

Abstract
Purpose
Acalabrutinib (ACP-196) is a novel, potent, and highly selective BTK inhibitor, which binds covalently to Cys481 in the ATP-binding pocket of BTK. We sought to evaluate the anti-tumor effects of acalabrutinib treatment in two established mouse models of chronic lymphocytic leukemia (CLL).
Experimental Design
Two distinct mouse models were used, the TCL1 adoptive transfer model where leukemic cells from Eμ-TCL1 transgenic mice are transplanted into C57BL/6 mice, and the human NSG primary CLL xenograft model. Mice received either vehicle or acalabrutinib formulated into the drinking water.
Results
Utilizing biochemical assays we demonstrate that acalabrutinib is a highly selective BTK inhibitor as compared to ibrutinib. In the human CLL NSG xenograft model, treatment with acalabrutinib demonstrated on-target effects including decreased phosphorylation of PLCγ2, ERK and significant inhibition of CLL cell proliferation. Further, tumor burden in the spleen of the mice treated with acalabrutinib was significantly decreased compared to vehicle treated mice. Similarly, in the TCL1 adoptive transfer model, decreased phosphorylation of BTK, PLCγ2 and S6 was observed. Most notably, treatment with acalabrutinib resulted in a significant increase in survival compared to mice receiving vehicle.
Conclusions
Treatment with acalabrutinib potently inhibits BTK in vivo, leading to on-target decreases in the activation of key signaling molecules (including BTK, PLCγ2, S6 and ERK). In two complementary mouse models of CLL acalabrutinib significantly reduced tumor burden and increased survival compared to vehicle treatment. Overall, acalabrutinib showed increased BTK selectivity compared to ibrutinib while demonstrating significant anti-tumor efficacy in vivo on par with ibrutinib.
Keywords: ibrutinib, acalabrutinib, BTK and chronic lymphocytic leukemia
INTRODUCTION
Bruton’s tyrosine kinase (BTK), a member of the TEC family of kinases, is a key node in the B-cell receptor (BCR) signaling pathway and is essential for normal B-cell development.(1, 2) BTK functions as a bridge between the BCR and the activation of key downstream survival signals, including NF-κB. Expression of BTK is limited to cells of the hematopoietic lineage, excluding T-cells, and is upregulated in CLL cells compared to normal B-cells.(3) Mutation of BTK in humans results in the development of X-linked agammaglobulinemia, characterized by a block in B-cell development resulting in the absence of mature B-cells. In a subset of B-cell malignancies BTK is also essential for proliferation and survival.(4, 5) In particular, knockdown of BTK induces tumor cell death in primary chronic lymphocytic leukemia (CLL) cells and lymphoma cell lines that are dependent on BCR signaling.(6, 7) Further, genetic ablation of BTK inhibits disease progression in mouse models of CLL, indicating its continued importance for B-cell malignancies.(7, 8)
Targeting the BCR pathway (through inhibition of SYK, BTK or PI3K) in patients with CLL has proven very effective.(9) Activation of BCR and NF-κB signaling in the lymph node microenvironment appears to be a central event in CLL pathogenesis and disease progression, and is effectively disrupted in vivo by the BTK inhibitor ibrutinib.(10, 11) Ibrutinib, as a single agent, has demonstrated a high rate of durable clinical responses in patients with CLL, irrespective of adverse prognostic features, including very high risk patients with deletion 17p.(12–14) Ibrutinib is now approved for the treatment of patients with CLL who have received at least one prior therapy or harbors a 17p deletion.(15)
Despite the impressive clinical results with ibrutinib, most patients do not experience a complete response and a subset of patients develop resistance. Resistance develops most commonly through mutations in BTK or PLCγ2, suggesting that BTK is indeed a critically important target for ibrutinib.(16, 17) Additionally, alternative targets of ibrutinib (including, but not limited to ITK, EGFR, and TEC) may account for some adverse effects such as diarrhea, rash, atrial fibrillation, and bruising.(18) Further, ibrutinib has been shown to inhibit NK-cell and macrophage function, likely due to inhibition of alternative kinases such as ITK, which could reduce the benefit of combinations of ibrutinib with anti-CD20 and potentially other therapeutic monoclonal antibodies dependent upon antibody-dependent cellular cytotoxicity.(19–22) Together, the observation that resistance develops through mutations in BTK and that inhibition of kinases other than BTK may have unwanted effects suggests that a more specific and potent BTK inhibitor may have therapeutic benefit.
CLL cells depend on survival and proliferation signals in the tissue microenvironment.(23, 24) Analysis of on-target effects in tumor cells residing in the microenvironment is particularly important in the age of BCR-directed inhibitors that induce only minimal cell death in vitro but demonstrate striking results in vivo. Mouse models have therefore gained additional importance to investigate drug effects dependent on tumor-host interactions. Currently, one of the most widely utilized models for CLL is the transgenic Eμ-TCL1 (TCL1) mouse, in which the human TCL1 gene is expressed under the control of the immunoglobulin heavy chain variable region promoter and enhancer.(25) These mice spontaneously develop a CD5+/CD19+ CLL-like leukemia with unmutated IGHV and have a response to CLL therapies analogous to humans.(26) To overcome the heterogeneity in presentation and the delay of tumor development of the spontaneous model, leukemic splenic lymphocytes from TCL1 mice can be engrafted into SCID or immunocompetent mice. In addition to this transgenic mouse model, xenograftment of mononuclear cells (MNCs) from CLL patients into NOD/scid/γc null (NSG) mice has also been shown to recapitulate the tumor-host interactions encountered in the diseased human lymph node, including activation of BCR and NF-кB signaling and tumor proliferation.(27)
We report herein on the potency, selectivity and on-target efficacy of acalabrutinib, a novel, potent and highly selective BTK inhibitor. Acalabrutinib binds covalently to Cys481 in the ATP-binding pocket of BTK, similarly to ibrutinib. It has been shown to have improved pharmacologic features, such as rapid oral absorption and a short plasma half-life.(28) Early results from the ongoing first in human phase 1–2 study demonstrate impressive clinical activity of acalabrutinib in CLL.(28) Here, we investigate pharmacodynamic properties of acalabrutinib in two complementary murine models of CLL demonstrating on-target effects on BCR signaling, tumor biology and anti-leukemic efficacy further justifying exploration of this treatment in CLL.
MATERIALS AND METHODS
Kinase Binding Selectivity Profiling
Acalabrutinib and ibrutinib (Acerta Pharma B.V., Redwood City, CA) were profiled at 1μM in an ATP site dependent competition binding assay for 395 wild-type human kinases at DiscoverRx (San Diego, CA).(29)
Potency and return of B-cell function assay
For potency assays, female C57BL/6 mice were administered vehicle, acalabrutinib or ibrutinib via oral gavage. After 3 hours, spleens were extracted and splenocytes were stimulated with goat anti-mouse IgM (Southern Biotech, Birmingham, AL) at 10μg/mL for 18 hours in a 37°C, 5% CO2 incubator. For the return of B-cell function experiments, mice were orally administered vehicle or 25mg/kg acalabrutinib or ibrutinib at time zero. Spleens were extracted at various time points ranging from 3 to 24 hours. Isolated splenocytes were then stimulated with anti-IgM at 10μg/mL for 18 hours in a 37°C, 5% CO2 incubator. In both experiments, cells were washed with FACS buffer (PBS 0.5% BSA) and Fc receptors were blocked (Fc Block; BD Biosciences, San Jose, CA). Cells were stained with fluorochrome-conjugated antibodies that recognize CD19 and one of the following CD69, CD86 or phospho-S6(S235/236) (BD Biosciences). 7-AAD (Life Technologies, Carlsbad, CA) was used to identify dead cells. Inhibition of BCR signaling was determined by measuring the expression of the activation markers, on the surface of CD19+ B-cells by flow cytometry. Greater than 5,000 gated CD19+ cells per sample were acquired on a FACSVerse flow cytometer using FACSuite 1.0.5 and analyzed using FCS Express (Version 4, De Novo, Glendale, CA).
