

Abstract
The RNA-guided endonuclease Cpf1 is a promising tool for genome editing in eukaryotic cells1–7. However, the utility of the commonly used Acidaminococcus sp. BV3L6 Cpf1 (AsCpf1) and Lachnospiraceae bacterium ND2006 Cpf1 (LbCpf1) is limited by their requirement of a TTTV protospacer adjacent motif (PAM) in the DNA substrate. To address this limitation, we performed a structure-guided mutagenesis screen to increase the targeting range of Cpf1. We engineered two AsCpf1 variants carrying the mutations S542R/K607R and S542R/K548V/N552R, which recognize TYCV and TATV PAMs, respectively, with enhanced activities in vitro and in human cells. Genome-wide assessment of off-target activity using BLISS7 assay indicated that these variants retain high DNA targeting specificity, which we further improved by introducing an additional non-PAM-interacting mutation. Introducing the identified mutations at their corresponding positions in LbCpf1 similarly altered its PAM specificity. Together, these variants increase the targeting range of Cpf1 by approximately three-fold in human coding sequences to one cleavage site per ~11 bp.
Programmable endonucleases from class 2 microbial CRISPR-Cas systems have enabled a wide range of applications in eukaryotic genome editing1–7. In addition to the widely-used type II-A Cas9, recent work has demonstrated that Cpf1, a type V-A system, can mediate efficient genome editing. Cpf1 has several distinct advantages compared to Cas9; for instance, it has low mismatch tolerance4–7, does not require a tracrRNA, and can process its own crRNA array into mature crRNAs to facilitate targeting of multiple genes concurrently2, 3.
We previously identified two orthologs of Cpf1 with robust activity in mammalian cells, Acidaminococcus sp. BV3L6 Cpf1 (AsCpf1) and Lachnospiraceae bacterium ND2006 Cpf1 (LbCpf1)1, both of which require a TTTV protospacer adjacent motif (PAM), where V can be A, C, or G. For applications for which the location of the target site is critical, e.g. homology-directed repair or generation of loss-of-function mutations at specific exonic positions, the requirement of a TTTV PAM may limit the availability of suitable target sites, reducing the practical utility of Cpf1. To address this limitation, we aimed to engineer variants of Cpf1 that can recognize alternative PAM sequences in order to increase its targeting range.
Previous work has shown that the PAM preference of Cas9 can be altered by mutations to residues in close proximity to the PAM DNA duplex8–11. We sought to investigate whether the PAM preference of Cpf1, despite its strong evolutionary conservation across different orthologs1, can also be modified. Based on the crystal structure of AsCpf1 in complex with crRNA and target DNA12, we selected 60 residues in AsCpf1 in proximity to the PAM duplex for targeted mutagenesis (Fig. 1a and Supplementary Table 1a). By randomizing the codons at each position using cassette mutagenesis, we constructed a plasmid library of AsCpf1 variants encoding most single amino acid substitutions at these residues. The use of codon randomization allowed us to attain greater mutational coverage than would have been expected with error-prone PCR, since it prevents representational bias caused by the template sequence.
