

Abstract
Large granular lymphocytes (LGLs) are large lymphocytes with azurophilic granules in their cytoplasm. LGLs are either natural killer (NK) cells or T lymphocytes. Expansions of the LGLs in the peripheral blood are seen in various conditions, including three clonal disorders: T-cell LGL (T-LGL) leukemia, chronic lymphoproliferative disorders of NK cells (CLPD-NK), and aggressive NK-cell leukemia (ANKL). However, the monoclonal and polyclonal expansion of LGLs has been associated with many other conditions. The present article describes these LGL disorders, with special emphasis on the clinical features, pathogenesis, and treatments of the three above-mentioned clonal disorders.
Keywords: LGL, T-LGL leukemia, chronic lymphoproliferative disorders of NK cells, aggressive NK-cell leukemia
Introduction
Large granular lymphocytes (LGLs) are large lymphocytes with round or reniform nuclei, a broad cytoplasm, and azurophilic granules in their cytoplasm (Fig. 1) (1). They account for 10-15% of the peripheral blood mononuclear cells. Most of the normal LGLs in the peripheral blood are natural killer (NK) cells, while some are T lymphocytes (2). Monoclonal or polyclonal LGL expansions are found in patients with various conditions. According to the current World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues, clonal LGL expansions are divided into three disorders (3): T-cell LGL (T-LGL) leukemia, chronic lymphoproliferative disorders of NK cells (CLPD-NK), and aggressive NK-cell leukemia (ANKL) (Fig. 2-1 to Fig. 2-4). T-LGL leukemia is the most common disorder, followed by CLPD-NK; ANLK is rare.
Figure 1.
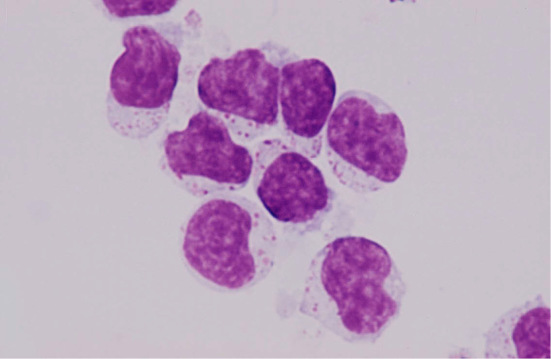
LGLs isolated from a normal individual.
Figure 2.

Peripheral blood smears of T-LGL leukemia, CLPD-NK, and ANKL patients. 1: T-LGL lukemia. 2: T-LGL leukemia, small-cell type. 3: CLPD-NK. 4: ANKL (the arrows indicate ANKL cells). T-LGL: T-cell large granular lymphocyte, CLPD-NK: chronic lymphoproliferative disorders of natural killer cells, ANKL: aggressive NK-cell leukemia
Case Description
A 40-year-old single physical education teacher presented to the author's clinic with T-LGL leukemia and pure red cell aplasia (PRCA). He had been diagnosed with CD3+CD4-CD8- T-LGL leukemia at another hospital 7 years previously. His hemoglobin level was 8 g/dL, and a bone marrow examination revealed 7% erythroblasts. Cyclophosphamide was recommended, but he refused it due to the possibility of infertility (4). Three years later, cyclosporine was administered, but was subsequently discontinued due to renal damage. One year later, the patient's hemoglobin level dropped to 2 g/dL, a bone marrow examination revealed 3% erythroblasts, and he became blood transfusion-dependent. The patient was diagnosed with PRCA and was referred to the author's clinic.
On a physical examination, the patient was pale, without apparent lymphadenopathy or hepatosplenomegaly. A laboratory analysis revealed the following: hemoglobin, 3 g/dL; reticulocytes, 1.3%; WBC count, 3.4×109/L with 68.2% lymphocytes. Most of the lymphocytes were “small” lymphocytes (approximate diameter: 12.2 μm) with numerous azurophilic granules in their narrow cytoplasm. The lymphocytes were CD3+CD4-CD8-CD16-CD56-CD57-, T-cell receptor (TCR)αβ- and TCRγδ+. The TCR Jγ and TCR Cβ1 genes displayed monoclonal rearrangement. STAT3 Y640F,D661Y or STAT5b exon16 mutations were not found.
Cyclosporine was restarted, but was discontinued again due to renal damage. Methotrexate was administered and then discontinued due to hepatotoxicity. The patient was then treated with azathioprine, which was also discontinued due to hepatotoxicity. After storing the patient's sperm, oral cyclophosphamide (100 mg, daily) was administered for two weeks; the dose was then reduced to 50 mg daily. The patient's reticulocyte count began to increase one month later, and his anemia disappeared eight months later. Cyclophosphamide was discontinued after the TCR genes were confirmed to show germ-line configurations by Southern blotting. In total, it was administered for 10 months. One year has passed since treatment was discontinued, and no relapse has been detected. As a physical education teacher, he is now able to run with his students.
The Clinical Features of T-LGL Leukemia
T-LGL leukemia is a rare lymphoproliferative disorder that usually follows an indolent clinical course. As shown in Table, it typically develops around 60 years of age (median), with males and females equally affected (females may be slightly more affected). T-LGL leukemia is associated with various types of cytopenia. In Western countries, severe neutropenia with infection is often seen (5-13). In contrast, neutropenia is not as common in Japan and China, while severe anemia (including PRCA) is more frequently found (14-18). In Western countries, T-LGL leukemia is commonly associated with rheumatoid arthritis and Felty's syndrome (5-13); in Asia, association with these diseases is not common (14-18).
Table.
The Clinical Features of T-LGL Leukemia and CLPD-NK Reported in Asian and Western Countries.
| Reference | 15, 16 | 18 | 8 | 10 | 11 | 12 | 13 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T or NK | T | NK | T | T | NK | T | NK | T | T | T | NK |
| Number of patients | 35 | 10 | 22 | 129 | 11 | 44 | 14 | 29 | 26 | 201 | 28 |
| Median age | 60 | 58 | 47 | 7 | 39 | 63 | 67 | 57 | 58 | 59 | 58 |
| Male/female | 16/19 | 4/6 | 14/8 | 57/71 | 6/5 | 22/22 | 7/7 | 12/17 | 13/13 | 90/111 | 14/14 |
| Splenomegaly (%) | 11 | 10 | 50 | 91 | 35 | 0 | 66 | 23 | 24 | 25 | |
| Hepatomegaly (%) | 3 | 0 | 23 | 64 | 10 | 12 | 10 | 14 | |||
| Adenopathy (%) | 3 | 0 | 1 | 27 | 5 | 0 | 10 | 4 | 6 | 7 | |
| Rheumatoid Arthritis (%) | 6 | 0 | 28 | 0 | 20 | 14 | 38 | 15 | 17 | 11 | |
| Neutropenia (%) | 54 | 20 | 36 | 84 | 64 | 52 | 69 | 77 | 61 | 48 | |
| <0.5×109/L (%) | 6 | 10 | 0 | 48 | 18 | 41 | 46 | 69 | 23 | 26 | 16 |
| Anemia (%) | 80 | 30 | 86 | 49 | 1 | 89 | 77 | 77 | 24 | 25 | |
| Hb<8.0 g/dL (%) | 46 | 0 | 50 | 36 | 31 | 15 | 7 | 4 | |||
| Thrombocytopenia (%) | 6 | 10 | 23 | 19 | 75 | 36 | 15 | 45 | 35 | 19 | 8 |
Our studies (15, 16) and Kwong’s study (18) are from Asia. The others (8, 10-13) are from Western countries. T-LGL: T-cell large granular lymphocyte, CLPD-NK: chronic lymphoproliferative disorders of natural killer cells
The surface phenotype of T-LGL leukemia is mostly CD3+CD4-CD8+, and less often CD3+CD4-CD8-, CD3+CD4+CD8-, or CD3+CD4+CD8+. The normal counterpart of CD3+CD4-CD8+ leukemic cells is thought to be in vivo-primed effector-type cytotoxic T lymphocytes (CTLs) of unknown antigen specificity, because T-LGLs are mostly CD45RA+CD27-CD28-CD57+/-CD94+NKG2+ (19,20), contain perforin and other cytotoxic molecules (21,22), and exhibit strong cytotoxicity in vitro when anti-CD3 antibodies are added to cultures of CD3+ LGLs and Fcγ receptor-bearing target cells (23), and when anti-CD3 × anti-CD10 bispecific antibodies are added to the cultures of CD3+ LGLs and CD10+ target cells (24). CD3+CD4+CD8-/+ leukemic cells are considered to be derived from T-LGLs with the CD4+CD56+TCRαβ+ phenotype found in small quantities in normal individuals (25). CD3+CD4-CD8- T-LGL leukemia is almost always of the TCRγδ+ phenotype, which is present in small amounts in the normal peripheral blood. Its clinical features are not very different from those of the TCRαβ+ phenotype (26-29). CD56+ T-LGL leukemia has been reported to follow an aggressive clinical course (30,31); however, the presence of the CD56 antigen does not always indicate an aggressive clinical course. In our series, the CD56 antigen was detected in 4 of 21 cases of indolent T-LGL leukemia. In contrast, the one patient with aggressive T-LGL leukemia was negative for the CD56 antigen (14-16).