Patient samples and xenotransplantation of NSG mice
Mononuclear cells (MNCs) were obtained from 12 patients with CLL: 83% treatment naïve, 67% male, 40% advanced Rai stage, 83% IGHV unmutated and 27% harboring a del17p. Cells were collected in accordance with the Declaration of Helsinki on an IRB approved protocol at the National Institutes of Health.(10) MNCs were prepared by density-gradient centrifugation (Ficoll Lymphocyte Separation Media; ICN Biomedicals, Irvine, CA) and viably frozen in 90% fetal bovine serum (FBS) and 10% DSMO (Sigma, St. Louis, MA) in liquid nitrogen. MNCs were treated in vitro or xenografted into NSG mice (NOD.Cg-Prkdcscid Il2rgtm1WjI/SzJ, Jackson Laboratory, Bar Harbor, ME) as described by Herman et al.(27) Briefly, MNCs harvested from CLL patients were adoptively transferred at 1 × 108 cells per mouse into multiple recipient mice. Acalabrutinib treatment was initiated on day −1 (at the time of busulfan priming) at 0.3 or 0.16mg/mL formulated into the drinking water (provided by Acerta Pharma B.V). In select experiments drinking water was formulated at 0.06mg/mL. Mice were sacrificed for pharmacodynamic endpoints after three weeks of treatment, then whole blood and spleens were collected. Splenocytes were obtained by homogenizing harvested spleens, and these samples were then filtered through 70μm nylon sieves (BD Falcon, Franklin Lakes, NJ). Erythrocytes were lysed using ACK buffer (Quality Biological, Inc., Gaithersburg, MD). Acalabrutinib and ibrutinib were drugged in vitro for 1 hour at 1μM concentrations. In a select experiment patient MNSs were pretreated in vitro for 3 hours with 1μM concentrations of acalabrutinib or ibrutinib prior to stimulation with anti-human IgM at 20μg/mL (Jackson ImmunoResearch, West Grove, PA) for 18 hours in a 37°C, 5% CO2 incubator simulating in vivo experimental conditions.
TCL1 adoptive transfer mouse model
The TCL1 adoptive transfer model was utilized in designated experiments and these experiments were performed under a protocol approved by The Ohio State University Institutional Laboratory Animal Care and Use Committee. TCL1 transgenic mice on a C57BL/6 strain have been previously described.(7) 1 × 107 leukemic cells from TCL1 transgenic mice were transplanted into C57BL/6 mice purchased from Jackson Laboratories, resulting in a CD5+/CD19+ leukemia with peripheral blood, spleen, and nodal involvement. Following adoptive transfer, 0.16mg/mL of acalabrutinib formulated into the drinking water (provided by Acerta Pharma B.V.) was started when recipient mice had ≥10% CD5+/CD19+ leukocytes in the peripheral blood as determined by flow cytometry. Mice were followed for survival or sacrificed for pharmacodynamic endpoints following one or four weeks of treatment.
Flow cytometry
MNCs treated in vitro or harvested from the spleens of xenografted NSG mice were stained as previously described (30) with panels of the following surface antibodies: anti- CD45, CD19, CD3, CD5, CD69 and CD86. In indicated experiments cells were fixed with 4% paraformaldehyde, permeabilized with 90% methanol (Thermo Fisher, Waltham, MA) and stained with the following antibodies: IgG1-isotype control, IgG2b-isotype control, anti- phospho-BTK(Y223), phospho-PLCγ2(Y759), phospho-S6(S235/236), phospho-p65(S529), phospho-ERK(T202/Y204) or Ki-67 (BD Biosciences). Cells were analyzed on a FACS Canto II flow cytometer (BD Biosciences) using FACS-DIVA 6.1.1 and FlowJo (Version 10, TreeStar, Ashland, OR). In select experiments, determination of absolute cell counts per μL peripheral blood was done by adding AccuCount blank particles (Spherotech, Lake Forest, IL). The abundance of human cells in the spleen was measured as the percentage of human cells (defined by CD45 staining) among all nucleated cells (defined by forward and side scatter properties). Splenocytes from the TCL1 adoptive transfer mice were stained with the following antibodies: anti- B220, CD3, phospho-S6(S235/236) (BD Biosciences), phospho-BTK(Y223) and phospho-PLCγ2(Y1217) (Abcam, Cambridge, UK). Cells were acquired on a FACSVerse flow cytometer using FACSuite (Version 1.0.5, BD Biosciences) and analyzed using FCS Express. The median fluorescent intensity of the phospho-specific antibodies was measured from the B-cell population (B220+CD3−).
BTK Occupancy Probe ELISA
This method was run as previously described.(28) 96-well OptiPlates (PerkinElmer; Waltham, MA) were coated overnight at 4°C with 125ng/well anti-Btk antibody (BD Biosciences) and blocked with bovine serum albumin the following day for 2–3 hours at room temperature. Lysis buffer containing protease inhibitor cocktail (Sigma-Aldrich; St. Louis, MO) were used to lyse frozen splenocyte cell pellets. Lysates were incubated for 1 hour on ice in the presence or absence of a saturating concentration of acalabrutinib (10−6M), followed by an incubation of a biotinylated derivate (ACP-4016; 10−7 M) serving as a probe. The equivalent of 5 ×105 cells of lysate/well, in replicates of three, were added to the coated OptiPlates and incubated for 2 hours. After a 1 hour incubation with Streptavidin-HRP (Invitrogen; ELISA grade) (120ng/mL), a SuperSignal ELISA Femto Substrate (Thermo Fisher Scientific) was added and chemiluminescence was measured on an EnVision® multiplate reader (PerkinElmer). The percent BTK occupancy for the drug-treated mice were calculated relative to the average signal from the vehicle control group. The sample without exogenous acalabrutinib represents 100% free BTK (or 0% occupied BTK), and the samples with exogenous acalabrutinib represents 0% free BTK (or 100% occupied BTK).
Statistical Evaluation
In order to compare parameters related to the donor CLL patient, groups of mice engrafted with cells from one patient were averaged per treatment group and a paired Student’s t-test was applied (Prism5, GraphPad, La Jolla, CA). To compare treatment effect across mice, an unpaired t-test was applied taking into account the random batch effect (JMP, SAS, Cary, NC). For survival analysis, estimates of overall survival were obtained using the Kaplan-Meier method, and the log-rank test was used to evaluate differences between curves.
