

Abstract
Background
In the heart acute injury induces a fibrotic healing response that generates collagen rich scarring that is at first protective but if inappropriately sustained can worsen heart disease. The fibrotic process is initiated by cytokines, neuroendocrine effectors and mechanical strain that promote resident fibroblast differentiation into contractile and extracellular matrix producing myofibroblasts. The mitogen-activated protein kinase (MAPK) p38α (Mapk14 gene) is known to influence the cardiac injury response, but its direct role in orchestrating programmed fibroblast differentiation and fibrosis in vivo is unknown.
Methods
A conditional Mapk14 allele was used to delete the p38α encoding gene specifically in cardiac fibroblasts or myofibroblasts using 2 different tamoxifen-inducible Cre recombinase expressing gene-targeted mouse lines. Mice were subjected to ischemic injury or chronic neurohumoral stimulation and monitored for survival, cardiac function and fibrotic remodeling. Antithetically, mice with fibroblast-specific transgenic overexpression of activated MAPK kinase 6 (MKK6), a direct inducer of p38, were generated to investigate if this pathway can directly drive myofibroblast formation and the cardiac fibrotic response.
Results
In mice loss of Mapk14 blocked cardiac fibroblast differentiation into myofibroblasts and ensuing fibrosis in response to ischemic injury or chronic neurohumoral stimulation. A similar inhibition of myofibroblast formation and healing was also observed in a dermal wounding model with deletion of Mapk14. Transgenic mice with fibroblast-specific activation of MKK6-p38 developed interstitial and perivascular fibrosis in the heart, lung and kidney due to enhanced myofibroblast numbers. Mechanistic experiments show that p38 transduces cytokine and mechanical signals into myofibroblast differentiation through the transcription factor serum-response factor (SRF) and the signaling effector calcineurin.
Conclusions
These findings suggest that signals from diverse modes of injury converge on p38α MAPK within the fibroblast to program the fibrotic response and myofibroblast formation in vivo, suggesting a novel therapeutic approach with p38 inhibitors for future clinical application.
Keywords: Fibrosis, myocardial infarction, fibroblast, transgenic mice, MAPK signaling
Introduction
The ability to generate a fibrotic scar after myocardial infarction (MI) injury through the accumulation of extracellular matrix (ECM) is critical for acute wound healing; yet excessive fibrosis worsens disease and accelerates the progression to heart failure 1-6. The inability to directly treat fibrosis is due to an incomplete understanding of the underlying molecular regulatory pathways that directly program process and the associated activation of tissue resident fibroblasts into myofibroblasts. This programmed conversion into myofibroblasts is associated with greater secretion of ECM and tissue remodeling as these cells likely become contractile due to newly acquired expression of genes such as smooth muscle α-actin (αSMA) 2, 7, 8. Injured tissues contain a variety of mechanical and cytokine/neurohumoral signals that contribute to the differentiation of fibroblasts into myofibroblasts, although effective pharmaco-therapies based on these regulatory events have yet to emerge 2, 6.
Transforming growth factor-β (TGFβ) signaling directly mediates myofibroblast differentiation and ECM production, although much of this work was performed in vitro 2. In fibroblasts, activation of TGFβ receptor 1 –TGFβ receptor 2 heterodimers causes the transcription factors SMAD2/3 to complex with SMAD4 in the nucleus in altering gene expression to promote myofibroblast differentiation 9. Studies in Smad3 null mice suggest its importance for injury-induced ECM secretion, although αSMA positive myofibroblasts were still present suggesting that multiple effectors are likely critical for the full fibrotic response downstream of TGFβ signaling 10. Similarly, overexpression of inhibitory SMADS failed to block TGFβ-dependent αSMA stress fiber formation in isolated cardiac fibroblasts, again suggesting that additional pathways are required for full myofibroblast differentiation and function 11.
TGFβ receptor signaling can also initiate myofibroblast differentiation through a non-canonical pathway mediated by mitogen-activated protein kinase (MAPK) effectors such as MAPK kinases 6 (MKK6) and its direct downstream target kinase, p38 2, 9. Activation of MKK6-p38 in fibroblasts and other mesenchymal cell types induces collagen and αSMA transcriptional activity as well as the morphologic appearance of αSMA positive stress fibers 11-13. Moreover, pharmacologic inhibitors of p38 activity reduce fibrotic remodeling in skeletal muscle of muscular dystrophy models and more broadly in diseased hearts, lungs and kidneys 12, 14-17.
Using newly engineered tamoxifen-inducible Cre recombinase gene-targeted mice for genetic manipulation of either tissue resident fibroblasts or differentiated myofibroblasts 18-21, we show that Mapk14 (p38α) deletion blocks myofibroblast formation in vivo in multiple tissues and in response to various disease causing stimuli. Moreover, induction of p38 activity by transgenesis with an activated MKK6 cDNA in fibroblasts was sufficient to drive fibrosis and myofibroblast formation in multiple tissues of the mouse.
Methods
Animal models
LoxP-targeted Mapk14 mice and mice containing a “knock-in” of a MerCreMer cDNA into either the Tcf21 or Postn genomic loci were previously described (C57BL/6 background) 17, 18, 20, 22. Constitutively active MKK6 transgenic (tg) mice (C57BL/6 background) were engineered using a cytomegalovirus (CMV)-β-actin (chicken β-actin)-LoxP promoter construct 23, in which an activated human MKK6 cDNA was cloned downstream of a LoxP-flanked chloramphenicol acetyl transferase (CAT)-polyA stop sequence. Tamoxifen was given by i.p. injection in control and experimental mice with pharmaceutical grade tamoxifen dissolved in peanut oil for 5 consecutive days (100 mg/kg body weight) followed by maintenance on tamoxifen-citrate chow (400 mg/kg body weight, Harlan Laboratories) for a minimum of 2 weeks or until the experiment was terminated.
MI injury was generated by permanently ligating the left coronary artery of ∼8 week-old mice 24, 25. To model ischemia – reperfusion (IR) injury, the left coronary artery was transiently ligated for 30 minutes followed by continuous reperfusion until the mice were euthanized and hearts harvested. For both MI and IR surgical procedures mice were perioperatively given a single s.c. injection of a mixture of lidocaine (1-4 mg/kg body weight) and bupivacaine (1-2 mg/kg body weight) at the site of surgical incision and anesthetized with inhaled 2% isoflurane, intubated through the mouth and ventilated throughout the procedure. A single post-operative dosage of buprenorphrine-sustained release formula (3.25 mg/kg body weight) was given by s.c injection at 0.05-0.1 mg/kg, as well as for all subsequent surgical procedures described below.
M-mode echocardiography with a Hewlett Packard SONOS 5500 instrument was used to assess ventricular geometry and function 10 and 28 days after surgery depending on the study in mice anesthetized with inhaled 2% isoflurane for the duration of the procedure. As this is a non-invasive procedure analgesia is not needed. Chronic angiotensin II / phenylephrine (AngII/PE) treatment (432 μg/kg/d /100 mg/kg/d, respectively) was delivered for 2 weeks by Alzet osmotic mini-pumps (Durect Corp), which were subcutaneously implanted in 8 week-old mice under inhaled 2% isoflurane anesthesia. For AngII/PE treatment cardiac function and tissue collection was performed 14 days after treatment. At the completion of all studies hearts were perfused in cardioplegic buffer to preserve diastolic dimensions and then fixed in 10% formalin, embedded in paraffin, and prepared for histological analysis. Fibrosis was assessed by Mason's trichrome staining at 10 and 28 days after IR injury, 14 days after AngII/PE treatment, or 4 months after MKK6 transgene induction (mice were then 5 months-old). The average area of fibrosis was calculated across 5 evenly distributed transverse sections of the heart running from apex to the base using NIH Image J software.
For dermal wound healing assays, fully anesthetized 12 week-old mice (induced and maintained on inhaled 2% isoflurane) were given two dorsal 6 mm skin excision punch biopsies, and wound closure rate and health was assessed daily for 1 week as previously described 11. Wound closure was calculated by measuring the change in wound area from the initial size of the biopsy and normalizing this value to the size of the initial wound. All animal experimentation was approved by the Institutional Animal Care and Use Committee of Cincinnati Children's Hospital Medical Center and the University of Washington. Number of mice used is given in the results section, figure legends or shown in the figures.
