

Letter to the Editor
Nakamura et al. have recently reported on an accumulation of phosphorylated α-synuclein in subpial and periventricular astrocytes in a subset of patients with multiple system atrophy (MSA) in Neuropathology.1 The authors revealed that this finding is specific for MSA patients and possibly related with a long disease duration. The morphology and location of this astrocytic pathology are reminiscent of aging-related tau astrogliopathy (ARTAG), which is characterized by thorn-shaped astrocytes (TSA) or granular/fuzzy astrocytes in subependymal, subpial, perivascular, gray matter, and white matter regions.2 We previously reported that 7% of MSA patients (10/139) had ARTAG and these patients had the older age at death than MSA patients without ARTAG.3 We aimed to determine whether the accumulation of α-synuclein in subpial and perivascular astrocytes was identified in our MSA cohort and to clarify the specificity of this pathology in patients with MSA.
For neuropathological analysis, 140 patients with MSA were available in the Mayo Clinic brain bank in Jacksonville. The cohort included 55 women and 85 men ranging in age at death from 47 to 89 years. Given that the striatum was the most affected region of ARTAG,3 brain tissue containing the striatum and basal forebrain was sampled, embedded in paraffin wax, and mounted on glass slides at 5 μm thickness. Tissue was immunostained with an antibody against α-synuclein (NACP, rabbit polyclonal, 1:3000, formic acid pretreatment) or phospho-tau (CP13, mouse monoclonal, 1:1000; a gift from Dr. Peter Davies, Feinstein Institute for Medical Research). In patients with astrocytic accumulation of α-synuclein, tissue was processed for immunofluorescence double-staining with antibodies against α-synuclein (NACP, 1:2000) and phospho-tau (CP13, 1:1000). In addition, we screened 100 patients with Lewy body disease (LBD),4 38 patients with incidental LBD (primary diagnoses were Alzheimer Disease in 19, progressive supranuclear palsy in 6, corticobasal degeneration in 3, primary age-related tauopathy in 3, Frontotemporal lobar degeneration with ubiquitin in 2, pathological aging in 2, neurologically normal in 2, and vascular disease in 1 patients),5 and 26 neurologically normal patients without any coexisting Lewy-related pathology.
Similar to previous research,1 the accumulation of α-synuclein in subpial or perivascular astrocytes in the striatum were observed in 11% (16/140) of MSA patients (Table 1). Numerous TSAs were observed around the lenticulostriate arteries (Fig. 1A) and subpial mediobasal region in a section of the basal forebrain. The morphology of these findings resembles ARTAG (Fig. 1B). Of 16 MSA patients with α-synuclein positive astrogliopathy, 3 patients had concurrent ARTAG and the accumulation of α-synuclein in astrocytes was co-localized with tau (Fig. 1C). It is noteworthy that not all MSA with ARTAG cases (3/10) have α-synuclein positive astrogliopathy. Since Nakamura et al. reported that α-synuclein positive astrogliopathy was specific finding for MSA, we also screened LBD, incidental LBD including tauopathies and vascular disease, and neurologically normal patients. We detected the accumulation of α-synuclein in the subpial or perivascular astrocytes in the striatum in 4 LBD patients (Fig. 1D), but none in incidental LBD or neurologically normal patients. Of these 4 LBD patients, 3 patients had also ARTAG in the lenticulostriate arteries (Fig. 1E) and subpial mediobasal region. We conclude that α-synuclein positive astrogliopathy is not specific for MSA, but could be specific for α-synucleinopathies.
Table 1.