RESULTS
Acalabrutinib is a potent and selective inhibitor of BTK
Acalabrutinib is a novel, potent inhibitor of BTK, which like ibrutinib binds covalently to Cys481 in the ATP-binding pocket of BTK. Acalabrutinib and ibrutinib inhibit BTK at similar concentrations (IC50 of 5.1 and 1.5nM, respectively) as previously reported.(28) Including BTK, ten kinases contain a conserved cysteine residue that aligns with the Cys481 in BTK (Supplementary Table S1).(18) Ibrutinib inhibits all of these kinases with IC50 values in the low nanomolar range.(18, 28, 31) In contrast, acalabrutinib inhibits only BTK, BMX, ERBB4 and TEC at concentrations <100nM.(28) To further investigate the specificity of acalabrutinib we assayed its potential noncovalent binding interaction with kinases by screening a comprehensive panel of 395 kinases, including mutants, in the DiscoverRx KINOMEscan™. Minimal binding outside of BTK was observed at 1μM acalabrutinib, further demonstrating a high degree of selectivity (Figure 1A and Supplementary Table S1). Of note, ITK, which is known to be inhibited by ibrutinib at therapeutic doses, was not significantly inhibited by acalabrutinib (Supplementary Table S1). Together, this suggests that acalabrutinib selectively inhibits BTK, while ibrutinib may elicit broader inhibition across multiple kinases.
Figure 1. Acalabrutinib is a potent and selective inhibitor of BTK.
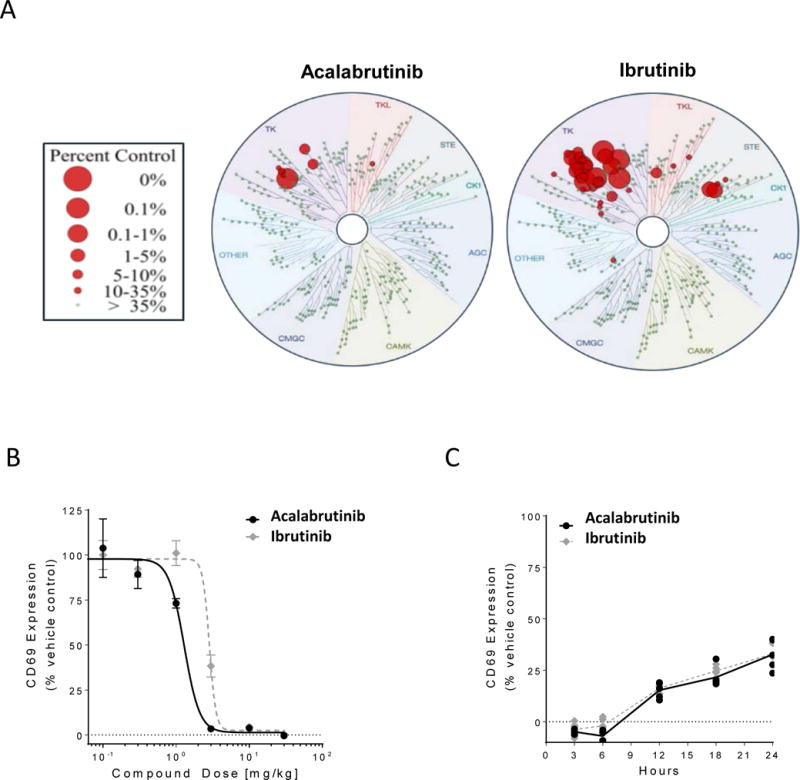
(A) Acalabrutinib and ibrutinib were profiled at 1 μM over a panel 395 wild-type human kinases, including mutants, at DiscoveRx kinase assays. The size of the red circles represents the extent of inhibition with larger circles meaning stronger inhibition compared to control signal as defined in the scale. (B) Mice (5/group/dose) were orally given vehicle, acalabrutinib or ibrutinib. After 3 hours, spleens were extracted and splenocytes stimulated with anti-IgM for 18 h, followed by CD69 expression analysis by flow cytometry. (C) Mice (5/group) received 25 mg/kg of vehicle, acalabrutinib or ibrutinib at time zero. Spleens were extracted at various time points and splenocytes stimulated with anti-IgM for 18 h, followed by CD69 expression analysis by flow cytometry. Black lines and symbols represent acalabrutinib, grey lines and symbols represent ibrutinib.
Next we sought to compare the potency of acalabrutinib and ibrutinib in vivo. C567BL/6 mice were gavaged orally with either ibrutinib or acalabrutinib at doses ranging from 0.1 to 30mg/kg. After 3 hours, splenocytes were collected and stimulated with anti-IgM and evaluated for expression of CD69 (an activation marker downstream of BCR signaling). We found that the effective doses of acalabrutinib and ibrutinib were similar (1.3 and 2.9mg/kg, respectively; Figure 1B). Expanding on this, we found that acalabrutinib inhibited BCR signaling (as determined by CD69 expression) significantly more than ibrutinib at both 1mg/kg and 3mg/kg concentrations (Supplementary Figure S1). To further compare the functional BTK inhibition between acalabrutinib and ibrutinib we evaluated the return of BCR signaling over time. Mice were orally gavaged with vehicle alone or acalabrutinib or ibrutinib at a saturating concentration of 25mg/kg. Splenocytes were collected at various time points ranging from 3 hours to 24 hours after treatment. The splenocytes were stimulated with anti-IgM and evaluated for expression of CD69 (Supplementary Figure S2). We found that the return of B-cell function after treatment with ibrutinib or acalabrutinib occurred in a similar fashion, with both demonstrating approximately 70% inhibition of B-cell function 24 hours after drug administration compared to vehicle control (Figure 1C). We further verified this by looking at changes in additional activation markers, specifically, CD86 and phospho-S6 (Supplementary Figure S3).
Acalabrutinib demonstrates equal in vitro on-target effects as ibrutinib
To confirm these findings, we evaluated on-target effects of BTK inhibition in vitro in primary CLL patient MNCs. We first sought to compare the ability of acalabrutinib and ibrutinib to block BCR stimulation in vitro, similar to what was shown in vivo. We found that pretreatment with acalabrutinib (P=0.01) or ibrutinib (P=0.004) significantly prevented BCR stimulation by anti-IgM to comparable levels (Supplementary Figure S4 and Figure 2A; P=0.84). To further evaluate the on-target effects of BTK inhibition, MNCs were treated in culture with or without 1μM of acalabrutinib or ibrutinib for one hour and then evaluated for changes in BCR signaling. We found that treatment with acalabrutinib led to a significant reduction in mean fluorescence intensity (MFI) of the autophosphorylation site of BTK (Figure 2B, P<0.05, median change −15%, IQR −28 to −6). In addition, we found a significant reduction in the phosphorylation of PLCγ2, a direct downstream target of BTK (P=0.005, median change −32%, IQR −69 to −15), S6, a component of the 40S ribosomal subunit expressed downstream of both the BCR and phosphoinositide 3-kinase (PI3K) signaling pathways, (P=0.03, median change −20%, IQR −40 to +24) and NF-κB, a more distal downstream target, (p65, P=0.01, median change −26%, IQR −37 to −18), after treatment with acalabrutinib (Figures 2C–E). No significant differences in inhibition of any of the evaluated BCR signaling molecules were observed between acalabrutinib and ibrutinib (Figures 2B–E). Lastly, we compared the reduction in surface activation markers CD69 and CD86 after 24 hours of in vitro acalabrutinib and ibrutinib treatment, and found no significant difference in the reduction of baseline expression between the two inhibitors (Figure 2F).
Figure 2. Acalabrutinib demonstrates equal in vitro on-target effects as ibrutinib.