Fibroblast cultures and treatments
Mouse embryonic fibroblasts (MEFs) were isolated from embryonic (E) day13.5 Mapk14fl/fl mice for primary culture as described previously 26. MEFs were cultured in Dulbecco's Minimal Essential Media (DMEM) supplemented with 10% fetal bovine serum (FBS) and non-essential amino (NEAs) acids, and passages 2 and 3 were used for experimentation. Myofibroblast differentiation was analyzed for αSMA stress fiber formation by immunocytochemistry 72 hours after induction with either recombinant porcine TGFβ (10 ng/ml, R&D Systems), AngII (100 nM, Sigma), or adenovirus delivering the following genes: ΔCnA (activated calcineurin truncation), βgal (β-galactosidase), Cre recombinase, TRPC6 (transient receptor potential canonical 6), or serum response factor (SRF)11, 24, 27, 28. These treatments were also used for collagen contraction assays. Here 40,000 MEFs were seeded into collagen gel matrices, induced to differentiate, and gel area measured 48 hours later. Stretch-induced differentiation was performed by seeding MEFs on 6-well Flexcell culture plates and imparting a 20% stretch to the membranes by loading the plates on large diameter circular posts (28 mm, Flexcell International). Stretch oscillations were performed continuously for 48 hours at 2 different frequencies, 0.5 and 1.0 Hz, and then fixed for immunofluorescence staining.
Immunofluorescence, flow cytometry and Western blot analysis
Cultured MEFs were fixed in 4% paraformaldehyde and stained with αSMA antibody (1:1000, mouse monoclonal, Clone 1A4, Sigma) to detect actin stress fibers and 4′,6-diamidino-2-phenylindole (DAPI, 1:1000, Invitrogen) to visualize the nucleus. Alexa 488 or Alexa 568 conjugated secondary antibody directed against mouse IgG was used to detect αSMA. In cardiac tissue sections myofibroblasts were identified in infarcted hearts 28 days after injury and in MKK6-Tg mice 4 months after tamoxifen induction. Hearts were dissected, fixed overnight in 10% formalin, and paraffin embedded. Five micron sections were subjected to antigen retrieval and subsequently stained with the combination of αSMA antibody and isolectin-B4 (Vector Biolabs) for endothelial cell identification, or for vimentin (1:250, rabbit monoclonal, Abcam).
For Western blotting of purified cardiac fibroblasts isolated from genetically modified hearts, a Langendorff apparatus was used to perfuse the heart with type II collagenase (2mg/ml) and liberase blendzyme (0.4mg/ml) in Krebs-Henseleit buffer. Hearts were processed and cells collected and prepared for flow cytometry as described previously 29. Cells were incubated for 1 hour with the fibroblast allophycocyanin (APC)-conjugated antibody MEFSK4 (1:10, Miltenyi Biotec), washed 3 times in blocking buffer, and incubated with 4′,6-diamidino-2-phenylindole (DAPI) to identify live cells. The isolated fibroblast fraction was lysed in RIPA buffer (Thermo Fisher Scientific) and prepared in Laemmli buffer (Sigma Aldrich) for SDS-PAGE gel and western blot. For each group, protein extract from 125,000 fibroblasts were loaded per lane and the following antibodies used: total p38α (1:1000, rabbit polyclonal, Cell Signaling), phosphorylated p38 (1:1000, rabbit polyclonal, Cell Signaling), MKK6 (1:500, rabbit polyclonal, Genetex), phosphorylated MK2 (1:100, rabbit polyclonal, Cell Signaling), αSMA (1:1000, mouse monoclonal, Sigma), vimentin (1:1000, rabbit monoclonal, Abcam), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 1:10,000, mouse monoclonal, Fitzgerald Industries).
Reverse transcriptase polymerase chain reaction (RT-PCR)
Cardiac fibroblasts were isolated as described above and RNA was isolated using Qiashredder homogenization and RNAeasy preparations (Qiagen). Total RNA was reverse transcribed using random hexamer primers and Superscript III first-strand synthesis kit (Invitrogen). Real-time PCR was performed using Sso Advanced SYBR Green (Biorad) and GAPDH expression was used for normalization. The following primer sets were used to identify the human MKK6 transgenic transcripts: 5′-CACTTGACCGAGAGCATTGATGAG, 5′-AGTCGAAAGGCAAGAAGCGA (also See Supplemental Table).
Statistics
One-way analysis of variance (ANOVA) was used to determine statistical significance for experiments with more than 2 groups, and statistically significant pairwise differences were further determined by Newman-Keuls methods (Prism software). For experiments in which 2 groups were compared unpaired t-tests were used to determine statistical significance. For survival assessments from the Kaplan Meier curves post-hoc t-test were performed at each time point between Mapk14fl/fl and Mapk14fl/fl Tcf21MCM genotypes. P-values < 0.05 were considered statistically significant and all the tests performed two-sided. To determine sample sizes for the proposed experiments the desired power was established as 80% and the criterion for significance (α) is 0.05, which was an n = 8-12 for both MI and IR injury and n = 5-8 AngII/PE models.
Results
Fibrotic signals require p38 MAPK for myofibroblast differentiation
Here primary MEFs were first isolated from homozygous LoxP-targeted Mapk14 mice (Mapk14fl/fl) and then adenovirally transduced with either β-galactosidase (Adβgal) as a control or Cre recombinase (AdCre) to delete Mapk14. Four days after adenoviral transduction p38α protein expression was reduced by over 90% as detected by Western blot (Figure 1A). Using these conditions, MEFs with and without p38α were treated with TGFβ or AngII to differentiate them into αSMA expressing myofibroblasts by immunocytochemistry (Figure 1B). We also measured mRNA levels for αSMA (Acta2), collagen 1a1 (Col1a1), fibronectin (Fn1), and EDA splice variant of fibronectin (Fn1-EDA) (Figure S1 A-D and Supplemental Table). Approximately 60% of WT MEFs differentiated into myofibroblasts 4 days after treatment with TGFβ or AngII; whereas Mapk14fl/fl MEFs infected with AdCre failed to appreciably differentiate using these same 2 agonists (Figure 1B).
Figure 1.
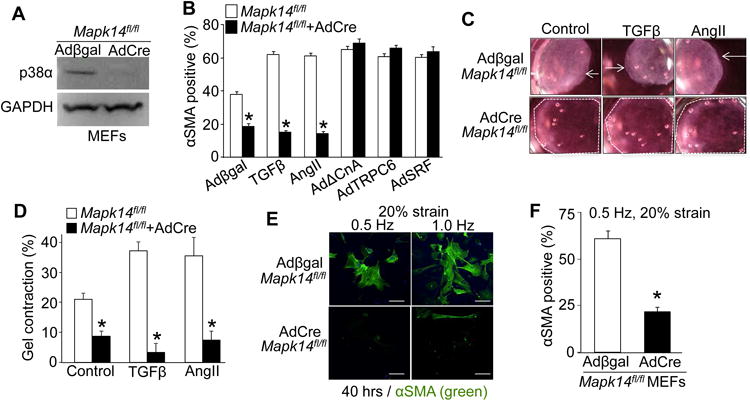
p38 transduces chemical and mechanical stimuli to initiate myofibroblast differentiation. A, Western blot analysis showing highly efficient deletion of p38 protein from Mapk14fl/fl MEFs 4 days after AdCre infection versus Adβgal control infection. GAPDH was used as a loading control. B, Immunofluorescent-based quantification of the percent of MEFs with αSMA+ stress fibers in Mapk14fl/fl MEFs versus Mapk14fl/fl MEFs with AdCre infection to delete p38α protein expression. MEFs were also infected with the additional adenoviruses shown along the x-axis or treated with TGFβ to alter signaling in these cells. Error bars are SEM, N≥530, *P<0.05 vs Mapk14fl/fl. C, Representative images and D, quantification of contracted collagen gel matrices seeded with Mapk14fl/fl MEFs virally transduced with Adβgal or AdCre and treated with TGFβ or AngII. The white arrows show the direction of contraction, while the dashed white outline shows the full uncontracted collagen gel. Average contraction ± SEM, N=3, *P<0.05 vs Mapk14fl/fl. The entire area of the picture is 28 mm across in panel C. E, Immunofluorescent images and F, quantification of the number of Mapk14fl/fl MEFs with αSMA+ stress fibers (green) after 48 hours of cyclic stretching (20% strain) at 0.5 and 1.0 Hz. Scale bar = 50 μm. Data shows the average number of MEFs with αSMA+ stress fibers normalized to the total number of nuclei ± SEM, N≥80, *P<0.05.
