

Abstract
N-glycanase deficiency, or NGLY1 deficiency, is an extremely rare human genetic disease. N-glycanase, encoded by the gene NGLY1, is an important enzyme involved in protein deglycosylation of misfolded proteins. Deglycosylation of misfolded proteins precedes the endoplasmic reticulum (ER)-associated degradation (ERAD) process. NGLY1 patients produce little or no N-glycanase (Ngly1), and the symptoms include global developmental delay, frequent seizures, complex hyperkinetic movement disorder, difficulty in swallowing/aspiration, liver dysfunction, and a lack of tears. Unfortunately, there has not been any therapeutic option available for this rare disease so far. Recently, a proposed molecular mechanism for NGLY1 deficiency suggested that endo-β-N-acetylglucosaminidase (ENGase) inhibitors may be promising therapeutics for NGLY1 patients. Herein, we performed structure-based virtual screening of FDA-approved drug database on this ENGase target to enable repurposing of existing drugs. Several Proton Pump Inhibitors (PPIs), a series of substituted 1 H-Benzo [d] imidazole, and 1H-imidazo [4,5-b] pyridines, among other scaffolds, have been identified as potent ENGase inhibitors. An electrophoretic mobility shift assay was employed to assess the inhibition of ENGase activity by these PPIs. Our efforts led to the discovery of Rabeprazole Sodium as the most promising hit with an IC50 of 4.47±0.44 μM. This is the first report that describes the discovery of small molecule ENGase inhibitors, which can potentially be used for the treatment of human NGLY1 deficiency.
Keywords: NGLY1, endo-β-N-acetylglucosaminidase (ENGase) inhibitors, drug repurposing, structure-based virtual screening, proton pump inhibitors
Graphical abstract
To create your abstract, type over the instructions in the template box below. Fonts or abstract dimensions should not be changed or altered.
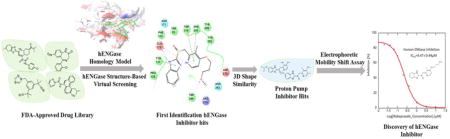
N-glycanase deficiency, also known as NGLY1 deficiency, is an extremely rare genetic disease. The symptoms of NGLY1 deficiency include global developmental delay, seizures, complex hyperkinetic movement disorder, difficulty in swallowing/aspiration, liver dysfunction, and a lack of tears.1 Due to NGLY1 gene mutations, NGLY1 patients produce negligible or no N-glycanase enzyme (Ngly1), an enzyme that is involved in non-lysosomal deglycosylation of mis-folded N-linked glycoproteins in the cytosol before being targeted for destruction by the proteosomal complex.2 However, the underlying mechanism has not been well understood since the first report of this genetic disorder in 2012.3 Recently, Tadashi’s group proposed a potential pathological mechanism linking NGLY1 deficiency to the endoplasmic reticulum (ER)-associated degradati on (ERAD) pathway. It is well known that N-glycoproteins can be degraded through both the lysosomal degradation and non-lysosomal degradation process. In non-lysosomal degradation pathway, the N-glycoproteins (mostly misfolded protein) are translocated from the ER to the cytosol and degraded by proteasomes through the ERAD process. Ngly1 is functionally important in ERAD as it cleaves the amide bond between the Asparagine residue (Asn) of the protein and the proximal GlcNAc, chemically bonded to Asn, in N-glycan.1 When NGLY1 gene was knocked out in a cellular model, endo-β-N-acetylglucosaminidase (ENGase) could act as a surrogate deglycosylation enzyme, resulting in the generation of unconventional proteins carrying partially deglycosylated residual N-GlcNAc sugar stubs. Proteins with N-GlcNAc residues were found to undergo aggregation, preventing or delaying the degradation of misfolded N-glycoproteins. Interestingly, this ERAD dysregulation could be reversed in cells by an additional ENGASE gene knockout.4 Furthermore, the most recent mouse model showed that the ENGASE gene