation. When postsynaptic intracellular chloride concentrations significantly increase, which occurs during seizure discharges, GABAA receptors become depolarizing instead of hyperpolarizing, and inhibitory interneurons effectively become excitatory; this mechanism is thought to induce or facilitate seizures (Staley et al., 1995; Bernard et al., 2000; Lillis et al., 2012; Trevelyan et al., 2015). In line with this hypothesis, under certain conditions, epileptiform discharges can be initiated in entorhinal cortex slices by selective optogenetic activation of individual classes of interneurons, including parvalbumin (PV)‐ and somatostatin (SOM)‐expressing neurons (Shiri et al., 2015; Yekhlef et al., 2015). For example, in the 4‐aminopyridine (4AP) in vitro slice model, stimulating PV‐expressing cells rapidly evokes epileptiform activity with a distinct low‐voltage fast‐onset pattern resembling those that occur spontaneously in the same model (Shiri et al., 2015). Similarly, Yekhlef and colleagues (2015) demonstrated that stimulating either PV‐ or SOM‐expressing neurons in medial entorhinal cortical slices perfused in 4AP triggers interictal and preictal spikes that rapidly lead to seizure‐like events. Furthermore, these stimulations were accompanied by a rapid and transient accumulation of extracellular potassium, an effect also found in spontaneous events, before the emergence of epileptiform activity. Such an accumulation in itself can contribute to these discharges and is consistent with the hypothesis that intense interneuronal activation can result in an excitatory drive resulting from elevated intracellular chloride and a consequent increased extracellular potassium mediated by the K+‐Cl– cotransporter KCC2 (Kaila et al., 1997; Viitanen et al., 2010; Yekhlef et al., 2015).
The role of inhibitory interneurons is likely to be nuanced, and researchers have suggested that their effect is apparent only when there is a background of pathological activity (Ellender et al., 2014; Sessolo et al., 2015; Yekhlef et al., 2015). Activating PV interneurons in vitro generated epileptiform discharges only in areas with concomitant localized hyperactivity (Sessolo et al., 2015). Furthermore, directly increasing the GABA reversal potential by loading hippocampal pyramidal cells with chloride using halorhodopsin was not enough on its own to induce epileptiform discharges; although overall network activity was affected, discharges emerged only after a low dose of 4AP was added to increase background activity (Alfonsa et al., 2015). These findings suggest that the preictal environment is crucial in determining the role played by different cell types in seizure onset. Also highlighting the importance of the preictal state, in an in vivo model of absence seizures generated by optogenetic stimulation of excitatory neocortical neurons, Wagner and colleagues (2015) found that the average LFP power before optogenetic stimulation appears to be significantly lower in trials that led to induced seizures compared with those that did not. Likewise, the influence of different cell types may change or wax and wane over the course of a seizure. For example, Ellender and colleagues (2014) found in their in vitro preparation that, although PV interneurons are inhibitory during the early stages of epileptiform discharges, during the later clonic stages they not only excite pyramidal cells because of high intracellular chloride levels but also directly act to synchronize pyramidal cells across the network, thereby potentiating and maintaining epileptiform discharges (Ellender et al., 2014).
EPILEPTOGENESIS
Epileptogenesis is the process in which neural circuit reorganization leads to epilepsy. Understanding how the circuit reorganizes and understanding the conditions required for the emergence of seizures remain critical, unsolved goals for epilepsy research (Buckmaster and Dudek, 1997; Kelley et al., 2009). One of the challenges to achieving these goals is that weeks or even years can pass between an epilepsy‐inducing brain injury and the onset of seizures, and during this time the insult will have induced a myriad of processes, including inflammation, neurogenesis, cell death, and gliosis (Mathern et al., 1995; Lukasiuk et al., 2003; Duffy et al., 2012; Choy et al., 2014a, 2014b). The degree of neuronal activity during development may play an important role by setting homeostatic limits that can influence subsequent seizure expression. In a genetic Drosophila model of epilepsy, researchers were able to prevent the emergence of the seizure phenotype by reducing neuronal activity with halorhodopsin during a critical period in embryonic development (Giachello and Baines, 2015). Conversely, using ChR to increase activity during this period, they were able to induce the seizure phenotype in wild‐type flies. If this property is conserved across species, identifying such a critical developmental window may have profound consequences for genetic epilepsies and could also inform our understanding and treatment of acquired epilepsies, in which significant neurogenesis is known to occur.
Even after epilepsy emerges, however, it is unclear which features of an epileptic circuit make it hyperexcitable and inherently prone to seizures. Epilepsy is often associated with widespread circuit changes, but it is difficult to separate epileptogenic changes from those that are compensatory or mere epiphenomena. In temporal lobe epilepsy, common structural changes include mossy fiber sprouting, granule cell dispersion, loss of pyramidal cells in CA1, and changes to inhibitory neurons (Thom and Bertram, 2012), but we do not yet know which of these changes, if any, makes the region prone to seizures. Directly manipulating individual cell populations will help to characterize epileptic circuits further and to reveal the roles of those cells (Buckmaster and Lew, 2011; Peng et al., 2013). For example, in rats with epilepsy induced by status epilepticus, there is substantial axonal reorganization in the hippocampus, including in dentate granule cells and in PV‐ and SOM‐expressing interneurons (Peng et al., 2013). By targeting SOM‐expressing CA1 neurons and using optogenetic stimulation in slices taken from these epileptic rats, Peng and colleagues (2013) demonstrated that these interneurons increase their functional territories in the reorganized circuit, extending their influence from the CA1 into the dentate gyrus. Recently, three separate studies have shown that hippocampal grafts of GABAergic interneurons reduce seizure frequency in chronic epilepsy (Hunt et al., 2013; Cunningham et al., 2014; Henderson et al., 2014). First, Hunt and colleagues (2013) showed that implanting inhibitory neurons into the mouse hippocampus dramatically reduced spontaneous seizures, whereas grafting these cells into the amygdala did not affect seizure frequency. Cunningham and colleagues (2014) transplanted ChR2‐expressing human‐derived maturing GABAergic interneurons into the hippocampi of epileptic mice and discovered extensive migration and integration with host circuitry. Channelrhodopsin‐2 stimulation led to robust postsynaptic responses in host hippocampal neurons. Similarly, Henderson and colleagues (2014) transplanted ChR2‐expressing fetal medial ganglionic eminence GABAergic progenitor cells into the mouse hippocampi 2 weeks after pilocarpine‐induced status epilepticus and found reduced seizure frequency between 61 and 80 days after the initial injury. These cells became functionally integrated into the network, and stimulating ChR2‐expressing cells from hippocampal slices collected during this period yielded responses in host granule cells, innervated by these transplanted interneurons. Histology indicated that the grafts differentiated into interneuron subtypes, including neuropeptide Y‐, PV‐, and SOM‐expressing cells. Additional functional mapping of these grafts may reveal the connectivity changes that reduce seizure activity and also help assess potential cell therapies for epilepsy.