Our past studies showed that p38 directly stimulates serum response factor (SRF)-mediated transcription of transient receptor potential canonical 6 (TRPC6) channel that in turn activates Ca2+-calcineurin signaling, a pathway necessary for myofibroblast formation 11. To confirm p38's placement in this pathway control MEFs (Mapk14fl/fl, Adβgal) and p38α protein deleted MEFs (Mapk14fl/fl, AdCre) were adenovirally transduced with either constitutively active calcineurin (AdΔCnA), TRPC6 (AdTRPC6) or SRF (AdSRF) (Figure 1B & S1 A-D). All three treatments elicited increased matrix gene expression and robust formation of αSMA expression and stress fibers in a majority of the fibroblast population independent of the presence of p38, suggesting that p38 lies upstream of these genetic inducers of myofibroblast differentiation (Figure 1B & S1 A-D).
We next employed a collagen-gel contraction assay, as the ability to contract is another signature of myofibroblasts (Figure 1C and 1D). Gels seeded with control MEFs replete with p38α (Mapk14fl/fl, Adβgal) contracted when treated with TGFβ or AngII, but loss of p38α protein (Mapk14fl/fl, AdCre) inhibited contraction (Figure 1C and 1D). However, proliferation rates in these same cells groups measured with 10 μM 5-ethynyl-2′-deoxyuridine (EDU) were not different in low serum but were slightly higher in the p38α deleted cells with high serum, collectively indicating that the observed inhibition of gel contraction was not due to diminished fibroblast numbers during the assay period (Figure S1 E). Interestingly, none of the chemical inducers of myofibroblast differentiation used here converted 100% of the MEFs in culture, likely due to heterogeneity in cell cycle and their phases of maturation during the differentiation process.
Mechanical rigidity and strain also trigger mesenchymal cells to undergo a change in fate 2, 30. Here Mapk14fl/fl control and deleted MEFs were cultured on silicon membranes and subjected to 40 hours of cyclic stretching with 20% strain (Figure 1E and 1F). Both 0.5 and 1.0 Hz frequency of stretching caused nearly 70% of the population of wildtype MEFs to develop αSMA-positive stress fibers while MEFs devoid of p38α showed significantly reduced levels (Figure 1E and 1F). These results indicate that p38α is also required for transducing mechanical signals underlying myofibroblast differentiation.
Deletion of Mapk14 in resident fibroblasts minimizes injury-induced cardiac fibrosis
The in vitro work described above suggests that fibroblast-specific inhibition of p38α signaling blocks stimulus-induced myofibroblast formation. Indeed, four days after ischemic injury p38α and p38γ gene expression increased specifically in cardiac fibroblasts (Figure S2 A-B), although p38α was the dominant isoform identified. Hence, we deleted the Mapk14 gene encoding p38α only in resident fibroblasts using a tamoxifen-inducible MerCreMer (MCM) cDNA expressed from the Tcf21 genetic locus18, 19 to examine cardiac injury responses and fibrosis in adult mice. Here tamoxifen was given until the completion of the experiment at 3 months of age (Figure 2A). Purified cardiac fibroblasts showed a 74% deletion of p38α protein as assessed by western blotting (Figure 2B). Fibroblasts were flow-sorted with the MEFSK4 antibody, which is a global fibroblast marker that selects nearly 95% of Tcf21+ fibroblasts, although it can also detect CD11b+ leukocytes 29.
Figure 2.
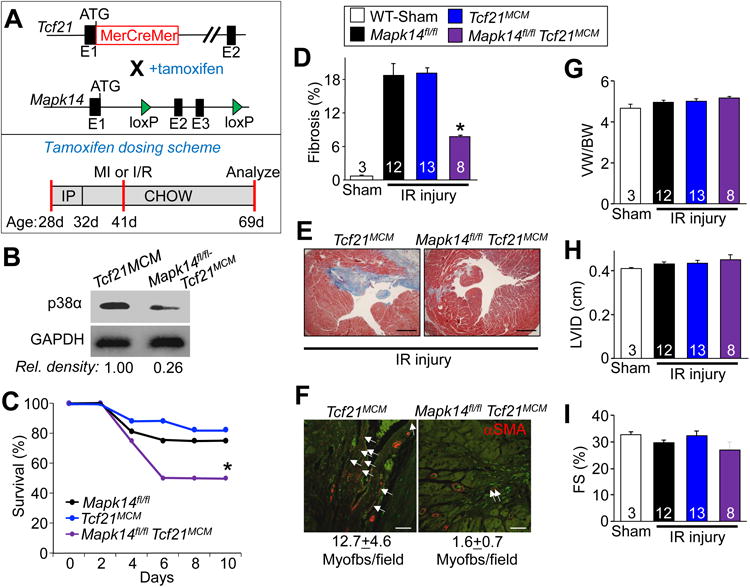
Mapk14 deletion in resident cardiac fibroblasts minimizes the fibrotic response to ischemic injury. A, Top panel: schematic showing the Tcf21 locus containing a tamoxifen-regulated MerCreMer (MCM) cDNA and a Mapk14-loxP targeted allele contained within mice so that with tamoxifen administration Cre-dependent recombination can inactivate the Mapk14 gene. Lower panel: schematic of the tamoxifen dosing scheme used for these experiments with age listed in days (d). B, Western blot analysis for p38α protein from purified cardiac fibroblasts isolated from hearts of Mapk14fl/fl Tcf21MCM mice after tamoxifen induction. GAPDH was used as a loading control. C, Kaplan-Meier plot of survival of the indicated genotypes of mice over 10 days after IR injury. D, Quantification and E, Masson's trichrome stained histological images from the hearts of the indicated mice 28 days after IR injury. Blue staining indicates fibrosis. Scale bar = 150 μm. Average fibrotic area ± SEM, N=3 for sham and N≥8, *P<0.05 vs Mapk14fl/fl and Tcf21MCM. F, Immunofluorescent staining and quantification of myofibroblast numbers (white arrows) in the cardiac infarction border zone 28 days after injury. Myofibroblasts are αSMA (red) positive and negative for isolectin-B4 (green). Scale bar = 50 μm. Data represent the average number of αSMA+/isolectin-B4- cells/section ± SEM, 5 sections were measured per heart, N=3 for sham and N≥8 mice per group. G, Quantification of ventricular weight (VW) normalized to body weight (BW) 28 days after ischemic injury, Average VW/BW ± SEM, *P<0.05 vs Mapk14fl/fl and Tcf21MCM. Echocardiography of H, left ventricle inner diameter (LVID) and I, fractional shortening (FS%) from Mapk14fl/fl-Tcf21MCM, Mapk14fl/fl and Tcf21MCM mice 28 days post IR injury. Data are averages ± SEM, *P<0.05 vs Mapk14fl/fl and Tcf21MCM. The legend above panels D and E represent all of the data in the figure. Number of mice analyzed is shown within the bars of panels D,G-I
To examine whether adult cardiac fibroblasts, like MEFs, require p38α to differentiate into myofibroblasts the above described flow cytometry method was used to isolate and culture cardiac fibroblasts from tamoxifen treated Mapk14fl/fl and Mapk14fl/fl Tcf21MCM mice. Fibroblasts were then treated with the same profibrotic agonists and assessed for myofibroblast differentiation. Again, p38α deficiency (Mapk14fl/fl Tcf21MCM) blocked TGFβ and AngII-dependent myofibroblast differentiation, but adenoviral transduction with the down-stream TRPC6-calcineurin signaling axis restored differentiation in vitro (Figure S2 C). Unlike what was observed in MEFs, p38α deficiency increased new DNA synthesis in Mapk14fl/fl Tcf21MCM cardiac fibroblasts in low serum media suggesting that these later cardiac fibroblasts are inherently more proliferative (Figure S2 D).
