

Abstract
Acute on chronic liver failure (ACLF) was first described in 1995 as a clinical syndrome distinct to classic acute decompensation. Characterized by complications of decompensation, ACLF occurs on a background of chronic liver dysfunction and is associated with high rates of organ failure and significant short-term mortality estimated between 45% and 90%. Despite the clinical relevance of the condition, it still remains largely undefined with continued disagreement regarding its precise etiological factors, clinical course, prognostic criteria and management pathways. It is concerning that, despite our relative lack of understanding of the condition, the burden of ACLF among cirrhotic patients remains significant with an estimated prevalence of 30.9%. This paper highlights our current understanding of ACLF, including its etiology, diagnostic and prognostic criteria and pathophysiology. It is evident that further refinement of the ACLF classification system is required in order to detect high-risk patients and improve short-term mortality rates. The field of metabolomics certainly warrants investigation to enhance diagnostic and prognostic parameters, while the use of granulocyte-colony stimulating factor is a promising future therapeutic intervention for patients with ACLF.
Keywords: acute liver failure, acute decompensation of cirrhosis, hepatorenal syndrome, chronic, hepatic encephalopathy, systemic inflammation
Introduction
Patients hospitalization for acute complications of liver cirrhosis, who also suffer from hepatic or extrahepatic organ failure (s), are at risk of high short-term mortality and are described as having acute-on-chronic liver failure (ACLF)[1–2]. Cirrhosis occurs due to chronic disease progression and loss of hepatic function; however, the condition is commonly asymptomatic until an acute episode of decompensation. Acute decompensation occurs due to complications of portal hypertension and hepatic dysfunction, presenting as variceal bleeding, infection, hepatic encephalopathy and ascites. With recent improvements in the medical management of acute decompensation, many patients are able to return to a compensated state. However, a proportion of patients suffer from significant pathological sequelae, characterized by hepatic and/or extrahepatic organ failure or multi-organ failure requiring management in intensive care and significant life support, such as extracorporeal membrane oxygenation.
As a clinical entity, the term ACLF was first coined in 1995 and described as an acute and rapid decline in liver function in patients with existing chronic liver disease, characterized by complications of decompensated liver failure, high rates of organ failure and significant short-term mortality[3]. ACLF is now considered a separate clinical entity to classic decompensated liver cirrhosis due to the presence of organ failure and systemic inflammation, a higher rate of mortality and specific precipitating factors, as well as its demographic; commonly a younger patient population with an alcoholic etiology of cirrhosis[4–5]. The condition may occur as a result of extrahepatic precipitating factors, such as infection, on a background of superimposed chronic liver dysfunction[4]. While the precise pathophysiology of ACLF remains to be elucidated, amplified and unopposed inflammation seems to play a vital role[6–7]. Due to profound inflammation that occurs with ACLF, as well as its rapid progression, multiple organ supportive therapy is often required and is associated with a short-term mortality rate of 45%-90%[4]. Although the underlying cirrhosis in ACLF remains irreversible, the condition itself is thought to possess a reversible component, as it is often associated with a specific precipitating factor[8]. There is currently a scarcity of data regarding the epidemiology of ACLF; however, the high mortality rates, prolonged periods of hospitalization and the profound burden on healthcare systems associated with the condition demonstrate the importance in improving our understanding of the condition.
Definitions
Due to the overall ambiguity of ACLF, its exact definition still remains undecided. However, two definitions have been proposed in the last decade, predominantly based on expert judgements, rather than evidence-based data[9]. Due to distinct clinical and pathophysiological differences noted by academic communities in different geographic locations worldwide, it has been difficult to construct a universally acceptable definition that encompasses these variations. The first definition was proposed by the Asia-Pacific Association for the Study of the Liver (APASL); "Acute hepatic insult manifesting as jaundice and coagulopathy, complicated within 4 weeks by ascites and/or encephalopathy in a patient with previously diagnosed or undiagnosed chronic liver disease"[10]. On the other hand, both The American Association for the Study of Liver Disease (AASLD) and European Association for the Study of the Liver (EASL) define ACLF as the "Acute deterioration of pre-existing, chronic liver disease, usually related to a precipitating event and associated with increased mortality at 3 months due to multi-system organ failure"[11]. The definitions of ACLF differ based on geographic location, emphasizing the etiological heterogeneity of the condition in Eastern and Western countries. In addition, it is difficult to produce homogenous diagnostic criteria for ACLF due to the imprecise nature of both definitions. The latter definition suggests that the presence of organ failure is a key component of ACLF, indicating that organs behave differently in ACLF compared to classic decompensated liver disease. This review will utilize the second definition unless otherwise stated.
Due to disagreement among researchers regarding the definition of ACLF, the EASL-Chronic Liver Failure (EASL-CLIF) Consortium recently conducted a large observational study entitled the EASL-CLIF Acute-on-Chronic Liver Failure in Cirrhosis (CANONIC) study[12]. The North American Consortium for the Study of End-Stage Liver Disease (NACSELD), a similar large observational study, was also conducted to try to harmonize the various definitions of ACLF and improve our understanding of the condition.
The CANONIC study was designed to produce a definition of ACLF that can determine patients with liver dysfunction that are at high risk of short-term mortality, defined as a 28-day mortality of more than 15%. The results of the multi-center CANONIC study, based on the clinical evaluation of 1343 patients with pre-existing cirrhosis and acute decompensation (complicated by variceal bleeding, ascites, bacterial infection of hepatic encephalopathy), revealed that ACLF is very common (30.9% prevalence) and is distinct from typical acute decompensation. This is not simply due to the higher mortality rate and organ failure associated with ACLF, but also based on precipitating factors, age and the presence of acute systemic inflammation.
Clinical and prognostic factors
There are limited data regarding the outcomes of patients with cirrhosis progressing toward multi-organ failure. However, recent studies have attempted to ascertain specific clinical and prognostic factors to predict short and long-term patient outcomes. A prospective study was conducted at University College London (UCL) that followed 500 patients with varying severities of cirrhosis that were admitted to a single liver intensive care unit over a period of 5.5 years and received treatment according to pre-defined management criteria[5]. The study found that one third of all patients developed a single organ failure, of whom over 50% died during their first hospital admission. These results suggest that approximately half the cases of single organ dysfunction are reversible. However, it has also been found that patients who improve in intensive care following organ failure and are subsequently discharged possess a three-year mortality of almost 100%, implying that organ failure also has the ability to change the predicted progression of cirrhosis[13].
The CANONIC study produced similar results, demonstrating that approximately 50% of patients presenting with ACLF improve and return to a compensated state, while 20% deteriorate; clinical deterioration of ACLF is associated with a mortality of<50%. According to the study, the main factor that contributes to a patient's clinical improvement (55%) is the presence of only one failing extrahepatic organ, with deterioration more likely with increasing organ failure. In addition, early recognition of ACLF and initial aggressive management is a key factor in determining patient survival and prognosis.
Although there are several prognostic factors for patients with hepatic dysfunction developing organ failure, prognostic indicators predicting the outcome of cirrhotic patients with organ failure remain to be elucidated. In clinical practice, there are two primary types of prognostic model; those that assess the severity of hepatic dysfunction and those that evaluate single or multi-organ dysfunction. As the clinical prognosis of patients with decompensation is primarily determined by organ dysfunction rather than severity of liver disease, liver scoring criteria possess inherent restrictions as prognostic indicators of clinical outcome in patients with ACLF. Classic liver scoring criteria that focus on the severity of liver dysfunction, such as Child-Pugh score or Model of End Stage Liver Disease (MELD), have been shown to be less effective at addressing the outcome of patients with ACLF than the Sequential Organ Failure Assessment (SOFA) score that focuses on organ failure[14] The SOFA score has been found to be a significantly better predictor of short-term prognosis in patients with ACLF than liver-specific scores[15–17].
Child-Pugh score was originally designed to predict mortality during surgery but is currently used to assess the prognosis of patients with chronic liver disease and ascertain the necessity for liver transplantation. The score employs five clinical measures of liver disease: total bilirubin, serum albumin, prothrombin time, presence of ascites and hepatic encephalopathy. In terms of liver transplantation for patients with ACLF, Child-Pugh score is not sensitive enough to be utilized as an exclusive prognostic scoring system in this subset of patients. When used in combination with MELD score, its sensitivity is enhanced and is more sensitive in predicting patients that will benefit from definitive transplantation.
MELD score was initially designed to predict mortality within three months of surgery in patients undergoing a transjugular intrahepatic portosystemic shunt (TIPS) procedure, but has since been found to be an important tool in determining patient prognosis and priority for receipt of a liver transplant. The scoring system incorporates three widely available laboratory variables, namely international normalized ratio (INR), serum creatinine, and serum bilirubin. MELD score has been shown to be a valid predictor of long-term survival in patients with cirrhosis, as well as other areas beyond end-stage liver disease such as variceal bleeding, fulminant hepatic failure, alcoholic hepatitis and hepatocellular carcinoma[18–22] However, its role as a prognostic scoring system for patients with ACLF is still disputed. Various studies have found that MELD score alone cannot predict the postoperative mortality or morbidity for patients with ACLF, indicating the necessity for an effective scoring system to determine which patients can benefit from liver transplantation[23–26] Despite this, evidence exists to suggest that MELD score may have some utility as a prognostic marker in ACLF. A Chinese modified MELD was found to reveal poor prognosis in pre-ACLF patients[27] The prognostic model identified 5 independent factors associated with survival among patients with pre-ACLF and ACLF: MELD score, age, hepatic encephalopathy, triglyceride level and platelet count. Therefore, it is possible that broadening the diagnostic criteria of ACLF may enable implementation of a novel model to predict ACLF-related mortality following comprehensive medical therapy. In addition, change in MELD score at 2 weeks may provide an early opportunity for prognostication in ACLF, as suggested by the finding that a MELD score that does not deteriorate by week 2 predicts a 93.8% chance of survival for the next 60 days[28]
The SOFA scoring system was adopted by the CANONIC study to produce the CLIF-SOFA score (later simplified to the CLIF Consortium [CLIF-C] organ failure score). According to a prognostic model to predict survival, described by Jalan et al. (2014), the CLIF-C organ failure score, white cell count and age are all independent predictors of mortality[24]. These factors were used to produce a new, more accurate scoring system entitled the CLIF-C ACLF score. The CLIF-C ACLF score was compared to the traditional systems for assessing organ allocation in liver transplantation, namely MELD and Child-Pugh scores. Similarly to the SOFA scoring system, it was found that the CLIF-C ACLF score was also significantly more accurate than the traditional models. The MELD and Child-Pugh systems were found to underestimate the risk of mortality in ACLF, while the CLIF-C ACLF score, which measures both hepatic and extrahepatic organ dysfunction, was found to discriminate between survivors and non-survivors significantly more accurately. It could, therefore, be suggested that, while MELD and Child-Pugh scores may be useful to assess patients' eligibility for transplantation, they do not have the accuracy, precision or prognostic value to act as the only systems to evaluate the fruitlessness of continued care[29].