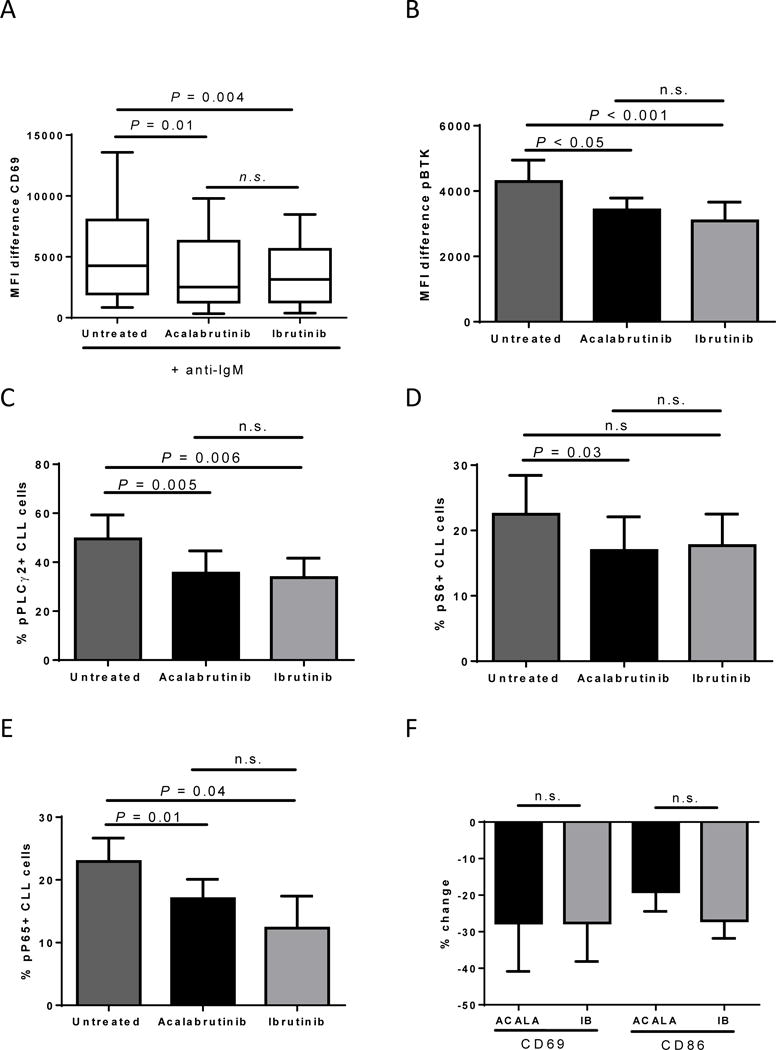
CLL patient MNCs from the peripheral blood or lymph node were treated in vitro with vehicle (untreated), or 1μM acalabrutinib or ibrutinib. (A) CLL patient MNCs (n=7) were pretreated with BCR-inhibitors for 3 hours and then stimulated with anti-IgM for 18 h, followed by CD69 expression analysis by flow cytometry. Results shown are for the CLL population. Shown is a min to max box and whisker plot. (B) CLL patient MNCs (n=5) were treated in vitro for 1 hour, fixed, permeabilized, and stained for phospho-BTK (pBTK). Results shown are for the CLL population. Shown is the mean (± SEM) MFI difference (pBTK-isotype control). (C-E) CLL patient MNCs (n=5) were treated in vitro for 1 hour, fixed, permeabilized, and stained for phospho-PLCγ2 (C), phospho-S6 (D) and phospho-NF-κB (E). Results shown are for the CLL population. The mean (± SEM) percent of CLL cells expressing the indicated readout is shown. (F) CLL patient MNCs (n=7) were treated for 24 hours followed by CD69 and CD86 expression analysis by flow cytometry. Results shown are for the CLL population. The mean (± SEM) percent change in CLL cells expressing the indicated readout is shown compared to untreated. ACALA: acalabrutinib, IB: ibrutinib. All Statistics were determined by paired student t-test.
Acalabrutinib demonstrates in vivo on-target effects and reduced proliferation and tumor burden in the CLL xenograft mouse model
Given the comparable efficacy but increased specificity of acalabrutinib compared to ibrutinib, we next sought to determine the effect of acalabrutinib treatment in an in vivo setting. We first used the CLL xenograft mouse model where primary CLL cells were engrafted into NSG mice. Briefly, mice were given either vehicle or acalabrutinib via drinking water on from day −1, at the time of busulfan priming, and CLL MNCs were injected on day 0. Three weeks post engraftment, all mice were sacrificed and whole blood and splenocytes were collected. We first evaluated the change in key proteins in the BCR signaling pathway ex vivo in CLL cells collected from NSG mice spleens (the site of the highest BCR activation (27)). We found a strong and significant reduction in the phosphorylation of PLCγ2 (P=0.01, median change −68%, IQR −74 to −41, Figure 3A–B) in mice treated with acalabrutinib compared to vehicle. Similarly, we evaluated the phosphorylation of ERK (a protein regulated by both the BCR and other key microenvironmental factors) and again found a significant inhibition in phosphorylation in acalabrutinib treated mice compared to vehicle (P=0.02, median change −79%, IQR −90 to −48, Figure 3C). Together these data demonstrate the pharmacodynamic impact of acalabrutinib on CLL cells in the tissues (spleen) where CLL is propagated.
Figure 3. Acalabrutinib demonstrates on target effects and reduced proliferation and tumor burden in the CLL xenograft mouse model.
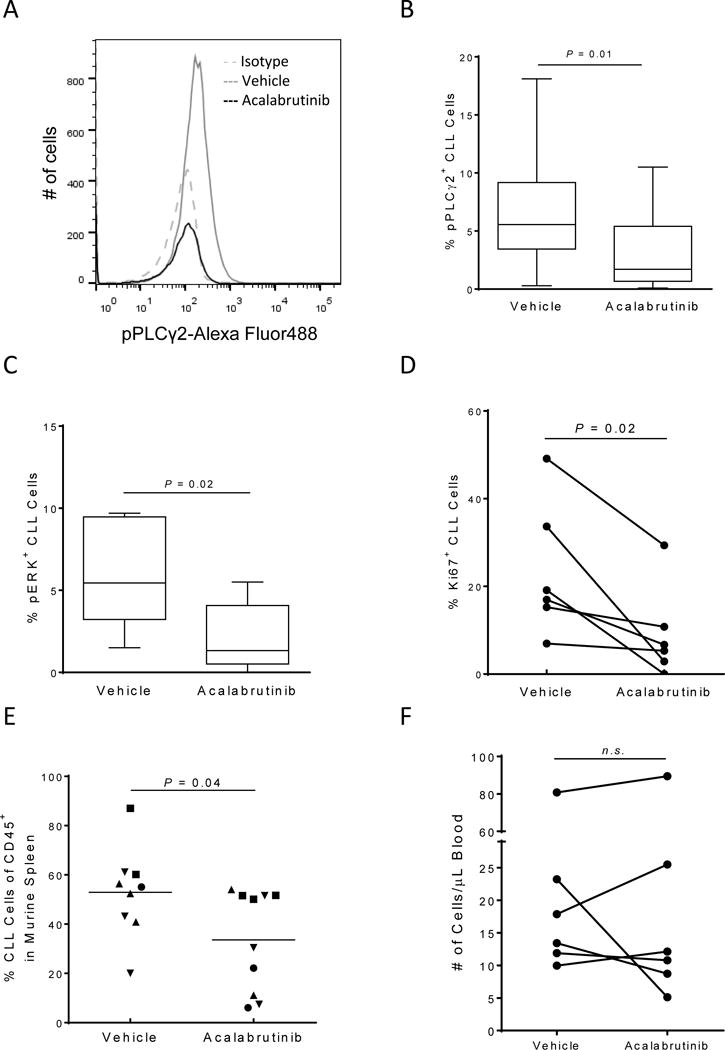
CLL MNCs (n=6) harvested from NSG mouse spleens (n=2–5 per patient) after 3 weeks of treatment were fixed, permeabilized, and stained with the indicated antibodies. Results shown are for the CLL population. (A) A representative histogram showing pPLCγ2 expression in a mouse treated with vehicle (solid grey line) compared to a mouse treated with acalabrutinib (solid black line). The dashed grey line represents the isotype control. (B-C) The median (± IQR) percent of pPLCγ2 (B) and pERK (C) is shown in a min to max box and whisker plot. (D) Percentage of CLL cells expressing KI67 is shown per patient for each treatment group. (E) Percentage of CLL cells among human CD45+ cells in the spleen. Each data point represents one mouse; symbols identify patients. (F) The absolute human CLL cell (CD45+/CD19+/CD5+) count in vehicle and acalabrutinib-treated mice per μl blood is shown. Data points represent the average measurements of 2–5 mice injected with MNCs from the same patient. All statistics were determined by paired student t-test.