deletion could partially rescue the lethality of Ngly1-deficient mice and mitigate the phenotypes.5 Therefore, ENGase is proposed as a promising drug target for treating human NGLY1 deficiency (Scheme 1). In earlier reports, the mechanism-based substrate mimics, such as Man9GlcNAc-thiazoline, were identified as human ENGase inhibitors with an IC50 of 0.42 μM.6 However, this N-glycan based oligosaccharide thiazolines may not be ideal for use as drugs because of the pharmacokinetic limitations of oligosaccharides. Furthermore, there are no published reports of ENGase small molecule inhibitors yet. Therefore, we aimed to develop small molecules, which target ENGase for the treatment of NGLY1 deficiency. Our approach relies on the repurposing of FDA-approved drugs for new uses as this approach possesses several advantages and has been gaining favor in the drug discovery field for the treatment of neglected rare diseases. It is superior to traditional high-throughput compound screening, from which only 0.01% of new drug leads were selected for clinical trials due to low hit rates and hits with only modest affinities. Our efforts led to the discovery of Proton Pump Inhibitors (PPIs), Lansoprazole, Rabeprazole, Omeprazole, Dexlansoprazole, and Tenatoprazole, as novel inhibitors of ENGase. The PPIs identified herein as ENGase inhibitors are clinically approved drugs and can therefore, be immediately considered for treating NGLY1 deficient patients due to their existing safety, tolerability and pharmacokinetics profile.
Scheme 1.
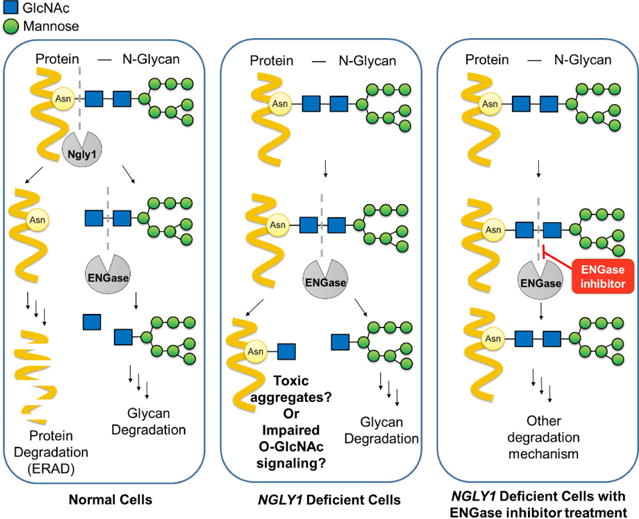
Proposed putative mechanism for NGLY1 deficiency.4 In normal cells, N-glycoproteins (mostly misfolded proteins) were first deglycosylated by Ngly1. Subsequently, while ENGase and other enzymes continuously degrade protein free N-glycans, the misfolded proteins were degraded through ERAD pathway. However, in NGLY1 deficient cells, ENGase-catalyzed hydrolysis of N-glycans on N-glycoproteins, resulting in the production of aggregation prone N-GlcNAc proteins, and this may potentially impair O-GlcNAc signaling. Therefore, by inhibiting ENGase enzyme activity, the intact N-glycoproteins can be degraded by other mechanisms without producing toxic, aggregation prone proteins carrying N-GlcNAc sugar stubs.
There were a few X-ray crystal structures of non-human ENGase (PDB ID: 3FHA7, 3FHQ7, and 2VTF8) published. We used the Arthrobacter protophormiae structure of endo-β-N-acetylglucosaminidase A (Endo-A) complex model with co-crystalized ligand GlcNAc-Asn or Man3GlcNAc-thiazoline (PDB ID: 3FHQ) coordinates in order to build a human ENGase (NP_001036038) homology model for the structure-based virtual screening. The active site sequences of human and Arthrobacter protophormiae ENGase were aligned. The essential catalytic residues — E173, N171, and Y205 — identified in Arthrobacter protophormiae are the same as the ones in human (Fig. S1). Furthermore, the important interacting residues W93, F125, W216, F243, W244, and Y299, showed in the crystal structure, have similar electrostatic properties to the corresponding amino acid residues in human: W93F, F125I, F243Y, W244N, and Y299F. Therefore, both the crystal structure and the human homology model were used in the binding pose evaluations (Fig. 1). Water molecules were then removed, and the missing bond order and geometries were edited. Hydrogen atoms were added, and the combined complex structure was submitted for protein preparation and energy minimization calculations using ICM9 and Schrödinger10. The fully refined structure with the bound ligand molecule was further submitted for grids calculation to define the active site as the collection of amino acids enclosed within an 8 Å radius sphere centered on the bound ligand. The target hENGase was optimized using Monte Carlo simulation and energy optimizations.