Figure 1.
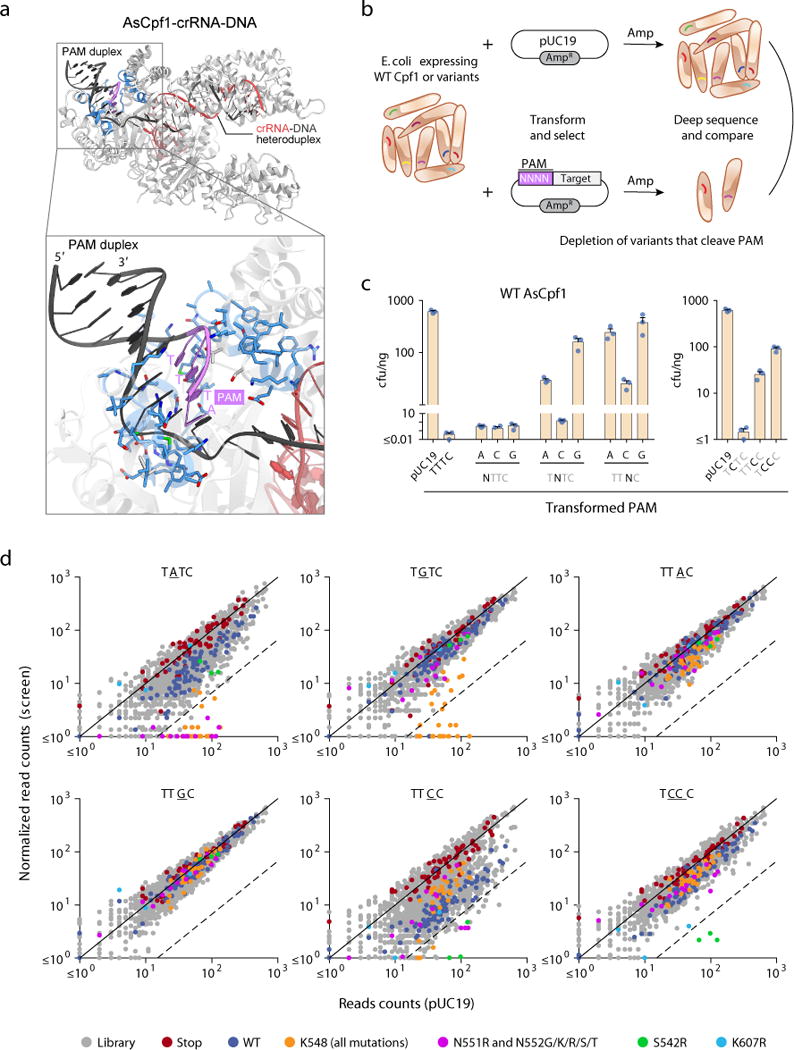
A bacterial interference-based negative selection screen identifies amino acid substitutions of AsCpf1 conferring activity at non-canonical PAMs. (a) Crystal structure of AsCpf1 (PDB ID: 5B43) in complex with crRNA and target DNA, highlighting the PAM nucleotides (magenta), and PAM-proximal residues selected for mutagenesis (blue). (b) Schematic of bacterial interference assay used to identify variants with altered PAM specificity. (c) Sensitivity of wild-type AsCpf1 to substitution mutations in the PAM as measured by bacterial interference. Bars show mean ± s.e.m. of n = 3 plated transformations. (d) Scatter plots of screen readout, highlighting depleted variants. Each dot represents a distinct wild-type or mutant codon. The dashed line indicates 15-fold depletion.
To identify variants within this library with cleavage activity at non-canonical PAMs, we adapted a plasmid interference-based depletion screen in E. coli1, 8, 13, 14 (Fig. 1b). In our modified assay, a pool of E. coli, each expressing crRNA and a variant of Cpf1 from a plasmid maintained with chloramphenicol, was transformed with a second plasmid carrying an ampicillin resistance gene and a target site bearing a mutated PAM. Successful cleavage of the second plasmid resulted in the loss of ampicillin resistance and subsequent cell death when grown on ampicillin selective media. By comparing the original library to the sequences of Cpf1-carrying plasmid DNA in surviving bacteria, we determined the variants that were depleted as a result of their novel cleavage activity of the mutated PAM.
To effectively use this approach to distinguish variants with non-canonical PAM activity from wild-type (WT) AsCpf1, we first determined PAM sequences at which WT AsCpf1 had minimal activity. We evaluated the sensitivity of WT AsCpf1 to substitution mutations in the PAM, as determined by E. coli death due to successful plasmid interference. We focused on PAMs with single nucleotide substitutions (i.e., NTTV, TNTV, and TTNV, where V was arbitrarily chosen to be C). When transformed with NTTC and TCTC PAMs, E. coli expressing WT AsCpf1 had negligible survival on ampicillin media (Fig. 1c), indicating that these PAM sequences supported AsCpf1-mediated DNA plasmid cleavage and were not usable for screening the variant library. By contrast, the other five PAMs with a single mutation (TATC, TGTC, TTAC, TTCC, and TTGC) had notable survival rates. We subsequently screened the variant library for activity at these five PAMs, as well as an additional PAM with a double mutation (TCCC) (Fig. 1d).
Following deep sequencing readout, ~86% of the possible variants at the targeted residue positions were represented with at least 15 reads in the pUC19-transformed negative control to allow assessment of their depletion. For TATC, TGTC, TTCC, and TCCC PAMs, at least one AsCpf1 variant in the library was highly depleted (≥15-fold) (Fig. 1d and Supplementary Table 1b). For TATC and TGTC, many of depleted variants were at Lys548, a conserved residue that forms hydrogen bonds with the PAM duplex12, 15. A number of hits were also observed for TTCC and TCCC, most notably an arginine substitution at Ser542, a non-conserved residue.
