

Abstract
Increasing incidence rates of invasive Streptococcus dysgalactiae subspecies equisimilis (SDSE) infections have been reported worldwide, but the evolutionary mechanisms underlying this development remain elusive. Through prospective surveillance of invasive SDSE infections in western Norway, we observed the emergence of a novel and virulent SDSE genotype, stG62647. This emm-type, rarely encountered as a cause of invasive disease during 1999–2012, emerged in 2013 as the predominant SDSE-genotype. The stG62647-infections were associated with an aggressive clinical course, including the occurrence of streptococcal toxic shock syndrome, necrotizing soft-tissue infections and endocarditis. All the invasive stG62647-isolates were subjected to whole genome sequencing, attempting to explore the genetic events underpinning its epidemicity. Although 10% of the genomes was unique for stG62647-genotype, notably 18 out of 19 isolates contained a disrupted streptococcal invasive locus (sil) due to the insertion of a transposase, IS1548, into the silB-gene. We postulate that the virulence of stG6267-isolates could be partly attributable to the abrogation of the attenuating control normally exerted by this regulon, although experimental verification was not performed. To the best of our knowledge, this is the first study employing large scale whole genome sequencing to illuminate the genetic landscape of epidemic lineages in SDSE.
Introduction
Streptococcus dysgalactiae subspecies equisimilis (SDSE) is an emerging human pathogen with a disease spectrum similar to the closely related Streptococcus pyogenes (S. pyogenes)1, 2. Although SDSE has been regarded as less virulent than S. pyogenes, severe clinical manifestations have been documented, including necrotizing soft tissue infection and streptococcal toxic shock syndrome3, 4. SDSE and S. pyogenes also share extensive genetic homology, including genes encoding conserved virulence factors (emm, streptolysin O, streptolysin S, streptokinase and c5a peptidase) and virulence regulators (control of virulence-regulon [covRS] and streptococcal invasive locus [sil])5, 6.
During the past decade, an upsurge in the incidence of invasive SDSE disease has been documented worldwide, in some geographic regions even surpassing the incidence of invasive S. pyogenes infections7–9. In our region of western Norway, SDSE is currently the leading cause of invasive beta-haemolytic disease2. The epidemiological and molecular events promoting this development, however, have yet to be elucidated. Several studies involving whole genome sequencing have investigated the genomic events leading to epidemic spread of hypervirulent S. pyogenes clones, revealing mutations in virulence regulators and virulence genes, as well as the acquisition of novel bacteriophages10, 11. Similar studies have not been conducted for SDSE.
Mutations in regulons normally exerting negative transcriptional control of virulence genes have been demonstrated to induce the transition from a mild to a more invasive phenotype, particularly in the covRS-regulon and the sil-locus12, 13. covRS is a two-component regulatory system, comprising the genes covR and covS, controlling the expression of approximately 10% of the S. pyogenes transcriptome14. covRS-mutants have an upregulated expression of many virulence factors, increased resistance to phagocytosis, and increased virulence potential in murine bacteraemia models14. sil is a virulence-regulon found in approximately 80% of SDSE and 25% of S. pyogenes, and S. pyogenes-isolates with mutations in this locus have been reported to cause infections pursuing an aggressive clinical course6, 13. sil comprises genes encoding a putative two component regulator (silA and silB), a transporter system (silD and silE), and two small pheromone-like peptides (silC and silCR) with differing functions; silC being linked to virulence, whereas silCR is associated with competence, bacterial quorum sensing, and attenuation of virulence in both SDSE and S. pyogenes-infections15.
Through prospective registration and genotyping of invasive SDSE-disease from 2012, we identified an outbreak of the SDSE emm-type stG62647 in our health region. Here, we present a detailed epidemiological and molecular characterization of the outbreak, and through whole genome sequencing we attempted to unravel the genetic rationale for this virulent genotype, with particular emphasis on acquired bacteriophages, the repertoire of virulence factors, and mutations in the virulence regulons covRS and sil.
Materials and Methods
Study setting and definitions
Health Region Bergen in Western Norway has a catchment area of 430 000 inhabitants, and comprises the tertiary care hospital Haukeland University Hospital, along with the two secondary care hospitals Haraldsplass Deaconess Hospital and Voss Hospital. Data on the epidemiology of SDSE-disease in this region in the period 1999–2013 has previously been reported2. From 2012 to 2015 SDSE isolates obtained from invasive disease were prospectively collected, and clinical information was abstracted from patient records. All stG62647-isolates during 1999–2015 were included in the study. Invasive disease was defined as isolation of SDSE from a sterile site, or from a non-sterile site in association with verified necrotizing soft tissue infections. Clinical entities and streptococcal toxic shock syndrome were defined as previously described2. Severe disease was defined as the presence of necrotizing soft tissue infection, endocarditis, streptococcal toxic shock syndrome or death ensuing within 30 days of admission.
Bacterial isolates, DNA extraction and sequencing
All isolates displayed large colony size (>0.5 mm in diameter after 24 hours) and β-haemolysis on 5% sheep blood agar. Serogroup specificity had previously been determined using a rapid agglutination test (Oxoid Streptococcal Grouping Kit, Hampshire, UK), and species identity had been confirmed by matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-ToF MS), using Microflex™ with the MALDI Biotyper database (Bruker Daltonik, Bremen, Germany). Bacterial suspensions were pre-treated with Mutanolysin (M9901, Sigma-Aldrich, Dorset, UK), Lysozyme (L6876, Sigma-Aldrich) and Proteinase K (19133, Qiagen, Hilden, Germany), and genomic DNA was extracted using MagNA Pure 96 (Roche Life science). Genomic libraries were made using Nextera XT Kit (Illumina, Essex, UK), and 150 basepairs paired-end sequencing was performed on HiSeq. 4000 (Illumina).