Fig. 1.
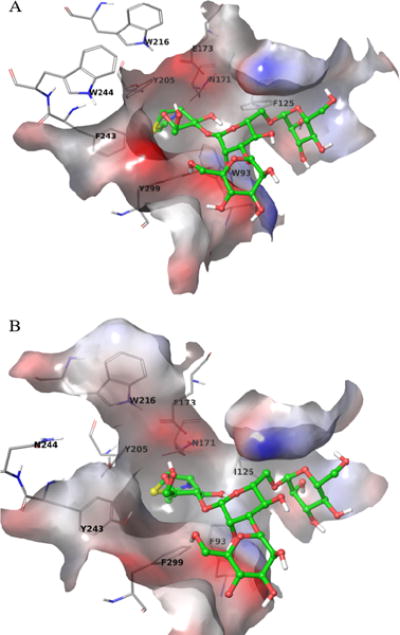
Active site comparison of A) Crystal structure of Arthrobacter protophormiae ENGase and B) Human homology model of ENGase. A) The crystal structure of the binding pocket is shown with the ligand Man3GlcNAc-thiazoline (shown in green sticks). Important interacting amino acid residues are labeled and shown in grey lines. B) The human homology model was built from the crystal structure by mutating residues in the active site.
The external source of FDA-approved drugs in the BindingDB11 (https://www.bindingdb.org/bind/ByFDAdrugs.jsp) database (in sdf format) was processed using the ligand preparation tools (Chemaxon12 and QikPro13). The final coordinates were stored in a single sdf file for a final library of 1338 molecules that are commercially available from 26 vendors. The ligand library was considered for virtual screening using Glide SP/XP14, ICM and GOLD15. Grid potentials, that accounted for the shape of the binding pocket, hydrophobicity, electrostatic potentials and hydrogen-bonding profile, were rapidly generated. The compounds were screened for ENGase binding properties using our own workflow that employs a rigid target and flexible ligands in the internal coordinate’s space. Docking calculations, in search for ENGase inhibitors, were performed using the ICM and Glide docking module with default setup and re-scoring with GOLD. The structures with the high scores were energy minimized in the same environment and saved in PDB format. These energy-minimized hits were then reposed into ICM and converted into ICM object, and MMFF charges were assigned for each of the ligands. Compounds having desired scores, hydrogen bond formation, and hydrophobic interactions that were estimated by interatomic distances were chosen for further analysis. The conformational stability of each candidate was also estimated by the force field energy difference between the complexes conformation and the freely minimized conformation, and the top-scoring candidates from this category were selected for further analysis. Compounds in each of the three categories were visually inspected to eliminate molecular candidates that do not have ideal hydrogen bond geometry, hydrophobic molecular surfaces, or torsion angles. The resulting 62 screening hit structures from the FDA database were further analyzed using molecular property filters in Schrödinger QikProp. The FDA approved drugs that are commercially available given in Table 1 and Fig. S3 were procured. The stability data, NMR and MS characterization data, and the HPLC data, demonstrating the purity of compounds purchased, are available from respective vendors.
Table 1.