We evaluated whether variants identified in the screen had activity in HEK293T cells by targeting them to endogenous sites in two genes (DNMT1 and VEGFA) (Fig. 2a and Supplementary Fig. 1a). Most of the variants we tested generated indels at target sites with their corresponding PAMs; in particular, K548V was most active at a TATC target site, whereas S542R markedly increased activity for two TTCC target sites as well as a TCCC site. Combining the top single amino acid mutations into double and triple mutants further improved activity (Fig. 2a and Supplementary Fig. 1b). We selected the variants with the highest activity, S542R/K607R (hereafter referred to as RR) and S542R/K548V/N552R (hereafter referred to as RVR), for further investigation.
Figure 2.
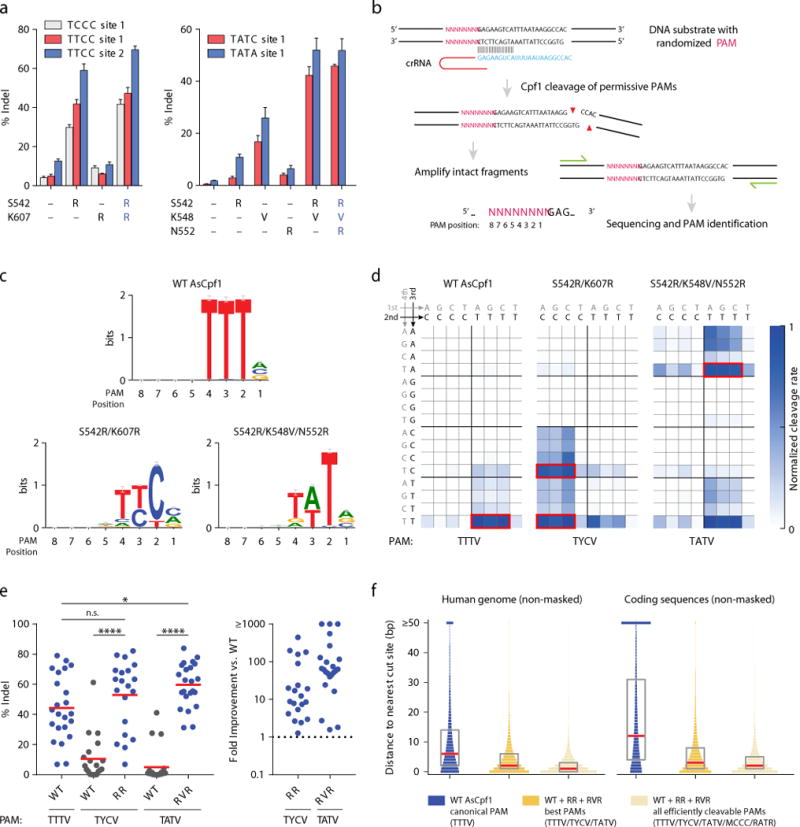
Construction and characterization of AsCpf1 variants with altered PAM specificities. (a) Combinatorial mutagenesis identifies AsCpf1 variants that cleave target sites with TYCV and TATV PAMs in HEK293T cells, where Y = C or T, and V = A, C, or G (see also Supplementary Fig. 1). Bars show mean ± s.e.m. for n = 4 transfected cell cultures. (b) Schematic of in vitro cleavage assay used to determine global PAM specificity (see also Supplementary Fig. 2–3). (c) Web logos of the most rapidly cleaved PAMs for wild-type (WT), S542R/K607R (RR), and S542R/K548V/N552R (RVR) variants. (d) Normalized cleavage rates for all 4-base PAMs for WT and variants. NNRN PAMs are not shown due to negligible cleavage. The most active PAMs are boxed in red. (e) Comparison of the activity of WT, RR, and RVR at their preferred PAMs at a diverse panel of target sites in HEK293T cells (see also Supplementary Fig. 5). For indel percentages, each dot represents the mean of n = 3 transfected cell cultures, and the red lines indicate the overall means within each group. For fold improvement, each dot represents the ratio of the means of the corresponding indel replicates. n.s. p > 0.05 (Mann-Whitney); *p < 0.05 (Mann-Whitney); ****p < 0.0001 (Wilcoxon signed-rank). (f) Targeting range of AsCpf1 variants in the human genome and in coding sequences (see also Supplementary Fig. 7). Plots show the probability mass function of the distance in base pairs to the nearest cleavage site. The boxplots indicate median and interquartile range. Genomic regions that contain Ns or masked repeats were ignored.