Assembly, annotation and molecular characterization
Reads quality was assessed using FastQC (bioinformatics.bbsrc.ac.uk), and filtered using Trimmomatic16. Trimmed reads were submitted to Center for Genomic Epidemiology for online assembly by Spades17, and subsequent assessment with Quast18. Annotation was performed using RAST19. emm-type was determined by BLAST-search against the emm-type database curated by Centers for Disease Control (http://www.cdc.gov). Multilocus sequence typing (MLST) profiles were identified using the Center for Genomic Epidemiology website20, and novel MLST alleles and profiles were submitted to pubMLST (www.pubMLST.org). To compare the strains for variations across the whole genome, a single nucleotide polymorphism (SNP) based phylogenetic analysis was performed as previously described21. Of the six full genomes deposited in GenBank, S. dysgalactiae subspecies equisimilis ATCC 12394 (GenBank accession number CP002215) had the closest genetic resemblance to our stG62647-genomes, and was selected as a reference-genome in the SNP-analysis. Geneious version 10.0 was used to screen for the presence and mutation of selected virulence genes and regulators (Table 1)22. The selection of genes was adapted from the S. pyogenes-virulence factors listed in the Virulence Factors Database (www.mgc.ac.cn/VFs), with some minor modifications. Genomes were screened for bacteriophages by Phaster, and for Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) using CRIPSRFinder23, 24.
Table 1.
Genetic repertoire of the invasive stG62647-isolates.
| Gene | Reference | CC20 | ST17 | Phage | Rep. SDSE |
|---|---|---|---|---|---|
| Adhesion | |||||
| dltABCD-operon | BAH81801–4 | + | + | — | + |
| ProteinF1/gfba | U31115 | — | + | — | + |
| ProteinF2/fbaB/PFBP | AB084272/AY612221 | — | — | — | − |
| fbaA | AB040536 | — | — | — | + |
| Sof | X83303 | — | — | — | − |
| sfbX | AF335322 | — | — | — | − |
| gapC | AF375662 | + | + | — | + |
| fbp54 | BAH81758 | + | + | — | + |
| Shr | BAH82346 | + | + | — | + |
| Lmb | BAH81434 | + | + | — | + |
| sclA | AY459361 | ||||
| Enzymes | |||||
| Streptolysin O | AB050249 | + | + | — | + |
| Streptolysin S | AY033399 | + | + | — | + |
| Streptokinase | BAH80750 | + | + | — | + |
| Streptodornase (sda) | AAL98274 | — | — | + | — |
| Streptodornase B (sdaB) | AAM80352 | — | — | − | — |
| Streptodornase D (sdaD) | WP_048327803 | — | — | + | + |
| Streptodornase (sdn) | BAH81602 | — | — | + | + |
| Mitogenic factor 2 | AAL97446 | — | — | + | — |
| Mitogenic factor 3 | KKC20252 | — | — | + | + |
| Mitogenic factor 4 | AAM79702 | — | — | + | — |
| Phospholipase A2 | AAM79811 | — | — | + | + |
| Immune evasion | |||||
| drsG | BAH81431 | — | + | — | + |
| Protein G | M13825 | + | + | — | + |
| C5a-peptidase | BAH81432 | + | + | — | + |
| hasA | WP_011055113 | — | — | — | — |
| hasB | WP_002992300 | — | — | — | — |
| hasC | BAH82462 | + | + | — | + |
| Superantigens | |||||
| speA | U40453 | — | — | + | + |
| speB (protease) | L26125 | — | — | — | — |
| speC | M35514 | — | — | + | + |
| speG | AB105080 | + | — | — | + |
| speH | AF124500 | — | — | + | + |
| speI | AF438524 | — | — | + | + |
| speJ | AF321000 | — | — | — | — |
| speK | WP_011054728 | — | — | + | — |
| speL | WP_011017837 | — | — | + | — |
| speM | AIT77602 | — | — | + | + |
| Ssa | U48792 | — | — | + | − |
| smeZ | AB046865 | — | — | — | + |
| Virulence regulon | |||||
| covRS | BAH80897/8 | + | + | — | + |
| Sil | GQ184566 | Mut | — | — | + |
CC20, stG62647-isolates belonging to clonal complex 20; ST17, stG62647-isolate belonging to MLST-profile 17; Phage, bacteriophage-associated; Rep. SDSE, previously reported in SDSE; Mut, mutated. Symbols + and–denote presence or absence of genetic feature, respectively.
Statistical analysis
Data were processed using SPSS PASW STATISTICS, version 21.0 (IBM SPSS Statistics for Windows, Version 21.0. Armonk, NY: IBM Corp). Categorical data were analysed using Fisher’s exact test, and non-parametric data were analysed with Mann Whitney U-test. A two-sided p-value ≤ 0.05 was considered statistically significant.
Ethical standards
The study underwent institutional ethics review and approval (2010/1406 Regional Ethics Committee West, Norway). Informed consent was obtained from participants. All experiments were carried out in full accordance with the approved ethics applications specified above.