While there are clear determinants of poor prognosis in cirrhotic patients, including the severity of liver dysfunction and presence of organ failure, mortality rates continue to vary widely; this is probably due to the heterogeneous nature of liver ICU admission criteria across different hospitals. Interestingly, however, a prospective study from India found that patients possessed a 30- and 90-day mortality of 50% and 63% respectively, statistics that are comparable to those in Western literature[30–31]. Due to the various interacting intrinsic and extrinsic factors in the pathophysiology of ACLF, the ideal system to identify cirrhotic patients who are at high risk of developing ACLF is yet to be defined.
Pathophysiology
The exact pathophysiology of ACLF is yet to be fully understood, however there are several features that are thought to play vital roles, including infection, systemic inflammation and an altered host response to injury[32]. A notion similar to the predisposition, injury, response and organ failure (PIRO) concept in sepsis has been developed in order to deconstruct the characteristic inflammatory response during ACLF (Fig. 1)[32–34]. Originally proposed in 1900, the PIRO score was designed to measure the clinical features and outcomes of patients with sepsis[34]. Despite this, it is also useful in interpreting the clinical progression of ACLF.
Fig.1.
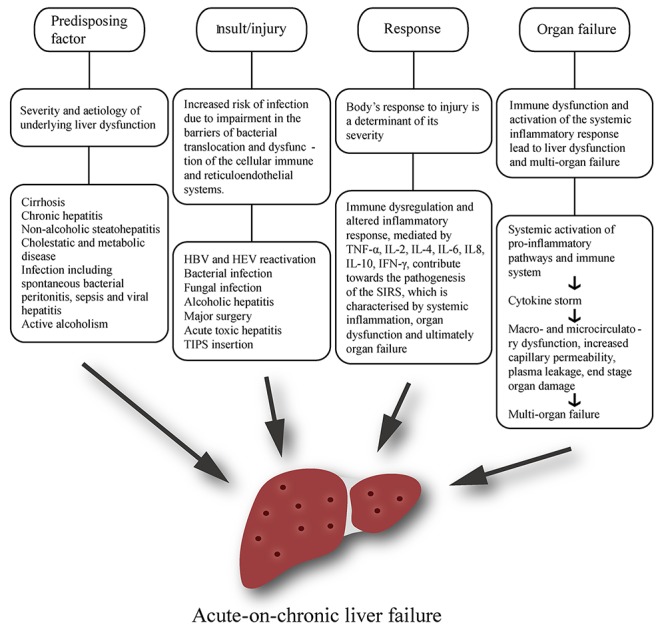
Pathophysiology of acute-on-chronic liver failure (ACLF).
Predisposing factors
The primary predisposing factor for ACLF is the severity and etiology of underlying liver dysfunction. Most underlying disease in ACLF is attributed to compensated cirrhosis of any etiology, while chronic hepatitis, non-alcoholic steatohepatitis and cholestatic and metabolic liver disease also qualify as precipitating causes that exaggerate underlying liver dysfunction. Steatosis, however, is not acknowledged as an underlying factor[10]. In the West, alcoholic cirrhosis is responsible for 45-70% of all predisposing liver diseases in patients with ACLF, while hepatitis-related cirrhosis constitutes for 10-30%[5,10,34–35]. In most Asian countries the etiological factors are reversed, with chronic hepatitis B infection accounting for 70% of cases and only 15% related to alcohol[10].
In a study of 102 patients with ACLF, the most common precipitating factor of ACLF was infection, which was observed in 53% of cases. Spontaneous bacterial peritonitis (SBP) and sepsis accounted for 47% of these cases, while viral hepatitis was seen in the remaining 6%[36].
The CANONIC study indicates that one of the primary predisposing factors in the pathophysiology of ACLF is active alcoholism during the previous three months, as demonstrated by the finding that alcohol consumption is present in 24.5% of patients with ACLF[12]. This suggests that ACLF may be more frequent in patients with alcoholic cirrhosis due to an overrepresentation of severe alcoholic hepatitis. The results of the CANONIC study provide an intriguing insight into this hypothesis. Patients with alcoholic cirrhosis and active alcohol consumption suffered from ACLF more frequently than patients with non-alcoholic cirrhosis or alcoholic cirrhosis without active consumption. Patients in this category also had a higher INR, serum bilirubin levels and required corticosteroid treatment more frequently. While ACLF was present in 42% of active drinkers, this group only comprised one third of all patients with alcoholic cirrhosis. In addition, there were no major differences between those with non-alcoholic cirrhosis and those with alcoholic cirrhosis that were not active drinkers. Overall, these findings indicate that an overrepresentation of severe alcoholic cirrhosis does not explain the high prevalence of ACLF in patients with alcoholic cirrhosis. Therefore, in those with alcoholic cirrhosis, severe alcoholic hepatitis is not required for the presence of ACLF.
Insult/injury
Regardless of the etiology, cirrhotic patients are intrinsically at increased risk of infection and sepsis, though the exact mechanism behind the development of this immunosuppression is not fully understood[37]. It is thought that this may be due to impairment in the barriers of bacterial translocation, neutrophil dysfunction and dysfunction of the cellular immune and reticuloendothelial systems; this shall be discussed in more detail[7]. These events occur on the background of an altered pro-inflammatory environment associated with elevated serum cytokine levels[10,38].
The general consensus among Western experts is that precipitating injury may be either hepatic or non-hepatic, while the APASL recommend that the acute primary precipitating event should be hepatic in origin[5,10,35]. It is important to note that this is not always easy to discern. In the Asian subcontinent, HBV reactivation is a significant precipitant of ACLF, while superimposed HEV is a major cause of ACLF in India[39–40]. As well as viral infection, bacterial and fungal infections may also result in bacterial translocation and therefore contribute toward the development of the SIRS.
There is a clear distinction between hepatic-ACLF and extrahepatic-ACLF based on the etiology of precipitating events. Hepatic insults include exacerbation of chronic hepatitis B, superimposed infection with hepatitis A virus (HAV) or hepatitis E virus (HEV), active alcoholism and hepatotoxic drug use, while extrahepatic insults commonly include bacterial infection and upper gastrointestinal hemorrhage. A study conducted by Shiet al. (2015) involving 405 patients with ACLF demonstrated that hepatic-ACLF precipitated by hepatic insults resulted in relatively well-compensated cirrhosis with frequent hepatic and coagulation dysfunction, while extrahepatic-ACLF precipitated by extrahepatic insults was associated with a greater severity of underlying cirrhosis and a higher incidence of remote organ failure, involving the cerebral, renal, circulatory and respiratory systems[41] Although both groups had high short-term mortality (28-day transplant-free mortality: 48.3%vs. 50.7%; P = 0.22), extrahepatic ACLF was associated with a significantly higher 90-day and 1-year mortality (90-day: 58.9% vs. 68.3%, P = 0.035; 1-year: 63.9% vs. 74.6%, P = 0.019).
HBV-induced ACLF commonly develops following either HBV reactivation on a background of chronic HBV infection and chronic liver disease, or following an acute HBV infection in the presence of co-existing chronic liver disease[42] Although the precise pathogenesis involved in HBV reactivation remains to be elucidated, it is thought to occur due to changes in immunological control of viral replication processes and dysfunctional host defenses. It has been found that acute decompensation is more common in patients infected with HBV genotypes B and D, although frequency is comparable in hepatitis B e antigen (HBeAg) positive or negative patients[43] The mechanism of liver injury in HBV-induced ACLF is predominantly due to the presence of an increased number of inflammatory T cells that cross-react with HBeAg and hepatitis B core antigen (HBcAg), resulting in the generation of HBeAg and HBcAg-specific T cells that are immunologically overactive. HBV reactivation also results in a reduced expression of programmed cell death protein 1 (PD-1), a protein that is involved in the dampening of pathogenic CD8+ T cell responses, thus precipitating further hepatic dysfunction[44–45]
In Asia and Africa, acute HEV infection is a leading cause of acute liver failure, with a median incidence of 21%, and is associated with a high mortality[40] In contrast, the prevalence of HEV in precipitating ACLF in the West is unknown due to a lack of routine testing for HEV and a sporadic incidence within the general population. Acute HEV infection results in cell-mediated immune injury and hepatocyte dysfunction, with a profound increase in cytokine production due to activation of type 1 and type 2 T helper cells [46] Regulatory T cells are also thought to have a role in the pathogenesis of HEV-induced ACLF, as evidenced by an increase in the percentage of CD4+CD25+Foxp3+ regulatory T (TREG) cells and CD4+CD25− Foxp3+effector TREG cells, suggesting that regulatory T cells are involved in hepatic injury and recovery[47] The TREG cell pool is depleted during HAV infection due to Fas-mediated cellular apoptosis, resulting in severe liver injury[48] As well as acute HEV infection, super-infection with HAV also results in the development of ACLF[49]
It is important to note that the study by EASL-CLIF demonstrated that, in a large number of ACLF cases, a causative agent cannot be identified, with no acute precipitating event recognized in 43% of patients with ACLF[12]. In fact, interestingly, even in the cases where a precipitating event could be identified, it was unrelated to the severity of ACLF or immediate- or short-term mortality[12]. This was demonstrated in the CANONIC study, where 23.2% of patients developed ACLF with no past incidents of acute decompensation (hepatic encephalopathy, ascites, variceal hemorrhage)[12]. Therefore, the ability to predict decompensation in the absence of classic precipitating factors through the development of novel biomarkers is necessary to identify individuals at high risk of ACLF[50].