The Diagnostic Criteria and Terminology of T-LGL Leukemia
The diagnostic criteria of T-LGL leukemia are somewhat confusing. In the 2008 WHO Classification (3), T-LGL leukemia is defined by a persistent (>6 months) increase in the number of peripheral blood LGLs (usually 2-20×109 LGLs/L), without a clear cause. However, patients with <2×109 LGLs/L of clonally expanded LGLs are often seen, and the clinical features of these patients have been reported to be similar to those in with >2×109 LGLs/L (32). The duration of LGL persistence is not necessarily required for the diagnosis, because patients with severe anemia or neutropenia must be treated immediately without observing them in a drug-free state for six months or more (15,16). It is the authors opinion that whenever a patient presents cytopenia (either anemia or neutropenia) of an unexplained etiology, the demonstration of the clonal expansion of LGLs should be sufficient to make a diagnosis of T-LGL leukemia, irrespective of whether more than 2×109/L LGLs are present (15,16). In the absence of cytopenia, we must carefully differentiate whether a reactive and transient process is being observed. The clonality of the LGLs is detected by the Southern blotting of the TCR genes, a PCR of the TCR genes, or by monoclonal antibodies against TCR Vβ gene repertoires. In daily clinical practice, Tanahashi et al. (33) recommended a simplified way of identifying the LGL leukemia of T- and NK-cell lineages when both of the following criteria are fulfilled: lymphocytes account for >52% of the WBC, and when >50% of the lymphocytes are LGLs.
In normal individuals, the absolute numbers of LGLs in the peripheral blood have been reported to be 223±99 /μL (mean±SD) (5), 198±112 /μL (34), and 210±20 /μL (35). The number in males is significantly higher than that in females (35). The author's examination revealed that the absolute numbers of peripheral blood LGLs in normal Japanese individuals were 630±261 /μL in males (n=20), and 350±176 /μL in females (n=20), with the number in males being significantly higher (14). In an examination of 35 normal Japanese individuals, Ishida et al. (personal communication) found that the number was 320±265 /μL. It is not clear whether the difference in the absolute LGL counts between Japanese individuals and individuals from other backgrounds stems from differences between races and/or environmental factors. The French study group accepted, with some exceptions, the diagnostic criteria of the peripheral expansion of LGLs (>0.5×109/L) for a period of >6 months, excluding transient or reactive LGL proliferation (13).
Historically, Brouet et al. (36) first described the expansion of LGLs in some patients with T-cell chronic lymphocytic leukemia in 1975. McKenna et al. (37) analyzed four such cases, and considered the process to be a distinct clinicopathological entity in 1977. Since then, similar case descriptions have been accumulated. LGL leukemia has had various names. Leukemia of LGL was first named by Loughran et al. (5,6), while the name T-LGL leukemia has been accepted worldwide, particularly since it was proposed in the 2001 and 2008 WHO classifications. Other names have included Tγ-lymphoproliferative disease (38), lymphoproliferative disease of granular lymphocytes (7), and granular lymphocyte-proliferative disorders (14-16). Some of the names use the term leukemia, while others do not. The reason why the term leukemia is not used is because it is unclear whether T-LGL leukemia represents a true lymphoid malignancy or a self-limited clonal proliferation. Because the number of LGLs stays stable over a period of time, and because spontaneous regression can occur, most T-LGL leukemia patients do not seem to have leukemia. Similarly to the situation of monoclonal gammopathy of undetermined significance (MGUS), the alternative name of “T-cell clonopathy of undetermined significance” has been proposed (9). The reason why the term granular lymphocytes (GLs), but not LGLs, is sometimes used is because some patients have small GLs (rather than large GLs) as described above in the Case Description. Tanahashi et al. (39) compared the diameter of normal GLs and found that the GLs from most patients with LGL leukemia were ≥14 μm, while 6 out of the 26 patients with LGL leukemia showed GLs of ≤12.5 μm. This small subtype was recognized among younger patients and was associated with STAT3 mutations (39). As shown in Fig. 2, most of our cases exhibited typical LGLs, while some exhibited small GLs.
Lymphocytes are described as “large” when they are more than twice the size of erythrocytes in diameter. Lymphocytes that are less than twice the size of erythrocytes are considered to be “small” (40). Then what is the diameter of T-LGL leukemia cells? Some are more than twice the size of erythrocytes, but many are less twice the size. Strictly speaking, the term LGL cannot be uniformly applied. However, it is not known whether the size of LGLs changes after forced drying when peripheral blood films are made. In Japan blood films are dried with cold air using a dryer. In Western countries, to the best of the author's knowledge, films are dried naturally without using a dryer. We have to compare the size differences between these two methods.
LGL Expansions in Various Conditions
The monoclonal expansion of nonmalignant B cells occurs frequently in elderly individuals. This is thought to be a consequence of the chronic stimulation of B cells by self-antigens or microbial antigens over the patient's lifetime. The similar clonal expansions of T cells occur in elderly individuals. The clonal CD3+CD8+CD28-TCRαβ+ cell populations are frequently found in normal subjects of >65 years of age; in such patients, they represent up to 37.5% of all CD8+ cells (41). CD3+CD8+CD28- cells have been shown to lack proliferative responses to CD3/TCR stimuli and to display high CTL activity. Thus, it is possible that repeated exposure to major histocompatibility complex (MHC) class I-restricted antigens results in the clonal accumulation of end-stage mature CTLs. However, it was not described whether the morphology of these CTLs was similar to that of LGLs. It is likely that these clonal CD8+ CTLs respond to the same antigenic stimuli, regain their proliferative capacity, and result in T-LGL leukemia.
LGL expansions are found in various conditions. Viral infections, including cytomegalovirus (CMV), have been reported to induce transient or persistent clonal CD3+CD4+ or CD3+CD8+ T-LGL expansions (42), and CMV-expanded T-LGLs have been proven to recognize CMV antigens (43). Serum reactivity to human T-cell leukemia virus (HTLV) type 1 proteins was described in LGL leukemia patients (44). Forty-four percent of 53 patients had anti-HTLV antibodies (45). However, HTLV-1 to -4 and their related viruses have not been proven to be causative agents of LGL leukemia (45).