To assess the global PAM preference of the RR and RVR variants and compare them with WT AsCpf1, we adapted an in vitro PAM identification assay described previously (Fig. 2b)1, 16. We incubated cell lysate from HEK293T cells expressing AsCpf1 (or an engineered variant) with in vitro-transcribed crRNA and a library of plasmid DNA containing a constant target preceded by a degenerate sequence (5′-NNNNNNNN-target). By amplifying and deep sequencing the intact substrates and comparing with the negative control, we determined the sequences that were successfully cleaved. For each Cpf1 variant, nine reactions were carried out in parallel, each incubated for a different amount of time, in order to assess cleavage kinetics (see Methods and Supplementary Fig. 2–3).
As expected, WT AsCpf1 was most active at TTTV PAMs (Fig. 2c–d) and had lower activity at TTTT, supporting the previously reported definition of the WT PAM as TTTV1, 6. WT also cleaved other sequences including NTTV, TCTV, and TTCV at low rates, consistent with our observations in HEK293T cells (Supplementary Fig. 4) and in E. coli. By contrast, the RR and RVR variants had the highest activity at TYCV (where Y can be C or T) and TATV PAMs, respectively, compared to little or no activity for WT Cpf1 at those PAMs (Fig. 2c–d). The variant PAMs were not as strictly defined as that of WT: The RR variant also cleaved ACCC and CCCC PAMs (and, to a lesser extent, VYCV), and the RVR variant also cleaved RATR PAMs (where R can be A or G).
To assess the robustness of the engineered PAM activity, we investigated the activity of the RR and RVR variants at their preferred PAMs (i.e., TYCV and TATV, respectively) across a diverse panel of endogenous target sites in HEK293T cells (Fig. 2e and Supplementary Fig. 5). The RR and RVR variants achieved > 50% indel for 14 out of 20 TYCV sites (70%) and 18 out of 23 TATV sites (78%), respectively, compared to little or no activity for WT AsCpf1 at most of these sites (p < 0.0001 for both variants; Wilcoxon signed-rank). By comparison, WT AsCpf1 achieved > 50% indel for 8 out of 23 TTTV sites (35%). These data suggest that, at their respective preferred PAMs, the variants have comparable or slightly higher activity than the WT nuclease (Fig. 2e). The RR variant also exhibited substantial rates of editing in mouse Neuro2a cells (> 20% indel for 6 out of 9 TYCV sites) (Supplementary Fig. 6).
Based on our observations that the RR variant also cleaves VYCV PAMs in vitro, albeit at a substantially lower rate than TYCV, we tested the activity of the RR variant at a separate panel of VYCV sites in HEK293T cells (Supplementary Fig. 4). Across the four genes assessed (CFTR, DNMT1, EMX1, and VEGFA), the RR variant achieved > 20% indel for 24 out of 36 VYCV sites (67%), suggesting that, when necessary, target sites with VYCV PAMs can also be considered for editing with the RR variant.
To quantify how these Cpf1 PAM variants affect the targeting range of the CRISPR-Cpf1 system, we performed a computational analysis of the distribution of PAM sequences in the human genome (Fig. 2f and Supplementary Fig. 7), excluding Ns and masked repeats. When considering only the most active PAMs, the variants and WT collectively expand the targeting range of Cpf1 to one target site per ~11 bp in human coding sequences (corresponding to approximately 3-fold increase relative to WT alone) and reduce the median distance to the nearest cleavage site to 3 bp. Moreover, when considering a more broadly defined set of efficiently cleavable PAMs (in particular, the preferred PAMs plus MCCC and RATR, where M can be A or C), the targeting range is further expanded to one site per ~7 bp in human coding sequences, with a median distance to the nearest cleavage site of 2 bp.
We evaluated the genome-wide editing specificity of the RR and RVR variants using BLISS (double-strand breaks labeling in situ and sequencing), which quantifies DNA double-stranded breaks (DSBs) across the genome7 (Supplementary Table 2). To fairly compare the variants to WT, we selected target sites bearing PAMs that can be reliably cleaved by all three enzymes; TTTV was the only PAM that met this criterion, although it has lower activity for the RR variant. For three of the four target sites evaluated (VEGFA, GRIN2B, and DNMT1), no off-target activity was detected from deep sequencing of the BLISS-identified loci (Fig. 3a) for any of the nucleases. For the fourth target site (EMX1), BLISS identified six off-target sites with detectable indels; all six sites had a TTCA PAM and no more than one mismatch in the first 19 nucleotides of the guide. As expected, both variants had increased activity at these off-target sites compared to WT, consistent with their increased ability to recognize TTCA PAMs. On the other hand, when targeting a site in the RPL32P3 gene with known TTTV off-target sites5, the variants exhibited similar or reduced off-target activity (Fig. 3b), which is also consistent with PAM preference. Collectively, these results indicate that the variants retain a high level of editing specificity that is comparable to WT AsCpf1. We note that a few of the off-target sites with low indel frequencies were not detected by BLISS at the time point we sampled, likely reflecting the dependence of BLISS on the timing of DSB formation7.