Clinical and pathological features of MSA and LBD with α-synuclein astrogliopathy
| Cases | Age at death | Duration | Sex | Clinical diagnosis | Pathological diagnosis | ARTAG |
|---|---|---|---|---|---|---|
| MSA-1 | 47 | 10 | F | MSA-P | SND/OPCA | − |
| MSA-2 | 55 | 12 | F | MSA | SND/OPCA | − |
| MSA-3 | 56 | 11 | F | MSA-P | SND/OPCA | − |
| MSA-4 | 59 | 2 | M | PSP | SND | + |
| MSA-5 | 63 | NA | M | PSP | SND/OPCA | − |
| MSA-6 | 67 | 6 | M | MSA-P | SND/OPCA | − |
| MSA-7 | 67 | NA | M | MSA | SND | − |
| MSA-8 | 68 | 10 | M | MSA | SND | − |
| MSA-9 | 68 | 5 | M | MSA-C | SND/OPCA | − |
| MSA-10 | 69 | 7 | M | MSA-P | SND | − |
| MSA-11 | 72 | 9 | M | MSA | SND | + |
| MSA-12 | 74 | 6 | F | PSP | SND | − |
| MSA-13 | 75 | 15 | M | MSA-P | OPCA | − |
| MSA-14 | 76 | 9 | F | MSA-P | SND | − |
| MSA-15 | 76 | 5 | F | MSA-P | SND/OPCA | − |
| MSA-16 | 83 | NA | M | PD | SND/OPCA | + |
| LBD-1 | 75 | 16 | M | DLB | TLBD | + |
| LBD-2 | 78 | NA | F | PD | LBD | + |
| LBD-3 | 88 | 10 | F | PD | TLBD | + |
| LBD-4 | 93 | 19 | M | PDD | DLBD | − |
Abbreviations: DLB, dementia with Lewy bodies; DLBD, diffuse type Lewy body disease; MSA-C, MSA with predominant cerebellar ataxia; MSA-mixed; MSA-P, MSA with predominant parkinsonism; NA, not available; OPCA, MSA with predominant olivopontocerebellar involvement; MSA with predominant; PD, Parkinson disease; PDD, PD with dementia; PSP, progressive supranuclear palsy; SND, MSA with predominant striatonigral involvement; TLBD, transition type Lewy body disease.
Figure 1.
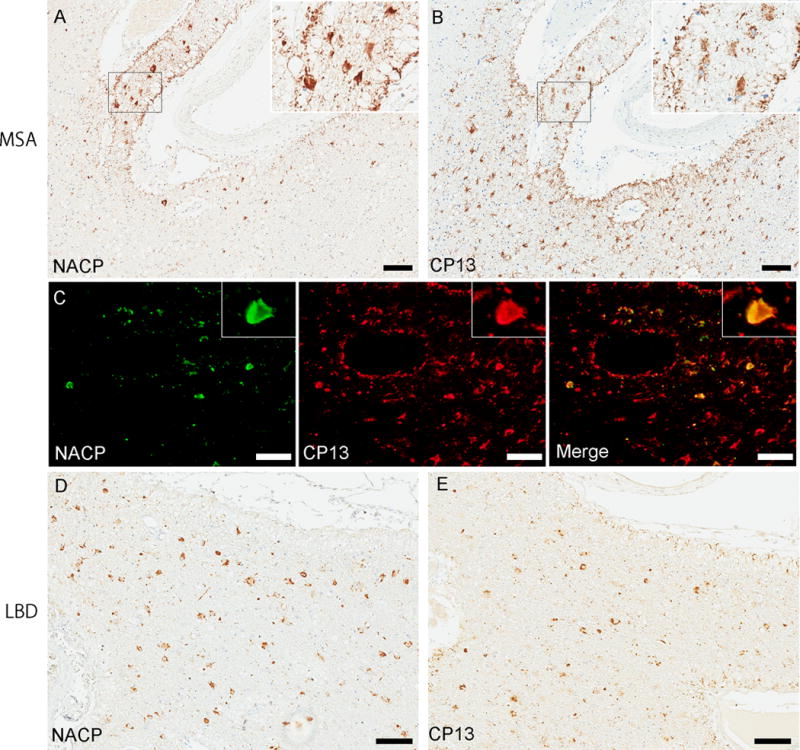
Immunohistochemistry for α-synuclein (NACP) or tau (CP13) and immunofluorescence double-staining on tissue from the basal forebrain in MSA-11 (A-C) or LBD-2 (D, E). A moderate amount of α-synuclein-positive thorn-shaped astrocytes (TSAs) and subpial astrocytes are observed surrounding the anterior perforating lenticulostriate arteries (A). The morphology of these findings resembles ARTAG; numerous tau-positive TSAs are seen surrounding the anterior perforating lenticulostriate arteries (B). Immunofluorescence double-staining for NACP and CP13 shows that a subset of astrocytes is positive for both α-synuclein and tau in the same MSA case (C). Similar findings are seen in LBD cases. A moderate amount of α-synuclein- or tau- positive TSAs are observed surrounding the anterior perforating lenticulostriate arteries (D, E). Bars = 100 μm in (A, B, D, and E) and 20 μm in (C).