The persistent elevation of monoclonal or oligoclonal CD3+CD8+CD57+TCRαβ+ CTL or T-cell LGLs has been reported in a significant proportion of recipients following allogeneic hematopoietic stem cell transplantation (HSCT) (46-49). These patients tend to have an overall survival advantage with a lower incidence of relapse, and a higher incidence of graft-versus-host disease and CMV reactivation; this implies that LGLs of donor origin may have a role in targeting the recipient's tumor cells and normal tissues as well as CMV. The absence of STAT3 gene mutations, and the declining longitudinal kinetics of LGLs in the late period support their benign nature (49). The clonal expansion of CD3+CD8+CD57+ LGLs has been reported after autologous HSCT (50,51). The number of infections or the clinical response to autologous HSCT did not appear to be correlated with the degree of the LGL expansions, and the expansions were indolent, were not associated with cytopenias, and did not require therapy. Studies are required to determine the functions of expanded T-LGLs. Interestingly, LGL expansions have also been reported in solid organ transplant recipients (52). A significant number of heart and renal transplant patients developed clonal CD3+CD57+ T-LGL expansions. None of the patients developed neutropenia or had active donor organ rejection, and most patients did not receive therapy for T-LGL expansion. Although the details were not shown, NK-cell expansions were also found in most of the patients (52).
Some patients with chronic myeloid leukemia or Philadelphia chromosome-positive acute lymphoblastic leukemia who are treated with the tyrosine kinase inhibitor dasatinib, but not with other tyrosine kinase inhibitors, show significant LGL expansions [reviewed in (53)]. This phenomenon was first described by Mustjoki et al. (54) and Kim et al. (55), and many reports thereafter. LGL expansions begin as early as two hours from the start of therapy, peak at 3 months, and persist throughout the therapy. The cessation of dasatinib treatment induces rapid decline of the LGL counts, almost to the baseline levels. Expanded LGLs are reported to be CD3+CD8+ monoclonal or polyclonal T cells, or NK cells. LGL lymphocytosis is often accompanied by fevers, colitis, and pleural effusions, which suggests that these LGLs are involved in the aberrant immune response. LGLs may participate in the elimination of residual leukemic cells, and LGL expansions are associated with excellent, long-lasting therapeutic responses in dasatinib-treated patients. LGL expansions may also participate in CMV inactivation.
T-LGL expansions are found in association with various B-cell dyscrasias, including MGUS, chronic lymphocytic leukemia, follicular lymphoma, and polyclonal hyper- or hypo-gammaglobulinemia (56,57). The co-association of the B-cell pathology with T-LGL suggests that either a common antigen drives the clonal B and T cells, or that a B-cell abnormality could serve as the stimulus for T-LGL expansion as a form of anti-tumor surveillance.
The Molecular Pathogenesis of T-LGL Leukemia
Leukemic LGLs were found to display high levels of activated signal transducer and activator of transcription 3 (STAT3), and the treatment of leukemic LGLs with the JAK-selective tyrosine kinase inhibitor AG-490 induced apoptosis with a corresponding decrease in STAT-DNA binding activity (58). Recently, recurrent somatic mutations in the Src homology (SH) 2 domain of the STAT3 gene have been found in 27-40% of patients with T-LGL leukemia (59,60) and 30% of patients with CLPD-NK (60), leading to the constitutive activation of STAT3 and the dysregulation of genes downstream of STAT3. These gain-of-function mutations enhance the transcriptional activity of the mutated proteins. Patients with STAT3 mutations presented with neutropenia and rheumatoid arthritis more frequently than patients without these mutations (59), and patients with symptomatic disease and a history of multiple treatments (60). The association between PRCA and neutropenia with STAT3 gene mutations was also reported in Asia (61,62). Activating mutations in the SH2 domain of the STAT5B gene were identified in 2% of LGL leukemia patients (63). The clinical course of the disease in patients with the STAT5B N642H mutation was aggressive and fatal, and was clearly different from typical LGL leukemia (63). Activating STAT5B mutations were also found in 6 of 11 cases (55%) of CD4+CD8-/+TCRαβ+ T-LGL leukemia, whereas STAT5B mutations were very rare among patients with CD4-CD8+ T-LGL leukemia or CLPD-NK (25). The disease course in CD4+ T-LGL leukemia cohort was indolent, and none of the patients with STAT5B mutations required therapy during the observation period (25). STAT3 and STAT5B mutations can be used as molecular markers for the diagnosis of LGL leukemia, and present novel therapeutic targets for STAT3 and STAT5b inhibitors, which are currently in development.
Other than the JAK/STAT pathways, multiple cell survival pathways, including sphingolipid signaling, RAS/MEK/ERK, and SFK/PI3K/Akt, have been found to be constitutively activated in LGL patients [reviewed in (64)]. A biological approach identified IL-15 and platelet-derived growth factor as master survival signaling switches that may have a profound effect on all known deregulations in T-LGL leukemia. The Fas-mediated apoptotic pathway is impaired, and this may support the accumulation of LGLs (65).
Taken together, the above findings strongly support the role of autologous or foreign antigenic stimulation as an initial event. This would lead to the expansion of fully differentiated effector/cytotoxic LGLs, which are not eliminated due to the impairment of the apoptotic pathway. The acquisition of STAT3/STAT5B mutations might occur late in the natural history of the disease, certainly later than the establishment of clonality (66).
The Pathogenesis of Associated Cytopenias
Anemia, neutropenia, and less often thrombocytopenia are associated alone or in combination with T-LGL leukemia (Table). Anemia develops from the T-LGL-mediated suppression of erythroid progenitor cells (26,38). Whether T-LGLs directly kill these progenitor cells or suppress erythropoiesis by means of humoral mediators is an intriguing question. In a patient with CD3+CD4-CD8-TCRγδ+ T-LGL leukemia, it was shown that the patient's LGLs directly killed erythroid progenitor cells (67). These LGLs, which express killer-cell inhibitory receptors, were shown to be able to kill target cells in an MHC-unrestricted manner, but their cytolytic activity was inhibited by signaling through killer-cell inhibitory receptors when target cells expressed HLA class I antigens. However, because the expression of HLA class I antigens on erythroid progenitor cells is physiologically downregulated, the T-LGLs could lyse erythroid progenitors. Immature CD34+ progenitors and myeloid cells express HLA class I antigens and they were not lysed by the T-LGLs. Regarding the pathogenesis of neutropenia, T-LGLs express Fas ligand (FasL), and mature neutrophils express Fas. Fas-FasL system-mediated neutrophil apoptosis is highly likely to explain the cause of neutropenia (reviewed in 68). Several studies have indicated the presence of anti-neutrophil autoantibodies in the serum of some patients, but in most of these cases the detected antibodies were anti-HLA antibodies, rather than true anti-neutrophil autoantibodies, and it is unlikely that anti-neutrophil autoantibodies cause neutropenia (68).
CD3+CD8+ T-LGL expansions are found in bone marrow failure syndromes, such as aplastic anemia (AA) (69,70), paroxysmal nocturnal hemoglobinuria (PNH) (70), and myelodysplastic syndromes (MDS) (71,72). The frequency with which T-LGLs and bone marrow failure syndromes coexists suggests an etiological relationship rather than simple coincidence. In a large cohort of 367 patients with MDS and 140 patients with AA (73), STAT3-mutated clones could be found, not only in the known LGL-concomitant cases, but also in a small proportion of unsuspected cases (AA, 7%; and MDS, 2.5%). AA patients with STAT3 mutations showed an interesting trend toward a better response to immunosuppressive therapy; an association with the presence of human leukocyte antigen-DR15 was also found. MDS patients harboring STAT3-mutated clones showed a lower degree of bone marrow cellularity and a higher frequency of developing chromosome 7 abnormalities. STAT3-mutated LGL clones may facilitate the persistent dysregulation of autoimmune activation, which is responsible for the primary induction of bone marrow failure in a subset of AA and MDS patients. Bone marrow failure patients and LGL leukemia patients have similarly elevated levels of antibodies to BA21 protein, which are located in the transmembrane region of the HTLV-1 envelope (74). This suggests that LGL leukemia, AA, MDS and PNH might share a common pathogenesis. LGL expansions are frequently found in patients with cytopenia of unknown etiology and should be thoroughly investigated in this patient group (75).