Using the same tamoxifen dosing strategy (Figure 2A) Mapk14fl/fl Tcf21MCM mice and the proper controls were subjected to an ischemia-reperfusion (IR) procedure because the standard MI injury protocol caused 100% lethality (data not shown). The IR procedure was better tolerated by Mapk14fl/fl Tcf21MCM mice, which now showed only 50% left ventricular wall rupture versus 100% with MI injury (Figure 2C). Despite this improved tolerance Mapk14fl/fl Tcf21MCM mice still showed significantly greater lethality when compared to IR injured littermate controls (Figure 2C). Inspection of the mice after I/R-induced lethality showed ventricular wall rupture and inefficient fibrotic scar formation, as observed in other mouse models lacking proper fibrotic matrix deposition 11, 20, 21, 25, 31-33. Indeed, surviving Mapk14fl/fl Tcf21MCM mice had a 54% reduction in ventricular fibrotic area when compared to Mapk14fl/fl and Tcf21MCM controls (Figure 2D and 2E). Cardiac sections were also quantified for myofibroblast numbers by scoring for αSMA positive and isolectin-B4 negative (endothelial marker) cells. The data show that while the border zone of hearts from IR injured Mapk14fl/fl and Tcf21MCM control mice had relatively large numbers of myofibroblasts, the same regions from hearts of Mapk14fl/fl Tcf21MCM mice had significantly fewer (Figure 2F). However, this injury model elicited no change in cardiac mass, left ventricular diastolic chamber dimension, or fractional shortening amongst any of the groups (Figure 2G-2I). Importantly, pulse wave and tissue Doppler imaging indicated that relative to IR injured Mapk14fl/fl controls, Mapk14fl/fl Tcf21MCM mice had lower isovolumetric relaxation times (IRVT) and E/e' ratios suggesting that decreasing fibrosis abrogates injured-induced diastolic dysfunction (Table 1).
Table 1.
Diastolic function measure by pulse wave and tissue Doppler.
| Genotype | N | Injury | IVRT‡ (ms) | Septal E/e′ |
|---|---|---|---|---|
| NTG | 7 | Sham | 14.05±1.16* | -28.53+1.8* |
| MKK6 Tcf21MCM | 7 | Sham | 19.79±1.47† | -39.63+2.32† |
| Mapk14fl/fl | 6 | IR | 19.38±1.96† | -37.46+1.89† |
| Mapk14fl/fl Tcf21MCM | 5 | IR | 12.06±1.00* | -23.66+1.73* |
| Mapk14fl/fl PostnMCM | 6 | IR | 18.22±1.19† | -38.02+1.69† |
Values represent the mean±sem, ANOVA,
p<0.05 vs IR injured Mapk14fl/fl and
p<0.05 vs NTG. Analysis was performed 28 days post sham or IR injury. Lateral wall measurements followed a similar trend but were not statistically different. Key:
Isovolumetric relaxation time (IVRT).
Mapk14 deletion in existing myofibroblasts partially reduces cardiac fibrosis
The Tcf21MCM allele allows for deletion of Mapk14 from only resident fibroblasts in the heart, which permits assessment of this signaling effector's role prior to an activating stimulus. However, we also assessed the importance of Mapk14 activity in maintaining myofibroblast function and differentiation during an injury event using a mouse model in which the MerCreMer cDNA was expressed the genetic locus of periostin (Postn). We have shown that PostnMCM mice can be used to mark essentially all newly activated fibroblasts (myofibroblasts) within the heart during an injury event, but not in “unactivated” fibroblasts 20, 21. Here, tamoxifen was given to 8 week old mice, 5 days prior to MI surgery, and then kept on tamoxifen until harvest 28 days later (Figure 3A). Separate cohorts of mice were also generated so that the Rosa26mT/mG reporter allele could be used to track and isolate newly generated myofibroblasts. The data showed a 66% reduction in p38α protein using this cell sorting approach and a near complete loss of phospho-p38 signal by western blotting (data not shown) in Mapk14fl/fl PostnMCM Rosa26mT/mG cells after injury (Figure 3B).
Figure 3.
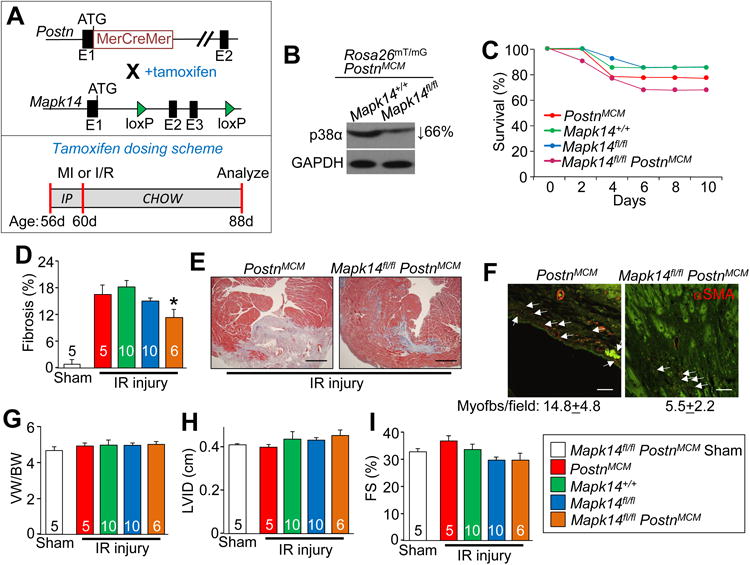
Mapk14 deletion in myofibroblasts reduces the fibrotic response to ischemia injury. A, Top panel: schematic showing the Postn locus containing a tamoxifen-regulated MerCreMer (MCM) cDNA and a Mapk14-LoxP targeted allele contained within mice so that with tamoxifen administration Cre-dependent recombination can inactivate the Mapk14 gene. Lower panel: schematic of the tamoxifen dosing scheme listed in days (d). B, Western blot analysis of p38α protein expression in eGFP sorted and purified fibroblasts from the hearts of the indicated mice after MI injury. GAPDH was used as a loading control. C, Kaplan-Meier plot of survival in the indicated genotypes of mice over 10 days after MI injury. D, Quantification and E, Masson's trichrome stained histological images from the hearts of the indicated mice 28 days after IR injury. Blue staining indicates fibrosis. Scale bar = 150 μm. Average fibrotic area ± SEM, N≥5, *P<0.05 vs Mapk14fl/fl and PostnMCM. F, Immunofluorescent staining and quantification of myofibroblast numbers (white arrows) in the cardiac infarction border zone 28 days after IR injury. Myofibroblasts are αSMA (red) positive and negative for the endothelial marker isolectin-B4 (green). Scale bar = 50 μm. Data represent the average number of αSMA+/isolectin-B4- cells/section ± SEM, 5 sections were measured per heart, N≥5 mice per group. G, Quantification of ventricular weight (VW) normalized to body weight (BW) 28 days after IR injury, Average VW/BW ± SEM, *P<0.05 vs Mapk14fl/fl and PostnMCM. Echocardiography of H, left ventricle inner diameter (LVID) and I, fractional shortening (FS%) from Mapk14fl/fl PostnMCM, Mapk14fl/fl and PostnMCM mice 28 days post IR injury. Data are averages ± SEM, *P<0.05 vs Mapk14fl/fl and PostnMCM. The legend next to panel I represent all of the data in the figure. Number of mice analyzed is shown within the bars of panels D,G-I
Unlike Mapk14fl/fl Tcf21MCM mice, Mapk14fl/fl PostnMCM animals had improved tolerance to MI injury such that 70% survived, which is slightly lower than the average survival rates in the littermate controls (Figure 3C). The underlying cause of death in Mapk14fl/fl PostnMCM mice was again ventricular wall rupture (Figure 3C). Mapk14fl/fl PostnMCM mice and their littermate controls were also subjected to the less severe IR injury protocol (approximately 20% infarct area in controls) described earlier. Here nearly 100% of both control and Mapk14fl/fl PostnMCM survived the injury, but we still observed a significant reduction in the scar and fibrotic area in Mapk14fl/fl PostnMCM mice compared to 3 different control groups (Figure 3D and 3E). Immunofluorescent staining indicated that myofibroblast number was also significantly reduced in hearts of injured Mapk14fl/fl PostnMCM mice compared to controls (Figure 3F). No change was observed in cardiac mass, left ventricular chamber dimension or fractional shortening amongst any of the groups (Figure 3G-3I). Despite the reduction in fibrosis, hearts from Mapk14fl/fl PostnMCM mice had comparable IVRT and E/e' measurements as Mapk14fl/fl (Table 1), suggesting that the remaining fibrotic area is still enough to trigger diastolic dysfunction. Overall, these results indicate that p38α is necessary for both generation of myofibroblasts with injury and their persistence (see discussion).