Next, we evaluated the anti-leukemic effect of acalabrutinib. We measured the expression of the proliferation marker Ki67 in harvested human CLL splenocytes from xenografted NSG mice. CLL cell proliferation was significantly inhibited in mice receiving acalabrutinib compared to vehicle (P=0.02, median change −50%, IQR −94 to −28, Figure 3D). Concurrently, we evaluated the tumor burden in the spleen by determining the proportion of CLL cells among all human cells of the harvested splenocytes. Acalabrutinib significantly reduced the number of human CLL cells in the murine spleen (P=0.04, median change −33%, IQR −64 to −29, Figure 3E). In a subset of patients we compared the anti-leukemic effect of acalabrutinib to ibrutinib in independent experiments. We found that acalabrutinib and ibrutinib demonstrated similar reductions in both Ki67 and splenic tumor burden (Supplementary Figure S5A–B). This further suggests that the anti-tumor effect of these agents is predominately through inhibition of BTK.
BCR-directed kinase inhibitors in patients with CLL characteristically induce a transient increase in lymphocytosis due to mobilization of cells out of the tissue microenvironment into the blood.(32, 33) In the NSG primary CLL xenograft model and in a modified adoptive transfer model using cells from TCL1 transgenic mice, ibrutinib caused a transient increase in the number of circulating tumor cells.(27, 34) We therefore sought to determine if acalabrutinib treatment would also lead to treatment-induced lymphocytosis. Using the whole blood collected after three weeks of treatment we compared CLL blood counts between vehicle and acalabrutinib treated mice. Across all experiments, no significant increase in the number of cells per μL of blood was observed (P=0.83, median change +0.76%, IQR −46% to +27%, Figure 3F). However, there was considerable inter-patient variability; with xenografted MNCs from half of the patients we evaluated demonstrating a decrease in the circulating CLL cell count in mice treated with acalabrutinib compared to vehicle, while in the other half we observed an increase in circulating cells on acalabrutinib. Together these data suggest treatment-induced lymphocytosis may be less pronounced after initiation of acalabrutinib than ibrutinib, which is in agreement with early reports in patients treated with acalabrutinib.(28)
Acalabrutinib inhibits BCR signaling and increases survival in the aggressive TCL1 model
Having demonstrated reductions in BCR signaling and anti-leukemic activity in the primary CLL xenograft model, we sought to evaluate acalabrutinib in the more aggressive TCL1 adoptive transfer model. Briefly, leukemic Eμ-TCL1 cells were transplanted into C57BL/6 mice and drug treatment was initiated after recipient mice had a tumor burden (CD5+/CD19+ cells) of ≥10% in the peripheral blood. Mice received either vehicle or acalabrutinib continually via drinking water. We first evaluated the occupancy levels in the TCL1 adoptive transfer model after either one or four weeks of treatment. Drug occupancy of murine BTK was found to be ≥90% with median occupancy of 96% and 97% after one or four weeks of treatment, respectively (Figure 4A). This indicates that BTK is fully inhibited in this mouse model and remains inhibited over time. We next sought to evaluate the changes in key signaling molecules downstream of BCR as a measure of on-target drug effects. At the indicated time points mice were sacrificed and splenocytes were collected. Leukemic splenocytes were stimulated ex vivo with anti-IgM and the phosphorylation status of BTK and down-stream kinases were evaluated. We found that the auto-phosphorylation of BTK was significantly reduced in mice treated with acalabrutinib compared to vehicle treated mice (P<0.001, median change −31%, IQR −42 to −21, Figure 4B). Similarly, the phosphorylation of PLCγ2 was also significantly reduced in mice treated with acalabrutinib compared to vehicle treated mice (P=0.001, median change −22%, IQR −25 to −16, Figure 4C). Lastly, we evaluated the phosphorylation of S6. We found that acalabrutinib-treated mice had significantly reduced induction of pS6 after anti-IgM stimulation compared to vehicle-treated mice (P<0.001, median change −35%, IQR −40 to −26, Figure 4D). Together this shows that even in an aggressive model of CLL, acalabrutinib treatment greatly reduced BCR-induced signaling.
Figure 4. Acalabrutinib demonstrates significant and sustained inhibition of BCR signaling in the TCL1 adoptive transfer model.
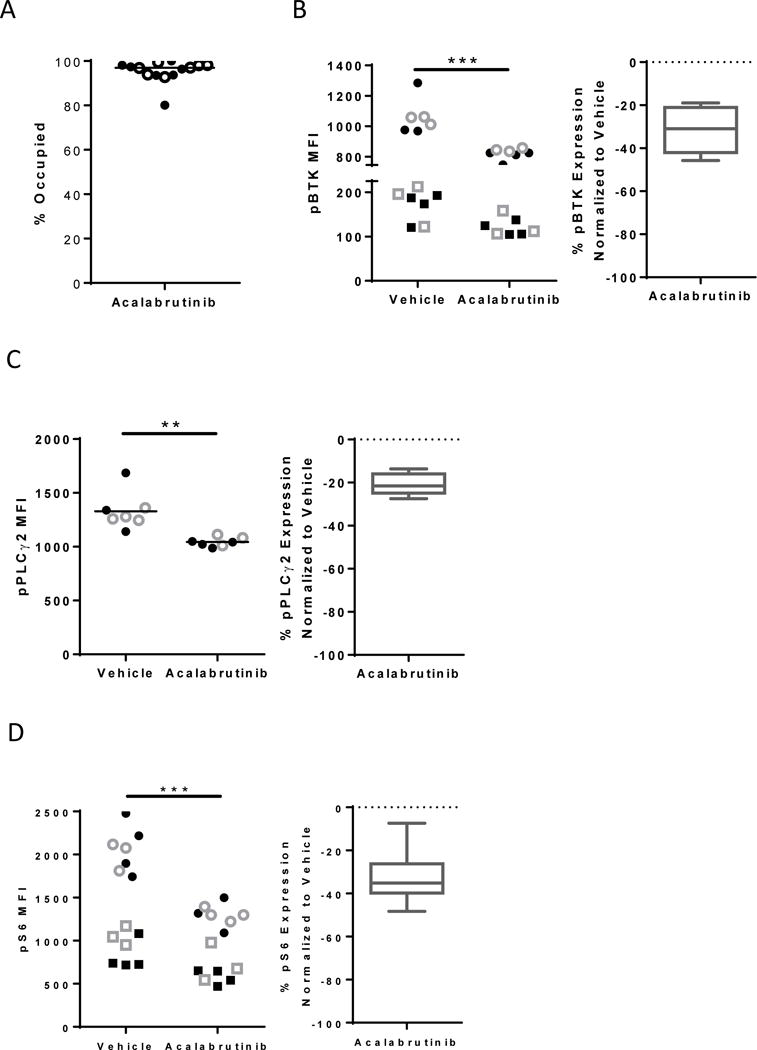
Mice were engrafted with leukemic TCL1 cells and treated with vehicle or acalabrutinib. (A) % occupancy of Btk (n=14 per group) was determined after acalabrutinib treatment. Filled circles represent splenocytes harvested after one week of treatment and open circles represent splenocytes harvested after four weeks of treatment. (B-D) MFI expression (left) or % change in expression compared to vehicle (right) of pBtk (n=15 per group) (B), pPLCγ2 (n=7 per group) (C) and pS6 (n=14 per group) (D) evaluated in B-cell population by flow cytometry is shown. Filled symbols represent splenocytes harvested after one week of treatment and open symbols represent splenocytes harvested after four weeks of treatment. Circles and Squares differentiate independent experiments. Box and whisker plots show to min to max values. Asterisks indicate statistical significance as determined by normalized unpaired t-test. **P<0.01 and ***P<0.001.