OPTOGENETIC SEIZURE CONTROL
Controlling seizures, ideally without side effects, is still an unattained goal; approximately 30% of patients with epilepsy do not respond to conventional therapies. However, it has been shown that optogenetics can be used to ameliorate seizures and that this can be achieved with only a brief targeted intervention at the beginning of the seizure (Krook‐Magnuson et al., 2013; Paz et al., 2013).
Since the first study, in 2009, that reported halorhodopsin could be used to control hippocampal epileptiform activity in vitro (Tønnesen et al., 2009), progress has been rapid, and in vivo studies have quickly followed (Wykes et al., 2012; Krook‐Magnuson et al., 2013; Paz et al., 2013; Sukhotinsky et al., 2013). Optogenetics has now been shown to be effective for ameliorating seizures in vivo across a spectrum of epilepsy models, including neocortical, temporal lobe, poststroke, and absence epilepsies as well as status epilepticus (see Table 1). For example, several studies have shown that halorhodopsin, expressed in excitatory neurons, can be used to stop seizures. Inhibiting neocortical CamKII‐expressing neurons with halorhodopsin reduced epileptiform activity in the tetanus rat model of neocortical epilepsy (Wykes et al., 2012), and suppressing hippocampal activity was effective in a mouse model of temporal lobe epilepsy (Krook‐Magnuson et al., 2013). After photothrombotic cortical stroke in rats, using halorhodopsin to inhibit CamKII‐expressing neurons in the ventrobasal thalamus halted thalamocortical seizures (Paz et al., 2013), and, in the lithium‐pilocarpine rat model of status epilepticus, inhibiting hippocampal CamKII‐expressing neurons with halorhodopsin delayed seizure onset (Sukhotinsky et al., 2013).
Table 1.
Summary of Regions Targeted for Optogenetic Control of Seizures In Vivoa
| Target region | Promoter | Cell type | Opsin | Model | Effect | Reference |
|---|---|---|---|---|---|---|
| Motor cortex | CamKII | Glutamatergic | NpHR | Intracortical tetanus toxin | Reduced frequency of epileptiform events | Wykes et al., 2012 |
| Ventrobasal thalamus | CamKII | Glutamatergic | NpHR | Photothrombotic stroke | Reduces power of epileptic events | Paz et al., 2013 |
| Hippocampus | CamKII | Glutamatergic | NpHR | Systemic pilocarpine‐induced SE | Delayed SE onset by 5 min | Sukhotinsky et al., 2013 |
| Ipsilateral hippocampus CA1 | PV | GABAergic | ChR2 | Intrahippocampal KA spontaneous seizures | Reduces seizure duration | Krook‐Magnuson et al., 2013 |
| Contralateral hippocampus CA1 | PV | GABAergic | ChR2 | Intrahippocampal KA spontaneous seizures | Reduces seizure duration | Krook‐Magnuson et al., 2013 |
| Ipsilateral hippocampus CA1 | CamKII | Glutamatergic | NpHR | Intrahippocampal KA spontaneous seizures | Reduces seizure duration | Krook‐Magnuson et al., 2013 |
| Ipsilateral cerebellum | PV | GABAergic | ChR2 | Intrahippocampal KA spontaneous seizures | Reduces seizure duration | Krook‐Magnuson et al., 2014 |
| Contralateral cerebellum | PV | GABAergic | ChR2 | Intrahippocampal KA spontaneous seizures | Reduces seizure duration | Krook‐Magnuson et al., 2014 |
| Ipsilateral cerebellum | PV | GABAergic | NpHR | Intrahippocampal KA spontaneous seizures | Reduces seizure duration | Krook‐Magnuson et al., 2014 |
| Contralateral cerebellum | PV | GABAergic | NpHR | Intrahippocampal KA spontaneous seizures | Reduces seizure duration | Krook‐Magnuson et al., 2014 |
| Midline cerebellum (vermis) | PV | GABAergic | ChR2 | Intrahippocampal KA spontaneous seizures | Reduces seizure duration and increase time to next seizure | Krook‐Magnuson et al., 2014 |
| Midline cerebellum (vermis) | PV | GABAergic | NpHR | Intrahippocampal KA spontaneous seizures | No effect on time to next seizure | Krook‐Magnuson et al., 2014 |
| Hippocampus | PV | GABAergic | NpHR | Intrahippocampal KA spontaneous seizures | No effect | Krook‐Magnuson et al., 2014 |
| Deep cerebellar nuclei | PV | GABAergic | ChR2 | Intrahippocampal KA spontaneous seizures | Reduces seizure duration and increase time to next seizure | Krook‐Magnuson et al., 2014 |
| Purkinje neurons in ipsilateral cerebellum | Pcp2 | Purkinje (GABAergic) | ChR2 | Intrahippocampal KA spontaneous seizures | Reduces seizure duration | Krook‐Magnuson et al., 2014 |
| Hippocampus | Thy1 | Panneuronal | ChR2 | 4‐AP intrahippocampal | Seizure activity (signal power) during stimulation reduced by 80% but does not stop events | Chiang et al., 2014 |
| Hippocampus | hSyn | Panneuronal | NpHR | Intrahippocampal Bicuculline | Reduced bursting frequency | Berglind et al., 2014 |
| Ventrobasal thalamus | CamKII | Glutamatergic | NpHR | Penicillin in somatosensory cortex | No effect | Han et al., 2015 |
| Ipsilateral dentate gyrus | Pomc | Granule cell | NpHR | Intrahippocampal KA spontaneous seizures | Reduces seizure duration | Krook‐Magnuson et al., 2015 |
| Contralateral dentate gyrus | Pomc | Granule cell | NpHR | Intrahippocampal KA spontaneous seizures | No effect | Krook‐Magnuson et al., 2015 |
| Ipsi/contralateral dentate gyrus | Pomc | Granule cell | ChR2 | Intrahippocampal KA spontaneous seizures | Increased seizure severity | Krook‐Magnuson et al., 2015 |
| Superior colliculus | hSyn | Panneuronal | ChR2 | Systemic PTZ | Reduced seizure severity | Soper et al., 2015 |
| Superior colliculus | hSyn | Panneuronal | ChR2 | Bicuculline in area tempestus | Reduced seizure severity and frequency | Soper et al., 2015 |