Results
Among 267 cases of invasive SDSE disease identified from 1999 to 2015, 19 cases were attributable to group C streptococci belonging to emm-type stG62647. The first patient was a German tourist presenting with bacteraemia, acute spondylodiscitis and streptococcal toxic shock syndrome in 2006. Two additional sporadic cases were identified in 2006 and 2010; the latter was referred from another health region. However, the last 16 cases were encountered within a relatively short time frame from March 2013 to August 2015. Consequently, stG62647 was the predominant emm-type during this period, accounting for >20% of all invasive SDSE-isolates. The temporal distribution of emm-types associated with invasive SDSE disease during 1999–2015 is depicted in Fig. 1.
Figure 1.

The temporal distribution of emm-types associated with 267 cases of invasive SDSE-disease encountered during 1999–2015. stG62647 was the predominant emm-type in the period 2013–2015, comprising approximately 20% of the invasive SDSE-isolates. The category “Other” includes stG166b (12), stG2078 (9), stG652 (8), stC5345 (4), stC36 (2), stC2574 (2), stC3852 (2), stG4831 (2), stG5420 (2), stC1400 (1), stC7901 (1), stG12 (1), stG120 (1), stG245 (1), stG4222 (1), stG507 (1), stG653 (1), stG6792 (1), stG840 (1) and 29 isolates not available for emm-typing.
The clinical characteristics of the 19 invasive infections associated with stG62647 are presented in Table 2. Median age was 70 years, and a male predominance was noted. Osteoarticular infection was the most frequent clinical entity (42%), followed by skin and soft-tissue infection (32%). Of note, approximately half of the patients experienced severe disease manifestations: two had necrotizing soft-tissue infections, one had endocarditis, five developed streptococcal toxic shock syndrome, and three patients died within a few days after admission. Severe disease was significantly more frequent in cases associated with stG62647 than among patients infected with SDSE-isolates belonging to other emm-types during the study period (47/248, 19%, P = 0.01).
Table 2.
Clinical characteristics of the invasive stG6247-cases.
| Case id. | Year | Sex | Age | SSTI | NSTI | OAI | Other | STSS | Mors30 |
|---|---|---|---|---|---|---|---|---|---|
| T267 | 2006 | M | 70 | — | — | + | — | + | — |
| T274 | 2006 | F | 93 | — | — | — | + | — | — |
| T437 | 2010 | F | 67 | — | — | + | — | — | — |
| T560 | 2013 | F | 87 | — | — | + | — | — | — |
| T562 | 2013 | M | 66 | — | — | + | — | — | — |
| T576 | 2013 | M | 28 | — | + | — | — | + | — |
| T599 | 2013 | M | 60 | — | — | + | — | — | — |
| T604 | 2014 | M | 61 | — | + | — | — | + | — |
| T606 | 2014 | M | 29 | — | — | + | — | — | — |
| T607 | 2014 | M | 86 | + | — | — | — | — | + |
| T619 | 2014 | F | 63 | — | — | + | — | + | — |
| T630 | 2014 | M | 77 | + | — | — | — | + | — |
| T642 | 2015 | M | 70 | — | — | — | + | — | — |
| T645 | 2015 | M | 88 | + | — | — | — | — | + |
| T653 | 2015 | F | 85 | + | — | — | — | — | — |
| T654 | 2015 | M | 63 | — | — | + | — | — | — |
| T655 | 2015 | F | 86 | — | — | — | + | — | + |
| T661 | 2015 | M | 77 | + | — | — | — | — | — |
| T666 | 2015 | F | 72 | + | — | — | — | — | — |
Case id., case identity; SSTI, skin and soft-tissue infection; NSTI, necrotizing soft-tissue infection; OAI, osteoarticular infection; STSS, streptococcal toxic shock syndrome; Mors30, mortality within 30 days of admission; M, male; F, female. Other comprises nosocomial pneumonia (T274), prosthetic valve endocarditis (T642) and primary bacteremia (T655). Symbols + and–denote presence or absence of clinical features, respectively.
Whole genome sequencing, MLST and SNP-analysis
The assembled genomes had an average genome-size of 2.1 Mbp, the mean coverage was ~50 fold, and the G + C-content was approximately 39%. MLST revealed a relatively uniform bacterial population, with 15 of the 19 invasive stG62647 isolates classified as ST20. Additionally, three isolates shared six out of seven alleles with ST20, and constituted the novel ST-profiles ST319, ST320 and ST321. These 18 isolates were designated Clonal Complex 20 (CC20). The T666-isolate, however, belonged to the unrelated MLST-profile ST17, an apparently promiscuous ST-profile shared by several other SDSE emm-types (pubMLST.org). The stG62647-genomes mapped to ~90% of the SDSE ATCC 12394 reference genome, and from a core genome of 1.86 Mbp, a total of 14 163 SNPs valid in all genomes were identified. The CC20-isolates differed by an average of 214 SNPs, ranging from 16 to 718. In comparison, the T666 isolate diverged from the CC20 isolates by ~8270 SNPs. The phylogenetic tree of the 19 outbreak isolates based on core genome SNP-analysis is depicted in Fig. 2.
Figure 2.

Phylogenetic tree of the stG62647-isolates based on core-genome SNP-analysis. (a) Phylogenetic tree of all 19 stG62647-isolates. (b) Phylogenetic tree of the 18 stg62647-isolates belonging to clonal complex 20. The ST17-isolate (T666) was omitted from the analysis to increase the phylogenetic resolution of the tree. Scales indicate substitutions per site. MLST-profile is indicated in parentheses.