The Treatment of T-LGL Leukemia
Because the clinical course of most patients with T-LGL leukemia is indolent, it is not always necessary to treat this disease. The patients who require treatment include those with symptomatic or transfusion-dependent anemia, and those with severe neutropenia (absolute neutrophil count <500 /μL) or moderate neutropenia (absolute neutrophil count >500 /μL) with recurrent infections. There is no standard treatment. Immunosuppressive therapy with agents including cyclophosphamide, methotrexate, cyclosporine and steroids have been used.
For patients with T-LGL leukemia who have severe anemia, the author prefers to use cyclophosphamide as a first-line therapy. This preference is based on a 1997 study in which the author was involved, in which hematological complete remission was obtained in all of the seven patients treated with cyclophosphamide (76). Five of the seven patients were in molecular complete remission, and did not experience a relapse for at least one year after the discontinuation of cyclophosphamide. Cyclophosphamide was initiated at a daily dose of 100 mg, orally. The dose was reduced to 50 mg per day after two weeks. For Japanese patients, a continued daily dose of 100 mg seems to be excessive. Cyclophosphamide was administered until the absolute lymphocyte count fell to <1,000 /μL and the TCR genes showed germ-line configurations. Hematological responses were first observed after 8 weeks of treatment, with the patients obtaining hematological complete remission after six months. Similar results describing the effectiveness of cyclophosphamide have been reported from Western countries (77,78) as well as from Japanese retrospective nationwide study (79). The treatment is continued until TCR genes are in germ-line configurations; however, because the complications of infertility and MDS/acute myeloid leukemia are dependent on the cumulative cyclophosphamide dose and the length of exposure, the duration of cyclophosphamide treatment should be as short as possible. It has not been shown, however, whether TCR Southern blotting is sufficient to confirm the absence of clonality, or whether the absence of minimal residual disease should be confirmed by a PCR. Considering the presence of small amounts of clonal T lymphocytes in old healthy individuals, it may not be necessary to obtain a PCR-based complete remission.
In patients with severe neutropenia, cyclophosphamide is also effective (78). Methotrexate has long been used as a first-line therapy for T-LGL leukemia with anemia or neutropenia (80). A prospective multicenter phase II study showed that the overall response rate to methotrexate was 38%, and the patients who failed methotrexate therapy and who were subsequently treated with cyclophosphamide showed an overall response rate of 64% (81). Prospective studies to compare cyclophosphamide and methotrexate are required. Successful responses to purine analogs have been reported, but the numbers of patients treated with these agents are still very low.
Chronic Lymphoproliferative Disorders of NK Cells (CLPD-NK)
CLPD-NK, which was proposed as a provisional entity in the 2008 WHO Classification (3), is characterized by the chronic expansion of mature-looking CD3-CD56+/-CD16+ NK cells in the peripheral blood (82-85). The condition develops at approximately 60 years of age (median), and males and females are equally affected (Table). In most cases, patients present with a chronic indolent course without cytopenias or symptoms, and treatment is not required. The disease is diagnosed when ≥2×109/L NK cells are present in the peripheral blood for at least 6 months, without a clearly identified cause (3). The number of NK cells may remain stable without treatment or show spontaneous regression.
Considering the role of NK cells in innate immunity, some etiological agents may stimulate and expand a subset of the NK-cell population (86). The possible association between chronic viral infections and skewed NK-cell proliferation, as evidenced by the skewed expression of killer immunoglobulin-like receptors (KIR), has been described (86,87). Such common foreign antigens may also stimulate T-cell LGLs, thereby leading to both disorders (66), as shown in a recent report, which described the detection of monoclonal T populations in patients with KIR-restricted CLPD-NK (88). Thus T-LGL leukemia and CLPD-NK are likely to represent two sides of the same coin, although they appear to be distinct, separate disorders (66).
Rare cases transform to ANKL. Patients with Epstein-Barr virus (EBV)-positive NK cells tend to transform. These EBV-positive patients usually have chronic active EBV infection, hypersensitivity to mosquito bites or hydroa vacciniforme, and should be carefully followed for the emergence of clonally expanding NK cells (83,89-91). The clonality of EBV-positive NK cells can be examined by Southern blotting using a probe recognizing the EBV terminal repeat. A clonal expansion of NK cells is noted in a substantial number of such patients, which indicates that patients with EBV-associated CLPD-NK may develop ANKL or extranodal NK-cell lymphoma (83,89-91).
Aggressive NK-cell Leukemia (ANKL)
ANKL was originally reported by Fernandez et al. (92) and Koizumi et al. (93), and was further described by Imamura et al. (94), Song et al. (95) and Suzuki et al. (96). Patients are affected at a relatively young age (median: 42 years). There is no sex predilection (or a slight male predominance). It seems to be highly prevalent in Asian people. Fever, hepatosplenomegaly and lymphadenopathy are common. The clinical course is rapidly progressive, and the prognosis is poor with an overall survival of 2 months (96,97). Liver dysfunction, disseminated intravascular coagulation, and hemophagocytic syndrome are often seen during the course of the disease, particularly at the terminal stage. This disease is characterized by the presence of NK cells of a slightly immature-looking morphology, mainly in the peripheral blood, bone marrow, liver and spleen. A diagnosis is made when the following findings are fulfilled (83,85,97,98): (i) morphologically, slightly immature-looking large lymphocytes, with a broad, pale cytoplasm and azurophilic granules, and somewhat fine nuclear chromatins and occasional nucleoli, are present in the peripheral blood and/or bone marrow; (ii) immunophenotypically, these lymphocytes are CD3-CD56+CD16-/+CD57-; and (iii) genetically, they have germ-line configurations of the TCR and IGH genes. The ANKL cells appear slightly immature in their morphology, but the presence of the CD94 antigen indicates their mature cell origin (99). Three types of leukemic cells were classified. Type I cells are LGLs, while type III cells are pleomorphic and atypical cells with prominent nucleoli. Type II cells show an intermediate morphology between types I and III (100). At presentation, this disease is sometimes indistinguishable from CLPD-NK, but from our experience, the presence of conspicuous nucleoli in NK cells strongly supports a diagnosis of ANKL. Furthermore, the diagnosis can be predicted by the following factors: <40 years of age, fever, lymph node swelling, and hepatosplenomegaly (15). Extranodal NK-cell lymphoma may also present as (or eventually develop) leukemic changes, and the distinction between ANKL and extranodal NK-cell lymphoma is sometimes difficult unless a nasal lesion is found. Few clinicopathological differences exist between younger NK-cell lymphoma and ANKL patients (101). The incidence of skin involvement is significantly higher among patients with extranodal NK-cell lymphoma (83). An array-based comparative genomic hybridization analysis demonstrated clear genetic differences between ANKL and extranodal NK-cell lymphoma, suggesting that these are two distinct diseases (102).
Oligo-array comparative genomic hybridization revealed that the most frequently deleted chromosomal regions in ANKL and extranodal NK-cell lymphoma were in 6q21, which includes tumor suppressor genes FOXO3 and PRDM1, which may be involved in the pathogenesis of NK-cell neoplasms (103). In most patients, clonal EBV is found in tumor cells, and EBV is considered to be the etiological agent (89,104,105). Rare cases of EBV-negative ANKL have been described (106). It tends to occur in older patients and is clinically and pathologically indistinguishable from EBV-positive ANKL, with a similar fulminant clinical course. The high prevalence of Asian patients seen with EBV-positive disease seems less evident with EBV-negative cases.