ARTAG is associated with aging, hence the name, and we previously confirmed this in MSA patients; the age at death was older in MSA with ARTAG than that in MSA without ARTAG.3 Unexpectedly, however, the α-synuclein positive astrogliopathy seemed to be unrelated with aging. The age at death was not significantly different between MSA patients with and without astrocytic accumulation of α-synuclein (mean ± SD: 67 ± 9 years vs 67 ± 8 years, P = 0.755). Also, we did not find significant difference in the disease duration between the two groups [median (25th, 75th %-tile): 9 (6, 10) years vs 7 (5, 9) years, P = 0.181], although Nakamura et al. found the possible relation between this astrocytic pathology and a long disease duration. The discrepancy of the results of disease duration could be due to the different screening site; we screened only sections of the striatum and basal forebrain.
MSA can be clinicopathologically divided into two subtypes: MSA with predominant parkinsonism and MSA with predominant cerebellar ataxia.6 Given the fact that the proportion of each subtype is different between Asian and Caucasian populations, ethnic and genetic factors could contribute the etiology of MSA.7 In the present study based on the Caucasian MSA cohort, we confirmed that α-synuclein positive astrogliopathy was commonly seen in both Asian and Caucasian population. Although the morphology of α-synuclein positive astrogliopathy is similar to ARTAG, only a subset of cases had both pathologies. Given that the α-synuclein positive astrogliopathy was seen in only MSA and LBD, this pathology seems to be specific for α-synucleinopathies. Further study is necessary to understand the origin of α-synuclein in astrocytes and pathological role of the α-synuclein positive astrogliopathy.
Acknowledgments
We would like to thank the patients and their families who donated brains to help further the scientific understanding of neurodegeneration. The authors would also like to acknowledge Dr. Peter Davies (Feinstein Institute for Medical Research, LIJ-North Shore Health System, NY) for the monoclonal anti-tau antibody CP13, Linda Rousseau (Mayo Clinic, Jacksonville, FL) and Virginia Phillips (Mayo Clinic) for histologic support, and Monica Castanedes-Casey (Mayo Clinic) for immunohistochemistry support. Supported by NIH grant P50 NS072187 and a Jaye F. and Betty F. Dyer Foundation Fellowship.
References
- 1.Nakamura K, Mori F, Kon T, Tanji K, Miki Y, Tomiyama M, et al. Accumulation of phosphorylated alpha-synuclein in subpial and periventricular astrocytes in multiple system atrophy of long duration. Neuropathology. 2016;36:157–67. doi: 10.1111/neup.12243. [DOI] [PubMed] [Google Scholar]
- 2.Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 2016;131:87–102. doi: 10.1007/s00401-015-1509-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koga S, Dickson DW, Bieniek KF. Chronic Traumatic Encephalopathy Pathology in Multiple System Atrophy. J Neuropathol Exp Neurol. 2016;75:963–70. doi: 10.1093/jnen/nlw073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kosaka K, Yoshimura M, Ikeda K, Budka H. Diffuse type of Lewy body disease: progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree–a new disease? Clin Neuropathol. 1984;3:185–92. [PubMed] [Google Scholar]
- 5.Dickson DW, Fujishiro H, DelleDonne A, Menke J, Ahmed Z, Klos KJ, et al. Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol. 2008;115:437–44. doi: 10.1007/s00401-008-0345-7. [DOI] [PubMed] [Google Scholar]
- 6.Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–6. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ozawa T, Revesz T, Paviour D, Lees AJ, Quinn N, Tada M, et al. Difference in MSA phenotype distribution between populations: genetics or environment? J Parkinsons Dis. 2012;2:7–18. doi: 10.3233/JPD-2012-11056. [DOI] [PubMed] [Google Scholar]