| Superior colliculus | hSyn | Panneuronal | ChR2 | GEPR‐3s (audiogenic seizure) | Reduced seizure severity and increased latency to onset | Soper et al., 2015 |
| Superior colliculus | hSyn | Panneuronal | ChR2 | Systemic γ‐butyrolactone (absence seizures) | Reduced seizure frequency and duration | Soper et al., 2015 |
| Hippocampus | Thy1 | Panneuronal | ChR2 | 4‐AP intrahippocampal | Seizure activity (signal power) during stimulation reduced but does not stop events | Ladas et al., 2015 |
| Cerebellar nuclei | hSyn | Panneuronal | ChR2 | Tg mice spontaneous SWDs | Reduced number of SWD events | Kros et al., 2015 |
| Cerebellar nuclei | hSyn | Panneuronal | ChR2 | C3H/HeOuJ mice spontaneous SWDs | Reduced number of SWD events | Kros et al., 2015 |
| Dentate gyrus | Vgat | Interneurons | ChR2 | KA intrahippocampal SE | Reduced seizure frequency post‐KA and stopped seizure propagation to MEC and motor cortex | Lu et al., 2016 |
| MEC | Vgat | Interneurons | ChR2 | KA intrahippocampal SE | Stopped seizure activity in MEC but did not stop seizure activity in dentate gyrus and M1 | Lu et al., 2016 |
| Dentate gyrus | Gad | Interneurons | ChR2 | KA intrahippocampal SE | Reduced seizure frequency post‐KA and stopped seizure propagation to MEC and motor cortex | Lu et al., 2016 |
| Dentate gyrus | Gad | Interneurons | NpHR | KA intrahippocampal SE | No effect on seizures | Lu et al., 2016 |
GEPR, genetically epilepsy‐prone rat; hSYN, human synapsin; KA, kainic acid; NpHR, halorhodopsin; Pcp2, purkinje cell protein 2; Pomc, proopiomelanocortin; PTZ, pentylenetetrazol; PV, parvalbumin; SE, status epilepticus; SWD, spike‐wave discharge; Thy1, thymocyte differentiation antigen 1; Vgat, vesicular GABA transporter.
Another approach for suppressing seizures in vivo is to activate interneurons selectively with ChR2 (Krook‐Magnuson et al., 2013; Lu et al., 2016). For example, Krook‐Magnuson and colleagues (2013) reported that selectively stimulating PV interneurons reduced seizure duration by 43% compared with within‐animal nonstimulated control. However, stimulating larger populations of interneurons in vivo may further improve efficacy as suggested by Ledri and colleagues (2014); stimulating glutamate decarboxylase (Gad)‐2‐ChR2 neurons, which include PV, SOM, neuropeptide Y, and cholecystokinin interneuronal subpopulations, was more effective for suppressing epileptiform activity in slice preparations than either PV or SOM interneurons alone. Although a direct comparison between targeting mixed and specific interneuron populations for in vivo seizure control has yet to be performed, stimulating mixed interneuron cell types in vivo has been shown to be effective for controlling intrahippocampal KA‐induced acute seizures (Lu et al., 2016). Also, both halorhodopsin and ChR may be less effective at the later stages of seizures because of the accumulation of intracellular chloride (Huberfeld et al., 2007; Ellender et al., 2014; Alfonsa et al., 2015; Soper et al., 2015). One potential means of suppressing activity without directly modulating intracellular chloride is the opsin archaeorhodopsin, an outward proton pump (Raimondo et al., 2012; Pavlov et al., 2013; Alfonsa et al., 2015). However, although archaeorhodopsin has been effective for stopping epileptiform activity in vitro, there are no published reports of its effects on seizures in vivo at the time of writing.
For each form of epilepsy, multiple regions may be good targets for seizure control, and it is important to understand which are the most effective with the fewest side effects. For example, with a mouse model of temporal lobe epilepsy, Krook‐Magnuson and colleagues (2014) reported that spontaneous seizures could be controlled in regions including the ipsilateral and contralateral hippocampi, ipsilateral dentate gyrus, and the cerebellum. However, targeting PV neurons in the vermis of the cerebellum not only successfully stopped seizures on demand but also increased the time between seizures. Furthermore, targeting the cerebellum may also be effective for controlling absence seizures (Kros et al., 2015). Whether the cerebellum or other regions, such as the superior colliculus (Soper et al., 2015), can generally alleviate seizure activity clinically remains to be seen, but identifying regions with general antiseizure properties would be particularly useful for treating refractory epilepsies, such as multifocal epilepsies, in which surgery is not an option (Chen et al., 2015; West et al., 2015). Optogenetic seizure control has exciting potential for epilepsy therapy, but it is important to note that optogenetics is still in its infancy, and there will be significant hurdles before translation to the clinic becomes a reality. In contrast, deep brain stimulation (DBS) has a longer clinical history and has been recently approved for seizure control in the clinic (Morrell, 2011; Fisher and Velasco, 2014; Bergey et al., 2015). Although the mechanisms that underlie DBS are not well understood, studies suggest that the regions chosen for stimulation and the choice of stimulation parameters determine its efficacy (Lozano and Lipsman, 2013). Thus, in the short‐term, one promising application of optogenetics research may be to inform the location and stimulation parameters that will be most effective for seizure control with DBS (Chiang et al., 2014; Fisher and Velasco, 2014; Bergey et al., 2015; Ladas et al., 2015).