The disease is refractory to chemotherapy and exhibits a relentless progressive course with a poor outcome (107-109). The types of chemotherapy that should be used, and whether HSCT should be combined are matters of great concern (107-111). As in extranodal NK-cell lymphoma, this poor outcome may be partially explained by the presence of P-glycoprotein (P-gp), a multidrug resistance gene-encoded protein on the NK-cell membrane, which extrudes various cytotoxic agents, such as vinca alkaloids and anthracyclines (112-114), and combination chemotherapies using P-gp-unrelated drugs are expected. As an initial therapy, L-asparaginase chemotherapy resulted in a better response. A retrospective Japan-Korea multicenter study (100) was conducted to better elucidate the clinicopathological features and therapeutic modalities for ANKL. A total of 34 patients were analyzed. Six patients received allogeneic HSCT and two received autologous HSCT, all of whom were in non-complete remission. After HSCT, 4 patients who received allogeneic HSCT and one who received autologous HSCT reached a complete remission. The median survival of all patients was 51 days. The median survival of the patients who were treated with and without HSCT was 266 and 36 days, respectively. Two patients who received allogeneic HSCT were alive and in complete remission. All patients without HSCT died of ANKL. The use of L-asparaginase was indicated as a factor for longer survival. An early diagnosis, treatment with L-asparaginase-based chemotherapy, and allogeneic HSCT might lead to improved patient outcomes. A Korean study also supported the above findings (115). In one patient, when ANKL relapsed after allogeneic HSCT, cyclosporine was rapidly tapered and chemotherapy was initiated. During hematopoietic recovery, the number of atypical lymphocytes increased, and they were found to be donor-derived EBV-specific CTL. The patient achieved a partial response and the EBV viral load decreased to the normal range (116). EBV-reactive immunotherapy may be promising.
Future Prospects
First, by increasing our understanding about the nature of T-LGL leukemia, we will recognize whether or not T-LGL leukemia is a true hematologic malignancy. Based on the findings, the name “T-LGL leukemia” may be changed to something more appropriate. Second, the diagnostic criteria of T-LGL leukemia should be more sophisticated. Third, further investigations are required to find the primary event of LGL proliferation, and the commitment of molecular changes to allow LGL expansions. Fourth, the treatment of T-LGL leukemia remains unsatisfactory, and molecularly targeted therapy should be extensively investigated in this era of precision cancer medicine. ANKL is very difficult to treat. This is mainly due to our limited knowledge on the molecular events leading to the development and progression of the disease. Extensive investigations are required.
Author's disclosure of potential Conflicts of Interest (COI).
Kazuo Oshimi: Advisory role, Eisai.
Acknowledgement
I thank Drs. Fumihiro Ishida, Junji Suzumiya, Takuto Miyagishima, and the staff members of Kushiro Rosai Hospital and Kushiro Central Hospital for their help and advice. I also thank Ms. Hiroko Mikuni for preparing the manuscript.
References
- 1. Timonen T, Ortaldo JR, Herberman RB. Characteristics of human large granular lymphocytes and relationship to natural killer and K cells. J Exp Med 153: 569-582, 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schmidt RE, Murray C, Daley JF, Schlossman SF, Ritz J. A subset of natural killer cells in peripheral blood displays a mature T-cell phenotype. J Exp Med 164: 351-356, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Swerdlow SH, Campo E, Harris NL, et al. , Eds. IARC Press, Lyon, 2008: 272-277. [Google Scholar]
- 4. Buchanan JD, Fairley KF, Barrie JU. Return of spermatogenesis after stopping cyclophosphamide therapy. Lancet 2: 156-157, 1975. [DOI] [PubMed] [Google Scholar]
- 5. Loughran TP, Marshall EK, Starkebaum G, et al. Leukemia of large granular lymphocytes: association with clonal chromosomal abnormalities and autoimmune neutropenia, thrombocytopenia, and hemolytic anemia. Ann Intern Med 102: 169-175, 1985. [DOI] [PubMed] [Google Scholar]
- 6. Loughran TP, Starkebaum G. Large granular lymphocyte leukemia. Report of 38 cases and review of the literature. Medicine 66: 397-405, 1987. [PubMed] [Google Scholar]
- 7. Semenzato G, Pandolfi F, Chisesi T, et al. The lymphoproliferative disease of granular lymphocytes. A heterogeneous disorder ranging from indolent to aggressive conditions. Cancer 60: 2971-2978, 1987. [DOI] [PubMed] [Google Scholar]
- 8. Loughran TP Jr. . Clonal diseases of large granular lymphocytes. Blood 82: 1-14, 1993. [PubMed] [Google Scholar]
- 9. Dhodapkar MV, Li CY, Lust JA, Tefferi A, Phyliky RL. Clinical spectrum of clonal proliferations of T-large granular lymphocytes: a T-cell clonopathy of undetermined significance? Blood 84: 1620-1627, 1994. [PubMed] [Google Scholar]
- 10. Neben MA, Morice WG, Tefferi A. Clinical features in T-cell vs. natural killer-cell variants of large granular lymphocyte leukemia. Eur J Haematol 71: 263-265, 2003. [DOI] [PubMed] [Google Scholar]
- 11. Osuji N, Matutes E, Tjonnfjord G, et al. T-cell large granular lymphocyte leukemia: a report on the treatment of 29 patients and a review of the literature. Cancer 107: 570-578, 2006. [DOI] [PubMed] [Google Scholar]
- 12. Aribi A, Huh Y, Keating M, et al. T-cell large granular lymphocytic (T-LGL) leukemia: experience in a single institution over 8 years. Leuk Res 31: 939-945, 2007. [DOI] [PubMed] [Google Scholar]
- 13. Bareau B, Rey J, Hamidou M, et al. Analysis of a French cohort of patients with large granular lymphocyte leukemia: a report on 229 cases. Haematologica 95: 1534-1541, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oshimi K. Granular lymphocyte proliferative disorders: report of 12 cases and review of the literature. Leukemia 2: 617-627, 1988. [PubMed] [Google Scholar]
- 15. Oshimi K, Yamada O, Kaneko T, et al. Laboratory findings and clinical courses of 33 patients with granular lymphocyte-proliferative disorders. Leukemia 7: 782-788, 1993. [PubMed] [Google Scholar]
- 16. Kawahara S, Sasaki M, Isobe Y, et al. Clinical analysis of 52 patients with granular lymphocyte proliferative disorder (GLPD) showed frequent anemia in indolent T-cell GLPD in Japan. Eur J Hematol 82: 308-314, 2009. [DOI] [PubMed] [Google Scholar]
- 17. Kwong YL, Wong KF. Association of pure red cell aplasia and T large granular lymphocyte leukemia. J Clin Pathol 51: 672-675, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kwong YL, Au YW, Leung AY, Tse EW. T-cell large granular lymphocyte leukemia: an Asian perspective. Ann Hematol 89: 331-339, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bigouret V, Hoffmann T, Arlettaz L, et al. Monoclonal T-cell expansions in asymptomatic individuals and in patients with large granular leukemia consist of cytotoxic effector T cells expressing the activating CD94:NKG2C/E and NKD2D killer cell receptors. Blood 101: 3198-3204, 2003. [DOI] [PubMed] [Google Scholar]