Here we also modeled a more chronic fibrotic injury response by continuous infusion of AngII with PE as previously described 21. Mapk14fl/fl PostnMCM mice and their controls were subjected to 2 weeks of AngII/PE infusion via osmotic mini-pumps, which significantly increased cardiac ventricular mass and fibrosis in Mapk14fl/fl and PostnMCM control mice (Figure 4A-4C). By comparison, myofibroblast-specific loss of Mapk14 blocked the hypertrophic response and significantly reduced cardiac fibrosis (Figure 4A-4C). Echocardiographic assessment revealed that left ventricular chamber dimensions and function were similar across all groups with 2 weeks of AngII/PE treatment (Figure 4D and 4E). However, the results show that p38α signaling is required in myofibroblasts to maintain the fibrotic function of these cells in response to AngII/PE neuroendocrine signaling, which also supports the hypertrophic response of the heart.
Figure 4.
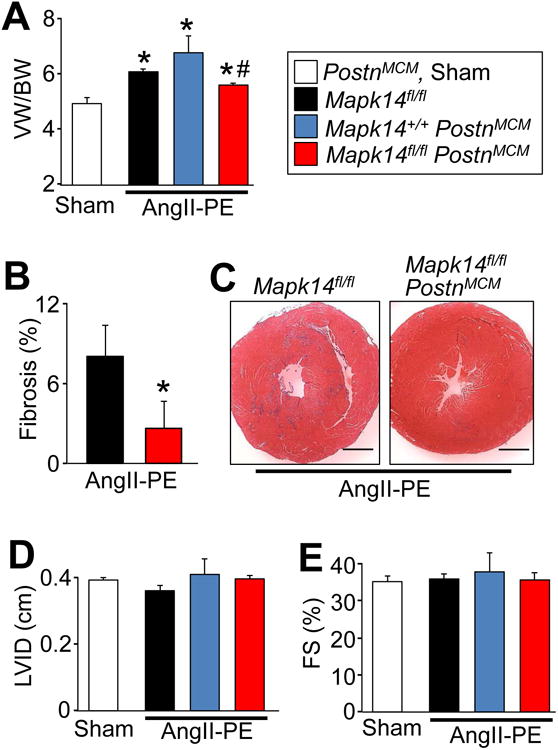
Mapk14 deletion in myofibroblasts reduces AngII/PE-induced fibrosis and cardiac hypertrophy. A, Quantification of ventricular weight (VW) normalized to body weight (BW) after 2 weeks of AngII/PE infusion by osmotic mini-pump (432 μg/kg/d / 100 mg/kg/d) and tamoxifen induction. Average VW/BW ± SEM, N≥5, *P<0.05 vs Sham, #P<0.05 vs Mapk14fl/fl and PostnMCM. B, Quantification and C, Masson's trichrome-stained transverse heart histological images for fibrosis (blue) in the indicated genotypes of mice after 2 weeks AngII/PE. Scale bar = 1 mm. Average fibrotic area ± SEM, N≥5, *P<0.05 vs Mapk14fl/fl. Echocardiography of D, left ventricle inner diameter (LVID) and E, fractional shortening (FS%). Data are averages ± SEM, N≥5, *P<0.05 vs Sham, # P<0.05 vs Mapk14fl/fl and PostnMCM. The legend next to panel A represent all of the data in the figure.
Fibroblast-specific activation of p38 signaling induces fibrosis in vivo
To determine whether activation of p38 MAPK signaling in fibroblasts is sufficient to drive myofibroblast differentiation in vivo, an inducible Cre-dependent system was used to express constitutively active MKK6 in Tg mice (Figure 5A). Real-time PCR of this MKK6 cDNA in cardiac fibroblasts isolated from MKK6-Tg mice and infected with AdCre showed that transgene expression is Cre dependent (Figure 5B). Similarly, dermal and pulmonary fibroblasts isolated from these Tg mice remained quiescence in culture without significant αSMA expression, yet AdCre infection induced de novo αSMA expression, stress fibers and myofibroblast differentiation as MKK6 was expressed (Figure 5C).
Figure 5.
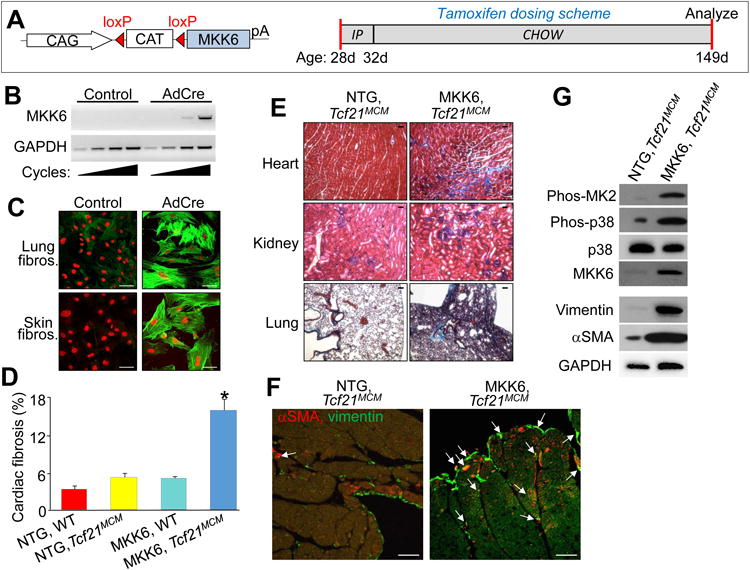
Constitutive activation of p38 in cardiac fibroblasts enhances fibrotic and hypertrophic remodeling. A, Left panel: schematic of the MKK6 LoxP-dependent and inducible transgene driven by a ubiquitous promoter (CMV β-actin). Right panel: schematic of the tamoxifen dosing scheme used for these experiments with age listed in days (d). B, RT-PCR for the activated MKK6 transgene from cardiac fibroblasts isolated from MKK6-Tg, Tcf21MCM mice and subjected to infection with Adβgal or AdCre. GAPDH expression was used for normalization. C, Immunofluorescent images of fibroblasts isolated from lung and skin of MKK6-Tg mice that were then adenovirally transduced with Adβgal (control) or AdCre and immunostained for αSMA+ stress fibers (green) and nuclei (red). Scale bar = 50 μm. D, Quantification of the area of fibrosis (blue) in transverse histological heart sections with Masson's trichrome staining in the indicated genotypes of mice. Average fibrotic area ± SEM, N≥8, *P<0.05 vs NTG and Tcf21MCM. E, Masson's trichrome-stained histological sections from the indicated tissues of either non-Tg (NTG) or MKK6-Tg mice with the Tcf21MCM allele after 4 months of continuous tamoxifen treatment. Blue staining indicates fibrosis. Scale bar = 25 μm. F, Immunofluorescent staining of hearts from the indicated genotypes of mice after 4 months of continuous tamoxifen treatment. Myofibroblasts are positive for both αSMA (red) and the fibroblast marker vimentin (green) are designated with the white arrows. Scale bar = 100 μm. G, Western blot analysis for the indicated proteins from cardiac fibroblasts purified from Tcf21MCM and MKK6-Tg, Tcf21MCM hearts following 1 month of tamoxifen induction. GAPDH antibody was used as a loading control.