IMAGING SEIZURE NETWORKS
Optogenetic control of seizures holds much promise as a potential therapy, but little is known about the consequences that these focal interventions may have on seizure networks and underlying brain function. Evaluating seizure activity and its effects across the whole brain in vivo will help optimize treatments and limit potential off‐target effects (Faingold and Blumenfeld, 2015). Combining optogenetics with imaging tools such as fMRI (Lee et al., 2010; Desai et al., 2011; Kahn et al., 2011; Duffy et al., 2015; Liu et al., 2015; Weitz et al., 2015) or positron emission tomography (Kolodziej et al., 2014) has significant potential as a means of understanding these seizure networks in greater detail.
Visualizing activity over the whole brain can be achieved with fMRI as a readout and optogenetic stimulation to modulate activity (Lee et al., 2010; Duffy et al., 2015; Weitz et al., 2015). For example, stimulating the intermediate and dorsal hippocampus has been shown to evoke activity in distinct networks (Weitz et al., 2015). Although intermediate hippocampal stimulation resulted in widespread activity that included cortical and subcortical regions, stimulating the dorsal hippocampus resulted in activity restricted to the limbic system. Notably, dorsal hippocampal stimulation resulted in decreased fMRI signal in the contralateral dentate gyrus. This response may indicate the presence of seizure modulating activity attributed to the dentate gyrus. Because seizures are often detected and classified with physiology, simultaneous measurement of electrophysiological signatures allow confirmation of seizures by using well‐accepted measures (Logothetis, 2002; Kim and Ogawa, 2012). By developing MRI‐compatible optrodes for simultaneous LFP and fMRI in rats sedated with dexmedetomidine, we were able to show that stimulating hippocampal CamKII‐expressing neurons with ChR2 (using a subseizure threshold light intensity) results in activity restricted to the ipsilateral hippocampus and septum. However, when the light intensity delivered to the brain was increased, seizure‐like afterdischarges were generated and were confirmed with fMRI and electrophysiology together. The fMRI time series results in a prolonged hemodynamic response during afterdischarges, and simultaneously acquired LFP measurements allow a more informed analysis of the fMRI data. During afterdischarges, the activated network extended to the contralateral hemisphere, recruiting the hippocampus, septum and cerebellum as well as discrete cortical and thalamic regions (Duffy et al., 2015; see Fig. 1). These networks are similar but distinct from those reported following electrical hippocampal stimulation (Englot et al., 2009). During electrically induced afterdischarges, fMRI signal changes are observed in the hippocampus and septal nuclei, whereas reduced neuronal activity results in the cingulate, retrosplenial, and orbital frontal cortices. Genetically targeting only neuronal cells has the advantage of being able to evoke a hemodynamic response that is more physiological and less affected by stimulation of nonneuronal cells. Notably, despite seizures being evoked under a nonepileptogenic background, the activated regions encompass those that have been shown to be effective targets for controlling temporal lobe seizures (Krook‐Magnuson et al., 2013, 2014, 2015), demonstrating the potential that imaging seizure networks may have for identifying regions for seizure control. Nevertheless, the aforementioned studies imaged evoked seizures in a normal nonepileptogenic network. Because epilepsy is defined by significant alterations in the underlying networks, imaging seizures in a pathological setting may identify additional changes that may not be apparent in seizures evoked in a naïve brain.
Figure 1.
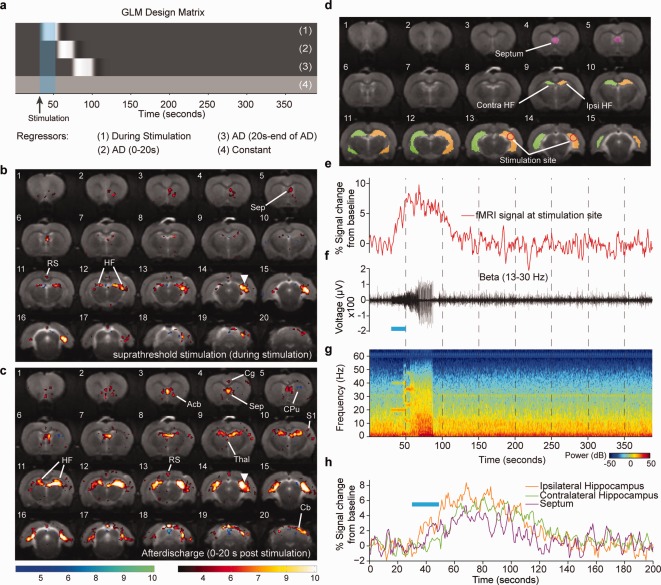
Single‐subject simultaneous LFP and optogenetic fMRI during seizure‐inducing (suprathreshold) stimulation of the hippocampus. a: GLM design matrix for the fMRI analysis. b: T‐statistic map showing regions of significant blood‐oxygen‐level‐dependent (BOLD) signal change during a seizure‐inducing stimulation (average of two trials). c: T‐statistic map showing regions of significant BOLD signal change during the first 20 sec of an epileptiform afterdischarge. Site of optical stimulation is marked by the arrowhead. d: Segmentation of four different regions of interest. e: fMRI time course for a single trial. f: Single trial simultaneously recorded LFP for the β band 13–30 Hz. g: Spectrogram of the LFP recording during fMRI acquisition. h: fMRI time course for the single trial from the ipsilateral hippocampus, septum, and contralateral hippocampus. Duration of optical stimulations is marked by blue bar. T‐statistic maps are thresholded at a significance level of P < 0.01, voxel‐wise false discovery rate corrected. Acb, accumbens nucleus; CPu, caudate putamen; RS, retrosplenial cortex; Thal, thalamus; Cg, cingulate cortex; HF, hippocampal formation; S1, primary somatosensory cortex; Sep, septum. Figure is reproduced from Duffy et al., 2015, with permission from Elsevier.