Figure 3.
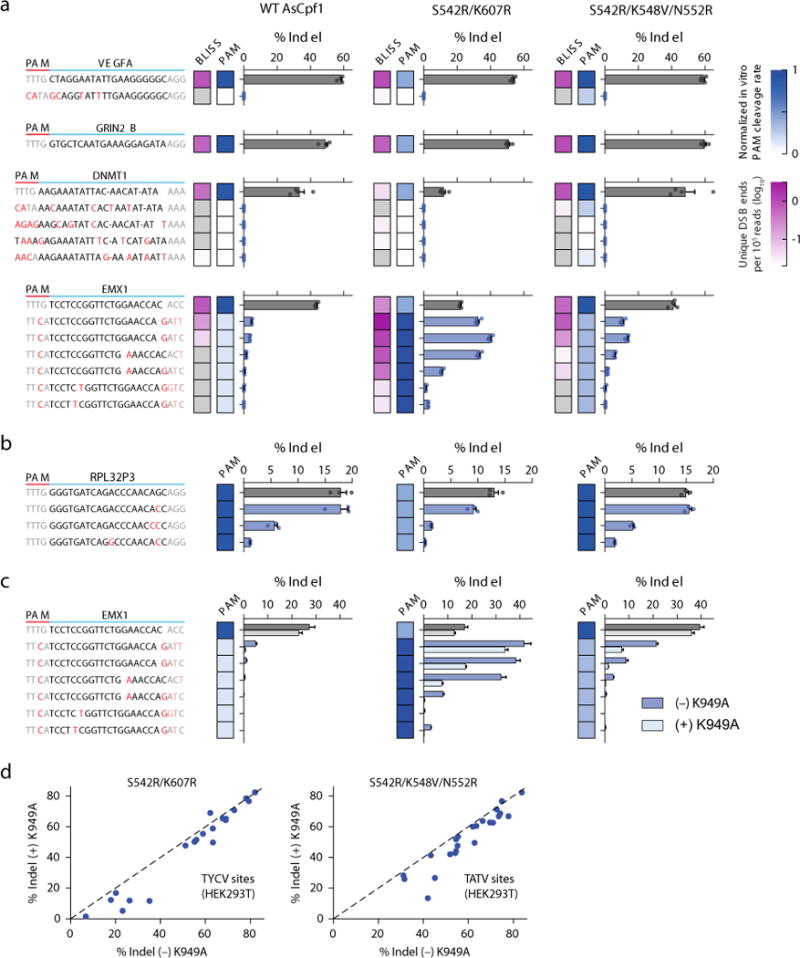
Specificity of AsCpf1 PAM variants. (a) DNA double-strand breaks labeling in situ and sequencing (BLISS) for 4 target sites (VEGFA, GRIN2B, EMX1, and DNMT1) in HEK293T cells. The log10 number of unique double-strand break (DSB) ends per 105 reads is indicated by the magenta heat map. The normalized PAM cleavage rates from the in vitro cleavage assay in Fig. 2d are indicated by the blue heat map. Each BLISS-identified cleavage site was independently assessed for indel formation (bar graphs). Bars show mean ± s.e.m. for n = 4 transfected cell cultures. Mismatches in bases 21–23 of the target are grayed as they have minimal impact on cleavage efficiency4, 5. (b) Evaluation of an additional target site in the RPL32P3 gene with known TTTV off-target sites5. (c) Addition of a K949A mutation improves the specificity of WT AsCpf1 and variants (see also Supplementary Fig. 8). For (b) and (c), bars show mean ± s.e.m. for n = 3 transfected cell cultures. (d) On-target efficiency of the RR and RVR variants ± K949A. Each dot represents the mean of n = 3 transfected cell cultures.
We investigated whether the specificity of AsCpf1 can be improved by removing non-specific contacts between positively-charged or polar residues and the target DNA, similar to strategies previously employed with Streptococcus pyogenes Cas9 (SpCas9)17, 18. We identified K949A, which is located in the cleft of the protein that is hypothesized to interact with the non-target DNA strand, as a candidate (Supplementary Fig. 8). When combined with the RR and RVR variants, K949A reduced cleavage at all off-target sites assessed (Fig. 3c) while maintaining high levels of on-target activity (Fig. 3d).