Finally, we assessed overall survival in leukemic mice treated with acalabrutinib compared to vehicle. Recipient mice with a tumor burden (CD5+/CD19+ cells) of ≥10% were treated with either vehicle or acalabrutinib continually via drinking water and followed until death or predetermined early removal criteria. We found that mice treated with acalabrutinib had significantly improved survival compared with vehicle-treated mice (median 81 vs 59 days, respectively; P=0.02, Figure 5).
Figure 5. Acalabrutinib increases survival in the TCL1 adoptive transfer model compared to vehicle.
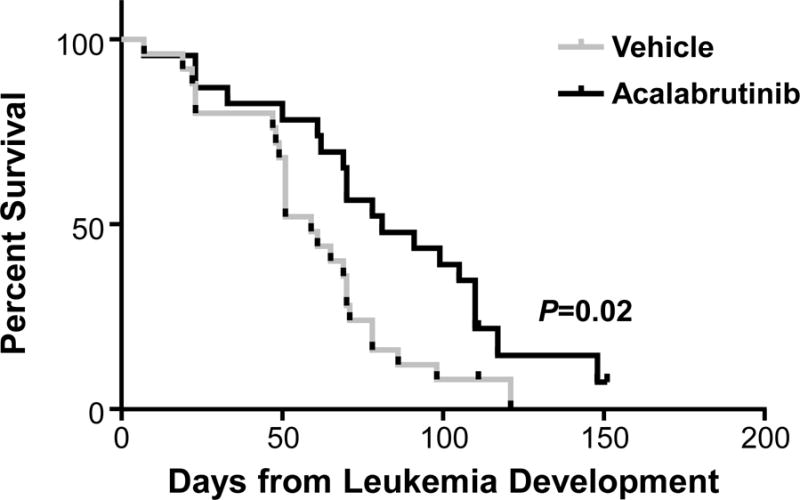
Mice engrafted with leukemic TCL1 cells were treated with vehicle (n=25) or acalabrutinib (n=23). Acalabrutinib increases survival compared to vehicle treatment. Median overall survival was 81 day vs 59 days, respectively, P=0.02.
DISCUSSION
Here we show that the covalent BTK inhibitor, acalabrutinib, is highly selective for BTK and potently inhibits BCR signaling, resulting in a significant anti-tumor response in two established mouse models of CLL. Covalent BTK inhibitors bind Cys481 in the active site of the kinase and restoration of enzyme function requires novel protein synthesis. Despite this shared mechanism of target inhibition there are relevant differences between the different BTK inhibitors. Notably, in addition to BTK, nine kinases have a cysteine residue in a homologous amino acid sequence context and different BTK inhibitors show remarkably different selectivity for these kinases. At concentrations of less than 10 nM acalabrutinib selectively inhibits only BTK, while ibrutinib inhibits eight out of nine additional kinases containing the conserved cysteine residue.(28) The apparently inferior clinical activity of an earlier highly selective BTK inhibitor, spebrutinib (CC-292, AVL-292), raised the question whether inhibition of kinases other than BTK are, in part, responsible for ibrutinib’s anti-tumor efficacy.(35, 36) Here, we report that acalabrutinib is as potent as ibrutinib at inhibiting BTK and pathways downstream, utilizing in vitro and ex vivo assays. Further, acalabrutinib showed potent in vivo activity in two complementary mouse models representing the disease spectrum of primary CLL in the xenograft model and a more aggressive disease in the transgenic mouse model. Thus, potent, highly selective inhibition of BTK seems sufficient to inhibit BCR signaling and induce an anti-tumor response in vivo.
The different kinase selectivity of inhibitors that rely on the same mechanism of action, namely covalent binding of a conserved cysteine homologous to Cys481 in BTK is at first surprising. However, an additional component of potent inhibition of the kinase is the ability of the inhibitor to access the cysteine residue and establish a covalent bond. While the specific determinants of this additional layer of selectivity have to be further investigated, it is likely that overall protein confirmation and ease of access to the active site are important. While acalabrutinib has been demonstrated to be highly selective for BTK, inhibition of BMX, TEC, and ERBB4 were inhibited at <100nM in biochemical kinase assays; albeit much less potently than with ibrutinib (28). Although, we cannot completely rule out contribution of alternative kinases, published data demonstrate that BTK is the critical target of ibrutinib in disease modulation in CLL. First, genetic inactivation of BTK alone when introduced into the TCL1 mouse delays the onset of leukemia and improves survival (7) showing that BTK inactivation is sufficient to alter the phenotype of TCL1-induced CLL. Secondly, a single mutation of C481 in BTK preventing covalent binding of ibrutinib results in relapse in CLL patients. (17). These results confirm the role of BTK as a critical target in the control of CLL.
The activities of acalabrutinib and ibrutinib in vitro and ex vivo were by and large comparable. Also, activity in the murine models described here is similar to the previously published data for ibrutinib.(7, 27, 34) Taken together with early data showing clinical activity of acalabrutinib in patients with relapsed/refractory CLL, that is a least similar to what has been reported with ibrutinib, we conclude that selective inhibition of BTK is sufficient for the anti-tumor response observed in CLL patients. Notably, in patients acalabrutinib can be dosed twice daily and may result in more sustained inhibition of BTK than once daily administration of ibrutinib.(28, 37) In this regard the murine models reflect a continuous inhibition of BTK that, in patients, requires twice daily dosing as has been investigated with acalabrutinib.