In terms of non-infective precipitants, alcoholic hepatitis is both a common precipitant and predisposing factor of ACLF, particularly in the West, while other less frequent precipitants (8%) include major surgery, acute toxic hepatitis and TIPS insertion[12]. Although the dose and duration of alcohol consumption is important in determining the severity of background chronic liver disease, recent alcohol intake is the key contributing factor in the development of acute liver failure[51] Liver dysfunction occurs due to ethanol-induced hepatotoxicity, increased apoptosis, activation of the innate and adaptive immune systems, and impairment of hepatic regeneration[52] The liver undergoes further injury due to release of endotoxins to the liver as a result of ethanol-mediated gut dysbiosis and an increase in intestinal permeability. Profound hepatocyte apoptosis occurs following the metabolism of ethanol and the subsequent generation of reactive oxygen species, which results in excess mitochrondrial endoplasmic reticulum stress[53] Various other processes are involved in hepatocyte cell death, including the inhibition of survival genes (Met), induction of pro-apoptotic signaling pathways (tumor necrosis factor- TNF), release of danger-associated molecular patterns (DAMPs), complement C3 and C5 activation, neutrophilic infiltration and activation of the Kupffer cell-mediated pro-inflammatory cascade[52,54] Levels of TNF and tumor necrosis factor receptor superfamily member 1B bear a close correlation with disease severity, endotoxaemia, hepatocyte apoptosis and overall mortality[55] Excess ethanol intake also results in impaired hepatic regeneration via the limitation of DNA synthesis and regular microRNA signaling in mature hepatocytes[56]
A key feature of ACLF is its reversible component, whereby acute deterioration may be reversed with early identification and treatment of the precipitating factor(s), while also providing multi-organ supportive care addressing the complex pattern of physiologic disturbance in patients suffering with critical liver dysfunction. For example, rapid identification of patients with chronic liver disease and superimposed bacterial infection can prevent a significant deterioration in their clinical condition. In addition, patients with acute deterioration due to alcohol abuse must have regular follow-ups, be strongly advised about lifestyle changes and given access to support groups to prevent further flare-ups.
Host response to injury/infection
The body's response to injury is a major determinant of its severity. In ACLF, hepatocytes are exposed to chronically elevated concentrations of inflammatory cytokines, including interferon- g, TNF-α and interleukin (IL)-2 and IL-6. Depending on levels of pro-apoptotic or anti-apoptotic activity, inflammatory cytokines are able to stimulate both survival and apoptosis pathways and modulate the relative equilibrium of pro- and anti-apoptotic proteins, such as Bad and Bcl-2 respectively[57]. Apoptosis, or 'programmed cell death' is one of two main patterns of cell death; namely apoptosis and necrosis. Although apoptosis is the predominant mechanism involved in the pathophysiology of both acute and chronic liver dysfunction, the term 'necroapoptosis' has been recently coined in order to demonstrate the commonality of both pathways and the relative non-exclusivity of the two cell death entities[58]. The fact that patterns of cell death can vary greatly in different cells of one individual suggests that programmable necrosis is a plausible notion[59]. In addition, inflammation is also associated with significant neutrophil dysfunction, which also plays a critical role in the pathogenesis of ACLF[60–61].
These processes all contribute toward the pathogenesis of the systemic inflammatory response syndrome (SIRS), which is characterized by systemic inflammation, organ dysfunction and, ultimately, organ failure. SIRS is a syndrome that clinically presents as hypo- or hyperthermia, tachycardia, tachypnea and abnormal white cell count; these features characterize worsening liver dysfunction, resulting in severe decompensation (ascites, encephalopathy etc.), renal failure, associated infection and poorer prognosis[62–63]. SIRS is characterized by a prominent pro-inflammatory cytokine profile and has been found to promote the progression from stable, compensated cirrhosis to ACLF[64]. It is likely that bacterial translocation plays a critical role in determining the progression from compensated to decompensated liver dysfunctionvia development of the SIRS. Kupffer cells, the resident macrophages of the liver are the first line of defense against gut-derived pathogens and are activatedvia the lipopolysaccharide (LPS) and TLR4, or complement C3 and C5 pathways following acute injury and an increase in intestinal permeability[65] Kupffer cells exist as two distinct phenotypes, M1 and M2, with M1 activation occurring via toll-like receptors (TLRs) and DAMPs[65] The subsequent signaling pathway is complicated and involves assembly of the TLR4–CD14–Lymphocyte antigen 96 receptor complex, recruitment of adaptor molecules including TIR domain-containing adaptor molecule 1 and myeloid differentiation factor 88, and the activation of downstream signaling cascades that ultimately result in a sustained inflammatory response[66] There is a net increase in TNF production, which is the first cytokine to be released, and the increased levels directly correlate with liver injury and the development of SIRS. Concurrently, M2 Kupffer cells are stimulatedvia an alternate pathway involving IL-4 and IL-10 release from Th2 cells. Kupffer cells are intrinsically involved in the transformation of quiescent hepatic stellate cells to activated myofibroblasts, thus acting as a pivotal bridge between immune activation and dysfunction of hepatic microcirculation[67] LPS acts on hepatic stellate cells to activate downstream signaling cascades, resulting in the production of endothelin-1, nitric oxide, prostaglandins and thromboxane A2, causing profound deleterious effects on hepatic microcirculation and an increase in portal pressure.
The reason that patients with ACLF have an increased susceptibility to infection, and thus a greater severity of liver failure, is partly due to neutrophil dysfunction. Although hepatic neutrophils are increased in patients with ACLF, these neutrophils are predominantly dysfunctional, with an increased resting respiratory burst but a reduction in phagocytic function[60] HLA-DR is involved in effective monocyte function, however long-term suppression of HLA-DR commonly occurs in those with ACLF and is associated with poor prognosis and sepsis, organ failure and death, while a rebound increase in HLA-DR expression correlates with clinical recovery[68].
There are a multitude of other cells and signaling pathways involved in the development of SIRS, including dendritic cells and tyrosine-protein kinase Mer dysfunction. In summary, patients with ACLF suffer from severe immune dysregulation due to dysfunction in effector cell function, downstream signaling pathways and cytokine release, while activated immune cells are dysfunctional and energy-depleted.
As SIRS progresses, there is activation of the early compensatory anti-inflammatory response (CARS), which aims to restore immunologic balance. CARS activation results in various physiologic alterations including the reduction of lymphocytesvia apoptosis, a dampened monocytic response to cytokine stimulation, decreased expression of human leukocyte antigen (HLA) in monocytes and cutaneous anergy. The reduction in HLA expression alters the LPS-mediated production of various pro-inflammatory cytokines, including TNF-α IL-1, IL-6, and IL-8. The overall result of these processes is "sepsis-like" immune paralysis and the subsequent increased susceptibility to infection[69]. The literature has demonstrated a clear association between infection and ACLF, suggesting that the immunosuppressive features of cirrhosis, combined with neutrophil dysfunction, are responsible for the increased susceptibility to infection in patients suffering from ACLF, as well as the subsequent deterioration in organ function[12,35,37]. The exaggerated SIRS and defective CARS ultimately results in a profound susceptibility to infection and, even in the absence of infection, patients still possess sepsis-like characteristics, including a reduction in monocyte HLA-DR expression, TNF secretion and an increase in IL-6 production[70] Therefore, due to the deleterious relationship between infection and ACLF, aggressive surveillance and timely management of infection is necessary[35]. It is the paradoxical relationship between the pro-inflammatory and anti-inflammatory state during SIRS, on the background of liver dysfunction, that results in both systemic inflammation and an increased susceptibility to infection, thus acting in a vicious self-perpetuating cycle. The persistence of SIRS results in a hyperdynamic circulation, dysfunctional microcirculation, low mean arterial pressure and subsequent capillary leak and tissue hypoperfusion[71–72] This is the primary mechanism in the development of organ dysfunction which, combined with an increased incidence of bacterial infections, can cause multiple organ failure and worse survival and prognosis than those without it.
Both ACLF and severe sepsis share similar clinical characteristics; namely vasodilatory shock and multiple organ failure. Wasmuthet al. attempted to explain these similarities and found that patients with ACLF had similar immunologic 'defects' to patients with SIRS and severe sepsis, indicating that immune dysregulation is a common feature of both conditions[69].
The prominent pro-inflammatory state of patients with ACLF is mediated by a broad cytokine profile, including tumor necrosis factor (TNF)-α, interleukin (IL)-2, IL-4, IL-6, IL-8, IL-10 and IFN-γ[10,38]. Overall, the progression from stable cirrhosis to decompensation is facilitated by SIRS and is predominantly mediated by cytokines.
Patients with cirrhosis are at increased susceptibility to infection, making it a common feature of patients with ACLF. It is estimated that approximately 50% of hospital admissions of patients with cirrhosis are for sepsis, with up to 40% of these patients developing further nosocomial infections[73]. The relationship between infection and SIRS indicates that an altered inflammatory response in patients with cirrhosis results in immune dysregulation, predisposing these patients to infection that results in a vicious cycle of inflammation[74].
Organ failure
Patients that have ACLF possess higher mortality rates at the same Child-Pugh and MELD scores as those without ACLF[35]. Regardless of etiology, immune dysfunction and the abnormal activation of systemic inflammatory pathways appear to play vital roles in further accentuating liver dysfunction and promoting the development of multi-organ failure[74]. These are discussed below and illustrated in Fig. 2.
Fig.2.

Common multiple organ dysfunctions in acute-on-chronic liver failure and their pathogenic processes.
It has been proposed that an initial 'cytokine storm' occurs due to the systemic exaggeration of pro-inflammatory pathways and overstimulation of the immune system, resulting in macro- and microcirculatory dysfunction, increased capillary permeability, plasma leakage and end stage organ damage; ultimately resulting in multi-organ failure[69,75]. Once the 'eye of the storm' resolves, there is an upregulation of compensatory anti-inflammatory processes, resulting in patients becoming immunocompromised and particularly susceptible to nosocomial infections, sepsis and further deterioration[35].
As previously mentioned, cirrhotic patients who improve following organ failure in hospital and are subsequently discharged have an almost universal three-year mortality rate[13]. In addition, it has been found that patients who have been recently hospitalised (within 6 months) possess a significantly worse mortality than those without recent hospitalization (78%vs. 34%; respectively). Findings such as these have resulted in the development of a "multi-hit" hypothesis, suggesting that patients do not return to their previous baseline following organ injury due to a reduction in functional cell mass and immune dysfunction, despite achieving clinical recovery[35].
It is important to note that a large proportion of patients with chronic liver dysfunction do survive an acute insult and are subsequently discharged from hospital. For survivors, regular monitoring of their condition is vital in order to allow timely detection and prevention of sepsis, offer early support for organ failure and provide artificial liver support systems following acute hepatic insult in patients that do not undergo liver transplantation. Patients with a high MELD score should be priority listed for deceased donor liver tissue or living donor transplant tissue, as liver transplantation remains the definitive therapy in this subset of patients. All patients that have suffered an acute deterioration of hepatic function on the background of chronic liver disease must be screened for risk factors in order to prevent further flare-ups, as well as receive regular monitoring of their hepatic reserve. Thorough monitoring must certainly be carried out in the immediate post-discharge period as there is a median survival of only 4 months if patient admission MELD score is high, while 72% of survivors beyond this period report a poor quality of life.
Hepato-adrenal axis dysfunction
The term "hepato-adrenal syndrome" was first coined due to the frequent association between cirrhosis and adrenal insufficiency in patients with cirrhosis during septic episodes. Patients with both compensation and decompensation commonly report symptoms of adrenal dysfunction and it has been found that adrenal insufficiency occurs more frequently in patients with more severe liver diseases, as indicated by a high MELD score and hemodynamic instability[76–77].