Although many regions may be activated during a seizure, it is unlikely that all of these regions are equally efficacious for seizure control (see Table 1). As a seizure starts and spreads through a network, targeting regions early in the propagation pathway may be more effective for abolishing the seizure than regions recruited later. Using a focal hippocampally evoked seizure mouse model and multiregional electrophysiological recordings, Lu and colleagues (2016) showed that seizures propagated from dentate gyrus to medial entorhinal cortex (MEC). When interneurons (by targeting vesicular GABA transporter or Gad2) in the dentate gyrus were stimulated, seizure activity in dentate gyrus, MEC, and motor cortex stopped. However, when interneurons in the MEC were stimulated, seizure activity was abolished in that region but persisted in the dentate gyrus and the motor cortex, demonstrating that successfully controlling seizure activity within a region does not necessarily indicate effective seizure control. Therefore imaging seizure networks and their interactions with proposed seizure control interventions can help identify critical nodes that are effective for completely stopping seizures.
Imaging seizure networks has also allowed researchers to identify a signature pattern in fMRI and electrophysiological recordings that is thought to be associated with the impaired consciousness that occurs during seizures (Blumenfeld, 2012; Motelow et al., 2015) or during stimulations that regulate arousal (Liu et al., 2015). Impaired consciousness during seizures is known to be associated with prominent frontoparietal slow‐wave activity on electrophysiological recordings in humans (Englot et al., 2010). Using fMRI in rats has allowed researchers to investigate the underlying circuit mechanisms of this phenomenon (Englot et al., 2009; Motelow et al., 2015). These data indicate a possible role for the lateral septum and anterior hypothalamus, which are activated during fMRI acquisitions and are known to have inhibitory connections with subcortical arousal systems. Using mice anesthetized with ketamine and xylazine, Furman and colleagues (2015) reduced this cortical slow‐wave activity during electrically evoked hippocampal seizures by optogenetically stimulating cholinergic brainstem neurons, suggesting that targeting these circuits may be effective for restoring consciousness. Thus, imaging seizure networks is important for allowing epilepsy researchers to go beyond stopping seizures to identifying regions that may not directly form part of the seizure network but have pathological implications for brain function.
SUMMARY
Optogenetics has proved to be a useful set of tools for epilepsy research, with powerful potential applications in the clinic. However, many questions remain. For example, little is known about many aspects of epilepsy circuits, including the role of subclasses of interneurons, glia, and G‐protein‐coupled receptors such as metabotropic glutamate or GABAB receptors (Craig and McBain, 2014; Kubota, 2014; Roux et al., 2014). The breadth of optogenetic tools continues to expand rapidly, including opsins that can be excited with different wavelengths and that have improved kinetics or that can be switched on for extended periods with a single light pulse and switched off with another (Mattis et al., 2012). Furthermore, engineered opsins now come as inhibitory chloride channels or G‐protein‐coupled receptors and can even directly modulate gene expression and epigenetic processes (Konermann et al., 2013; Levitz et al., 2013; Berndt et al., 2014). Targeting specific cell populations with optogenetics can help us to understand the generation and propagation of seizures as well as the development of epilepsy. Modulation of circuits to interrupt seizure activity has exposed the possibility of highly specific antiepileptic strategies. Finally, these new techniques can be combined with whole‐brain imaging methods to observe seizure dynamics over the whole brain and during modulation of distinct neural circuits. Such imaging approaches can be used to identify specific nodes in networks as well as time points that might be crucial for seizure propagation or even the development of epilepsy. Optogenetics, in combination with the many other new technologies being developed, likely will pave the way for significant strides in our understanding and treatment of epilepsy in the near future.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no competing interests.
ROLE OF AUTHORS
MC, BAD, and JHL equally researched, wrote, and edited the Review.
SIGNIFICANCE Optogenetics is a relatively new technique that allows researchers to control brain cells and neural circuits by genetically engineering them to respond to light. Using optogenetic tools, epilepsy researchers have made breakthroughs in our understanding of seizures and the development of epilepsy. Recent studies have also highlighted a potential new therapeutic avenue. Furthermore, by combining optogenetics with whole‐brain imaging, seizure circuits can now be monitored in their entirety and manipulated with cell type specificity.
REFERENCES
- Alfonsa H, Merricks EM, Codadu NK, Cunningham MO, Deisseroth K, Racca C, Trevelyan AJ. 2015. The contribution of raised intraneuronal chloride to epileptic network activity. J Neurosci 35:7715–7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergey GK, Morrell MJ, Mizrahi EM, Goldman A, King‐Stephens D, Nair D, Srinivasan S, Jobst B, Gross RE, Shields DC, Barkley G, Salanova V, Olejniczak P, Cole A, Cash SS, Noe K, Wharen R, Worrell G, Murro AM, Edwards J, Duchowny M, Spencer D, Smith M, Geller E, Gwinn R, Skidmore C, Eisenschenk S, Berg M, Heck C, Van Ness P, Fountain N, Rutecki P, Massey A, O'Donovan C, Labar D, Duckrow RB, Hirsch LJ, Courtney T, Sun FT, Seale CG. 2015. Long‐term treatment with responsive brain stimulation in adults with refractory partial seizures. Neurology 84:810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Cossart R, Hirsch JC, Esclapez M, Ben‐Ari Y. 2000. What is GABAergic inhibition? How is it modified in epilepsy? Epilepsia 41:S90–S95. [DOI] [PubMed] [Google Scholar]