PPI compounds that inhibited hENGase.a
| No. | Drug Name | Structure | IC50 (μM) |
|---|---|---|---|
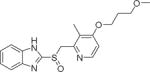
| 1 | Rabeprazole |
|
4.47±0.44 |
| 2 | Lansoprazole |
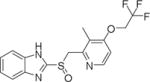
|
24.38±1.24 |
| 3 | Tenatoprazole |
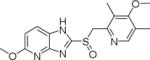
|
~20.0 |
| 4 | Dexlansoprazole |
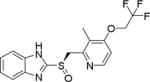
|
~25.0 |
| 5 | Omeprazole |
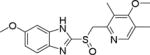
|
~25.0 |
The IC50 values of Rabeprazole and Lansoprazole were determined based on 8 dose IC50 curve obtained from three independent experiments; The IC50 values of Dexlansoprazole, Tenatoprazole, and Omeprazole were estimated using four different concentrations from two independent experiments (Fig. S4B).
The ENGase inhibition assay was designed and further optimized using bovine pancreatic ribonuclease B as substrate.7 Ribonuclease B has a single N-Glycan site at Asn34, with 5–9 mannose residues attached to the chitobiose core. Due to the heterogeneity in the glycosylation, ribonuclease B has a molecular weight of ~17 kDa. After Ngly1- or ENGase-mediated deglycosylation, the molecular weight of ribonuclease B decreases to ~14 kDa.16 In earlier studies, the activity of PNGase F (the yeast version of hNgly1) was assayed by examining the mobility shift of ribonuclease B in SDS-PAGE gel after deglycosylation.17 Therefore, we envisioned the use of the SDS-PAGE based mobility shift assay to selectively detect residual human ENGase activity after treatment with potential human ENGase inhibitors in our screening efforts. Recombinant human ENGase (Origene, USA) was incubated with ribonuclease B and the potential inhibitor. After incubation, all samples were heat inactivated and detected on 12% SDS-PAGE gel. Protein bands were stained using Coomassie blue R-250 and further quantified using the ImageJ software18. The final IC50 curve plot was generated using KaleidaGragh 4.1.3. The details of the optimized assay conditions are provided in the supporting information (Fig. S4).
From our preliminary ENGase inhibitor virtual screening, 13 drugs were predicted to be potential inhibitors of ENGase: Lansoprazole, Bortezomib, Ixazomib, MG132, Dexrazoxane HCl, Edoxaban, Atomoxetine HCl, Clonidine HCl, Dyphylline, Buspirone HCl, Guanfacine HCl, Acetazolamide, Tiamet G (The 2D structure of Lansoprazole is shown in Table 1 and the structure of other 12 drugs are shown in the s). The results of the preliminary ENGase inhibitor screening at 100 μM drug concentration showed that only Lansoprazole inhibited ENGase enzyme activity among all tested compounds (Fig. 2A, Fig. S4A). Because Lansoprazole has been in use as a proton-pump inhibitor (PPI), we further modeled and tested other PPI drugs to find more promising inhibitors. The results of the second round of ENGase inhibitor screening are summarized in Table 1 (active) and Fig.S3 (not active).
Fig. 2.
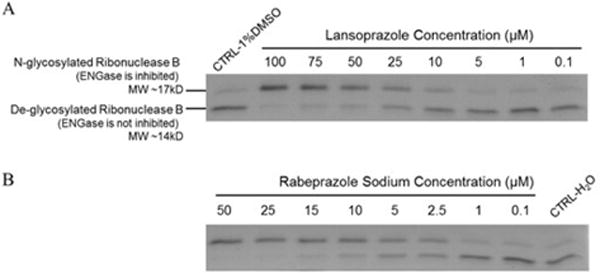
A) Lansoprazole and B) Rabeprazole mediated inhibition of human ENGase activity were tested at various concentrations using the electrophoretic mobility shift assay.
All ENGase inhibitors herein tested have substituted 1 H-benzo[d] imidazole or lH-imidazo [4,5-b] pyridines scaffold. Rabeprazole Sodium (IC50 = 4.47±0.44 μM) was found to be the best hit (Fig. 2B, Fig. 3 and Fig. S4B). The docking poses for PPIs are shown in Fig. S2. The molecular interactions are shown using Rabeprazole and crystal structure active site as an example (Fig. 4A and 4B). The hydrogen atoms from the imidazole and pyridine rings form a salt bridge with Glu173, the catalytic residue, of the ENGase enzyme. Also, strong π-π stacking interactions were found between the imidazole ring and Tyr131, and the pyridine ring with Trp216 and Phe243. This analysis strongly suggested that the 1H-benzo [d] imidazole and 1H-imidazo [4,5-b] pyridines core scaffold play significant roles in the drug-enzyme interactions.