Because Cpf1-family endonucleases have strong sequence and structural homology, the 542, 548, 552, and 607 positions in AsCpf1 have clear correspondences in other Cpf1 orthologs (Supplementary Fig. 9 and Supplementary Table 3). Based on sequence alignment and the crystal structure19, we hypothesized that LbCpf1 could also be engineered to recognize TYCV and TATV PAMs by introducing the mutations G532R/K595R and G532R/K538V/Y542R, respectively (Supplementary Fig. 10a). These mutations altered the PAM specificity of LbCpf1 in the predicted manner (Supplementary Fig. 10b and Supplementary Fig. 11), suggesting that this approach may be generally applicable across Cpf1 orthologs.
In summary, we have demonstrated that despite its evolutionary conservation, the PAM preference of Cpf1-family endonucleases can be altered by suitable mutations to residues close to the PAM duplex. Using a structure-guided mutagenesis screen, we engineered two variants, RR and RVR, which can robustly cleave target sites with TYCV and TATV PAMs, respectively, in mammalian cells. We extended this approach to similarly modify a second Cpf1 ortholog. Finally, we introduced an additional mutation that enhanced Cpf1 specificity. Collectively, these engineered Cpf1 variants increase the targeting range of Cpf1 to one cleavage site for every ~11 bp in human coding sequences and provide useful additions to the CRISPR-Cas genome engineering toolbox.
Methods
Library construction
Human codon-optimized AsCpf1 driven by a T7 promoter was cloned into a modified pACYC backbone, and unique restriction sites were introduced flanking the selected PAM-proximal AsCpf1 residues via suitable silent mutations. For each residue, a mutagenic insert was synthesized as short complementary oligonucleotides (Integrated DNA Technologies), with the mutated codon replaced by a degenerate NNK mixture of bases (where K = G or T). Each degenerate codon position was also barcoded by creating a unique combination of silent mutations in non-mutated neighboring codons in order to correct for sequencing errors during screen readout. The variant library was assembled by cassette mutagenesis, mini-prepped, pooled, and precipitated with isopropanol.
E. coli negative selection screen
NovaBlue(DE3) E. coli (Novagen) were transformed with the variant library and plated on LB agar containing 25 μg/mL chloramphenicol. Surviving colonies were scraped and cultured in ZymoBroth with 25 μg/mL chloramphenicol to an O.D. of 0.4–0.6 and made competent using a Mix & Go kit (Zymo). For each mutant PAM screened, the competent E. coli pool was transformed with 100 ng target plasmid containing the mutant PAM, incubated on ice for 15–30 min, heat shocked at 42 °C for 30s, and plated on LB agar (Affymetrix) containing 100 μg/mL ampicillin and 25 μg/mL chloramphenicol in the absence of IPTG. A negative control was obtained by transforming the E. coli with pUC19, which lacks the target site. Plasmid DNA from surviving colonies was isolated by midi-prep (Qiagen). The regions containing mutations were amplified with custom primers containing Illumina adaptors and paired-end sequenced with a 600-cycle MiSeq kit (Illumina). Reads were filtered by requiring perfect matches to silent codon barcodes; a Phred quality (Q score) of at least 30 for each of the three NNK bases; and consistency between forward and reverse reads, when applicable. The read count for each variant was normalized assuming that the mean abundance of TAG (stop) codons was equivalent to the negative control.
In vitro PAM identification assay
Plasmids encoding the AsCpf1 variants were transfected into HEK293T cells as described below. Cell lysate was prepared with lysis buffer (20 mM HEPES, 100 mM KCl, 5 mM MgCl2, 1 mM DTT, 5% glycerol, 0.1% Triton X-100) supplemented with ETDA-free cOmplete Protease Inhibitor Cocktail (Roche). crRNA was transcribed in vitro using custom oligonucleotides and HiScribe T7 in vitro Transcription Kit (NEB) following the manufacturer’s recommended protocol. For the PAM library, a degenerate 8 bp sequence preceding a 33 bp target site1 was cloned into the MCS in pUC19, and the library was digested with AatII and LguI and gel extracted prior to use. Each in vitro cleavage reaction consisted of 1 μL 10× CutSmart buffer (NEB), 25 ng PAM library, 250 ng in vitro transcribed crRNA, 0.5 μL of cell lysate, and water for a total volume of 10 μL. Reactions were incubated at 37 °C and quenched by adding 50 μL Buffer PB (Qiagen) followed by column purification. Purified DNA was amplified with two rounds of PCR over 29 total cycles using custom primers containing Illumina adaptors and sequenced with a 75-cycle NextSeq kit (Illumina). For each Cpf1 variant, separate in vitro cleavage reactions were carried out for 1.15 min, 4 min, 10 min, 15 min, 20 min, 30 min, 40 min, 90 min, and 175 min. The unmodified library of degenerate sequences was used as the 0 min time point. A negative control, using lysate from unmodified HEK293T cells, was taken at 10 min.