- 20. Wlodarski MW, Nearman Z, Jankowska A, et al. Phenotypic differences between healthy effector CTL and leukemic LGL cells support the notion of antigen-triggered clonal transformation in T-LGL leukemia. J Leukoc Biol 83: 589-601, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oshimi K, Shinkai Y, Okumura K, Oshimi Y, Mizoguchi H. Perforin gene expression in granular lymphocyte proliferative disorders. Blood 75: 704-708, 1990. [PubMed] [Google Scholar]
- 22. Kothapalli R, Bailey RD, Kusmartseva I, Mane S, Epling-Burnette PK, Loughran TP Jr. Constitutive expression of cytotoxic proteases and down-regulation of protease inhibitors in LGL leukemia. Int J Oncol 22: 33-39, 2003. [PubMed] [Google Scholar]
- 23. Oshimi K, Oshimi Y, Akahoshi M, et al. Role of T-cell antigens in the cytolytic activities of large granular lymphocytes (LGLs) in patients with LGL lymphocytosis. Blood 71: 473-479, 1988. [PubMed] [Google Scholar]
- 24. Kaneko T, Oshimi K, Seto T, Okumura K, Mizoguchi H. A bispecific antibody detects cytotoxic T lymphocytes of unknown antigen specificity in patients with granular lymphocyte-proliferative disorders. Br J Haematol 80: 151-156, 1992. [DOI] [PubMed] [Google Scholar]
- 25. Andersson EI, Tanahashi T, Sekiguchi N, et al. High incidence of activating STAT5B mutations in CD4-positive T-cell large granular lymphocyte leukemia. Blood 128: 2465-2468, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oshimi K, Hoshino S, Takahashi M, et al. Ti (WT31)-negative, CD3-positive, large granular lymphocyte leukemia with nonspecific cytotoxicity. Blood 71: 923-931, 1988. [PubMed] [Google Scholar]
- 27. Sandberg Y, Almeida J, Gonzalez M, et al. TCRγδ+ large granular lymphocyte leukemias reflect the spectrum of normal antigen-selected TCRγδ+ T-cells. Leukemia 20: 505-513, 2006. [DOI] [PubMed] [Google Scholar]
- 28. Bourgault-Rouxel AS, Loughran TP Jr, Zambello R, et al. Clinical spectrum of γδ+ T cell LGL leukemia: Analysis of 20 cases. Leuk Res 32: 45-48, 2008. [DOI] [PubMed] [Google Scholar]
- 29. Chen YH, Chadburn A, Evens AM, et al. Clinical, morphologic, immunophenotypic, and molecular cytogenetic assessment of CD4-/CD8-γδ T-cell large granular lymphocytic leukemia. Am J Clin Pathol 136: 289-299, 2011. [DOI] [PubMed] [Google Scholar]
- 30. Gentile TC, Uner AH, Hutchison RE, et al. CD3+, CD56+ aggressive variant of large granular lymphocyte leukemia. Blood 84: 2315-2321, 1994. [PubMed] [Google Scholar]
- 31. Alekshun TJ, Tao J, Sokol L. Aggressive T-cell large granular lymphocyte leukemia: a case report and review of the literature. Am J Hematol 82: 481-485, 2007. [DOI] [PubMed] [Google Scholar]
- 32. Semenzato G, Zambello R, Starkebaum G, Oshimi K, Loughran TP Jr. The lymphoproliferative disease of granular lymphocytes: updated criteria for diagnosis. Blood 89: 256-260, 1997. [PubMed] [Google Scholar]
- 33. Tanahashi T, Sekiguchi N, Matsuda K, et al. A screening method with lymphocyte percentage and proportion of granular lymphocytes in the peripheral blood for large granular lymphocyte (LGL) leukemia. Int J Hematol 105: 87-91, 2017. [DOI] [PubMed] [Google Scholar]
- 34. Chan WC, Link S, Mawle A, Check I, Brynes RK, Winton EF. Heterogeneity of large granular lymphocyte proliferations: delineation of two major subtypes. Blood 68: 1142-1153, 1986. [PubMed] [Google Scholar]
- 35. Kelemen E, Gergely P, Lehoczky D, Triska E, Demeter J, Vargha P. Permanent large granular lymphocytosis in the blood of splenectomized individuals without concomitant increase of in vitro natural killer cell cytotoxicity. Clin Exp Immunol 63: 696-702, 1986. [PMC free article] [PubMed] [Google Scholar]
- 36. Brouet JC, Sasportes M, Flandrin G, Preud'Homme JL, Seligmann M. Chronic lymphocytic leukaemia of T-cell origin. Immunological and clinical evaluation in eleven patients. Lancet 2: 890-893, 1975. [DOI] [PubMed] [Google Scholar]
- 37. McKenna RW, Parkin J, Kersey JH, Gajl-Peczalska KJ, Peterson L, Brunning RD. Chronic lymphoproliferative disorder with unusual clinical, morphologic, ultrastructural and membrane surface marker characteristics. Am J Med 62: 588-596, 1977. [DOI] [PubMed] [Google Scholar]
- 38. Reynolds CW, Foon KA. Tγ-lymphoproliferative disease and related disorders in humans and experimental animals: a review of the clinical, cellular, and functional characteristics. Blood 64: 1146-1158, 1984. [PubMed] [Google Scholar]
- 39. Tanahashi T, Sekiguchi N, Matsuda K, et al. Cell size variations of large granular lymphocyte leukemia: implication of a small cell subtype of granular lymphocyte leukemia with STAT3 mutations. Leuk Res 45: 8-13, 2016. [DOI] [PubMed] [Google Scholar]
- 40. Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of chronic (mature) B and T lymphoid leukaemias. French-American-British (FAB) Cooperative Group. J Clin Pathol 42: 567-584, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Posnett DN, Sinha R, Kabak S, Russo C. Clonal populations of T cells in normal elderly humans: the T cell equivalent to “benign monoclonal gammopathy”. J Exp Med 179: 609-618, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rossi D, Franceschetti S, Capello D, et al. Transient monoclonal expansion of CD8+/CD57+ T-cell large granular lymphocytes after primary cytomegalovirus infection. Am J Hematol 82: 1103-1105, 2007. [DOI] [PubMed] [Google Scholar]
- 43. Rodríguez-Caballero A, García-Montero AC, Bárcena P, et al. Expanded cells in monoclonal TCR-αβ+/CD4+/NKa+/CD8-/+dim T-LGL lymphocytosis recognize hCMV antigens. Blood 112: 4609-4616, 2008. [DOI] [PubMed] [Google Scholar]
- 44. Starkebaum G, Loughran TP Jr, Kalyanaraman VS, et al. Serum reactivity to human T-cell leukaemia/lymphoma virus type I proteins in patients with large granular lymphocytic leukaemia. Lancet 1: 596-599, 1987. [DOI] [PubMed] [Google Scholar]
- 45. Thomas A, Perzova R, Abbott L, et al. LGL leukemia and HTLV. AIDS Res Hum Retroviruses 26: 33-40, 2010. [DOI] [PubMed] [Google Scholar]
- 46. Dolstra H, Freijers F, Van De Wiel-Van Kemenade E, Schattenberg A, Galama J, De Witte T. Expansion of CD8+CD57+ T cells after allogeneic BMT is related with a low incidence of relapse and with cytomegalovirus infection. Br J Haematol 90: 300-307, 1995. [DOI] [PubMed] [Google Scholar]
- 47. Mohty M, Faucher C, Vey N, et al. Features of large granular lymphocytes (LGL) expansion following allogeneic stem cell transplantation: a long-term analysis. Leukemia 16: 2129-2133, 2002. [DOI] [PubMed] [Google Scholar]
- 48. Kim D, Al-Dawsari G, Chang H, et al. Large granular lymphocytosis and its impact on long-term clinical outcomes following allo-SCT. Bone Marrow Transplant 48: 1104-1111, 2013. [DOI] [PubMed] [Google Scholar]
- 49. Muñoz-Ballester J, Chen-Liang TH, Hurtado AM, et al. Persistent cytotoxic T lymphocyte expansions after allogeneic haematopoietic stem cell transplantation: kinetics, clinical impact and absence of STAT3 mutations. Br J Haematol 172: 937-946, 2016. [DOI] [PubMed] [Google Scholar]
- 50. Narumi H, Kojima K, Matsuo Y, et al. T-cell large granular lymphocytic leukemia occurring after autologous peripheral blood stem cell transplantation. Bone Marrow Transplant 33: 99-101. [DOI] [PubMed] [Google Scholar]
- 51. Wolniak KL, Goolsby CL, Chen Y-H, et al. Expansion of a clonal CD8+CD57+ large granular lymphocyte population after autologous stem cell transplant in multiple myeloma. Am J Clin Pathol 139: 231-241, 2013. [DOI] [PubMed] [Google Scholar]