Bacteriophages and CRISPRs
Bacteriophages were detected in five isolates and none of them harboured more than one phage. Four out of five had similarities to bacteriophages previously documented in Streptococcus pyogenes: Two isolates possessed a 35.2 kb phage displaying 67% homology to Φ315.3, one isolate contained a 41.8 kb phage bearing 70% resemblance to Φ315.2, and one isolate accommodated a 49.6 kb phage showing 60% identity to Φ315.5, all Φ315-phage-variants originally described in the Streptococcus pyogenes emm3 isolate, MGAS31525. Of note, these phages have been associated with the virulence genes mitogenic factor 4 (mf4), streptococcal superantigen (ssa) and streptococcus pyogenes exotoxin A (speA), respectively, but no phage-mediated virulence genes were detected in any of our isolates. The final bacteriophage consisted of a 49.6 kb element without significant similarity to previously published phages of S. pyogenes. CRISPR-systems were detected in all 19 stG62647-genomes, the majority harbouring two discrete loci. The number of CRISPR-spacers did not differ significantly between isolates containing bacteriophages (median of 14 spacers, range 11 to 23) and isolates without phages (median of 16 spacers, range 11–22), P = 1.0. The distribution of bacteriophages and CRISPRs is presented in Table 3.
Table 3.
Distribution of bacteriophages and CRISPRs.
| Isolate identity | ST-type | Bacteriophages | Number of CRISPR-regions | Total number of CRISPR- repeats |
|---|---|---|---|---|
| T267 | ST20 | — | 2 | 15 |
| T274 | ST20 | — | 2 | 20 |
| T437 | ST20 | — | 2 | 20 |
| T560 | ST20 | — | 2 | 17 |
| T562 | ST20 | — | 3 | 19 |
| T576 | ST20 | — | 2 | 14 |
| T599 | ST20 | — | 3 | 21 |
| T604 | ST20 | Φ315.3-like | 2 | 11 |
| T606 | ST20 | — | 2 | 13 |
| T607 | ST20 | Φ315.3-like | 2 | 14 |
| T619 | ST321 | — | 2 | 22 |
| T630 | ST319 | — | 2 | 15 |
| T642 | ST20 | — | 3 | 20 |
| T645 | ST20 | — | 2 | 11 |
| T653 | ST20 | — | 2 | 11 |
| T654 | ST20 | — | 2 | 14 |
| T655 | ST320 | Φ315.5-like | 3 | 23 |
| T661 | ST20 | Φ315.2-like | 2 | 12 |
| T666 | ST17 | ΦT666 - New | 2 | 22 |
CRISPR, Clustered Regularly Interspaced Short Palindromic Repeats. The symbol – denotes the absence of bacteriophages.
Virulence gene profiling
Whole genome screening of the 19 stG62647-isolates confirmed the presence of several established and putative virulence factors, shown in Table 1. The antiphagocytic hyaluronic acid capsule-operon has not previously been reported in SDSE, and accordingly only hasC was detected among our isolates. All 18 stG62647-isolates belonging to CC20 possessed speG, but lacked other differentially distributed virulence genes, such as gfba and drsG. Conversely, the ST17-isolate harboured both gfba and drsG, but lacked speG. No other streptococcal superantigens were identified.
A broad range of genes involved in adhesion were identified, including dltABCD-operon producing d-alanyl lipoteichoic acids mediating the initial contact between pathogen and host. Furthermore, two pilus-islands were discovered. These islands are also known as Fibronectin/Collagen/T-antigen-region (FCT) in S. pyogenes, and have been categorized into nine different FCT-regions according to their content and genomic arrangement26. The first pilus-island in the stG62647-isolates displayed a genetic structure similar to FCT-6-region, and the overall genetic identity to the FCT-6-region in the M2 S. pyogenes-isolate MGAS10270 (GenBank accession number CP000260) was 95%. The second pilus island did not concord with any of the nine reported FCT-regions of S. pyogenes. It had a structure resembling FCT-2, but had lost the terminal two transposases and the sortaseB-gene. However, it was structurally identical, and 95% homologous to an FCT-region found in the M58 S. pyogenes-strain GA40884 in GenBank (Accession number AWUA01000008).
Virulence regulatory systems
covRS was conserved among all the stG62647-isolates, and all alleles were 100% identical. Compared to the covRS regulon in GGS124 (GenBank accession number AP010935), a single non-synonymous substitution in covS was noted, resulting in I421V.
The streptococcal invasive locus (sil) was absent in the ST17-isolate, but was detected in the remaining stG62647-isolates. However, in all the CC20-isolates, the silB-gene was truncated due to the insertion of a transposable element after nucleotide 243 (Fig. 3). The transposase was 99% homologous to IS1548, a transposable element of the ISaS1-family, previously reported in several beta-haemolytic streptococci27. To investigate if this was a common feature in SDSE, the sil-locus in the 23 SDSE-genomes currently in Genbank were examined (ncbi.nlm.nih.gov/genome). None of them contained a transposase within the silB-gene, nor was this identified in any of the 333S. pyogenes-genomes deposited in GenBank. Lastly, from our sample of 267 invasive SDSE isolates identified between 1999 and 2015, an additional 76 isolates, chosen to represent the diversity of clinical and molecular characteristics, have been subjected to whole genome sequencing (Unpublished data). A transposable element within the silB-gene was not detected in any of these genomes, indicating that this is not a common genetic feature of SDSE-isolates within our geographical confines.
Figure 3.