Computational analysis of PAM cleavage kinetics
See also Supplementary Fig. 2–3. Sequencing reads were filtered by Phred quality (≥ 30 for all of the 8 degenerate PAM bases). For each cleavage reaction, a depletion ratio for each of the 48 PAM sequences was calculated as (normalized read count in cleavage reaction)/(normalization read count in negative control). Each depletion ratio was then divided by the median depletion ratio of all NNNNVRRT sequences, which were not cleaved by WT AsCpf1 or either of the variants. The depletion ratios of each PAM sequence (48 total) across time points for each Cpf1 variant were fit using non-linear least squares to an exponential decay model , where is the depletion ratio at time , and the terms , , and (the rate constant in min−1) are parameters. For each variant, the estimated cleavage rate k of each 4-base PAM was computed as the median cleavage rate of the 256 8-base sequences corresponding to that PAM; for instance, the cleavage rate of TTTA was computed as the median cleavage rate of the 256 sequences of the form NNNNTTTA. Finally, all cleavage rates were adjusted such that the highest rate of any 4-base PAM was equal to 1 for each variant.
Cell culture and transfection
Human embryonic kidney 293 and Neuro2a cell lines were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS (Gibco) at 37°C with 5% CO2 incubation. Cells were seeded one day prior to transfection in 24- or 96-well plates (Corning) at a density of approximately 1.2 × 105 cells per 24-well or 2.4 × 104 cells per 96-well and transfected at 50–80% confluency using Lipofectamine 2000 (Life Technologies), according to the manufacturer’s recommended protocol. For cell lysates, 500 ng of Cpf1 plasmid was delivered per 24-well. For indel analysis in HEK293T cells, a total of 400ng of Cpf1 plasmid plus 100ng crRNA plasmid was delivered per 24-well, or 100ng Cpf1 plus 50ng crRNA plasmid per 96-well. For BLISS and for indel analysis in Neuro2a cells, 500 ng of a plasmid with both Cpf1 and crRNA were delivered per 24-well. All indel and BLISS experiments used a guide length of 23 nucleotides.
Indel quantification
All indel rates were quantified by targeted deep sequencing (Illumina). For indel library preparation, cells were harvested approximately 3 days after transfection, and genomic DNA was extracted using a QuickExtract DNA extraction kit (Epicentre) by re-suspending pelleted cells in QuickExtract (80 μL per 24-well, or 20 μL per 96-well), followed by incubation at 65°C for 15 min, 68°C for 15 min and 98°C for 10 min. Amplicons for deep sequencing were generated using two rounds of PCR to attach Illumina handles. Indels were counted computationally by searching each amplicon for exact matches with strings delineating the ends of a 50–70 bp window around the cut site. The distance in bp between these strings was then compared to the corresponding distance in the reference genome, and the amplicon was counted as an indel if the two distances differed. For each sample, the indel rate was determined as (number of reads with an indel)/(number of total reads). Samples with fewer than 1000 total reads were excluded. Where negative control data is not shown, indel percentages represent background-subtracted maximum likelihood estimates. In particular, for a sample with R total reads, of which n ≤ R are indels, and false positive rate 0 ≤ α ≤ 1 (as determined by the negative control), the true indel rate was estimated as .
Computational analysis of Cpf1 targeting range
The complete GRCh38 human genome assembly and coding sequences, with repeats and low complexity regions masked, were downloaded from Ensembl and analyzed as described in Supplementary Fig. 7.
BLISS
All BLISS experiments and analysis were performed as previously described7. The data analysis for the staggered cut sites of Cpf1 was slightly modified from prior analysis7, 16 to increase sensitivity. Previously, to distinguish bona-fide nuclease induced events from the background DSBs in DSB hotspots, centromeres, and telomeres, we had used a cutoff based on the fraction of the pairwise reads that overlapped less than −6bp. This cutoff was set at 0.95 based on empirical data from Cas9 off-target analysis, but to accommodate the variation produced by the staggered cut sites of Cpf1, we found that greater sensitivity to bona-fide Cpf1 off-targets could be found by relaxing this cutoff to 0.85. All other analyses, such as the guide homology score calculations, were as described7.