While the exact mechanism of adrenal dysfunction remains to be elucidated, several hypotheses have been suggested, including: cytokine-mediated amplification of the hypothalamus-pituitary-adrenal axis, reduced levels of apoprotein-1/high-density lipoproteins and endotoxemia[76,78].
Despite the clinical relevance of hepato-adrenal syndrome, there is currently no agreement on relevant tests and normal ranges to evaluate adrenal function in cirrhotic patients, indicating the necessity to establish standardized diagnostic and prognostic markers of adrenal insufficiency in patients with liver dysfunction. Treatment is currently limited to hydrocortisone therapy in order to restore circulating glucocorticoid concentrations and improve hemodynamic stability. However, while hydrocortisone demonstrates initial improvements in hemodynamics, there is limited evidence to suggest it improves overall outcomes[79]. Routine steroid use must always be avoided in patients with ACLF, according to a recent controlled trial, and should be limited to patients with adrenal insufficiency[80].
Kidney dysfunction
In cirrhotic patients, pre-renal acute kidney injury and intrinsic kidney injury, including tubular necrosis, are the most common types of renal failure. Bacterial infection, such as SBP, is the most frequent precipitant of renal dysfunction in cirrhotic patients (30-40%), followed by hypovolemia due to gastrointestinal hemorrhage[81]. Renal failure is the most common organ failure in patients with ACLF and is closely related to the presence of SIRS. It has also been found to be a major prognostic indicator of remote organ dysfunction and short- and long-term mortality.
Renal failure following ACLF may be due to acute kidney injury (AKI), hepatorenal syndrome or pre-existing renal disease. According to the EASL-CLIF Consortium, kidney dysfunction is defined as serum creatinine between 1.5 and 1.9 mg/dL and kidney failure as serum creatinine of greater than 2 mg/dL or requirement of renal replacement therapy. AKI manifests as an abrupt decline in renal function, characterized by an increase in serum creatinine of 0.3 mg/dL within 48h or an increase of greater than 50% from baseline levels within the previous 7 days. Studies have demonstrated that the rate of kidney dysfunction in patients with ACLF varies between 22.8% and 51%, considerably higher than the reported prevalence of 20% in hospitalised patients with cirrhosis[82–83]. Patients with advanced cirrhosis are predisposed to chronic renal hypoperfusion and are therefore susceptible to AKI when exposed to precipitating factors such as bacterial infection, variceal hemorrhage and large volume paracentesis[84–85]. Two-thirds of patients develop iatrogenic AKI due to renal hypoperfusion, with most cases being iatrogenic secondary to diuretic use or lactulose associated diarrhea; these causes are reversible in almost 45% of cases. The remaining patients have either volume-nonresponsive AKI or hepatorenal syndrome (HRS). Structural AKI, including acute tubular necrosis and glomerulonephritis, accounts for the remaining one-third of AKI in patients with cirrhosis[84]. However, paradoxically, patients with ACLF develop structural and tubular dysfunction as a result of predisposition to infection and inflammation[5,83]
Various biomarkers of AKI have been recently developed in order to discriminate functional kidney dysfunction from structural kidney damage. Serum cystatin is a low molecular weight (13 kDa) nonglycosylated protein that is freely filtered at the glomerulus and subsequently reabsorbed by the proximal tubules and catabolized, but not secreted, by the renal tubules, therefore acting as a sensitive and early marker of glomerular injury[86]. In contrast to serum creatinine, serum cystatin is independent of age, gender and muscle mass. Biomarkers of proximal and distal tubular damage have also been investigated, with kidney-injury molecule (KIM-1), liver fatty acid binding protein (L-FABP) and neutrophil gelatinase-associated lipocalin (NGAL) appearing to be the most promising options[87–89]
The management of AKI in patients with ACLF involves identification of the cause and the discontinuation of nephrotoxic drugs, vasodilators, diuretics and non-steroidal anti-inflammatory drugs, while patients with co-existing bacterial infections must receive antibiotics according to local guidelines. A proportion of patients may require dialysis depending on various factors such as the presence of significant fluid overload, metabolic acidosis and hyperkalemia refractory to medical management.
In terms of mortality, patients with ACLF and kidney dysfunction and any single 'extrarenal' organ failure possess a short-term mortality of approximately 20%, similar to patients with single renal failure and serum creatinine greater than 2mg/dL[12] Patients with kidney failure and another organ failure have a short-term mortality of 30%, while patients with kidney failure and multiple organ failure (≥2 organs) have a short-term mortality of 77%. It is, therefore, evident that renal dysfunction has a large role in determining the short-term mortality of patients with ACLF. Due to the grim prognosis of patients with ACLF and AKI, certain measures must be adhered to in order to prevent the progression of AKI. These include the astute use of diuretics and avoidance of nephrotoxic drugs, as well as repletion of intravascular volume by albumin during large volume paracentesis in order to prevent paracentesis induced circulatory dysfunction. As a large proportion of patients often have co-existing bacterial infections, judicious antibiotic dose modifications must be considered according to creatinine clearance.
Hepatorenal syndrome (HRS) is a functional disorder that occurs due to hemodynamic dysfunction comprising of splanchnic vasodilation, systemic hypotension, renal vasoconstriction, cardiac dysfunction and abnormal autonomic and neurohormonal activation[81,90]. The hallmark of HRS is renal vasoconstriction, however development of the condition involves multiple mechanisms and a complex interplay between disturbances of hemodynamics, vasoconstrictor systems and vasodilator systems[91] HRS is commonly thought of as a diagnosis of exclusion, entertained only once other potential causes of kidney injury have been ruled out, including tubular injury, glomerulonephritis and prerenal disease. Patients with HRS characteristically have low arterial pressure, reduced systemic vascular resistance and increased cardiac output, however the presentation of renal failure in cirrhotic patients varies greatly, with some having features of circulatory dysfunction and others possessing a more prominent pro-inflammatory profile[92] Type 1 HRS is characterized by rapid and progressive renal dysfunction and is commonly precipitated by SBP, while type 2 HRS is associated with a moderate and stable reduction in glomerular filtration rate (GFR) and most commonly occurs in patients with relatively stable hepatic function. Despite resolution of the infection with antibiotics, type 1 HRS occurs in one-quarter of patients with SBP, while virtually all patients die within 10 weeks of the onset of renal failure if untreated[93] Patients with type 2 HRS are often diuretic-resistant and have a median survival of 3-6 months, which is considerably shorter than patients with cirrhosis and ascites in the absence of renal failure.
Terlipressin and albumin administration remains the gold standard of treatment for patients with hepatorenal syndrome, despite being unsuccessful in over 50% of patients[94]. In patients that do not respond to terlipressin, it is likely that other factors predominate in the development of renal dysfunction, such as SBP. Although it is traditionally suggested that renal dysfunction is reversible by undergoing transplantation, evidence demonstrates that cirrhotic patients are more likely to need aggressive renal replacement therapy post-transplantation compared to those being transplanted due to hepatorenal syndrome[95] This indicates that the basis of renal failure in ACLF differs to hepatorenal syndrome.
It has been suggested that inflammation is the key mediator of renal failure in patients with ACLF, as evidenced by the efficacy of anti-inflammatory agents such as N-acetylcysteine and albumin which are associated with an improvement in renal function in patients with alcoholic hepatitis[96–97]
It is interesting to note that there is a relationship between the cause of AKI and mortality in patients with cirrhosis. A study by Fagundeset al. demonstrates that AKI secondary to miscellaneous causes has the best survival rate, followed by hypervolemia and infection related-AKI, with AKI related to hepatorenal syndrome having the worst prognosis[98]
The CANONIC study reports that kidney failure is the most common organ failure in ACLF grade 1, indicating the increasing necessity for effective biomarkers in order to detect patients at risk of kidney dysfunction, as well as novel therapeutic strategies to address the poor prognosis of patients with ACLF and co-existing renal impairment.
Haematological dysfunction
Impaired coagulation is the most common hematological complication in patients with ACLF, as suggested by abnormal coagulation tests due to both compromised synthesis and depletion of coagulation factors. On hematological profiling, an increased prothrombin time is a frequent finding, while an international normalized ratio (INR) of>2.5, or platelet count<20000/ mL would meet the criteria for coagulation failure according to CLIF-SOFA[12].
Patients suffering from chronic liver disease have an increase in both anti-thrombotic and pro-thrombotic clotting factors[99]. Due to abnormal inflammation associated with ACLF, the condition is able to promote either state, manifesting as either hemorrhagic or thrombotic complications[100].
Sepsis, a relatively common complication of chronic liver disease, worsens the coagulopathy due to the presence of endogenous heparinoids and further increases the risk of hemorrhagic complications as a result of the development of portal hypertension and portacaval anastomoses[101].
Neurological failure
Hepatic encephalopathy is a frequent manifestation of ACLF, as both a precipitant of ACLF or a result of it. Neurological failure itself is defined as the development of Grade III/IV encephalopathy[102]. Both local and systemic alterations on the background of cirrhosis are responsible for the pathogenesis of encephalopathy. Patients with hepatic encephalopathy commonly develop an array of neuropsychiatric symptoms, including but not limited to psychomotor dysfunction, impaired memory, sensory abnormalities and reduced attention.
Hyperammonemia is linked to the development of hepatic encephalopathy, with additional hepatic insults on the background of hyperammonemia resulting in significant cerebral edema and inflammation. Bearing in mind that patients with chronic liver dysfunction often suffer from functional immunoparesis, a relationship between ammonia and inflammation has been proposed[103]. While the exact synergistic mechanism behind hyperammonemia and inflammation is yet to be elucidated, it is thought to be mediated by increased cytokine generation and iNOS expression, oxidative stress, abnormalities in GABA-ergic transmission and decreased cerebral blood flow[104,105]. Astrocyte-mediated metabolism of ammonia results in the production and accumulation of osmotically active glutamine, which can subsequently cause brain edema and intracranial hypertension[106–108] Induced hyperammonemia results in significant neuropsychological features in patients with both cirrhosis and SIRS, suggesting a synergistic relationship between hyperammonemia and inflammation[109] However, it is important to note that it is unclear whether it is the background neurological state in cirrhosis or the associated hyperammonemia that predisposes the brain to the deleterious effects of superimposed inflammation. Various animal studies involving LPS-challenged cirrhotic rats have demonstrated that inflammation, hyperammonemia and subsequent brain edema all contribute to the effects on consciousness in patients with hepatic encephalopathy. For example, Wrightet al. demonstrated that LPS-challenged cirrhotic rats experienced a significant increase in cerebral edema, with astrocytic and perivascular edema, thus mimicking ACLF[103] Interestingly, naïve non-cirrhotic rats with induced hyperammonemia actually experienced more severe LPS-induced brain edema than cirrhotic rats. Various studies support the notion that hyperammonia is responsible for the induction of microglial cells and other cerebral inflammatory mediators, thus priming the brain to the detrimental effects of superimposed inflammation[103,105,110] Interventional studies also support this notion, with administration of the novel therapy ornithine phenylactetate resulting in the reduction of circulating ammonia and subsequent protection against cerebral edema associated with liver failure[111].