- Berndt A, Lee SY, Ramakrishnan C, Deisseroth K. 2014. Structure‐guided transformation of channelrhodopsin into a light‐activated chloride channel. Science 344:420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H. 2012. Impaired consciousness in epilepsy. Lancet Neurol 11:814–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. 2005. Millisecond‐timescale, genetically targeted optical control of neural activity. Nat Neurosci 8:1263–1268. [DOI] [PubMed] [Google Scholar]
- Brodie MJ. 2010. Antiepileptic drug therapy: the story so far. Seizure 19:650–655. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. 1997. Neuron loss, granule cell axon reorganization, and functional changes in the dentate gyrus of epileptic kainate‐treated rats. J Comp Neurol 385:385–404. [PubMed] [Google Scholar]
- Buckmaster PS, Lew FH. 2011. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J Neurosci 31:2337–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin JA, Carlén M, Meletis K, Knoblich U, Zhang F, Deisseroth K, Tsai L‐H, Moore CI. 2010. Targeted optogenetic stimulation and recording of neurons in vivo using cell‐type‐specific expression of channelrhodopsin‐2. Nat Protoc 5:247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PC, Baumgartner J, Seo JH, Korostenskaja M, Lee KH. 2015. Bilateral intracranial EEG with corpus callosotomy may uncover seizure focus in nonlocalizing focal epilepsy. Seizure 24:63–69. [DOI] [PubMed] [Google Scholar]
- Chiang C‐C, Ladas TP, Gonzalez‐Reyes LE, Durand DM. 2014. Seizure suppression by high‐frequency optogenetic stimulation using in vitro and in vivo animal models of epilepsy. Brain Stim 7:890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy M, Dubé CM, Ehrengruber M, Baram TZ. 2014a. Inflammatory processes, febrile seizures, and subsequent epileptogenesis. Epilepsy Curr 14:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy M, Dubé CM, Patterson K, Barnes SR, Maras P, Blood AB, Hasso AN, Obenaus A, Baram TZ. 2014b. A novel, noninvasive, predictive epilepsy biomarker with clinical potential. J Neurosci 34:8672–8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig MT, McBain CJ. 2014. The emerging role of GABAB receptors as regulators of network dynamics: fast actions from a “slow” receptor? Curr Opin Neurobiol 26:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham M, Cho J‐H, Leung A, Savvidis G, Ahn S, Moon M, Lee PKJ, Han JJ, Azimi N, Kim K‐S, Bolshakov VY, Chung S. 2014. hPSC‐derived maturing GABAergic interneurons ameliorate seizures and abnormal behavior in epileptic mice. Cell Stem Cell 15:559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K. 2015. Optogenetics: 10 years of microbial opsins in neuroscience. Nat Neurosci 18:1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai M, Kahn I, Knoblich U, Bernstein J, Atallah H, Yang a, Kopell N, Buckner RL, Graybiel AM, Moore CI, Boyden ES. 2011. Mapping brain networks in awake mice using combined optical neural control and fMRI. J Neurophysiol 105:1393–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy BA, Choy M, Riegler J, Wells JA, Anthony DC, Scott RC, Lythgoe MF. 2012. Imaging seizure‐induced inflammation using an antibody targeted iron oxide contrast agent. Neuroimage 60:1149–1155. [DOI] [PubMed] [Google Scholar]
- Duffy BA, Choy M, Chuapoco MR, Madsen M, Lee JH. 2015. MRI compatible optrodes for simultaneous LFP and optogenetic fMRI investigation of seizure‐like afterdischarges. Neuroimage 123:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellender TJ, Raimondo J V, Irkle A, Lamsa KP, Akerman CJ. 2014. Excitatory effects of parvalbumin‐expressing interneurons maintain hippocampal epileptiform activity via synchronous afterdischarges. J Neurosci 34:15208–15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englot DJ, Mishra AM, Mansuripur PK, Herman P, Hyder F, Blumenfeld H. 2008. Remote effects of focal hippocampal seizures on the rat neocortex. J Neurosci 28:9066–9081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englot DJ, Modi B, Mishra AM, DeSalvo M, Hyder F, Blumenfeld H. 2009. Cortical deactivation induced by subcortical network dysfunction in limbic seizures. J Neurosci 29:13006–13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englot DJ, Yang L, Hamid H, Danielson N, Bai X, Marfeo A, Yu L, Gordon A, Purcaro MJ, Motelow JE, Agarwal R, Ellens DJ, Golomb JD, Shamy MC, Zhang H, Carlson C, Doyle W, Devinsky O, Vives K, Spencer DD, Spencer SS, Schevon C, Zaveri HP, Blumenfeld H. 2010. Impaired consciousness in temporal lobe seizures: role of cortical slow activity. Brain 133:3764–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faingold CL, Blumenfeld H. 2015. Targeting neuronal networks with combined drug and stimulation paradigms guided by neuroimaging to treat brain disorders. Neuroscientist 21:460–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RS, Velasco AL. 2014. Electrical brain stimulation for epilepsy. Nat Rev Neurol 10:261–270. [DOI] [PubMed] [Google Scholar]
- Furman M, Zhan Q, McCafferty C, Lerner BA, Motelow JE, Meng J, Ma C, Buchanan GF, Witten IB, Deisseroth K, Cardin JA, Blumenfeld H. 2015. Optogenetic stimulation of cholinergic brainstem neurons during focal limbic seizures: effects on cortical physiology. Epilepsia 56:e198–e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giachello CNG, Baines RA. 2015. Inappropriate neural activity during a sensitive period in embryogenesis results in persistent seizure‐like behavior. Curr Biol 25:2964–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard GV, McIntyre DC, Leech CK. 1969. A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol 25:295–330. [DOI] [PubMed] [Google Scholar]
- Henderson KW, Gupta J, Tagliatela S, Litvina E, Zheng X, Van Zandt MA, Woods N, Grund E, Lin D, Royston S, Yanagawa Y, Aaron GB, Naegele JR. 2014. Long‐term seizure suppression and optogenetic analyses of synaptic connectivity in epileptic mice with hippocampal grafts of GABAergic interneurons. J Neurosci 34:13492–13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashima M, Kinoshita H, Yamaguchi N, Koshino Y. 1996. Activation of GABAergic function necessary for afterdischarge generation in rat hippocampal slices. Neurosci Lett 207:101–104. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. 2007. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci 27:9866–9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberfeld G, Menendez de la Prida L, Pallud J, Cohen I, Le Van Quyen M, Adam C, Clemenceau S, Baulac M, Miles R. 2011. Glutamatergic preictal discharges emerge at the transition to seizure in human epilepsy. Nat Neurosci 14:627–634. [DOI] [PubMed] [Google Scholar]