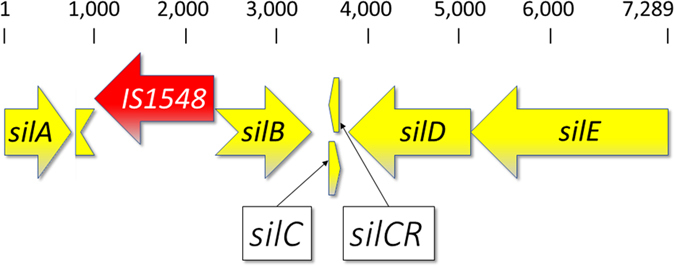
Genetic arrangement of the streptococcal invasive locus in the CC20-isolates. The sil-locus in the CC20 isolates has a transposase, IS1548, inserted into the silB-gene. silB encodes a sensory kinase activating the putative DNA response regulator encoded by silA. This leads to increased transcription of the virulence attenuating pheromone encoded by silCR, and suppression of the virulence associated silC-gene. The SilC-peptide is exported from the cell by the transporter-proteins SilD and SilE. Scale indicates nucleotide position. This disrupted sil-locus is deposited in GenBank under the accession code KY807567.
Discussion
Streptococcus dysgalactiae subspecies equisimilis (SDSE) is increasingly implicated in invasive infections worldwide, but the evolutionary events underlying this upsurge are unknown. With the present work, we have documented the recent emergence of a virulent SDSE genotype, stG62647, in western Norway, and delineated its clinical and genetic characteristics. To the best of our knowledge, this is the first study employing large scale whole genome sequencing to explore the genetic landscape of epidemic lineages in SDSE. Wang et al. characterized an outbreak of tonsillopharyngitis caused by SDSE in Hong Kong, but they sequenced only one isolate28.
stG62647-isolates did not cause invasive SDSE-infections in our region until a few sporadic cases were encountered from 2006. However, in the period from March 2013 to August 2015 it became the predominant emm-type in our health-region, responsible for more than 20% of all invasive SDSE-disease, and comprising 80% of the invasive group C streptococcal-isolates. Although this emm-type has previously been reported from several countries and continents, its relative proportion has generally been below 5%, indicating mainly sporadic encounters8, 29, 30. Interestingly, in a recent study from our neighbouring country, Sweden, stG62647 was one of the most frequently occurring emm-types during 2008–2011, comprising 25% of the invasive SDSE-isolates identified31. Differently, Rantala et al. found only one stG62647-isolate among 140 invasive SDSE cases encountered in Finland between 1995 and 200432. Taken together, this might indicate that the emergence of a virulent stG62647-type in the Nordic countries is a fairly recent event.
Based on MLST data, the stG62647-isolates constituted a relatively homogenous group, with 15 out of 19 belonging to ST20, and 18 out of 19 grouped together as CC20. However, SNP-analysis revealed a more diverse population, emphasizing the superior epidemiological resolution of a SNP-analysis compared to traditional methods. Moreover, it suggests that the genetic events resulting in the epidemiological spread of this genotype have occurred some time ago. The expected rates of spontaneous mutations and recombination events in SDSE have not been well defined, though, making conclusions on evolutionary relationships and clonality difficult.
Notably, half the patients in our study infected with stG62647-isolates experienced a fulminant clinical course, including streptococcal toxic shock syndrome and necrotizing soft-tissue infections. Such disease manifestations are usually associated with S. pyogenes, although our findings are in accordance with previous reports on stG62647. In Sweden, this emm-type was significantly associated with an invasive phenotype, and in a recent report from Austria stG62647 was linked to severe clinical manifestations31, 33. However, most epidemiological reports on SDSE genotypes comprise too few stG62647-isolates to evaluate its correlation with disease severity.
The streptococcal invasive locus (sil) has been demonstrated to repress bacterial virulence through the production of the pheromone like peptide silCR13. This locus was initially described in an M14 S. pyogenes isolate procured from a patient with necrotizing soft-tissue infections, and the strain was discovered to have a missense-mutation in the silCR start-codon13. Subsequent experimental murine skin-infection models revealed these silCR-mutants to confer increased mortality and extensive skin necrosis, whereas concomitant administration of synthetic silCR resulted in attenuated virulence13, 34. Surprisingly, all our CC20-isolates harboured a truncated silB-allele due to the insertion of an IS1548-transposon. This feature appears to be rare, and was neither identified among any of the SDSE- or S. pyogenes-genomes currently available in GenBank, nor in the genomes of contemporary invasive SDSE isolates from our health region. The silB-gene encodes a sensory kinase that activates the transcriptional regulator silA, leading to an upregulation of silCR-production and exportation, and concurrent suppression of silC 15, 35. In a recent murine model study, SDSE grown in the presence of silCR caused a milder disease than the control group. Inversely, mice vaccinated against silCR developed a significantly more severe illness than non-vaccinated mice6. Although not experimentally verified, it is plausible that the lack of silB in our stG62647-isolates would impair the silCR-levels, and favour the production of the virulence associated silC-peptide. This would likely constitute a significant contribution to the aggressive phenotype associated with our stG62647-isolates belonging to CC20. Experimentally induced mutations in silB in M14 S. pyogenes-strains resulted in reduced virulence13, 36. However, those isolates concurrently contained a truncated silCR-gene, and the results are thus not readily transferable.