One difference in the in vivo models between the two BTK inhibitors was that treatment-induced lymphocytosis in the human xenograft model was less prominent than previously noted with ibrutinib.(27, 34) Interestingly, a less pronounced treatment-induced lymphocytosis has also been reported for patients treated with acalabrutinib.(28) While mobilization of CLL from the lymphoid tissue into the blood reflects BTK inhibition(32, 33, 38) only a minor fraction of the tumor is mobilized into the periphery, and most tumor cell death is thought to occur within the tissue.(11, 33, 39) Thus, a blunting of the treatment-induced lymphocytosis may reflect increased apoptosis within the tissue with or without more rapid clearing of dead cells from the periphery. This is consistent with the high degree of nodal responses observed on acalabrutinib.(28) Unfortunately, it has proven difficult to reliably assess the fraction of dying cells in tissues as methods of obtaining single cells have been associated with a high level of background noise.(11, 33)
BTK inhibition appears to be well tolerated clinically, consistent with the observation that BTK loss of function in patients with XLA is associated with a selective deficiency of B-cell maturation and immunoglobulin production.(2) In contrast, some of the side effects observed with ibrutinib are thought to result from inhibition of other kinases such as TEC that might contribute to bleeding complications,(40–42) or EGFR that may contribute to rash and diarrhea.(43) Thus, more selective inhibition of BTK could be associated with a different, more favorable safety profile. Early data from the ongoing first phase 1–2 study of acalabrutinib in CLL indeed suggests a lower rate of rash and bleeding-related adverse events than previously observed with ibrutinib.(28) On the other hand some of the alternative targets of ibrutinib may be contributing to favorable effects of this agent; inhibition of ITK reportedly shifts CD4+ T-cells to the Th1 phenotype which could contribute to improvements in immune function noted for ibrutinib,(44, 45) and inhibition of TEC may inhibit interactions of CLL cells with tumor associated macrophages.(46) It remains to be seen whether these alternative targets are clinically important in CLL.
In summary, our data in vitro, ex vivo, and in vivo using the human xenograft model suggests that highly specific BTK inhibition is effective in disrupting BCR signaling, inhibits tumor proliferation and reduces tumor burden and significantly extends the survival of mice with adoptively transferred TCL1 leukemic cells similar to what has previously been shown with ibrutinib.(7, 27) Differences in specificity between BTK inhibitors may also prove to be important in the search for clinical combination regimens, as differences in the terms of synergy and adverse events due to off target effects may appear. Comparative studies will be needed to fully appreciate differences in potency, clinical efficacy and adverse event profiles between different BTK inhibitors; one such study has recently been initiated (NCT02477696).
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
B-cell Receptor (BCR)-directed kinase inhibitors are changing treatment paradigms of chronic lymphocytic leukemia (CLL). With the first-in-class BTK inhibitor ibrutinib, durable responses are common but complete responses are relatively rare, and some patients develop resistance. Additionally, adverse effects and impaired NK-cell and platelet function, with implications for combination therapy and safety, have been attributed to inhibition of kinases other than BTK. Conversely, it is unclear whether inhibition of additional kinases contributes to clinical efficacy of ibrutinib. We therefore investigated the novel, highly selective BTK inhibitor, acalabrutinib, in vivo, utilizing two established mouse models of CLL. We show that BCR signaling and proliferation of tumor cells is effectively inhibited in mice treated with acalabrutinib. Acalabrutinib also reduced tumor burden and increased survival compared to vehicle treated mice. Thus, while showing better overall kinase selectivity, acalabrutinib exerts potent anti-tumor effects on par with ibrutinib, supporting the critical role of BTK in CLL.
Acknowledgments
We thank our patients for participating and donating the blood and tissue samples to make this research possible. This work was supported by the Intramural Research Program the National, Heart, Lung and Blood Institute, the National Institutes of Health, R35 CA197734, R01 CA197870, K23 CA178183, R01 CA177292 and Acerta Pharma. A.M. was recipient of a pre-doctoral internship grant FPI from Spanish Ministry of Economy and Competitiveness & European Regional Development Fund (ERDF).
Research support: This research was supported by the Intramural Research Program of the National, Heart, Lung and Blood Institute, R35 CA197734, R01 CA197870, K23 CA178183, R01 CA177292 and Acerta Pharma.
Footnotes
Conflicts of Interest: M.G., F.K., R.I., T.C., A.K, R.U and B.L. are employees and equity holders of Acerta Pharma. J.B. A.J. and J.W. received research funding from Acerta Pharma. R.U. is an officer, equity holder and member of the board of directors of Acerta Pharma. A.W. received research funding from Pharmacylics and Acerta Pharma. C.U.N received funding from the Danish Cancer Society and has received travel grants/consultancy fees from Abbvie, Janssen, Gilead, Roche and Novartis. All other authors declare no competing financial interests.
AUTHORS’ CONTRIBUTIONS
Concept and design: S.H., C.N., D.C., T.C., A.K. B.L., J.B., A.W., A.J. and J.W.
Development of methodology: S.H., A.K., C.N., T.C., R.I., R.U., A.H., B.L., A.W., A.J. and J.W.
Acquisition of data: A.M., H.M-J., M.G., F.K., F.M., B.H., R.M., L.S. and T.C.
Analysis and interpretation of data: S.H., A.M., C.N., H.M-J and T.C
Writing, review and/or revision of manuscript: S.H., A.M., C.N., H.M-J., M.G., F.K., R.M., L.S., F.M., B.H., D.C., T.C., J.B., R.I., A.K., R.U., A.J., B.L., A.W. and J.W.