To assess the ability of activated MKK6 to induce fibrosis in vivo, we crossed mice containing the MKK6 Tg with Tcf21MCM mice and tamoxifen was given at weaning until 5 months of age when the experiment was terminated (Figure 5A). After this treatment protocol the hearts of MKK6-Tg, Tcf21MCM mice had significant accumulation of interstitial and perivascular fibrosis not observed in tamoxifen-fed controls (Figure 5D and 5E). Moreover, other organs and known sites of Tcf21 expression such as the kidney and lung were also fibrotic in these mice, a result again not seen in non-Tg littermate controls only given tamoxifen (Figure 5E). Indices of diastolic dysfunction were also examined by pulse wave and tissue Doppler imaging. MKK6-Tg, Tcf21MCM mice had significantly higher IVRT and E/e' relative to their NTG littermates and a similar level of diastolic dysfunction as that observed in IR injured Mapk14fl/fl controls (Table 1). Cardiac histological sections were also examined for myofibroblasts by immunofluorescent staining, which showed widespread myofibroblasts in interstitial spaces, within the endocardium, near the epicardial borders, and in perivascular niches with MKK6 transgene expression (Figure 5F). In support of these data, fibroblasts purified from MKK6-Tg, Tcf21MCM hearts using the MEFSK4 antibody showed high levels of myofibroblast markers like αSMA and vimentin compared with isolated fibroblasts from Tcf21MCM only hearts, suggesting that p38 activation promoted expansion of cardiac fibroblasts and their conversion to myofibroblasts (Figure 5G). Importantly, we also observed increased MKK6 protein expression in these purified fibroblasts from the hearts of MKK6-Tg, Tcf21MCM mice, as well as activation of p38 and MAPKAP2 (MK2) using phospho-specific antibodies and Western blotting (Figure 5G). These results indicate that increased p38 signaling in tissue-resident fibroblasts induces de novo myofibroblast formation and tissue fibrosis in otherwise healthy organs.
We also examined MKK6-Tg PostnMCM mice subjected IR injury in which tamoxifen was given as shown in Figure 3A. The mice were then examined 1-month post IR injury by echocardiography and by histologic and morphologic assessments (Figure 6A-6C). We observed that cardiac mass and fibrotic area were significantly increased in MKK6-Tg, PostnMCM mice in comparison to control littermates (Figure 6B and C), but that cardiac function was not different between the groups (Figure 6C). Similarly, fibrotic injury induced by 2 weeks of AngII/PE infusion significantly elevated cardiac mass and fibrosis in the hearts of MKK6-Tg, PostnMCM mice in comparison to littermate controls (Figure 6D and 6E). There was also a non-significant trend towards reduced ventricular performance with AngII/PE infusion in MKK6-Tg, PostnMCM mice compared with control mice (Figure 6F). Taken together, these data suggest that MKK6-p38 signaling enhances the fibrotic response through priming and accentuating myofibroblast function during injury, which can also enhance the hypertrophic response of the heart.
Figure 6.

p38 activation in myofibroblasts exacerbates cardiac fibrosis. A, Quantification of ventricular weight (VW) normalized to body weight (BW) 28 days after IR injury in the genotypes of mice shown. Tamoxifen was given 5 days prior to surgery and maintained for the experiment's duration. Data are averages ± SEM, *P<0.05 vs NTG and PostnMCM. B, Quantification of the area of fibrosis (blue) in transverse histological heart sections stained with Masson's trichrome 28 days after IR injury. Data are averages ± SEM, *P<0.05 vs NTG and PostnMCM. C, Echocardiography measured ventricular fractional shortening (FS%) 28 days after IR injury. Data are averages ± SEM. D, Quantification of VW/BW 14 days after AngII/PE infusion. Data are averages ± SEM, *P<0.05 vs NTG; #P<0.05 vs PostnMCM. E, Quantification of the area of fibrosis (blue) in transverse histological heart sections stained with Masson's trichrome 14 days after AngII/PE infusion. Data are averages ± SEM, *P<0.05 vs PostnMCM. F, Echocardiography of ventricular fractional shortening (FS%) 14 days after AngII/PE infusion. Number of mice used is shown in the bars of each panel for the entire figure.
We also examined whether MKK6-p38 signaling affects the fibrotic response in other conditions and tissues. In vitro wound closure rates were first assessed by creating microscale circular cutout wounds in monolayers of Mapk14fl/fl and Mapk14fl/fl+AdCre MEFs tracking the rate of individual fibroblast migration into the wound with continuous live cell imaging. Loss of p38α (Mapk14fl/fl+AdCre) caused a significant slowing of wound closure relative to MEFs expressing p38α (Figure 7A). To extend this in vitro assay we also directly assessed wound healing in a skin model of injury. Here MKK6-Tg, PostnMCM, and Mapk14fl/fl PostnMCM mice, along with their appropriate controls, were subjected to dermal punch biopsies and monitored for the rate of skin wound closure as previously described 11, 21. Similar to the results obtained with cardiac injury, mice devoid of p38 signaling in their myofibroblasts (Mapk14fl/fl PostnMCM) had significantly slower wound closure rates relative to controls, while MKK6-Tg, PostnMCM mice had significantly faster rates of skin wound closure (Figure 7B-7C). These results again support the hypothesis that p38 signaling is both necessary and sufficient for myofibroblast-mediated tissue wound closure (healing), scar formation and tissue fibrosis in vivo.
Figure 7.
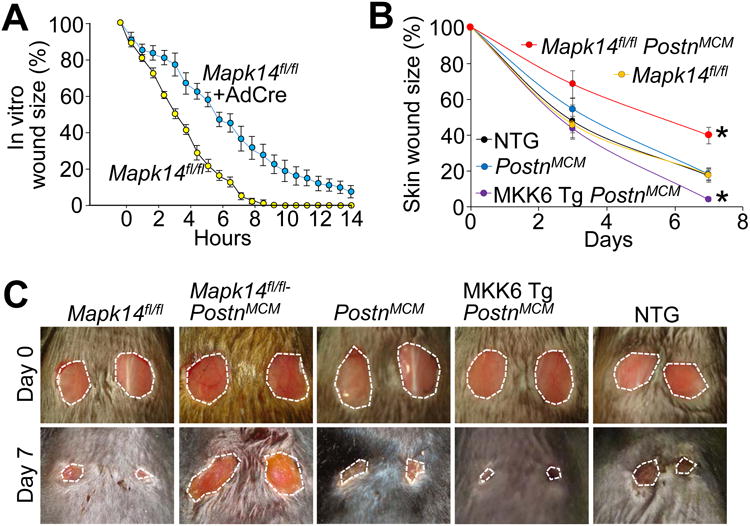
p38 regulates skin wound healing. A, Quantification of in vitro wound closure rates in Mapk14fl/fl and Mapk14fl/fl-AdCre MEF monolayers in serum free culture conditions using live cell imaging. B,C, Quantification of dermal would closure over 7 days measured relative to initial day 0 wound size and corresponding photographs of these punch biopsies from tamoxifen-treated adult NTG, MKK6-Tg, PostnMCM, and Mapk14fl/fl PostnMCM mice at day 0 and 7 of injury. Average closure ± SEM, N≥5, *P<0.05 vs Mapk14fl/fl or PostnMCM. The biopsies in panels C are 6 mm at day 0, which shows sizing of the entire micrograph.