IS1548 is a transposable element of the ISAS1-family, initially described in invasive Streptococcus agalactiae strains, but subsequently also detected in S. pyogenes and SDSE27, 37. It encodes two putative proteins of 377 and 62 amino acids, respectively, and has 19 bp imperfect inverted repeats at its termini. Interestingly, IS1548 has been reported to modulate the expression of several virulence factors in Streptococcus agalactiae. Al Safadi et al. demonstrated that the insertion of IS1548 immediately upstream of the laminin binding gene lmb, was associated with an ~5-fold increase both in the transcription of lmb, and in the phenotypic bacterial adhesion to laminin38. Conversely, insertion of IS1548 into the hylB and cpsD genes, abrogated the production of hyaluronidase and bacterial capsule, respectively37, 39. Of note, IS1548 has also been documented in the covRS-system in S. pyogenes. Several S. pyogenes-strains were passed through an experimental murine model, and spontaneous insertion of IS1548 into the covS-gene was observed in two isolates. These genetic alterations in the covRS-system rendered the bacteria more virulent and led to more extensive necrotic skin lesions12. Although mutational transition to a more virulent phenotype undoubtedly renders the bacteria more capable of invasive disease, it does not necessarily represent an evolutionary advantage. Trevino et al. showed that highly virulent covRS-mutated S. pyogenes-isolates indeed were significantly outcompeted by wild-type strains during growth in human saliva, indicating that different phenotypic properties might be required for a commensal state in the upper respiratory tract14. IS1548 has been suggested to mediate adaptive mutations, allowing the bacteria to switch between pathogenicity and commensalism27. Insertion of a transposable element is conceivably a more dynamic evolutionary transition than adaptation through point-mutations and deletions. We speculate that the insertion of IS1548 into the silB-gene confers a more aggressive and invasive bacterial phenotype through the downregulation of silCR. Furthermore, it is plausible that the process is reversible under certain environmental conditions, facilitating the return to a commensal state. The pathogenic mechanisms and bacterial signalling involved in this hypothesized transition have yet to be explored, and deserves further investigation.
A genetic dissection of the stG62647-isolates revealed the presence of several important virulence factors, including genes involved in immune evasion (emm, protein G and c5a-peptidase), adhesion (dltABCD, fbp54, gapC, shr and lmb), tissue penetration (streptokinase) and toxins (streptolysin O and streptolysin S). However, these genes appear to be ubiquitous in SDSE, and are thus unlikely to have a pivotal role in the successful epidemic spread and observed disease severity of stG62647 5. The differentially distributed speG was identified in all the CC20-isolates. speG does not confer superantigen-activity, though, and a correlation with disease severity has not been demonstrated40, 41. The covRS-regulon was conserved among the stG62647-isolates, and all showed a single amino acid substitution in covS, I421V, as compared to GGS124. However, a valine residue in this position is also present in wild type S. pyogenes-isolates, thus unlikely to be of clinical significance42.
Although bacteriophages were encountered in a subset of the isolates, no established or putative phage-associated virulence genes were detected. The CRISPR-cas system constitutes a part of the adaptive immune system in prokaryotic cells, conferring resistance to mobile genetic elements and bacteriophages43. Previous encounters with bacteriophages are deposited in the CRIPSR-locus as short sequence-specific DNA-fragments termed spacers. For S. pyogenes, an inverse relationship between the number of spacers and susceptibility to bacteriophages has been reported43. We did not observe such an association among our SDSE-isolates, albeit the frequency of bacteriophages was very low. We found our SDSE-isolates to contain markedly fewer bacteriophages and a higher number of spacers than S. pyogenes-genomes currently deposited in GenBank. Similar observations were reported by Shimomura et al., and a CRISPR-mediated increased resistance to bacteriophage-infection among SDSE compared to S. pyogenes is conceivable44.
The present study has some limitations. First, the partial retrospective design increased the risk of diagnostic misclassification, potentially leading to the underestimation of invasive SDSE and stG62647-infections. However, a scrutinous review of medical records likely led to the inclusion of all relevant SDSE cases in our region during the study period. Secondly, we acknowledge that the rise and fall of epidemic lineages of microbes is multifactorial, and although bacterial genetic events undoubtedly are contributory, knowledge on the prevalence of the outbreak genotype in asymptomatic carriers, along with the protective specific immunity in our population during the study period, would have increased our understanding of this stG62647-outbreak. Lastly, several of our proposed pathogenic mechanisms are based on extrapolation of experimental data from closely related species, as knowledge on virulence properties in SDSE is scarce. A substantial fraction of the genetic repertoire specific for the stG62647-lineage encodes hypothetical proteins, and their pathogenic contribution is difficult to ascertain within the confines of current knowledge. Hence, the possibility that the observed virulence potential of the stG62647-isolates emanates from any of these genetic elements cannot be ruled out. Furthermore, the significance of an insertional element in the silB-gene has not been verified experimentally, and its contribution to disease invasiveness or microenvironmental adaptation has yet to be elucidated. Ideally, the sil-locus should be examined from stG62647-isolates collected from different ecological niches, including asymptomatic carriers. A final conclusion could be obtained after the deletion of IS1548 from the silB gene of one of the stG62647-strain and the comparison of the virulence of the deletion mutant and the wild-type strain in an animal model. Adaptive virulence regulation through insertional elements constitutes an intriguing concept, and further studies are warranted.
Conclusions
We report the emergence of a novel and virulent SDSE genotype, stG62647, in our health region. The infections associated with stG62647 appeared to pursue a severe clinical course, including the occurrence of streptococcal toxic shock syndrome, necrotizing soft-tissue infections and endocarditis. We speculate that its virulence could be partly attributable to the insertion of a transposable element, IS1548, into the streptococcal invasive locus (sil), thus abrogating the virulence attenuating control exerted by this regulon. Invasive SDSE disease is currently three times more common than invasive S. pyogenes infections in our region, and sustained epidemiological attentiveness is indicated.