References
- 1.Buggy JJ, Elias L. Bruton Tyrosine Kinase (BTK) and Its Role in B-cell Malignancy. Int Rev Immunol. 2012;31:119–32. doi: 10.3109/08830185.2012.664797. [DOI] [PubMed] [Google Scholar]
- 2.Hendriks RW, Bredius RG, Pike-Overzet K, Staal FJ. Biology and novel treatment options for XLA, the most common monogenetic immunodeficiency in man. Expert Opin Ther Targets. 2011;15:1003–21. doi: 10.1517/14728222.2011.585971. [DOI] [PubMed] [Google Scholar]
- 3.Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117:6287–96. doi: 10.1182/blood-2011-01-328484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hendriks RW, Yuvaraj S, Kil LP. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat Rev Cancer. 2014;14:219–32. doi: 10.1038/nrc3702. [DOI] [PubMed] [Google Scholar]
- 5.Wiestner A. Targeting B-Cell receptor signaling for anticancer therapy: the Bruton’s tyrosine kinase inhibitor ibrutinib induces impressive responses in B-cell malignancies. J Clin Oncol. 2013;31:128–30. doi: 10.1200/JCO.2012.44.4281. [DOI] [PubMed] [Google Scholar]
- 6.Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woyach JA, Bojnik E, Ruppert AS, Stefanovski MR, Goettl VM, Smucker KA, et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL) Blood. 2014;123:1207–13. doi: 10.1182/blood-2013-07-515361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kil LP, de Bruijn MJ, van Hulst JA, Langerak AW, Yuvaraj S, Hendriks RW. Bruton’s tyrosine kinase mediated signaling enhances leukemogenesis in a mouse model for chronic lymphocytic leukemia. American journal of blood research. 2013;3:71–83. [PMC free article] [PubMed] [Google Scholar]
- 9.Wiestner A. The role of B-cell receptor inhibitors in the treatment of patients with chronic lymphocytic leukemia. Haematologica. 2015;100:1495–507. doi: 10.3324/haematol.2014.119123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117:563–74. doi: 10.1182/blood-2010-05-284984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herman SE, Mustafa RZ, Gyamfi JA, Pittaluga S, Chang S, Chang B, et al. Ibrutinib inhibits BCR and NF-kappaB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. 2014;123:3286–95. doi: 10.1182/blood-2014-02-548610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus Ofatumumab in Previously Treated Chronic Lymphoid Leukemia. N Engl J Med. 2014;371:213–23. doi: 10.1056/NEJMoa1400376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Coleman M, et al. Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125:2497–506. doi: 10.1182/blood-2014-10-606038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farooqui MZ, Valdez J, Martyr S, Aue G, Saba N, Niemann CU, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16:169–76. doi: 10.1016/S1470-2045(14)71182-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Claro RA, McGinn KM, Verdun N, Lee SL, Chiu HJ, Saber H, et al. FDA Approval: Ibrutinib for Patients with Previously Treated Mantle Cell Lymphoma and Previously Treated Chronic Lymphocytic Leukemia. Clin Cancer Res. 2015;21:3586–90. doi: 10.1158/1078-0432.CCR-14-2225. [DOI] [PubMed] [Google Scholar]
- 16.Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A, et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014;370:2352–4. doi: 10.1056/NEJMc1402716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–94. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burger JA, Buggy JJ. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) Leuk Lymphoma. 2013;54:2385–91. doi: 10.3109/10428194.2013.777837. [DOI] [PubMed] [Google Scholar]
- 19.Borge M, Belen Almejun M, Podaza E, Colado A, Fernandez Grecco H, Cabrejo M, et al. Ibrutinib impairs the phagocytosis of rituximab-coated leukemic cells from chronic lymphocytic leukemia patients by human macrophages. Haematologica. 2015;100:e140–2. doi: 10.3324/haematol.2014.119669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Da Roit F, Engelberts PJ, Taylor RP, Breij EC, Gritti G, Rambaldi A, et al. Ibrutinib interferes with the cell-mediated anti-tumor activities of therapeutic CD20 antibodies: implications for combination therapy. Haematologica. 2015;100:77–86. doi: 10.3324/haematol.2014.107011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohrt HE, Sagiv-Barfi I, Rafiq S, Herman SE, Butchar JP, Cheney C, et al. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood. 2014;123:1957–60. doi: 10.1182/blood-2014-01-547869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skarzynski M, Niemann CU, Lee YS, Martyr S, Maric I, Salem D, et al. Interactions between Ibrutinib and Anti-CD20 Antibodies: Competing Effects on the Outcome of Combination Therapy. Clin Cancer Res. 2015 doi: 10.1158/1078-0432.CCR-15-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caligaris-Cappio F, Bertilaccio MT, Scielzo C. How the microenvironment wires the natural history of chronic lymphocytic leukemia. Semin Cancer Biol. 2014;24:43–8. doi: 10.1016/j.semcancer.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 24.Herishanu Y, Katz BZ, Lipsky A, Wiestner A. Biology of chronic lymphocytic leukemia in different microenvironments: clinical and therapeutic implications. Hematol Oncol Clin North Am. 2013;27:173–206. doi: 10.1016/j.hoc.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A. 2002;99:6955–60. doi: 10.1073/pnas.102181599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson AJ, Lucas DM, Muthusamy N, Smith LL, Edwards RB, De Lay MD, et al. Characterization of the TCL-1 transgenic mouse as a preclinical drug development tool for human chronic lymphocytic leukemia. Blood. 2006;108:1334–8. doi: 10.1182/blood-2005-12-011213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herman SE, Sun X, McAuley EM, Hsieh MM, Pittaluga S, Raffeld M, et al. Modeling tumor-host interactions of chronic lymphocytic leukemia in xenografted mice to study tumor biology and evaluate targeted therapy. Leukemia. 2013;27:1769–73. doi: 10.1038/leu.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2015 doi: 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fabian MA, Biggs WH, 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–36. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 30.Herman SE, Barr PM, McAuley EM, Liu D, Wiestner A, Friedberg JW. Fostamatinib inhibits B-cell receptor signaling, cellular activation and tumor proliferation in patients with relapsed and refractory chronic lymphocytic leukemia. Leukemia. 2013;27:1769–73. doi: 10.1038/leu.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107:13075–80. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012;119:2590–4. doi: 10.1182/blood-2011-11-390989. [DOI] [PubMed] [Google Scholar]
- 33.Herman SE, Niemann CU, Farooqui M, Jones J, Mustafa RZ, Lipsky A, et al. Ibrutinib-induced lymphocytosis in patients with chronic lymphocytic leukemia: correlative analyses from a phase II study. Leukemia. 2014;28:2188–96. doi: 10.1038/leu.2014.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182–9. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346:219–28. doi: 10.1124/jpet.113.203489. [DOI] [PubMed] [Google Scholar]
- 36.Brown JR, Harb WA, Hill BT, Gabrilove J, Sharman JP, Schreeder MT, et al. Phase 1 Study Of Single Agent CC-292, a Highly Selective Bruton’s Tyrosine Kinase (BTK) Inhibitor, In Relapsed/Refractory Chronic Lymphocytic Leukemia (CLL) Blood. 2013;122:1630. [Google Scholar]
- 37.Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31:88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herman SE, Mustafa RZ, Jones J, Wong DH, Farooqui M, Wiestner A. Treatment with Ibrutinib Inhibits BTK- and VLA-4-Dependent Adhesion of Chronic Lymphocytic Leukemia Cells In Vivo. Clin Cancer Res. 2015;21:4642–51. doi: 10.1158/1078-0432.CCR-15-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wodarz D, Garg N, Komarova NL, Benjamini O, Keating MJ, Wierda WG, et al. Kinetics of CLL cells in tissues and blood during therapy with the BTK inhibitor ibrutinib. Blood. 2014;123:4132–5. doi: 10.1182/blood-2014-02-554220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamel S, Horton L, Ysebaert L, Levade M, Burbury K, Tan S, et al. Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia. 2015;29:783–7. doi: 10.1038/leu.2014.247. [DOI] [PubMed] [Google Scholar]
- 41.Levade M, David E, Garcia C, Laurent PA, Cadot S, Michallet AS, et al. Ibrutinib treatment affects collagen and von Willebrand factor-dependent platelet functions. Blood. 2014;124:3991–5. doi: 10.1182/blood-2014-06-583294. [DOI] [PubMed] [Google Scholar]
- 42.Lipsky AH, Farooqui MZ, Tian X, Martyr S, Cullinane AM, Nghiem K, et al. Incidence and risk factors of bleeding-related adverse events in patients with chronic lymphocytic leukemia treated with ibrutinib. Haematologica. 2015;100:1571–8. doi: 10.3324/haematol.2015.126672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melosky B, Hirsh V. Management of Common Toxicities in Metastatic NSCLC Related to Anti-Lung Cancer Therapies with EGFR-TKIs. Front Oncol. 2014;4:238. doi: 10.3389/fonc.2014.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539–49. doi: 10.1182/blood-2013-06-507947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun C, Tian X, Lee YS, Gunti S, Lipsky A, Herman SE, et al. Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood. 2015;126:2213–9. doi: 10.1182/blood-2015-04-639203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niemann CU, Herman SE, Maric I, Gomez-Rodriguez J, Biancotto A, Chang BY, et al. Disruption of in vivo chronic lymphocytic leukemia tumor-microenvironment interactions by ibrutinib - findings from an investigator initiated phase 2 study. Clin Cancer Res. 2015 doi: 10.1158/1078-0432.CCR-15-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.