- 52. Sabnani I, Zucker MJ, Tsang P, Palekar S. Clonal T-large granular lymphocyte proliferation in solid organ transplant recipients. Transplant Proc 38: 3437-3440, 2006. [DOI] [PubMed] [Google Scholar]
- 53. Qiu Z-Y, Li J-Y. Large granular lymphocytosis during dasatinib therapy. Cancer Biol Therapy 15: 247-255, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mustjoki S, Ekblomb M, Arstila TP, et al. Clonal expansion of T/NK-cells during tyrosine kinase inhibitor dasatinib therapy. Leukemia 23: 1398-1405, 2009. [DOI] [PubMed] [Google Scholar]
- 55. Kim DH, Kamel-Reid S, Chang H, et al. Natural killer or natural killer/T cell lineage large granular lymphocytosis associated with dasatinib therapy for Philadelphia chromosome positive leukemia. Haematologica 94: 135-139, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Papadaki T, Stamatopoulos K, Kosmas C, et al. Clonal T-large granular lymphocyte proliferations associated with clonal B cell lymphoproliferative disorders: report of eight cases. Leukemia 16: 2167-2169, 2002. [DOI] [PubMed] [Google Scholar]
- 57. Viny AD, Lichtin A, Pohlman B, Loughran T, Maciejewski J. Chronic B-cell dyscrasias are an important clinical feature of T-LGL leukemia. Leuk Lymphoma 49: 932-938, 2008. [DOI] [PubMed] [Google Scholar]
- 58. Epling-Burnette PK, Liu JH, Catlett-Falcone R, et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest 107: 351-362, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Koskela HLM, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 366: 1905-1913, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jerez A, Clemente MJ, Makishima H, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood 120: 3048-3057, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Qiu ZY, Fan L, Wang L, et al. STAT3 mutations are frequent in T-cell large granular lymphocytic leukemia with pure red cell aplasia. J Hematol Oncol 6: 82, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ishida F, Matsuda K, Sekiguchi N, et al. STAT3 gene mutations and their association with pure red cell aplasia in large granular lymphocyte leukemia. Cancer Sci 105: 342-346, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rajala HL, Eldfors S, Kuusanmäki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood 121: 4541-4550, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Steinway SN, LeBlanc F, Loughran TP Jr. The pathogenesis and treatment of large granular lymphocyte leukemia. Blood Rev 28: 87-94, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lamy T, Liu JH, Landowski TH, William S, Dalton WS, Loughran TP. Dysregulation of CD95/CD95 ligand-apoptotic pathway in CD3+ large granular lymphocyte leukemia. Blood 92: 4771-4777, 1998. [PubMed] [Google Scholar]
- 66. Zambello R, Teramo A, Gattazzo C, Semenzato G. Are T-LGL leukemia and NK-chronic lymphoproliferative disorder really two distinct diseases? Transl Med UniSa 8: 4-11, 2014. [PMC free article] [PubMed] [Google Scholar]
- 67. Handgretinger R, Geiselhart A, Moris A, Grau R, Teuffel O, Bethge W. Pure red-cell aplasia associated with clonal expansion of granular lymphocytes expressing killer-cell inhibitory receptors. N Engl J Med 340: 278-284, 1999. [DOI] [PubMed] [Google Scholar]
- 68. Pontikoglou C, Kalpadakis C, Papadaki HA. Pathophysiologic mechanisms and management of neutropenia associated with large granular lymphocytic leukemia. Expert Rev Hematol 4: 317-328, 2011. [DOI] [PubMed] [Google Scholar]
- 69. Go RS, Tefferi A, Li C-Y, Lust JA, Phyliky RL. Lymphoproliferative disease of granular T lymphocytes presenting as aplastic anemia. Blood 96: 3644-3646, 2000. [PubMed] [Google Scholar]
- 70. Karadimitris A, Li K, Notaro R, et al. Association of clonal T-cell large granular lymphocyte disease and paroxysmal nocturnal haemoglobinuria (PNH): further evidence for a pathogenetic link between T cells, aplastic anaemia and PNH. Br J Haematol 115: 1010-1014, 2001. [DOI] [PubMed] [Google Scholar]
- 71. Saunthararajah Y, Molldrem JJ, Rivera M, et al. Coincident myelodysplastic syndrome and T-cell large granular lymphocytic disease: clinical and pathophysiological features. Br J Haematol 112: 195-200, 2001. [DOI] [PubMed] [Google Scholar]
- 72. Huh YO, Medeiros LJ, Ravandi F, Konoplev S, Jorgensen JL, Miranda RN. T-cell large granular lymphocyte leukemia associated with myelodysplastic syndrome: a clinicopathologic study of nine cases. Am J Clin Pathol 131: 347-356, 2009. [DOI] [PubMed] [Google Scholar]
- 73. Jerez A, Clemente MJ, Makishima H, et al. STAT3 mutations indicate the presence of subclinical T-cell clones in a subset of aplastic anemia and myelodysplastic syndrome patients. Blood 122: 2453-2459, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Nyland SB, Krissinger DJ, Clemente MJ, et al. Seroreactivity to LGL leukemia-specific epitopes in aplastic anemia, myelodysplastic syndrome and paroxysmal nocturnal hemoglobinuria: results of a bone marrow failure consortium study. Leuk Res 36: 581-587, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bektas O, Uner A, Aydin SM, et al. High frequency of autonomous T-cell proliferation compatible with T-cell large granular lymphocytic leukemia in patients with cytopenia of unknown etiology. Int J Hematol 102: 211-217, 2015. [DOI] [PubMed] [Google Scholar]
- 76. Yamada O, Mizoguchi H, Oshimi K. Cyclophosphamide therapy for pure red cell aplasia associated with granular lymphocyte-proliferative disorders. Br J Haematol 97: 392-399, 1997. [DOI] [PubMed] [Google Scholar]
- 77. Go RS, Li C-Y, Tefferi A, et al. Acquired pure red cell aplasia associated with lymphoproliferative disease of granular T lymphocytes. Blood 98: 483-485, 2001. [DOI] [PubMed] [Google Scholar]
- 78. Moignet A, Hasanali Z, Zambello R, et al. Cyclophosphamide as a first-line therapy in LGL leukemia. Leukemia 28: 1134-1136, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fujishima N, Sawada K, Hirokawa M, et al. Long-term responses and outcomes following immunosuppressive therapy in large granular lymphocyte leukemia-associated pure red cell aplasia: a Nationwide Cohort Study in Japan for the PRCA Collaborative Study Group. Haematologica 93: 1555-1559, 2008. [DOI] [PubMed] [Google Scholar]
- 80. Loughran TP Jr, Kidd PG, Starkebaum G. Treatment of large granular lymphocyte leukemia with oral low-dose methotrexate. Blood 84: 2171-2174, 1994. [PubMed] [Google Scholar]
- 81. Loughran Jr TP, Zickl L, Olson TL, et al. Immunosuppressive therapy of LGL leukemia: prospective multicenter phase II study by the Eastern Cooperative Oncology Group (E5998). Leukemia 29: 886-894, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tefferi A, Li CY, Witzig TE, Dhodapkar MV, Okuno SH, Phyliky RL. Chronic natural killer cell lymphocytosis: a descriptive clinical study. Blood 84: 2721-2725, 1994. [PubMed] [Google Scholar]
- 83. Oshimi K, Kawa K, Nakamura S, et al. NK-cell neoplasms in Japan. Hematology 10: 237-245, 2005. [DOI] [PubMed] [Google Scholar]
- 84. Oshimi K. Progress in understanding and managing natural killer-cell malignancies. Br J Haematol 139: 532-544, 2007. [DOI] [PubMed] [Google Scholar]