Ethical considerations
The study underwent institutional ethics review and approval (2010/1406 Regional Ethics Committee West, Norway). Informed consent was obtained from participants. All experiments were carried out in full accordance with the approved ethics applications specified above.
Availability of data and materials
The genomes of the stG62647-isolates have been deposited in the SRA GenBank database under the study accession number SRP103012 (www.ncbi.nlm.nih.gov/sra). Isolate T642 has been deposited at DDBJ/ENA/GenBank under the accession NBUZ00000000, the version described in this paper is version NBUZ01000000. The streptococcal invasive locus containing a truncated silB-gene due to the insertion of IS1548, is deposited in GenBank under the accession number KY807567.
Acknowledgements
We thank the staff at the microbiological department at Haukeland University Hospital for excellent technical assistance and for providing access to their laboratory facilities. This study received support from Haraldsplass Deaconess Hospital, the University of Bergen, and the European Union (FP7/2007–2013) under the grant agreement 305340.
Author Contributions
B.K. and O.O. conceived the study. S.S. and H.M. contributed to the study design. O.O. and P.C.L. performed D.N.A. preparation and computational analysis. O.O. extracted clinical data and drafted the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Brandt CM, Spellerberg B. Human infections due to Streptococcus dysgalactiae subspecies equisimilis. Clin Infect Dis. 2009;49:766–772. doi: 10.1086/605085. [DOI] [PubMed] [Google Scholar]
- 2.Oppegaard O, Mylvaganam H, Kittang BR. Beta-haemolytic group A, C and G streptococcal infections in Western Norway: a 15-year retrospective survey. Clin Microbiol Infect. 2015;21:171–178. doi: 10.1016/j.cmi.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 3.Bruun T, et al. Necrotizing soft tissue infections caused by Streptococcus pyogenes and Streptococcus dysgalactiae subsp. equisimilis of groups C and G in western Norway. Clin Microbiol Infect. 2013;19:E545–550. doi: 10.1111/1469-0691.12276. [DOI] [PubMed] [Google Scholar]
- 4.Hashikawa S, et al. Characterization of group C and G streptococcal strains that cause streptococcal toxic shock syndrome. J Clin Microbiol. 2004;42:186–192. doi: 10.1128/JCM.42.1.186-192.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies MR, et al. Virulence profiling of Streptococcus dysgalactiae subspecies equisimilis isolated from infected humans reveals 2 distinct genetic lineages that do not segregate with their phenotypes or propensity to cause diseases. Clin Infect Dis. 2007;44:1442–1454. doi: 10.1086/516780. [DOI] [PubMed] [Google Scholar]
- 6.Michael-Gayego A, Dan-Goor M, Jaffe J, Hidalgo-Grass C, Moses AE. Characterization of sil in invasive group A and G streptococci: antibodies against bacterial pheromone peptide SilCR result in severe infection. Infect Immun. 2013;81:4121–4127. doi: 10.1128/IAI.00359-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambertsen LM, Ingels H, Schonheyder HC, Hoffmann S, Danish Streptococcal Surveillance Collaboration, G. Nationwide laboratory-based surveillance of invasive beta-haemolytic streptococci in Denmark from 2005 to 2011. Clin Microbiol Infect. 2014;20:O216–223. doi: 10.1111/1469-0691.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wajima T, et al. Molecular Characterization of Invasive Streptococcus dysgalactiae subsp. equisimilis, Japan. Emerg Infect Dis. 2016;22:247–254. doi: 10.3201/eid2202.141732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anonymous. Surveillance of pyogenic and non-pyogenic streptococcal bacteraemia in England, Wales and Northern Ireland: 2015. Health Protection Report, Public Health England10 (2016).
- 10.Nasser W, et al. Evolutionary pathway to increased virulence and epidemic group A Streptococcus disease derived from 3,615 genome sequences. Proc Natl Acad Sci USA. 2014;111:E1768–1776. doi: 10.1073/pnas.1403138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanson M, et al. A naturally occurring single amino acid replacement in multiple gene regulator of group A Streptococcus significantly increases virulence. Am J Pathol. 2015;185:462–471. doi: 10.1016/j.ajpath.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engleberg NC, Heath A, Miller A, Rivera C, DiRita VJ. Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J Infect Dis. 2001;183:1043–1054. doi: 10.1086/319291. [DOI] [PubMed] [Google Scholar]
- 13.Hidalgo-Grass C, et al. A locus of group A Streptococcus involved in invasive disease and DNA transfer. Mol Microbiol. 2002;46:87–99. doi: 10.1046/j.1365-2958.2002.03127.x. [DOI] [PubMed] [Google Scholar]
- 14.Trevino J, et al. CovS simultaneously activates and inhibits the CovR-mediated repression of distinct subsets of group A Streptococcus virulence factor-encoding genes. Infect Immun. 2009;77:3141–3149. doi: 10.1128/IAI.01560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belotserkovsky I, et al. Functional analysis of the quorum-sensing streptococcal invasion locus (sil) PLoS Pathog. 2009;5:e1000651. doi: 10.1371/journal.ppat.1000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nurk S, et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol. 2013;20:714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aziz RK, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larsen MV, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50:1355–1361. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaas RS, Leekitcharoenphon P, Aarestrup FM, Lund O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLoS One. 2014;9:e104984. doi: 10.1371/journal.pone.0104984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kearse M, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arndt D, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016;44:W16–21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beres SB, et al. Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci USA. 2002;99:10078–10083. doi: 10.1073/pnas.152298499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Falugi F, et al. Sequence variation in group A Streptococcus pili and association of pilus backbone types with lancefield T serotypes. J Infect Dis. 2008;198:1834–1841. doi: 10.1086/593176. [DOI] [PubMed] [Google Scholar]