Fig. 3.
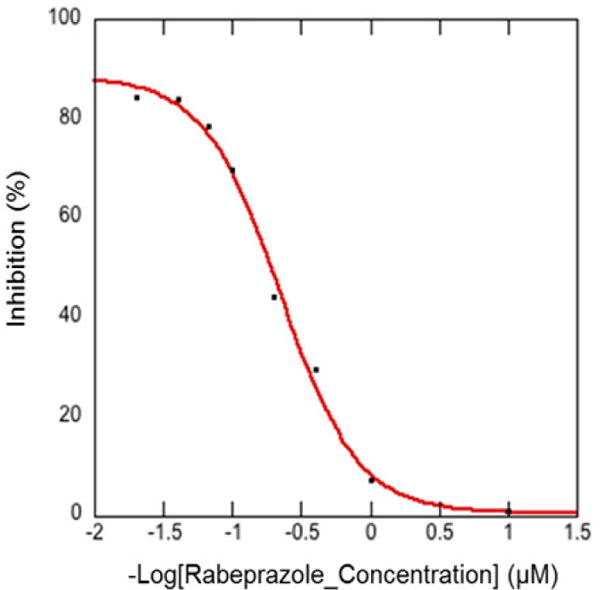
Inhibition of hENGase by Rabeprazole Sodium in 8 dose IC50 curve (IC50 = 4.47±0.44 μM) was obtained from the electrophoretic mobility shift assay. Data represent an average of three independent experiments.
Fig. 4.
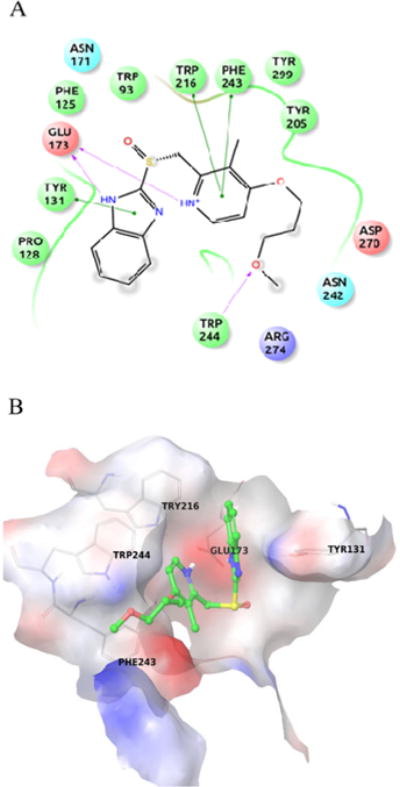
Rabeprazole binding interactions are shown in A) Ligplot. The important interacting residues are depicted using SiteMap20 and B) Crystal structure binding pocket of Arthrobacter protophormiae ENGase. Rabeprazole is shown in green sticks. The important interacting residues are shown in grey lines.
Our work is the first to identify small molecule ENGase inhibitors that have a potential to treat pathogenesis associated with NGLY1 deficiency. Here, we used the FDA-approved drug database for computational virtual screening based on the ENGase crystal structure and homology modeling. Among 13 FDA-approved drugs that were screened in the first round, Lansoprazole, a proton pump inhibitor, was identified as a potential ENGase inhibitor. Subsequently, seven additional proton pump inhibitors were tested and four of them were shown to inhibit ENGase activity. Among them, Rabeprazole Sodium has the best inhibitory effect with an IC50 of 4.47 μM. However, the target for Rabeprazole Sodium was H+/K+ ATPase, which had a target affinity of ~70nM.19 Based on the molecular interaction analysis, we propose that the imidazole ring and the pyridine ring are important for the affinity and binding. For linker region optimization, a sulfonyl group or carbon spacer could be introduced to optimize linker length and interactions. The C4-substituents on the pyridine ring, such as 3-methoxypropoxyl and other linear chains with amide bond, can be considered as potential chemical modifications for future SAR and pharmacophore studies to identify a novel series of more potent ENGase inhibitors. At the same time, finding a mass spectrometry based secondary high-throughput assay will greatly increase the screening efficiency and will be suitable for analysis of more complicated glycoprotein substrates carrying multiple glycoforms and N-glycans. Besides Ribonuclease B, we will also test the activity of ENGase inhibitors using other N-glycoproteins as substrates in our future work. Current efforts are focused on scaffold optimization to discover more potent and selective ENGase inhibitors for the treatment of human NGLY1 deficiency.