- Hunt RF, Girskis KM, Rubenstein JL, Alvarez‐Buylla A, Baraban SC. 2013. GABA progenitors grafted into the adult epileptic brain control seizures and abnormal behavior. Nat Neurosci 16:692–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn I, Desai M, Knoblich U, Bernstein J, Henninger M, Graybiel AM, Boyden ES, Buckner RL, Moore CI. 2011. Characterization of the functional MRI response temporal linearity via optical control of neocortical pyramidal neurons. J Neurosci 31:15086–15091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J. 1997. Long‐lasting GABA‐mediated depolarization evoked by high‐frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network‐driven, bicarbonate‐dependent K+ transient. J Neurosci 17:7662–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley MS, Jacobs MP, Lowenstein DH. 2009. The NINDS epilepsy research benchmarks. Epilepsia 50:579–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S‐G, Ogawa S. 2012. Biophysical and physiological origins of blood oxygenation level‐dependent fMRI signals. J Cereb Blood Flow Metab 32:1188–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziej A, Lippert M, Angenstein F, Neubert J, Pethe A, Grosser OS, Amthauer H, Schroeder UH, Reymann KG, Scheich H, Ohl FW, Goldschmidt J. 2014. SPECT‐imaging of activity‐dependent changes in regional cerebral blood flow induced by electrical and optogenetic self‐stimulation in mice. Neuroimage 103:171–180. [DOI] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Hsu PD, Heidenreich M, Cong L, Platt RJ, Scott DA, Church GM, Zhang F. 2013. Optical control of mammalian endogenous transcription and epigenetic states. Nature 500:472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook‐Magnuson E, Armstrong C, Oijala M, Soltesz I. 2013. On‐demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun 4:1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook‐Magnuson E, Szabo GG, Armstrong C, Oijala M, Soltesz I. 2014. Cerebellar directed optogenetic intervention inhibits spontaneous hippocampal seizures in a mouse model of temporal lobe epilepsy. eNeuro 1:e.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook‐Magnuson E, Armstrong C, Bui A, Lew S, Oijala M, Soltesz I. 2015. In vivo evaluation of the dentate gate theory in epilepsy. J Physiol 593:2379–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kros L, Eelkman Rooda OHJ, Spanke JK, Alva P, van Dongen MN, Karapatis A, Tolner EA, Strydis C, Davey N, Winkelman BHJ, Negrello M, Serdijn WA, Steuber V, van den Maagdenberg AMJM, De Zeeuw CI, Hoebeek FE. 2015. Cerebellar output controls generalized spike‐and‐wave discharge occurrence. Ann Neurol 77:1027–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y. 2014. Untangling GABAergic wiring in the cortical microcircuit. Curr Opin Neurobiol 26:7–14. [DOI] [PubMed] [Google Scholar]
- Ladas TP, Chiang C‐C, Gonzalez‐Reyes LE, Nowak T, Durand DM. 2015. Seizure reduction through interneuron‐mediated entrainment using low frequency optical stimulation. Exp Neurol 269:120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledri M, Madsen MG, Nikitidou L, Kirik D, Kokaia M. 2014. Global optogenetic activation of inhibitory interneurons during epileptiform activity. J Neurosci 34:3364–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Durand R, Gradinaru V, Zhang F, Goshen I, Kim D‐S, Fenno LE, Ramakrishnan C, Deisseroth K. 2010. Global and local fMRI signals driven by neurons defined optogenetically by type and wiring. Nature 465:788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung L‐WS. 1987. Hippocampal electrical activity following local tetanization. I. Afterdischarges. Brain Res 419:173–187. [DOI] [PubMed] [Google Scholar]
- Levitz J, Pantoja C, Gaub B, Janovjak H, Reiner A, Hoagland A, Schoppik D, Kane B, Stawski P, Schier AF, Trauner D, Isacoff EY. 2013. Optical control of metabotropic glutamate receptors. Nat Neurosci 16:507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillis KP, Kramer MA, Mertz J, Staley KJ, White JA. 2012. Pyramidal cells accumulate chloride at seizure onset. Neurobiol Dis 47:358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lee HJ, Weitz AJ, Fang Z, Lin P, Choy M, Fisher R, Pinskiy V, Tolpygo A, Mitra P, Schiff N, Lee JH. 2015. Frequency‐selective control of cortical and subcortical networks by central thalamus. Elife 4:e09215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis NK. 2002. The neural basis of the blood‐oxygen‐level‐dependent functional magnetic resonance imaging signal. Philos Trans R Soc Lond B Biol Sci 357:1003–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano AM, Lipsman N. 2013. Probing and regulating dysfunctional circuits using deep brain stimulation. Neuron 77:406–424. [DOI] [PubMed] [Google Scholar]
- Lu Y, Zhong C, Wang L, Wei P, He W, Huang K, Zhang Y, Zhan Y, Feng G, Wang L. 2016. Optogenetic dissection of ictal propagation in the hippocampal–entorhinal cortex structures. Nat Commun 7:10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasiuk K, Kontula L, Pitkänen A. 2003. cDNA profiling of epileptogenesis in the rat brain. Eur J Neurosci 17:271–279. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Pretorius JK, Babb TL. 1995. Influence of the type of initial precipitating injury and at what age it occurs on course and outcome in patients with temporal lobe seizures. J Neurosurg 82:220–227. [DOI] [PubMed] [Google Scholar]
- Mattis J, Tye KM, Ferenczi EA, Ramakrishnan C, O'Shea DJ, Prakash R, Gunaydin LA, Hyun M, Fenno LE, Gradinaru V, Yizhar O, Deisseroth K. 2012. Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat Methods 9:159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrell MJ. 2011. Responsive cortical stimulation for the treatment of medically intractable partial epilepsy. Neurology 77:1295–1304. [DOI] [PubMed] [Google Scholar]
- Motelow JE, Li W, Zhan Q, Mishra AM, Sachdev RNS, Liu G, Gummadavelli A, Zayyad Z, Lee HS, Chu V, Andrews JP, Englot DJ, Herman P, Sanganahalli BG, Hyder F, Blumenfeld H. 2015. Decreased subcortical cholinergic arousal in focal seizures. Neuron 85:561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissinen J, Halonen T, Koivisto E, Pitkänen A. 2000. A new model of chronic temporal lobe epilepsy induced by electrical stimulation of the amygdala in rat. Epilepsy Res 38:177–205. [DOI] [PubMed] [Google Scholar]