Discussion
Our results show that inductive mechanical and paracrine signals converge on p38 to initiate programmed fibroblast to myofibroblast differentiation and the fibrotic response within the mouse heart. Indeed, genetic activation of p38 signaling in what would otherwise be termed as “quiescent” fibroblasts produced both αSMA positive myofibroblasts as well as fulminant interstitial and perivascular fibrosis, in the absence of an injury signal. Our results also demonstrate that genetic deletion of Mapk14 markedly inhibited the fibrotic response to ischemic injury, chronic neurohumoral stimulation and skin wound closure, further indicating its necessity as a mediator of myofibroblast formation and tissue fibrosis in vivo, which also impacts the cardiac hypertrophic response.
The high cardiac rupture rates observed in Mapk14fl/fl-Tcf21MCM mice post-MI or -IR injury demonstrates how vital both p38 signaling and myofibroblast function are to the structural integrity of the myocardium. Indeed cardiac ventricular wall rupture has been associated with poor scar formation due to inefficient or incomplete ECM protein production and/or lack of myofibroblast formation in other mouse models 11, 21, 25, 31-33. Similarly, mice carrying a diphtheria toxin gene that is turned on in myofibroblasts also had reduced scarring and poor survival post-MI 20, 34.
There may also be intermediate stages of myofibroblast differentiation. For example, large amounts of fibrotic matrix can be secreted from less mature activated fibroblasts in which focal adhesions are immature and actin stress fibers contain cytoplasmic β-actin rather than αSMA 35. Nonetheless these immature myofibroblasts still contribute to fibrotic scarring. There may also be less fulminant injury states where regions of myocardium lack the full range of neurohumoral and mechanical signals needed to induce αSMA positive myofibroblasts 35-37. Another interesting previous observation is that p38 is mechanically sensitive and that inhibition p38 activity blocked aspects of stretch-induced myofibroblast differentiation 38 (Figure 1F). Finally, we observed in vitro that differentiating fibroblasts had increased gene expression of matrix proteins including the EDA splice variant of fibronectin that is uniquely secreted by myofibroblasts35 despite the fact that less than 100% of the fibroblast population show αSMA stress fibers when treated with a profibrotic agonist (Figure 1B, S1 A-D, and S2 C).
Previous in vitro experiments in MEFs and other mesenchymal cells have shown that inhibition of p38 signaling, including downstream effectors like MK2, block the expression of myofibroblast signature proteins with the reciprocal result observed when upstream p38 activators like MKK6 and MKK3 are overexpressed 11, 13, 38-40. Cultured fibroblasts treated with AngII showed that p38 activation was necessary for fibrosis while TGFβ prolonged p38-dependent increases in the expression of matrix proteins like collagen and fibronectin, as well as αSMA 11, 41, 42. Our in vitro assays place p38 downstream of extracellular signals like TGFβ, AngII, but upstream of known myofibroblast regulatory factors such as calcineurin, TRPC6, MK2, and SRF 11, 21, 39, 40. The temporal, lineage, and injury dependent activation of these combined molecular regulators will thus ultimately determine fibroblast function and hence the degree of fibrosis and its temporal persistence. However, our results are the first to define a fibroblast-specific role for MKK6-p38 signaling in vivo as an inducer of programmed myofibroblast differentiation and cardiac fibrosis.
The identification of a nodal role for p38 in transducing signals within the fibroblast suggests a pharmacologically tractable approach for anti-fibrotic therapies. Indeed, there are a number of well characterized pharmacologic inhibitors of p38 available, some of which have already shown the ability to broadly blunt fibrotic remodeling in mouse models of heart, lung and skeletal muscle disease 16, 17, 43-45. While such agents are clearly attractive to consider in treating human cardiac fibrotic disease states, we also need to consider the timing of anti-fibrotic therapies so that the initial wound healing phase can be effectively maintained.
Supplementary Material
Clinical Perspective.
What is new?
This was one of the first studies to directly target a gene and associated signaling pathway within the fibroblast of a mammalian heart (mouse) and show a direct role in regulating wound healing rafter myocardial infarction and effects on cardiac fibrosis.
This study showed that the gene encoding p38α mitogen-activated protein kinase is required to mediate fibroblast activation in the mouse heart following injury, and also that forced activation of p38 within fibroblasts using a transgenic approach was sufficient to drive fibrosis in multiple tissues of the mouse, including the heart.
What are the clinical implications?
This study suggests that p38 mitogen-activated protein kinase is a nodal signaling effector within the cardiac fibroblast that drives wound healing and long-term fibrosis in heart failure.
This study also suggests that pharmacologic inhibition of p38 mitogen-activated protein kinase in human heart failure patients could reduce progressive fibrotic burden and help maintain cardiac ventricular performance as a novel treatment.
However, this study also suggests that pharmacologic inhibition of p38 mitogen-activated protein kinase during acute myocardial infarction injury would inhibit wound healing and be detrimental to patients.
Acknowledgments
None
Sources of Funding: This work was supported by grants from the National Institutes of Health (JDM and JD) and by the Howard Hughes Medical Institute to JDM.
Disclosures: J.D.M. received significant research support from the National Institutes of Health as a program project grant headed by Dr Katherine Yutzey, which investigates cardiac fibrotic disease mechanisms (P01HL69779). Jennifer Davis received significant research support from the National Institutes of Health that is also related to the subject matter of cardiac fibrosis (R00 HL119353)
References
- 1.Schuetze KB, McKinsey TA, Long CS. Targeting cardiac fibroblasts to treat fibrosis of the heart: focus on HDACs. J Mol Cell Cardiol. 2014;70:100–107. doi: 10.1016/j.yjmcc.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stempien-Otero A, Kim DH, Davis J. Molecular networks underlying myofibroblast fate and fibrosis. J Mol Cell Cardiol. 2016;97:153–161. doi: 10.1016/j.yjmcc.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Hanlon R, Grasso A, Roughton M, Moon JC, Clark S, Wage R, Webb J, Kulkarni M, Dawson D, Sulaibeekh L, Chandrasekaran B, Bucciarelli-Ducci C, Pasquale F, Cowie MR, McKenna WJ, Sheppard MN, Elliott PM, Pennell DJ, Prasad SK. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010;56:867–874. doi: 10.1016/j.jacc.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, Mahon NG, McKenna WJ. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212–2218. doi: 10.1016/s0735-1097(00)01003-2. doi: [DOI] [PubMed] [Google Scholar]
- 5.Bruder O, Wagner A, Jensen CJ, Schneider S, Ong P, Kispert EM, Nassenstein K, Schlosser T, Sabin GV, Sechtem U, Mahrholdt H. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010;56:875–887. doi: 10.1016/j.jacc.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 6.Davis J, Molkentin JD. Myofibroblasts: trust your heart and let fate decide. J Mol Cell Cardiol. 2014;70:9–18. doi: 10.1016/j.yjmcc.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 10.Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, Frangogiannis NG. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. 2010;107:418–428. doi: 10.1161/CIRCRESAHA.109.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell. 2012;23:705–715. doi: 10.1016/j.devcel.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stambe C, Atkins RC, Tesch GH, Masaki T, Schreiner GF, Nikolic-Paterson DJ. The role of p38alpha mitogen-activated protein kinase activation in renal fibrosis. J Am Soc Nephrol. 2004;15:370–379. doi: 10.1097/01.asn.0000109669.23650.56. doi: [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Ma R, Flavell RA, Choi ME. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for activation of p38alpha and p38delta MAPK isoforms by TGF-beta 1 in murine mesangial cells. J Biol Chem. 2002;277:47257–47262. doi: 10.1074/jbc.M208573200. [DOI] [PubMed] [Google Scholar]