While this may be the case, experimental data suggest that ammonia levels, in fact, show no direct correlation with advanced hepatic encephalopathy and outcomes in patients with clinical ACLF. It has been found that non-cirrhotic hyperammonemic rats developed significant astrocytic edema with increased glutamine, which is expected and correlates with the ammonia-glutamine-edema hypothesis[103] Interestingly, however, only mild astrocytic edema is seen in saline-treated rats following bile-duct ligation compared to the LPS-treated group, despite similar levels of serum and cerebral ammonia in both cohorts. Overall, it is unclear the precise mechanisms that are responsible for astrocytic edema in cirrhotic rats, however it is thought to be due to underlying inflammatory processes. Although hepatic encephalopathy commonly occurs in patients with ACLF, overt cerebral edema is an infrequent finding, suggesting that it is hyperammonemia on the background of an upregulated systemic inflammatory state in cirrhosis that contributes to the features of encephalopathy, rather than the presence of edema itself. An important factor to bear in mind in the pathogenesis of hepatic encephalopathy is the role of cerebral blood flow (CBF), which is integral to the development of the condition. CBF is likely to be directly related to alterations in both ammonia and inflammation, as they independently interact to influence cerebral hemodynamics[112] Both factors, as discussed, appear to act in a synergistic fashion and to such an extent that they can even cause alterations in CBF in non-cirrhotic rat models.
It is important to note that patients with acute decompensation and hepatic encephalopathy have a different clinical course compared to those with ACLF. Isolated encephalopathy in patients with acute decompensation commonly occurs with long-term diuretic treatment, rather than as a result of liver dysfunction[113]. These patients often have a good prognosis, in the absence of significant inflammation and organ dysfunction. However, patients with ACLF and encephalopathy have an extremely high mortality rate due to the presence of SIRS, resulting in neurological dysfunction and multi-organ failure. As well as liver dysfunction, hepatic encephalopathy in the context of ACLF is often concomitant with active alcoholic and bacterial infection[104].
Future perspectives
Despite attempts to improve the ACLF bioclinical classifications and prognostic scoring systems, much work is still required to precisely define and characterize the systemic syndrome. Although it has become evident that a single ACLF biomarker is unlikely to be encountered for use in routine clinical practice, it is important to continue researching new biomarkers or biologic fingerprints and combine these novel indicators with updated ACLF scoring systems in order to accurately stratify patients in terms of their prognosis and necessity for transplantation.
Metabolomics is a newly emerging field of 'omics' research that may aid the identification of novel biomarkers of progression from compensated chronic liver dysfunction to acute decompensation and help with characterizing the metabolic profile of ACLF. The field of metabolomics is predominantly concerned with the comprehensive identification of small molecule metabolites within biologic systems. Metabolomics, therefore, provides more functional and phenotypical information regarding disease and allows a greater understanding of disease patterns in order to give more accurate disease profiling. An example of its application in modern medicine is in determining the prognosis of patients with bowel cancer, with the recent identification of the Kras gene as a strong predictor of poor response to chemotherapy. As a result, Kras screens are now routinely performed. In addition, metabolomics may also help evaluate metabolic modifications following the initiation of treatment, as well as potentially aiding the discovery of novel targets for the therapeutic management of ACLF. In terms of specific applications in liver dysfunction, metabolomics using proton nuclear magnetic resonance spectroscopy (NMRS) has demonstrated an intrinsic relationship between metabolic abnormalities and disease severity within hepatic sera and parenchyma[114–115]. The results from these studies suggest that metabolites correlate significantly with abnormalities in liver metabolism, such as fatty acids and glucose, while phospholipid precursors have a prognostic relationship with hepatic lesions. By a similar method, a specific serum metabolite fingerprint for ACLF has also been identified[116]. The main metabolites with increased signaling in ACLF have been found; these include lactate, pyruvate, ketone bodies, glutamine and several others, while high-density lipids are lower in patients with ACLF than chronic liver dysfunction. Several metabolites have, therefore, been identified and reflect significant pathophysiological changes in liver function associated with amino acid metabolism, energy metabolism and urea metabolism. Metabolites for remote organ dysfunction, such as renal impairment, have also been recognized. While these findings are positive, a large trial involving a multicentric population of compensated and ACLF patients of various etiologies is required to provide further support for the metabolomics hypothesis. It is therefore possible that metabolomics profiling may aid the clinical evaluation of decompensated cirrhotic patients in intensive care with ACLF, as well as provide new information regarding the metabolic processes in acute hepatic dysfunction and establish novel biomarkers to accurately diagnose and determine patient prognosis. Further investigation of the hepatic metabolomic profile in patients with ACLF may be useful in assessing highly sensitive hepatic metabolic alterations that correlate with disease severity and clinical prognosis.
As well as metabolomics, a small number of studies have also demonstrated the potential efficacy of granulocyte colony-stimulating factor (G-CSF) in patients with ACLF[117–119]. Originally developed in 1985 to reduce the incidence of neutrophenic fever after myelosuppressive chemotherapy in patients with non-myeloid malignancy, G-CSF therapy has since demonstrated the ability to stimulate the proliferation and differentiation of neutrophil progenitor cells, as well as activation of mature neutrophils. The hypothesis behind the use of G-CSF is that it may mobilise CD34+ bone marrow-derived stem cells and promote hepatic regeneration, as well as improve neutrophil function. The precise mechanism for these changes remains to be elucidated, however severalin vitro and in vivo studies have suggested that the neutrophil-activating effect of G-CSF is important in mediating its therapeutic benefits[120–121] G-CSF is considered a potent activator of mature circulating cells with the capability of priming respiratory burst, while simultaneously inducing the activation and release of secretory vesicles and cytoplasmic granules, as well as mediating the expression of surface adhesion molecules such as polymorphonuclear surface antigen CD11b/CD18. It is thought that these neutrophil-activating effects may be responsible for the reduction in sepsis, multiple organ failure and improved survival associated with its administration. The administration of colony-stimulating factor 1 has been shown to provide bone-marrow derived macrophages which promote early chemokine upregulation with hepatic recruitment of endogenous macrophages and neutrophils, resulting in an increased level of anti-inflammatory cytokine, IL-10, and reduction in hepatic myofibroblasts, ultimately reducing hepatic fibrosis and promoting regeneration[122] These processes may also be responsible for the regenerative effects of G-CSF.
Two randomized studies have indicated that G-CSF administration reduces the risk of developing deleterious extrahepatic sequelae such as renal and neurological dysfunction, as well as attenuating sepsis and improving short-term survival rates[117–118]. Garg et al. (2012) recruited consecutive patients with ACLF, which were randomly assigned to groups given 5 mg/kg G-CSF subcutaneously (12 doses; group A, n = 23) or placebo (group B, n = 24) plus standard medical therapy[117] The authors found that G-CSF therapy resulted in more than twice as many patients with ACLF surviving for 2 months, while also reducing the MELD and SOFA scores and preventing the development of sepsis, hepatorenal syndrome and hepatic encephalopathy. It has also been demonstrated that patients treated with G-CSF experience a 44% reduction in short-term mortality (60-90 days) compared to controls, as well as an improvement in liver function, increase in peripheral and intrahepatic CD34+ count and an increase in peripheral neutrophil/leukocyte counts[123] Although evidence is still limited, there is an apparent benefit observed on short-term mortality and, as the therapy is associated with only mild adverse effects, it may be a reasonable alternative in situations when liver transplantation is contraindicated or unavailable. Reported adverse effects include fever, rash, headache, nausea and herpes zoster. However, despite these promising findings, all patients included in these studies had the initial features of ACLF, rather than more advanced clinical characteristics such as sepsis and multiorgan failure.
As liver transplantation is often unavailable due to high costs and a lack of donor grafts, regeneration may be a viable option in the future. Further clinical studies are certainly required in order to ascertain the precise efficacy of G-CSF therapy in stimulating the reversal of liver dysfunction, as well as determine any long-term adverse effects.
Patients with ACLF may also benefit from cell transplantation of either hepatocytes of stem cells by stimulating cell repopulation of the cirrhotic and dysfunctional liver. Several studies have reported an improvement in liver function with cell transplantation, while mesenchymal stem cells may have additional beneficial actions, such as anti-inflammatory effects[124–127].
Conclusion
ACLF is a severe condition that occurs on the background of chronic liver dysfunction and results in the development of organ failure and is associated with inordinately high mortality. Although the condition is pathophysiologically, clinically and prognostically distinct to classic acute decompensation, it is yet to be defined and standardization of diagnostic criteria is still required. The generation of ACLF is intrinsically linked to an abnormal host response to precipitating injury, such as SIRS, and the prognosis of the patient is primarily dependent on the degree of immunoparesis and severity of organ failure.
Currently, treatment strategies are limited to symptomatic relief and organ support, indicating the necessity for novel biomarkers, drugs and devices for the management of ACLF. A multidisciplinary management strategy is required to ensure early detection of organ dysfunction, while engagement with the liver transplant community is needed to define a new allocation policy for this subset of patients. Future methods to further define and characterize ACLF involve additional investigation of the metabolomics profile of patients with ACLF, while G-CSF administration and cell transplantation seem to be novel and promising therapeutic interventions to improve patient outcomes. Further prospective clinical studies are required to fully elucidate the modifiable factors that predispose patients to ACLF and improve our understanding of this devastating syndrome, while also developing a personalized management plan for individual patients based on their clinical and genetic characteristics.
Contributor Information
Azeem Alam, Email: azeem.alam@kcl.ac.uk, Anaesthetics, Pain Medicine and Intensive Care, Department of Surgery and Cancer, Faculty of Medicine, Imperial College London, Chelsea & Westminster Hospital, London, SW7 2AZ, UK..
Ka Chun Suen, Anaesthetics, Pain Medicine and Intensive Care, Department of Surgery and Cancer, Faculty of Medicine, Imperial College London, Chelsea & Westminster Hospital, London, SW7 2AZ, UK..
Daqing Ma, Email: d.ma@imperial.ac.uk, Anaesthetics, Pain Medicine and Intensive Care, Department of Surgery and Cancer, Faculty of Medicine, Imperial College London, Chelsea & Westminster Hospital, London, SW7 2AZ, UK..