- Osawa S‐I, Iwasaki M, Hosaka R, Matsuzaka Y, Tomita H, Ishizuka T, Sugano E, Okumura E, Yawo H, Nakasato N, Tominaga T, Mushiake H. 2013. Optogenetically induced seizure and the longitudinal hippocampal network dynamics. PLoS One 8:e60928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov I, Kaila K, Kullmann DM, Miles R. 2013. Cortical inhibition, pH, and cell excitability in epilepsy: what are optimal targets for antiepileptic interventions? J Physiol 591:765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz JT, Davidson TJ, Frechette ES, Delord B, Parada I, Peng K, Deisseroth K, Huguenard JR. 2013. Closed‐loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nat Neurosci 16:64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Zhang N, Wei W, Huang CS, Cetina Y, Otis TS, Houser CR. 2013. A reorganized GABAergic circuit in a model of epilepsy: evidence from optogenetic labeling and stimulation of somatostatin interneurons. J Neurosci 33:14392–14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinel JP, Rovner LI. 1978. Electrode placement and kindling‐induced experimental epilepsy. Exp Neurol 58:335–346. [DOI] [PubMed] [Google Scholar]
- Pitkänen A, Schwartzkroin PA, Moshé SL, Mareš P, Kubová H. 2006. Electrical stimulation: induced models of seizures In: Pitkänen A, Schwartzkroin PA, Moshé SL, editors. Models of seizures and epilepsy. Cambridge, MA: Academic Press; p 153–159. [Google Scholar]
- Putnam TJ, Merritt HH. 1937. Experimental determination of the anticonvulsant properties of some phenyl derivatives. Science 85:525–526. [DOI] [PubMed] [Google Scholar]
- Racine RJ. 1972. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr Clin Neurophysiol 32:281–294. [DOI] [PubMed] [Google Scholar]
- Raimondo J V, Kay L, Ellender TJ, Akerman CJ. 2012. Optogenetic silencing strategies differ in their effects on inhibitory synaptic transmission. Nat Neurosci 15:1102–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux L, Stark E, Sjulson L, Buzsáki G. 2014. In vivo optogenetic identification and manipulation of GABAergic interneuron subtypes. Curr Opin Neurobiol 26:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessolo M, Marcon I, Bovetti S, Losi G, Cammarota M, Ratto GM, Fellin T, Carmignoto G. 2015. Parvalbumin‐positive inhibitory interneurons oppose propagation but favor generation of focal epileptiform activity. J Neurosci 35:9544–9557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiri Z, Manseau F, Lévesque M, Williams S, Avoli M. 2015. Interneuron activity leads to initiation of low‐voltage fast‐onset seizures. Ann Neurol 77:541–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorvon SD. 2009a. Drug treatment of epilepsy in the century of the ILAE: the first 50 years, 1909–1958. Epilepsia 50(Suppl 3):69–92. [DOI] [PubMed] [Google Scholar]
- Shorvon SD. 2009b. Drug treatment of epilepsy in the century of the ILAE: the second 50 years, 1959–2009. Epilepsia 50(Suppl 3):93–130. [DOI] [PubMed] [Google Scholar]
- Soper C, Wicker E, Kulick CV, N'Gouemo P, Forcelli PA. 2015. Optogenetic activation of superior colliculus neurons suppresses seizures originating in diverse brain networks. Neurobiol Dis 87:102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel E. 1937. Quantitative determination of reactivity by electric stimulation of the brain with the skull intact. J Lab Clin Med 22:1274–1276. [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR. 1995. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science 269:977–981. [DOI] [PubMed] [Google Scholar]
- Sukhotinsky I, Chan AM, Ahmed OJ, Rao VR, Gradinaru V, Ramakrishnan C, Deisseroth K, Majewska AK, Cash SS. 2013. Optogenetic delay of status epilepticus onset in an in vivo rodent epilepsy model. PLoS One 8:e62013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom M, Bertram EH. 2012. Temporal lobe epilepsy. Handb Clin Neurol 107:225–240. [DOI] [PubMed] [Google Scholar]
- Tønnesen J, Sørensen AT, Deisseroth K, Lundberg C, Kokaia M. 2009. Optogenetic control of epileptiform activity. Proc Natl Acad Sci U S A 106:12162–12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda I, Fujita S, Thamattoor AK, Buckmaster PS. 2015. Unit activity of hippocampal interneurons before spontaneous seizures in an animal model of temporal lobe epilepsy. J Neurosci 35:6600–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Muldoon SF, Merricks EM, Racca C, Staley KJ. 2015. The role of inhibition in epileptic networks. J Clin Neurophysiol 32:227–234. [DOI] [PubMed] [Google Scholar]
- Viitanen T, Ruusuvuori E, Kaila K, Voipio J. 2010. The K+‐Cl cotransporter KCC2 promotes GABAergic excitation in the mature rat hippocampus. J Physiol 588:1527–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner FB, Truccolo W, Wang J, Nurmikko A V. 2015. Spatiotemporal dynamics of optogenetically induced and spontaneous seizure transitions in primary generalized epilepsy. J Neurophysiol 113:2321–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitz AJ, Fang Z, Lee HJ, Fisher RS, Smith WC, Choy M, Liu J, Lin P, Rosenberg M, Lee JH. 2015. Optogenetic fMRI reveals distinct, frequency‐dependent networks recruited by dorsal and intermediate hippocampus stimulations. Neuroimage 107:229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West S, Nolan SJ, Cotton J, Gandhi S, Weston J, Sudan A, Ramirez R, Newton R. 2015. Surgery for epilepsy. Cochrane Database Syst Rev 7:CD010541. [DOI] [PubMed] [Google Scholar]
- Wykes RC, Heeroma JH, Mantoan L, Zheng K, MacDonald DC, Deisseroth K, Hashemi KS, Walker MC, Schorge S, Kullmann DM. 2012. Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med 4:161ra152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekhlef L, Breschi GL, Lagostena L, Russo G, Taverna S. 2015. Selective activation of parvalbumin‐ or somatostatin‐expressing interneurons triggers epileptic seizure‐like activity in mouse medial entorhinal cortex. J Neurophysiol 113:1616–1630. [DOI] [PubMed] [Google Scholar]
- Ziburkus J, Cressman JR, Barreto E, Schiff SJ. 2006. Interneuron and pyramidal cell interplay during in vitro seizure‐like events. J Neurophysiol 95:3948–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]