Supplementary Material
Acknowledgments
We acknowledge the funding support from NHLBI sponsored Programs of Excellence in Glycosciences (P01 HL107152), CCTS grant from the University of Utah and Huntsman Cancer Institute’s Computational support grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary material
Supplementary data associated with this article can be found, in the online version, at.
References and notes
- 1.Suzuki T, Huang C, Fujihira H. Gene. 2016;577:1–7. doi: 10.1016/j.gene.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suzuki T. Mol Aspects Med. 2016;51:89–103. doi: 10.1016/j.mam.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Need AC, Shashi V, Hitomi Y, Schoch K, Shianna KV, McDonald MT, Meisler MH, Goldstein DB. J Med Genet. 2012;49:353–61. doi: 10.1136/jmedgenet-2012-100819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang C, Harada Y, Hosomi A, Masahara-Negishi Y, Seino J, Fujihira H, Funakoshi Y, Suzuki T, Dohmae N, Suzuki T. Proc Natl Acad Sci U S A. 2015;112:1398–403. doi: 10.1073/pnas.1414593112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujihira H, Masahara-Negishi Y, Tamura M, Huang C, Harada Y, Wakana S, Takakura D, Kawasaki N, Taniguchi N, Kondoh G, Yamashita T, Funakoshi1 Y, Suzuki T. PLoS Genet. 2017;13(4):e1006696. doi: 10.1371/journal.pgen.1006696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li B, Takegawa K, Suzuki T, Yamamoto K, Wang LX. Bioorg Med Chem. 2008;16:4670–4675. doi: 10.1016/j.bmc.2008.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin J, Li L, Shaw N, Li Y, Song JK, Zhang W, Xia C, Zhang R, Joachimiak A, Zhang HC, Wang LX, Liu ZJ, Wang P. PLoS One. 2009;4(3):e4658. doi: 10.1371/journal.pone.0004658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ling Z, Suits MDL, Bingham RJ, Bruce NC, Davies GJ, Fairbanks AJ, Moir JWB, Taylor EJ. J Mol Biol. 2009;389:1–9. doi: 10.1016/j.jmb.2009.03.050. [DOI] [PubMed] [Google Scholar]
- 9.Abagyan R, Totrov M, Koznetsov D. J Comput Chem. 1994;15:488–506. [Google Scholar]
- 10.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. J Comput Aided Mol Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 11.Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. Nucleic Acids Res. 2007;35:198–201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MarvinSketch (version 6.2.2), calculation module developed by ChemAxon, (http://www.chemaxon.com/products/marvin/marvinsketch/).
- 13.QikProp (v. 1.6; http://www.schrodinger.com/Products/qikprop.html).
- 14.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 15.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 16.Plummer TH, Hirs CHW. J Biol Chem. 1963;238:1396–1401. [PubMed] [Google Scholar]
- 17.Plummer TH, Elder JH, Alexander S, Phelan AW, Tarentino AL. J Biol Chem. 1984;259:10700–10704. [PubMed] [Google Scholar]
- 18.Schneider Ca, Rasband WS, Eliceiri KW. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morii M, Takeguchi N. J Biol Chem. 1993;268:21553–21559. [PubMed] [Google Scholar]
- 20.Halgren TA. J Chem Inf Model. 2009;49:377–389. doi: 10.1021/ci800324m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.