References
- 1. Lee WM, Stravitz RT, Larson AM. Introduction to the revised American Association for the Study of Liver Diseases Position Paper on acute liver failure 2011[J]. Hepatology, 2012, 55(3): 965–967 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moore KP, Wong F, Gines P, et al. The management of ascites in cirrhosis: report on the consensus conference of the International Ascites Club[J]. Hepatology, 2003, 38(1): 258–266 . [DOI] [PubMed] [Google Scholar]
- 3. Ohnishi H, Sugihara J, Moriwaki H, Muto Y. Acute-on-chronic liver failure[J]. Ryoikibetsu shokogun shirizu 1995;( 7)( 7): 217–219. [PubMed] [Google Scholar]
- 4. Jalan R, Williams R. Acute-on-chronic liver failure: pathophysiological basis of therapeutic options[J]. Blood Purif, 2002, 20(3): 252–261 . [DOI] [PubMed] [Google Scholar]
- 5. Jalan R, Gines P, Olson JC, et al. Acute-on chronic liver failure[J]. J Hepatol, 2012, 57(6): 1336–1348 . [DOI] [PubMed] [Google Scholar]
- 6. Jalan R, Sen S, Williams R. Prospects for extracorporeal liver support[J]. Gut, 2004, 53(6): 890–898 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim TY, Kim DJ. Acute-on-chronic liver failure[J]. Clin Mol Hepatol, 2013, 19(4): 349–359 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Graziadei IW. The clinical challenges of acute on chronic liver failure[J]. Liver international: official journal of the International Association for the Study of the Liver 2011; 31 Suppl 324–26. [DOI] [PubMed] [Google Scholar]
- 9. Wlodzimirow KA, Eslami S, Abu-Hanna A, Nieuwoudt M, Chamuleau RA. A systematic review on prognostic indicators of acute on chronic liver failure and their predictive value for mortality[J]. Liver international: official journal of the International Association for the Study of the Liver 2013; 33(1): 40–52. [DOI] [PubMed] [Google Scholar]
- 10. Sarin SK, Kumar A, Almeida JA, et al. Acute-on-chronic liver failure: consensus recommendations of the Asian Pacific Association for the study of the liver (APASL)[J]. Hepatol Int, 2009, 3(1): 269–282 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olson JC, Wendon JA, Kramer DJ, et al. Intensive care of the patient with cirrhosis[J]. Hepatology, 2011, 54(5): 1864–1872 . [DOI] [PubMed] [Google Scholar]
- 12. Moreau R, Jalan R, Gines P, Pavesi M, Angeli P, Cordoba J, et al. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis[J]. Gastroenterology 2013; 144(7): 1426–37, 1437.e1–9. [DOI] [PubMed] [Google Scholar]
- 13. Jalan R, Stadlbauer V, Sen S, et al. Role of predisposition, injury, response and organ failure in the prognosis of patients with acute-on-chronic liver failure: a prospective cohort study[J]. Crit Care, 2012, 16(6): R227 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pugh RN, Murray-Lyon IM, Dawson JL, et al. Transection of the oesophagus for bleeding oesophageal varices[J]. Br J Surg, 1973, 60(8): 646–649 . [DOI] [PubMed] [Google Scholar]
- 15. Wehler M, Kokoska J, Reulbach U, et al. Short-term prognosis in critically ill patients with cirrhosis assessed by prognostic scoring systems[J]. Hepatology, 2001, 34(2): 255–261 . [DOI] [PubMed] [Google Scholar]
- 16. Das V, Boelle PY, Galbois A, et al. Cirrhotic patients in the medical intensive care unit: early prognosis and long-term survival[J]. Crit Care Med, 2010, 38(11): 2108–2116 . [DOI] [PubMed] [Google Scholar]
- 17. Levesque E, Hoti E, Azoulay D, et al. Prospective evaluation of the prognostic scores for cirrhotic patients admitted to an intensive care unit[J]. J Hepatol, 2012, 56(1): 95–102 . [DOI] [PubMed] [Google Scholar]
- 18. Chalasani N, Kahi C, Francois F, et al. Model for end-stage liver disease (MELD) for predicting mortality in patients with acute variceal bleeding[J]. Hepatology, 2002, 35(5): 1282–1284 . [DOI] [PubMed] [Google Scholar]
- 19. D'Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies[J]. J Hepatol, 2006, 44(1): 217–231 . [DOI] [PubMed] [Google Scholar]
- 20. Kremers WK, van IJperen M, Kim WR, et al. MELD score as a predictor of pretransplant and posttransplant survival in OPTN/UNOS status 1 patients[J]. Hepatology, 2004, 39(3): 764–769 . [DOI] [PubMed] [Google Scholar]
- 21. Srikureja W, Kyulo NL, Runyon BA, et al. MELD score is a better prognostic model than Child-Turcotte-Pugh score or Discriminant Function score in patients with alcoholic hepatitis[J]. J Hepatol, 2005, 42(5): 700–706 . [DOI] [PubMed] [Google Scholar]
- 22. Dunn W, Jamil LH, Brown LS, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis[J]. Hepatology, 2005, 41(2): 353–358 . [DOI] [PubMed] [Google Scholar]
- 23. Kim HY, Chang Y, Park JY, et al. Characterization of acute-on-chronic liver failure and prediction of mortality in Asian patients with active alcoholism[J]. J Gastroenterol Hepatol, 2016, 31(2): 427–433 . [DOI] [PubMed] [Google Scholar]
- 24. Jalan R, Saliba F, Pavesi M, et al. , and the CANONIC study investigators of the EASL-CLIF Consortium. Development and validation of a prognostic score to predict mortality in patients with acute-on-chronic liver failure[J]. J Hepatol, 2014, 61(5): 1038–1047 . [DOI] [PubMed] [Google Scholar]
- 25. Duan BW, Lu SC, Wu JS, et al. Model for End-Stage Liver Disease (MELD) score does not predict outcomes of hepatitis B-induced acute-on-chronic liver failure in transplant recipients[J]. Transplant Proc, 2014, 46(10): 3502–3506 . [DOI] [PubMed] [Google Scholar]
- 26. Zhang Q, Guo X, Zhao S, et al. Prognostic performance of clinical indices and model scorings for acute-on-chronic liver failure: A study of 164 patients[J]. Exp Ther Med, 2016, 11(4): 1348–1354 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xia Q, Dai X, Zhang Y, et al. A modified MELD model for Chinese pre-ACLF and ACLF patients and it reveals poor prognosis in pre-ACLF patients[J]. PLoS One, 2013, 8(6): e64379 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kumar R, Krishnamoorthy TL, Tan HK, et al. Change in model for end-stage liver disease score at two weeks, as an indicator of mortality or liver transplantation at 60 days in acute-on-chronic liver failure[J]. Gastroenterol Rep (Oxf), 2015, 3(2): 122–127 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McPhail MJ, Shawcross DL, Abeles RD, Chang A, Patel V, Lee GH, et al. Increased Survival for Patients With Cirrhosis and Organ Failure in Liver Intensive Care and Validation of the Chronic Liver Failure-Sequential Organ Failure Scoring System[J]. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association 2015; 13(7): 1353–1360.e8. [DOI] [PubMed] [Google Scholar]
- 30. Garg H, Kumar A, Garg V, Sharma P, Sharma BC, Sarin SK. Clinical profile and predictors of mortality in patients of acute-on-chronic liver failure[J]. Digestive and liver disease: official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver 2012; 44(2): 166–171. [DOI] [PubMed] [Google Scholar]
- 31. Bajaj JS, O'Leary JG, Reddy KR, et al. , and the NACSELD. Second infections independently increase mortality in hospitalized patients with cirrhosis: the North American consortium for the study of end-stage liver disease (NACSELD) experience[J]. Hepatology, 2012, 56(6): 2328–2335 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marshall J, Sweeney D. Microbial infection and the septic response in critical surgical illness[J]. Sepsis, not infection, determines outcome. Archives of surgery (Chicago, Ill.: 1960) 1990; 125(1): 17–22; discussion 22–3. [DOI] [PubMed] [Google Scholar]
- 33. Moreno RP, Metnitz B, Adler L, et al. , and the SAPS 3 Investigators. Sepsis mortality prediction based on predisposition, infection and response[J]. Intensive Care Med, 2008, 34(3): 496–504 . [DOI] [PubMed] [Google Scholar]
- 34. Levy MM, Fink MP, Marshall JC, et al. , and the International Sepsis Definitions Conference. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference[J]. Intensive Care Med, 2003, 29(4): 530–538 . [DOI] [PubMed] [Google Scholar]
- 35. Olson JC, Kamath PS. Acute-on-chronic liver failure: concept, natural history, and prognosis[J]. Curr Opin Crit Care, 2011, 17(2): 165–169 . [DOI] [PubMed] [Google Scholar]
- 36. Duseja A, Chawla YK, Dhiman RK, et al. Non-hepatic insults are common acute precipitants in patients with acute on chronic liver failure (ACLF)[J]. Dig Dis Sci, 2010, 55(11): 3188–3192 . [DOI] [PubMed] [Google Scholar]
- 37. Borzio M, Salerno F, Piantoni L, Cazzaniga M, Angeli P, Bissoli F, et al. Bacterial infection in patients with advanced cirrhosis: a multicentre prospective study[J]. Digestive and liver disease: official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver 2001; 33(1): 41–48. [DOI] [PubMed] [Google Scholar]
- 38. Ambrosino G, Naso A, Feltracco P, Carraro P, Basso SM, Varotto S, et al. Cytokines and liver failure: modification of TNF- and IL-6 in patients with acute on chronic liver decompensation treated with Molecular Adsorbent Recycling System (MARS)[J]. Acta bio-medica: Atenei Parmensis 2003; 74 Suppl27–9. [PubMed] [Google Scholar]
- 39. Kumar A, Saraswat VA. Hepatitis E and Acute-on-Chronic Liver Failure[J]. J Clin Exp Hepatol, 2013, 3(3): 225–230 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kumar Acharya S, Kumar Sharma P, Singh R, et al. Hepatitis E virus (HEV) infection in patients with cirrhosis is associated with rapid decompensation and death[J]. J Hepatol, 2007, 46(3): 387–394 . [DOI] [PubMed] [Google Scholar]
- 41. Shi Y, Yang Y, Hu Y, et al. Acute-on-chronic liver failure precipitated by hepatic injury is distinct from that precipitated by extrahepatic insults[J]. Hepatology, 2015, 62(1): 232–242 . [DOI] [PubMed] [Google Scholar]