- 85. Poullot E, Zambello R, Leblanc F, et al. Chronic natural killer lymphoproliferative disorders: characteristics of an international cohort of 70 patients. Ann Oncol 25: 2030-2035, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zambello R, Loughran Jr TP, Trentin L, et al. Serologic and molecular evidence for a possible pathogenetic role of viral infection in CD3-negative natural killer-type lymphoproliferative disease of granular lymphocytes. Leukaemia 9: 1207-1211, 1995. [PubMed] [Google Scholar]
- 87. Zambello R, Trentin L, Ciccone E, et al. Phenotypic diversity of natural killer (NK) populations in patients with NK-type lymphoproliferative disease of granular lymphocytes. Blood 81: 2381-2385, 1993. [PubMed] [Google Scholar]
- 88. Gattazzo C, Teramo A, Passeri F, et al. Detection of monoclonal T populations in patients with KIR-restricted chronic lymphoproliferative disorder of NK cells. Haematologica 99: 1826-1833, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kawa-Ha K, Ishihara S, Ninomiya T, et al. CD3-negative lymphoproliferative disease of granular lymphocytes containing Epstein-Barr viral DNA. J Clin Invest 84: 51-55, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kawa K. Diagnosis and treatment of Epstein-Barr virus-associated natural killer cell lymphoproliferative disease. Int J Haematol 78: 24-31, 2003. [DOI] [PubMed] [Google Scholar]
- 91. Kimura H, Ito Y, Kawabe S, et al. EBV-associated T/NK-cell lymphoproliferative diseases in nonimmunocompromised hosts: prospective analysis of 108 cases. Blood 119: 673-686, 2012. [DOI] [PubMed] [Google Scholar]
- 92. Fernandez LA, Pope B, Lee C, Zayed E. Aggressive natural killer cell leukaemia in an adult with establishment of an NK cell line. Blood 67: 925-930, 1986. [PubMed] [Google Scholar]
- 93. Koizumi S, Seki H, Tachinami T, et al. Malignant clonal expansion of large granular lymphocytes with a Leu-11+, leu-7- surface phenotype: in vitro responsiveness of malignant cells to recombinant human interleukin 2. Blood 68: 1065-1073, 1986. [PubMed] [Google Scholar]
- 94. Imamura N, Kusunoki Y, Kawa-Ha K, et al. Aggressive natural killer cell leukemia/lymphoma: report of four cases and review of the literature. Possible existence of a new clinical entity originating from the third lineage of lymphoid cells. Br J Haematol 75: 49-59, 1990. [DOI] [PubMed] [Google Scholar]
- 95. Song S-Y, Kim WS, Ko Y-H, et al. Aggressive natural killer cell leukemia: clinical features and treatment outcome. Haematologica 87: 1343-1345, 2002. [PubMed] [Google Scholar]
- 96. Suzuki R, Suzumiya J, Nakamura S, et al. Aggressive natural killer-cell leukemia revisited: large granular lymphocyte leukemia of cytotoxic NK cells. Leukemia 18: 763-770, 2004. [DOI] [PubMed] [Google Scholar]
- 97. Suzuki R, Suzumiya J, Yamaguchi M, et al. Prognostic factors for mature natural killer (NK) cell neoplasms: aggressive NK cell leukemia and extranodal NK cell lymphoma, nasal type. Ann Oncol 21: 1032-1040, 2010. [DOI] [PubMed] [Google Scholar]
- 98. Oshimi K. Leukemia and lymphoma of natural killer lineage cells. Int J Hematol 78: 18-23, 2003. [DOI] [PubMed] [Google Scholar]
- 99. Mori KL, Egashira M, Oshimi K. Differential stage of natural killer cell-lineage lymphoproliferative disorders based on phenotypic analysis. Br J Haematol 115: 225-228, 2001. [DOI] [PubMed] [Google Scholar]
- 100. Ishida F, Ko YH, Kim WS, et al. Aggressive natural killer cell leukemia: therapeutic potential of L-asparaginase and allogeneic hematopoietic stem cell transplantation. Cancer Sci 103: 1079-1083, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Takahashi E, Ohshima K, Kimura H, et al. Clinicopathological analysis of the age-related differences in patients with Epstein-Barr virus (EBV)-associated extranasal natural killer (NK)/T-cell lymphoma with reference to the relationship with aggressive NK cell leukaemia and chronic active EBV infection-associated lymphoproliferative disorders. Histopathology 59: 660-671, 2011. [DOI] [PubMed] [Google Scholar]
- 102. Nakashima Y, Tagawa H, Suzuki R, et al. Genome-wide array-based comparative genomic hybridization of natural killer cell lymphoma/leukemia: different genomic alteration patterns of aggressive NK-cell leukemia and extranodal NK/T-cell lymphoma, nasal type. Genes, Chromosomes Cancer 44: 247-255, 2005. [DOI] [PubMed] [Google Scholar]
- 103. Karube K, Nakagawa M, Tsuzuki S, et al. Identification of FOXO3 and PRDM1 as tumor-suppressor gene candidates in NK-cell neoplasms by genomic and functional analyses. Blood 118: 3195-3204, 2011. [DOI] [PubMed] [Google Scholar]
- 104. Hart DN, Baker BW, Inglis MJ, et al. Epstein-Barr viral DNA in acute large granular lymphocyte (natural killer) leukemic cells. Blood 79: 2116-2123, 1992. [PubMed] [Google Scholar]
- 105. Siu LLP, Chan JKC, Kwong YL. Natural killer cell malignancies: clinicopathological and molecular features. Histol Histopathol 17: 539-554, 2002. [DOI] [PubMed] [Google Scholar]
- 106. Nicolae A, Ganapathi KA, Pham TH, et al. EBV-negative aggressive NK-cell leukemia/lymphoma: clinical, pathologic, and genetic features. Am J Surg Pathol 41: 67-74, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ishida F, Kwong YL. Diagnosis and management of natural killer-cell malignancies. Expert Rev Hematol 3: 593-602, 2010. [DOI] [PubMed] [Google Scholar]
- 108. Suzuki R. Treatment of advanced extranodal NK/T cell lymphoma, nasal-type and aggressive NK-cell leukemia. Int J Hematol 92: 697-701, 2010. [DOI] [PubMed] [Google Scholar]
- 109. Kwong YL. The diagnosis and management of extranodal NK/T-cell lymphoma, nasal-type and aggressive NK-cell leukemia. J Clin Exp Hematop 51: 21-28, 2011. [DOI] [PubMed] [Google Scholar]
- 110. Ito T, Makishima H, Nakazawa H, et al. Promising approach for aggressive NK cell leukaemia with allogeneic haematopoietic cell transplantation. Eur J Haematol 81: 107-111, 2008. [DOI] [PubMed] [Google Scholar]
- 111. Kwong YL. Hematopoietic stem cell transplantation in natural killer cell lymphoma and leukemia. Int J Hematol 92: 702-707, 2010. [DOI] [PubMed] [Google Scholar]
- 112. Yamamoto T, Iwasaki T, Watanabe N, et al. Expression of multidrug resistance P-glycoprotein on peripheral blood mononuclear cells of patients with granular lymphocyte-proliferative disorders. Blood 81: 1342-1346, 1993. [PubMed] [Google Scholar]
- 113. Yamaguchi M, Kita K, Miwa H, et al. Frequent expression of P-glycoprotein/MDR1 by nasal T-cell lymphoma cells. Cancer 76: 2351-2356, 1995. [DOI] [PubMed] [Google Scholar]
- 114. Egashira M, Kawamata N, Sugimoto K, Kaneko T, Oshimi K. P-glycoprotein expression on normal and abnormally expanded natural killer cells and inhibition of P-glycoprotein function by cyclosporine A and its analogue, PSC833. Blood 93: 599-606, 1999. [PubMed] [Google Scholar]
- 115. Jung KS, Cho SH, Kim SJ, Ko YH, Kang ES, Kim WS. L-asparaginase-based regimens followed by allogeneic hematopoietic stem cell transplantation improve outcomes in aggressive natural killer cell leukemia. J Hematol Oncol 9: 41-44, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Haji S, Shiratsuchi M, Matsushima T, et al. Achievement of disease control with donor-derived EB virus-specific cytotoxic T cells after allogeneic peripheral blood stem cell transplantation for aggressive NK-cell leukemia. Int J Hematol 105: 540-544, 2017. [DOI] [PubMed] [Google Scholar]