Sample size and statistics
The sample sizes for each measurement were n = 3 for bacterial colony counts (Fig. 1c); n = 4 for combinatorial mutagenesis (Fig. 2a) and for indel analysis of BLISS loci (Fig. 3a); and n = 2 or n = 3 for all other indel data. The error bars in all figures show standard error of the mean.
Plasmids and guide sequences
A list of the plasmids and guide sequences used in this study can be found in Supplementary Table 4.
Data availability
Deep sequencing data is available on the NCBI Sequence Read Archive.
Supplementary Material
Acknowledgments
We thank A. Magnell for experimental assistance; R. Macrae for a critical review of the manuscript; and the entire Zhang laboratory for support and advice. W.X.Y. is supported by T32GM007753 from the National Institute of General Medical Sciences and a Paul and Daisy Soros Fellowship. H.N. is supported by JST, PRESTO (JPMJPR13L8), JSPS KAKENHI (Grant Numbers 26291010 and 15H01463). O.N. is supported by the Basic Science and Platform Technology Program for Innovative Biological Medicine from the Japan Agency for Medical Research and Development, AMED, and the Council for Science, and Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology. N.C. is supported by the Karolinska Institutet, the Swedish Research Council (521-2014-2866), the Swedish Cancer Research Foundation (CAN 2015/585), and the Ragnar Söderberg Foundation. F.Z. is a New York Stem Cell Foundation-Robertson Investigator. F.Z. is supported by the NIH through NIMH (5DP1-MH100706 and 1R01-MH110049), NSF, Howard Hughes Medical Institute, the New York Stem Cell, Simons, Paul G. Allen Family, and Vallee Foundations; and James and Patricia Poitras, Robert Metcalfe, and David Cheng. Reagents and further information will be available to the academic community through Addgene and the Zhang laboratory website (www.genome-engineering.org).
Footnotes
Author Contributions
L.G., D.C., and F.Z. conceived this study. L.G. and D.C. performed experiments with help from all authors. J.M. contributed to the bacterial selection screen. M.S. processed BLISS samples, and W.Y. analyzed BLISS data. T.Y., H.N., and O.N. provided unpublished AsCpf1 crystal structure information. N.C. provided an unpublished BLISS protocol. F.Z. supervised research. L.G. and F.Z. wrote the manuscript with input from all authors.
References
- 1.Zetsche B, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zetsche B, et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol. 2017;35:31–34. doi: 10.1038/nbt.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fonfara I, Richter H, Bratovič M, Le Rhun A, Charpentier E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016;532:517–521. doi: 10.1038/nature17945. [DOI] [PubMed] [Google Scholar]
- 4.Kim D, et al. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol. 2016;34:863–868. doi: 10.1038/nbt.3609. [DOI] [PubMed] [Google Scholar]
- 5.Kleinstiver BP, et al. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol. 2016;34:869–874. doi: 10.1038/nbt.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HK, et al. In vivo high-throughput profiling of CRISPR-Cpf1 activity. Nat Methods. 2017;14:153–159. doi: 10.1038/nmeth.4104. [DOI] [PubMed] [Google Scholar]
- 7.Yan WX, et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat Commun. 2017;8:15058. doi: 10.1038/ncomms15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleinstiver BP, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kleinstiver BP, et al. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol. 2015;33:1293–1298. doi: 10.1038/nbt.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirano S, Nishimasu H, Ishitani R, Nureki O. Structural Basis for the Altered PAM Specificities of Engineered CRISPR-Cas9. Mol Cell. 2016;61:886–894. doi: 10.1016/j.molcel.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 11.Anders C, Bargsten K, Jinek M. Structural Plasticity of PAM Recognition by Engineered Variants of the RNA-Guided Endonuclease Cas9. Mol Cell. 2016;61:895–902. doi: 10.1016/j.molcel.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamano T, et al. Crystal Structure of Cpf1 in Complex with Guide RNA and Target DNA. Cell. 2016;165:949–962. doi: 10.1016/j.cell.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esvelt KM, et al. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao P, Yang H, Rajashankar KR, Huang Z, Patel DJ. Type V CRISPR-Cas Cpf1 endonuclease employs a unique mechanism for crRNA-mediated target DNA recognition. Cell research. 2016;26:901–913. doi: 10.1038/cr.2016.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ran FA, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slaymaker IM, et al. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleinstiver BP, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong D, et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature. 2016;532:522–526. doi: 10.1038/nature17944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Deep sequencing data is available on the NCBI Sequence Read Archive.