- 42. Sheen IS, Liaw YF, Tai DI, et al. Hepatic decompensation associated with hepatitis B e antigen clearance in chronic type B hepatitis[J]. Gastroenterology, 1985, 89(4): 732–735 . [DOI] [PubMed] [Google Scholar]
- 43. Yuen MF, Wong DK, Zheng BJ, et al. Difference in T helper responses during hepatitis flares in hepatitis B e antigen (HBeAg)-positive patients with genotypes B and C: implication for early HBeAg seroconversion[J]. J Viral Hepat, 2007, 14(4): 269–275 . [DOI] [PubMed] [Google Scholar]
- 44. Aoki J, Kowazaki Y, Ohtsuki T, et al. Kinetics of peripheral hepatitis B virus-specific CD8+ T cells in patients with onset of viral reactivation[J]. J Gastroenterol, 2013, 48(6): 728–737 . [DOI] [PubMed] [Google Scholar]
- 45. Zhang Z, Zhang JY, Wherry EJ, Jin B, Xu B, Zou ZS, et al. Dynamic programmed death 1 expression by virus-specific CD8 T cells correlates with the outcome of acute hepatitis B[J]. Gastroenterology 2008; 134(7): 1938–49, 1949.e1–3. [DOI] [PubMed] [Google Scholar]
- 46. Saravanabalaji S, Tripathy AS, Dhoot RR, et al. Viral load, antibody titers and recombinant open reading frame 2 protein-induced TH1/TH2 cytokines and cellular immune responses in self-limiting and fulminant hepatitis e[J]. Intervirology, 2009, 52(2): 78–85 . [DOI] [PubMed] [Google Scholar]
- 47. Tripathy AS, Das R, Rathod SB, Gurav YK, Arankalle VA. Peripheral T regulatory cells and cytokines in hepatitis E infection[J]. European journal of clinical microbiology & infectious diseases: official publication of the European Society of Clinical Microbiology 2012; 31(2): 179–184. [DOI] [PubMed] [Google Scholar]
- 48. Choi YS, Lee J, Lee HW, et al. Liver injury in acute hepatitis A is associated with decreased frequency of regulatory T cells caused by Fas-mediated apoptosis[J]. Gut, 2015, 64(8): 1303–1313 . [DOI] [PubMed] [Google Scholar]
- 49. Zhang X, Ke W, Xie J, et al. Comparison of effects of hepatitis E or A viral superinfection in patients with chronic hepatitis B[J]. Hepatol Int, 2010, 4(3): 615–620 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bajaj JS. Defining acute-on-chronic liver failure: will East and West ever meet[J]? Gastroenterology, 2013, 144(7): 1337–1339 . [DOI] [PubMed] [Google Scholar]
- 51. Sarin SK, Kumar A, Almeida JA, et al. Acute-on-chronic liver failure: consensus recommendations of the Asian Pacific Association for the study of the liver (APASL)[J]. Hepatol Int, 2009, 3(1): 269–282 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets[J]. Gastroenterology, 2011, 141(5): 1572–1585 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease[J]. J Hepatol, 2011, 54(4): 795–809 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gustot T, Lemmers A, Moreno C, et al. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver[J]. Hepatology, 2006, 43(5): 989–1000 . [DOI] [PubMed] [Google Scholar]
- 55. Tilg H, Moschen AR, Kaneider NC. Pathways of liver injury in alcoholic liver disease[J]. J Hepatol, 2011, 55(5): 1159–1161 . [DOI] [PubMed] [Google Scholar]
- 56. Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment[J]. Nat Rev Gastroenterol Hepatol, 2015, 12(4): 231–242 . [DOI] [PubMed] [Google Scholar]
- 57. Elsing C, Harenberg S, Stremmel W, et al. Serum levels of soluble Fas, nitric oxide and cytokines in acute decompensated cirrhotic patients[J]. World J Gastroenterol, 2007, 13(3): 421–425 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury[J]. Gut, 2005, 54(7): 1024–1033 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lemasters JJV. V. Necrapoptosis and the mitochondrial permeability transition: shared pathways to necrosis and apoptosis[J]. Am J Physiol, 1999, 276(1 Pt 1): G1–G6 . [DOI] [PubMed] [Google Scholar]
- 60. Mookerjee RP, Stadlbauer V, Lidder S, et al. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome[J]. Hepatology, 2007, 46(3): 831–840 . [DOI] [PubMed] [Google Scholar]
- 61. Stadlbauer V, Mookerjee RP, Hodges S, et al. Effect of probiotic treatment on deranged neutrophil function and cytokine responses in patients with compensated alcoholic cirrhosis[J]. J Hepatol, 2008, 48(6): 945–951 . [DOI] [PubMed] [Google Scholar]
- 62. Rolando N, Wade J, Davalos M, et al. The systemic inflammatory response syndrome in acute liver failure[J]. Hepatology, 2000, 32(4 Pt 1): 734–739 . [DOI] [PubMed] [Google Scholar]
- 63. Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure[J]. Gastroenterology, 2003, 125(3): 755–764 . [DOI] [PubMed] [Google Scholar]
- 64. Jalan R, Olde Damink SW, Hayes PC, et al. Pathogenesis of intracranial hypertension in acute liver failure: inflammation, ammonia and cerebral blood flow[J]. J Hepatol, 2004, 41(4): 613–620 . [DOI] [PubMed] [Google Scholar]
- 65. Wan J, Benkdane M, Teixeira-Clerc F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease[J]. Hepatology, 2014, 59(1): 130–142 . [DOI] [PubMed] [Google Scholar]
- 66. Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update[J]. Hepatology, 2008, 48(1): 322–335 . [DOI] [PubMed] [Google Scholar]
- 67. Rockey DC, Fouassier L, Chung JJ, et al. Cellular localization of endothelin-1 and increased production in liver injury in the rat: potential for autocrine and paracrine effects on stellate cells[J]. Hepatology, 1998, 27(2): 472–480 . [DOI] [PubMed] [Google Scholar]
- 68. Xing T, Li L, Cao H, et al. Altered immune function of monocytes in different stages of patients with acute on chronic liver failure[J]. Clin Exp Immunol, 2007, 147(1): 184–188 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wasmuth HE, Kunz D, Yagmur E, et al. Patients with acute on chronic liver failure display "sepsis-like" immune paralysis[J]. J Hepatol, 2005, 42(2): 195–201 . [DOI] [PubMed] [Google Scholar]
- 70. Wasmuth HE, Kunz D, Yagmur E, et al. Patients with acute on chronic liver failure display "sepsis-like" immune paralysis[J]. J Hepatol, 2005, 42(2): 195–201 . [DOI] [PubMed] [Google Scholar]
- 71. Laleman W, Verbeke L, Meersseman P, et al. Acute-on-chronic liver failure: current concepts on definition, pathogenesis, clinical manifestations and potential therapeutic interventions[J]. Expert Rev Gastroenterol Hepatol, 2011, 5(4): 523–537., quiz 537 . [DOI] [PubMed] [Google Scholar]
- 72. Kumar A, Das K, Sharma P, et al. Hemodynamic studies in acute-on-chronic liver failure[J]. Dig Dis Sci, 2009, 54(4): 869–878 . [DOI] [PubMed] [Google Scholar]
- 73. Linderoth G, Jepsen P, Schønheyder HC, et al. Short-term prognosis of community-acquired bacteremia in patients with liver cirrhosis or alcoholism: A population-based cohort study[J]. Alcohol Clin Exp Res, 2006, 30(4): 636–641 . [DOI] [PubMed] [Google Scholar]
- 74. Malik R, Mookerjee RP, Jalan R. Infection and inflammation in liver failure: two sides of the same coin[J]. J Hepatol, 2009, 51(3): 426–429 . [DOI] [PubMed] [Google Scholar]
- 75. Russell JA. Management of sepsis[J]. N Engl J Med, 2006, 355(16): 1699–1713 . [DOI] [PubMed] [Google Scholar]
- 76. Anastasiadis SN, Giouleme OI, Germanidis GS, et al. Relative adrenal insufficiency in cirrhotic patients[J]. Clin Med Insights Gastroenterol, 2015, 8: 13–17 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tsai MH, Peng YS, Chen YC, et al. Adrenal insufficiency in patients with cirrhosis, severe sepsis and septic shock[J]. Hepatology, 2006, 43(4): 673–681 . [DOI] [PubMed] [Google Scholar]
- 78. Karagiannis AK, Nakouti T, Pipili C, et al. Adrenal insufficiency in patients with decompensated cirrhosis[J]. World J Hepatol, 2015, 7(8): 1112–1124 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fernández J, Escorsell A, Zabalza M, et al. Adrenal insufficiency in patients with cirrhosis and septic shock: Effect of treatment with hydrocortisone on survival[J]. Hepatology, 2006, 44(5): 1288–1295 . [DOI] [PubMed] [Google Scholar]
- 80. Arabi YM, Aljumah A, Dabbagh O, Tamim HM, Rishu AH, Al-Abdulkareem A, et al. Low-dose hydrocortisone in patients with cirrhosis and septic shock: a randomized controlled trial[J]. CMAJ: Canadian Medical Association journal= journal de l'Association medicale canadienne 2010; 182(18): 1971–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Martín-Llahí M, Guevara M, Torre A, et al. Prognostic importance of the cause of renal failure in patients with cirrhosis[J]. Gastroenterology, 2011, 140(2): 488–496.e4 . [DOI] [PubMed] [Google Scholar]
- 82. Garg H, Kumar A, Garg V, Sharma P, Sharma BC, Sarin SK. Clinical profile and predictors of mortality in patients of acute-on-chronic liver failure[J]. Digestive and liver disease: official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver 2012; 44(2): 166–171. [DOI] [PubMed] [Google Scholar]
- 83. Maiwall R, Kumar S, Chandel SS, et al. AKI in patients with acute on chronic liver failure is different from acute decompensation of cirrhosis[J]. Hepatol Int, 2015, 9(4): 627–639 . [DOI] [PubMed] [Google Scholar]
- 84. Garcia-Tsao G, Parikh CR, Viola A. Acute kidney injury in cirrhosis[J]. Hepatology, 2008, 48(6): 2064–2077 . [DOI] [PubMed] [Google Scholar]
- 85. Ginès P, Schrier RW. Renal failure in cirrhosis[J]. N Engl J Med, 2009, 361(13): 1279–1290 . [DOI] [PubMed] [Google Scholar]
- 86. Tsigou E, Psallida V, Demponeras C, et al. Role of new biomarkers: functional and structural damage[J]. Crit Care Res Pract, 2013, 2013: 361078 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ichimura T, Bonventre JV, Bailly V, et al. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury[J]. J Biol Chem, 1998, 273(7): 4135–4142 . [DOI] [PubMed] [Google Scholar]