- 27.Flechard M, Gilot P. Physiological impact of transposable elements encoding DDE transposases in the environmental adaptation of Streptococcus agalactiae. Microbiology. 2014;160:1298–1315. doi: 10.1099/mic.0.077628-0. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Zhang X, Zong Z. Genome sequence and virulence factors of a group G Streptococcus dysgalactiae subsp. equisimilis strain with a new element carrying erm(B) Sci Rep. 2016;6:20389. doi: 10.1038/srep20389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pinho MD, Melo-Cristino J, Ramirez M. Clonal relationships between invasive and noninvasive Lancefield group C and G streptococci and emm-specific differences in invasiveness. J Clin Microbiol. 2006;44:841–846. doi: 10.1128/JCM.44.3.841-846.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmad Y, et al. Genetic relationships deduced from emm and multilocus sequence typing of invasive Streptococcus dysgalactiae subsp. equisimilis and S. canis recovered from isolates collected in the United States. J Clin Microbiol. 2009;47:2046–2054. doi: 10.1128/JCM.00246-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trell K, Nilson B, Rasmussen M. Species and emm-type distribution of group C and G streptococci from different sites of isolation. Diagn Microbiol Infect Dis. 2016;86:467–469. doi: 10.1016/j.diagmicrobio.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 32.Rantala S, Vahakuopus S, Vuopio-Varkila J, Vuento R, Syrjanen J. Streptococcus dysgalactiae subsp. equisimilis Bacteremia, Finland, 1995-2004. Emerg Infect Dis. 2010;16:843–846. doi: 10.3201/eid1605.080803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leitner E, et al. Prevalence of emm types and antimicrobial susceptibility of Streptococcus dysgalactiae subsp. equisimilis in Austria. Int J Med Microbiol. 2015;305:918–924. doi: 10.1016/j.ijmm.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 34.Hidalgo-Grass C, et al. Effect of a bacterial pheromone peptide on host chemokine degradation in group A streptococcal necrotising soft-tissue infections. Lancet. 2004;363:696–703. doi: 10.1016/S0140-6736(04)15643-2. [DOI] [PubMed] [Google Scholar]
- 35.Eran Y, et al. Transcriptional regulation of the sil locus by the SilCR signalling peptide and its implications on group A streptococcus virulence. Mol Microbiol. 2007;63:1209–1222. doi: 10.1111/j.1365-2958.2007.05581.x. [DOI] [PubMed] [Google Scholar]
- 36.Kizy AE, Neely MN. First Streptococcus pyogenes signature-tagged mutagenesis screen identifies novel virulence determinants. Infect Immun. 2009;77:1854–1865. doi: 10.1128/IAI.01306-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Granlund M, Oberg L, Sellin M, Norgren M. Identification of a novel insertion element, IS1548, in group B streptococci, predominantly in strains causing endocarditis. J Infect Dis. 1998;177:967–976. doi: 10.1086/515233. [DOI] [PubMed] [Google Scholar]
- 38.Al Safadi R, et al. Enhanced expression of lmb gene encoding laminin-binding protein in Streptococcus agalactiae strains harboring IS1548 in scpB-lmb intergenic region. PLoS One. 2010;5:e10794. doi: 10.1371/journal.pone.0010794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sellin M, Olofsson C, Hakansson S, Norgren M. Genotyping of the capsule gene cluster (cps) in nontypeable group B streptococci reveals two major cps allelic variants of serotypes III and VII. J Clin Microbiol. 2000;38:3420–3428. doi: 10.1128/jcm.38.9.3420-3428.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao J, et al. Cloning, expression, and characterization of the superantigen streptococcal pyrogenic exotoxin G from Streptococcus dysgalactiae. Infect Immun. 2007;75:1721–1729. doi: 10.1128/IAI.01183-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kittang BR, Skrede S, Langeland N, Haanshuus CG, Mylvaganam H. emm gene diversity, superantigen gene profiles and presence of SlaA among clinical isolates of group A, C and G streptococci from western Norway. Eur J Clin Microbiol Infect Dis. 2011;30:423–433. doi: 10.1007/s10096-010-1105-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tatsuno I, Okada R, Zhang Y, Isaka M, Hasegawa T. Partial loss of CovS function in Streptococcus pyogenes causes severe invasive disease. BMC Res Notes. 2013;6:126. doi: 10.1186/1756-0500-6-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nozawa T, et al. CRISPR inhibition of prophage acquisition in Streptococcus pyogenes. PLoS One. 2011;6:e19543. doi: 10.1371/journal.pone.0019543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimomura Y, et al. Complete genome sequencing and analysis of a Lancefield group G Streptococcus dysgalactiae subsp. equisimilis strain causing streptococcal toxic shock syndrome (STSS) BMC Genomics. 2011;12:17. doi: 10.1186/1471-2164-12-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The genomes of the stG62647-isolates have been deposited in the SRA GenBank database under the study accession number SRP103012 (www.ncbi.nlm.nih.gov/sra). Isolate T642 has been deposited at DDBJ/ENA/GenBank under the accession NBUZ00000000, the version described in this paper is version NBUZ01000000. The streptococcal invasive locus containing a truncated silB-gene due to the insertion of IS1548, is deposited in GenBank under the accession number KY807567.