- 14.Kompa AR, See F, Lewis DA, Adrahtas A, Cantwell DM, Wang BH, Krum H. Long-term but not short-term p38 mitogen-activated protein kinase inhibition improves cardiac function and reduces cardiac remodeling post-myocardial infarction. J Pharmacol Exp Ther. 2008;325:741–750. doi: 10.1124/jpet.107.133546. [DOI] [PubMed] [Google Scholar]
- 15.See F, Kompa A, Martin J, Lewis DA, Krum H. Fibrosis as a therapeutic target post-myocardial infarction. Curr Pharm Des. 2005;11:477–487. doi: 10.2174/1381612053382098. doi: [DOI] [PubMed] [Google Scholar]
- 16.Matsuoka H, Arai T, Mori M, Goya S, Kida H, Morishita H, Fujiwara H, Tachibana I, Osaki T, Hayashi S. A p38 MAPK inhibitor, FR-167653, ameliorates murine bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2002;283:L103–112. doi: 10.1152/ajplung.00187.2001. [DOI] [PubMed] [Google Scholar]
- 17.Wissing ER, Boyer JG, Kwong JQ, Sargent MA, Karch J, McNally EM, Otsu K, Molkentin JD. P38alpha MAPK underlies muscular dystrophy and myofiber death through a Bax-dependent mechanism. Hum Mol Genet. 2014;23:5452–5463. doi: 10.1093/hmg/ddu270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, Banfi S, Sauer MF, Olsen GS, Duffield JS, Olson EN, Tallquist MD. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139:2139–2149. doi: 10.1242/dev.079970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acharya A, Baek ST, Banfi S, Eskiocak B, Tallquist MD. Efficient inducible Cre-mediated recombination in Tcf21 cell lineages in the heart and kidney. Genesis. 2011;49:870–877. doi: 10.1002/dvg.20750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD, Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis J, Salomonis N, Ghearing N, Lin SC, Kwong JQ, Mohan A, Swanson MS, Molkentin JD. MBNL1-mediated regulation of differentiation RNAs promotes myofibroblast transformation and the fibrotic response. Nat Commun. 2015;6:10084. doi: 10.1038/ncomms10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishida K, Yamaguchi O, Hirotani S, Hikoso S, Higuchi Y, Watanabe T, Takeda T, Osuka S, Morita T, Kondoh G, Uno Y, Kashiwase K, Taniike M, Nakai A, Matsumura Y, Miyazaki J, Sudo T, Hongo K, Kusakari Y, Kurihara S, Chien KR, Takeda J, Hori M, Otsu K. p38alpha mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol Cell Biol. 2004;24:10611–10620. doi: 10.1128/MCB.24.24.10611-10620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krenz M, Gulick J, Osinska HE, Colbert MC, Molkentin JD, Robbins J. Role of ERK1/2 signaling in congenital valve malformations in Noonan syndrome. Proc Natl Acad Sci U S A. 2008;105:18930–18935. doi: 10.1073/pnas.0806556105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 25.Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, Lorts A, Brunskill EW, Dorn GW, 2nd, Conway SJ, Aronow BJ, Robbins J, Molkentin JD. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res. 2007;101:313–321. doi: 10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 27.Xu J, Gong NL, Bodi I, Aronow BJ, Backx PH, Molkentin JD. Myocyte enhancer factors 2A and 2C induce dilated cardiomyopathy in transgenic mice. J Biol Chem. 2006;281:9152–9162. doi: 10.1074/jbc.M510217200. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Cseresnyes Z, Randall WR, Schneider MF. Activity-dependent nuclear translocation and intranuclear distribution of NFATc in adult skeletal muscle fibers. J Cell Biol. 2001;155:27–39. doi: 10.1083/jcb.200103020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D'Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD. Revisiting Cardiac Cellular Composition. Circ Res. 2016;118:400–409. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Sridhar B, Leinwand LA, Anseth KS. Characterization of cell subpopulations expressing progenitor cell markers in porcine cardiac valves. PLoS One. 2013;8:e69667. doi: 10.1371/journal.pone.0069667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma Y, de Castro Bras LE, Toba H, Iyer RP, Hall ME, Winniford MD, Lange RA, Tyagi SC, Lindsey ML. Myofibroblasts and the extracellular matrix network in post-myocardial infarction cardiac remodeling. Pflugers Arch. 2014;466:1113–1127. doi: 10.1007/s00424-014-1463-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K, Olson EN. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107:294–304. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, Saito M, Fukuda K, Nishiyama T, Kitajima S, Saga Y, Fukayama M, Sata M, Kudo A. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med. 2008;205:295–303. doi: 10.1084/jem.20071297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Mollmann H, Looso M, Troidl C, Offermanns S, Wettschureck N. Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ Res. 2016;118:1906–1917. doi: 10.1161/CIRCRESAHA.116.308643. [DOI] [PubMed] [Google Scholar]
- 35.Hinz B. The myofibroblast: paradigm for a mechanically active cell. J Biomech. 2010;43:146–155. doi: 10.1016/j.jbiomech.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Tibbitt MW, Langer SJ, Leinwand LA, Anseth KS. Hydrogels preserve native phenotypes of valvular fibroblasts through an elasticity-regulated PI3K/AKT pathway. Proc Natl Acad Sci U S A. 2013;110:19336–19341. doi: 10.1073/pnas.1306369110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sandbo N, Dulin N. Actin cytoskeleton in myofibroblast differentiation: ultrastructure defining form and driving function. Transl Res. 2011;158:181–196. doi: 10.1016/j.trsl.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J, Chen H, Seth A, McCulloch CA. Mechanical force regulation of myofibroblast differentiation in cardiac fibroblasts. Am J Physiol Heart Circ Physiol. 2003;285:H1871–1881. doi: 10.1152/ajpheart.00387.2003. [DOI] [PubMed] [Google Scholar]
- 39.Xu L, Yates CC, Lockyer P, Xie L, Bevilacqua A, He J, Lander C, Patterson C, Willis M. MMI-0100 inhibits cardiac fibrosis in myocardial infarction by direct actions on cardiomyocytes and fibroblasts via MK2 inhibition. J Mol Cell Cardiol. 2014;77:86–101. doi: 10.1016/j.yjmcc.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vittal R, Fisher A, Gu H, Mickler EA, Panitch A, Lander C, Cummings OW, Sandusky GE, Wilkes DS. Peptide-mediated inhibition of mitogen-activated protein kinase-activated protein kinase-2 ameliorates bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2013;49:47–57. doi: 10.1165/rcmb.2012-0389OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim SI, Kwak JH, Zachariah M, He Y, Wang L, Choi ME. TGF-beta-activated kinase 1 and TAK1-binding protein 1 cooperate to mediate TGF-beta1-induced MKK3-p38 MAPK activation and stimulation of type I collagen. Am J Physiol Renal Physiol. 2007;292:F1471–1478. doi: 10.1152/ajprenal.00485.2006. [DOI] [PubMed] [Google Scholar]
- 42.Meyer-Ter-Vehn T, Gebhardt S, Sebald W, Buttmann M, Grehn F, Schlunck G, Knaus P. p38 inhibitors prevent TGF-beta-induced myofibroblast transdifferentiation in human tenon fibroblasts. Invest Ophthalmol Vis Sci. 2006;47:1500–1509. doi: 10.1167/iovs.05-0361. [DOI] [PubMed] [Google Scholar]
- 43.Kyoi S, Otani H, Matsuhisa S, Akita Y, Tatsumi K, Enoki C, Fujiwara H, Imamura H, Kamihata H, Iwasaka T. Opposing effect of p38 MAP kinase and JNK inhibitors on the development of heart failure in the cardiomyopathic hamster. Cardiovasc Res. 2006;69:888–898. doi: 10.1016/j.cardiores.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 44.Park JK, Fischer R, Dechend R, Shagdarsuren E, Gapeljuk A, Wellner M, Meiners S, Gratze P, Al-Saadi N, Feldt S, Fiebeler A, Madwed JB, Schirdewan A, Haller H, Luft FC, Muller DN. p38 mitogen-activated protein kinase inhibition ameliorates angiotensin II-induced target organ damage. Hypertension. 2007;49:481–489. doi: 10.1161/01.HYP.0000256831.33459.ea. [DOI] [PubMed] [Google Scholar]
- 45.Wilde JM, Gumucio JP, Grekin JA, Sarver DC, Noah AC, Ruehlmann DG, Davis ME, Bedi A, Mendias CL. Inhibition of p38 mitogen-activated protein kinase signaling reduces fibrosis and lipid accumulation after rotator cuff repair. J Shoulder Elbow Surg. 2016;25:1501–1508. doi: 10.1016/j.jse.2016.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.