- 88. Wang G, Gong Y, Anderson J, et al. Antioxidative function of L-FABP in L-FABP stably transfected Chang liver cells[J]. Hepatology, 2005, 42(4): 871–879 . [DOI] [PubMed] [Google Scholar]
- 89. Belcher JM, Garcia-Tsao G, Sanyal AJ, et al. , and the TRIBE-AKI Consortium. Association of AKI with mortality and complications in hospitalized patients with cirrhosis[J]. Hepatology, 2013, 57(2): 753–762 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ginès P, Schrier RW. Renal failure in cirrhosis[J]. N Engl J Med, 2009, 361(13): 1279–1290 . [DOI] [PubMed] [Google Scholar]
- 91. Turban S, Thuluvath PJ, Atta MG. Hepatorenal syndrome[J]. World J Gastroenterol, 2007, 13(30): 4046–4055 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Arroyo V, Terra C, Ginès P. Advances in the pathogenesis and treatment of type-1 and type-2 hepatorenal syndrome[J]. J Hepatol, 2007, 46(5): 935–946 . [DOI] [PubMed] [Google Scholar]
- 93. Appenrodt B, Zielinski J, Brensing KA, et al. Degree of hepatic dysfunction and improvement of renal function predict survival in patients with HRS type I: a retrospective analysis[J]. Eur J Gastroenterol Hepatol, 2009, 21(12): 1428–1432 . [DOI] [PubMed] [Google Scholar]
- 94. Nazar A, Pereira GH, Guevara M, et al. Predictors of response to therapy with terlipressin and albumin in patients with cirrhosis and type 1 hepatorenal syndrome[J]. Hepatology, 2010, 51(1): 219–226 . [DOI] [PubMed] [Google Scholar]
- 95. Nadim MK, Genyk YS, Tokin C, Fieber J, Ananthapanyasut W, Ye W, et al. Impact of the etiology of acute kidney injury on outcomes following liver transplantation: acute tubular necrosis versus hepatorenal syndrome[J]. Liver Transplantation: official publication of the American Association for the Study of Liver Diseases and the International Liver Transplantation Society 2012; 18(5): 539–548. [DOI] [PubMed] [Google Scholar]
- 96. Holt S, Goodier D, Marley R, et al. Improvement in renal function in hepatorenal syndrome with N-acetylcysteine[J]. Lancet, 1999, 353(9149): 294–295 . [DOI] [PubMed] [Google Scholar]
- 97. Akriviadis E, Botla R, Briggs W, et al. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial[J]. Gastroenterology, 2000, 119(6): 1637–1648 . [DOI] [PubMed] [Google Scholar]
- 98. Fagundes C, Barreto R, Guevara M, et al. A modified acute kidney injury classification for diagnosis and risk stratification of impairment of kidney function in cirrhosis[J]. J Hepatol, 2013, 59(3): 474–481 . [DOI] [PubMed] [Google Scholar]
- 99. Mackavey CL, Hanks R. Hemostasis, coagulation abnormalities, and liver disease. [v.][J]. Crit Care Nurs Clin North Am, 2013, 25(4): 435–446., v . [DOI] [PubMed] [Google Scholar]
- 100. Caldwell SH, Hoffman M, Lisman T, et al. , and the Coagulation in Liver Disease Group. Coagulation disorders and hemostasis in liver disease: pathophysiology and critical assessment of current management[J]. Hepatology, 2006, 44(4): 1039–1046 . [DOI] [PubMed] [Google Scholar]
- 101. Montalto P, Vlachogiannakos J, Cox DJ, et al. Bacterial infection in cirrhosis impairs coagulation by a heparin effect: a prospective study[J]. J Hepatol, 2002, 37(4): 463–470 . [DOI] [PubMed] [Google Scholar]
- 102. Lee GH. Hepatic encephalopathy in acute-on-chronic liver failure[J]. Hepatol Int, 2015, 9(4): 520–526 . [DOI] [PubMed] [Google Scholar]
- 103. Wright G, Davies NA, Shawcross DL, et al. Endotoxemia produces coma and brain swelling in bile duct ligated rats[J]. Hepatology, 2007, 45(6): 1517–1526 . [DOI] [PubMed] [Google Scholar]
- 104. Shawcross DL, Sharifi Y, Canavan JB, et al. Infection and systemic inflammation, not ammonia, are associated with Grade 3/4 hepatic encephalopathy, but not mortality in cirrhosis[J]. J Hepatol, 2011, 54(4): 640–649 . [DOI] [PubMed] [Google Scholar]
- 105. Pedersen HR, Ring-Larsen H, Olsen NV, et al. Hyperammonemia acts synergistically with lipopolysaccharide in inducing changes in cerebral hemodynamics in rats anaesthetised with pentobarbital[J]. J Hepatol, 2007, 47(2): 245–252 . [DOI] [PubMed] [Google Scholar]
- 106. Häussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy[J]. Gastroenterology, 1994, 107(5): 1475–1480 . [DOI] [PubMed] [Google Scholar]
- 107. Cordoba J, Gottstein J, Blei AT. Glutamine, myo-inositol, and organic brain osmolytes after portocaval anastomosis in the rat: implications for ammonia-induced brain edema[J]. Hepatology, 1996, 24(4): 919–923 . [DOI] [PubMed] [Google Scholar]
- 108. Tofteng F, Hauerberg J, Hansen BA, Pedersen CB, Jorgensen L, Larsen FS. Persistent arterial hyperammonemia increases the concentration of glutamine and alanine in the brain and correlates with intracranial pressure in patients with fulminant hepatic failure[J]. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism 2006; 26(1): 21–27. [DOI] [PubMed] [Google Scholar]
- 109. Shawcross DL, Davies NA, Williams R, et al. Systemic inflammatory response exacerbates the neuropsychological effects of induced hyperammonemia in cirrhosis[J]. J Hepatol, 2004, 40(2): 247–254 . [DOI] [PubMed] [Google Scholar]
- 110. Rodrigo R, Cauli O, Gomez-Pinedo U, et al. Hyperammonemia induces neuroinflammation that contributes to cognitive impairment in rats with hepatic encephalopathy[J]. Gastroenterology, 2010, 139(2): 675–684 . [DOI] [PubMed] [Google Scholar]
- 111. Davies NA, Wright G, Ytrebø LM, et al. L-ornithine and phenylacetate synergistically produce sustained reduction in ammonia and brain water in cirrhotic rats[J]. Hepatology, 2009, 50(1): 155–164 . [DOI] [PubMed] [Google Scholar]
- 112. Wright GA, Sharifi Y, Newman TA, Davies N, Vairappan B, Perry HV, et al. Characterisation of temporal microglia and astrocyte immune responses in bile duct-ligated rat models of cirrhosis[J]. Liver international: official journal of the International Association for the Study of the Liver 2014; 34(8): 1184–1191. [DOI] [PubMed] [Google Scholar]
- 113. Córdoba J, García-Martinez R, Simón-Talero M. Hyponatremic and hepatic encephalopathies: similarities, differences and coexistence[J]. Metab Brain Dis, 2010, 25(1): 73–80 . [DOI] [PubMed] [Google Scholar]
- 114. Amathieu R, Nahon P, Triba M, et al. Metabolomic approach by 1H NMR spectroscopy of serum for the assessment of chronic liver failure in patients with cirrhosis[J]. J Proteome Res, 2011, 10(7): 3239–3245 . [DOI] [PubMed] [Google Scholar]
- 115. Martínez-Granados B, Morales JM, Rodrigo JM, et al. Metabolic profile of chronic liver disease by NMR spectroscopy of human biopsies[J]. Int J Mol Med, 2011, 27(1): 111–117 . [DOI] [PubMed] [Google Scholar]
- 116. Amathieu R, Triba MN, Nahon P, et al. Serum 1H-NMR metabolomic fingerprints of acute-on-chronic liver failure in intensive care unit patients with alcoholic cirrhosis[J]. PLoS One, 2014, 9(2): e89230 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Garg V, Garg H, Khan A, et al. Granulocyte colony-stimulating factor mobilizes CD34(+) cells and improves survival of patients with acute-on-chronic liver failure[J]. Gastroenterology, 2012, 142(3): 505–512.e1 . [DOI] [PubMed] [Google Scholar]
- 118. Duan XZ, Liu FF, Tong JJ, et al. Granulocyte-colony stimulating factor therapy improves survival in patients with hepatitis B virus-associated acute-on-chronic liver failure[J]. World J Gastroenterol, 2013, 19(7): 1104–1110 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Khanam A, Trehanpati N, Garg V, Kumar C, Garg H, Sharma BC, et al. Altered frequencies of dendritic cells and IFN-gamma-secreting T cells with granulocyte colony-stimulating factor (G-CSF) therapy in acute-on- chronic liver failure[J]. Liver international: official journal of the International Association for the Study of the Liver 2014; 34(4): 505–513. [DOI] [PubMed] [Google Scholar]
- 120. Anderlini P, Przepiorka D, Champlin R, et al. Biologic and clinical effects of granulocyte colony-stimulating factor in normal individuals[J]. Blood, 1996, 88(8): 2819–2825 . [PubMed] [Google Scholar]
- 121. de Haas M, Kerst JM, van der Schoot CE, et al. Granulocyte colony-stimulating factor administration to healthy volunteers: analysis of the immediate activating effects on circulating neutrophils[J]. Blood, 1994, 84(11): 3885–3894 . [PubMed] [Google Scholar]
- 122. Thomas JA, Pope C, Wojtacha D, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function[J]. Hepatology, 2011, 53(6): 2003–2015 . [DOI] [PubMed] [Google Scholar]
- 123. Chavez-Tapia NC, Mendiola-Pastrana I, Ornelas-Arroyo VJ, et al. Granulocyte-colony stimulating factor for acute-on-chronic liver failure: systematic review and meta-analysis[J]. Ann Hepatol, 2015, 14(5): 631–641 . [PubMed] [Google Scholar]
- 124. Shi M, Zhang Z, Xu R, et al. Human mesenchymal stem cell transfusion is safe and improves liver function in acute-on-chronic liver failure patients[J]. Stem Cells Transl Med, 2012, 1(10): 725–731 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Yuan SF, Jiang T, Sun LH, et al. Use of bone mesenchymal stem cells to treat rats with acute liver failure[J]. Genet Mol Res, 2014, 13(3): 6962–6980 . [DOI] [PubMed] [Google Scholar]
- 126. Zhang Y, Chen XM, Sun DL. Effects of coencapsulation of hepatocytes with adipose-derived stem cells in the treatment of rats with acute-on-chronic liver failure[J]. Int J Artif Organs, 2014, 37(2): 133–141 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Tögel FE, Westenfelder C. Mesenchymal stem cells: a new therapeutic tool for AKI[J]. Nat Rev Nephrol, 2010, 6(3): 179–183 . [DOI] [PubMed] [Google Scholar]