

Infectious diseases caused by drug-resistant fungi are an increasing threat to public health because of the high mortality rates and high costs associated with treatment. Thus, understanding of the molecular mechanisms of drug resistance is of crucial interest for the medical community. Here we investigated the role of regulated protein mistranslation, a characteristic mechanism used by C. albicans to diversify its proteome, in the evolution of fluconazole resistance. Such codon ambiguity is usually considered highly deleterious, yet recent studies found that mistranslation can boost adaptation in stressful environments. Our data reveal that CUG ambiguity diversifies the genome in multiple ways and that the full spectrum of drug resistance mechanisms in C. albicans goes beyond the traditional pathways that either regulate drug efflux or alter the interactions of drugs with their targets. The present work opens new avenues by which to understand the molecular and genetic basis of microbial drug resistance.
KEYWORDS: Candida albicans, fluconazole, LOH, aneuploidy, codon ambiguity, drug resistance evolution, phenotypic variability, protein mistranslation
ABSTRACT
Regulated erroneous protein translation (adaptive mistranslation) increases proteome diversity and produces advantageous phenotypic variability in the human pathogen Candida albicans. It also increases fitness in the presence of fluconazole, but the underlying molecular mechanism is not understood. To address this question, we evolved hypermistranslating and wild-type strains in the absence and presence of fluconazole and compared their fluconazole tolerance and resistance trajectories during evolution. The data show that mistranslation increases tolerance and accelerates the acquisition of resistance to fluconazole. Genome sequencing, array-based comparative genome analysis, and gene expression profiling revealed that during the course of evolution in fluconazole, the range of mutational and gene deregulation differences was distinctively different and broader in the hypermistranslating strain, including multiple chromosome duplications, partial chromosome deletions, and polyploidy. Especially, the increased accumulation of loss-of-heterozygosity events, aneuploidy, translational and cell surface modifications, and differences in drug efflux seem to mediate more rapid drug resistance acquisition under mistranslation. Our observations support a pivotal role for adaptive mistranslation in the evolution of drug resistance in C. albicans.
IMPORTANCE Infectious diseases caused by drug-resistant fungi are an increasing threat to public health because of the high mortality rates and high costs associated with treatment. Thus, understanding of the molecular mechanisms of drug resistance is of crucial interest for the medical community. Here we investigated the role of regulated protein mistranslation, a characteristic mechanism used by C. albicans to diversify its proteome, in the evolution of fluconazole resistance. Such codon ambiguity is usually considered highly deleterious, yet recent studies found that mistranslation can boost adaptation in stressful environments. Our data reveal that CUG ambiguity diversifies the genome in multiple ways and that the full spectrum of drug resistance mechanisms in C. albicans goes beyond the traditional pathways that either regulate drug efflux or alter the interactions of drugs with their targets. The present work opens new avenues to understand the molecular and genetic basis of microbial drug resistance.
INTRODUCTION
Fungal infections are an increasingly serious health problem because of immune deficiencies caused by diseases like HIV/AIDS and cancer therapies, as well as prolonged antibiotic treatments (1). Deep-seated and disseminated infections are difficult and extremely costly to treat and are normally associated with high mortality rates. In the United States alone, the costs of antifungal therapy have reached $8 billion/annum (2). Resistance to the commonly used azoles is increasing, and alternative antifungals, such as encapsulated amphotericin B or echinocandins, are expensive and increase the cost of antifungal therapy dramatically.
Antifungal resistance has been studied intensively and involves DNA mutations, genome plasticity, cell signaling, and gene expression alterations (3–5). In the specific case of azoles, resistance is due mainly to mutations in the ergosterol biosynthesis pathway targeted by the drug and drug efflux. Lanosterol 14α-demethylase (encoded by ERG11) is a key enzyme in the ergosterol biosynthesis pathway and the direct target of azole antifungals. Overexpression of UPC2, the transcription factor regulating ERG11, or aneuploidy events that increase the ERG11 copy number lead to increased azole resistance (6, 7). Gain-of-function mutations in TAC1 and MRR1 (7, 8), transcription factors that positively regulate the transcription of efflux pumps encoded by the CDR1/CDR2 or MDR1 genes (5, 9), also result in increased drug resistance. Types of mutations that result in acquired resistance include genome rearrangements, protein sequence changes (4, 5), aneuploidy and loss of heterozygosity (LOH) (6, 10–13), and increased mutagenesis due to loss of mismatch repair or elevated levels of DNA double-strand break repair (3).
Candida albicans, the most prevalent pathogen of humans, has a unique codon usage system whereby translational fidelity is modulated by environmental cues (14). A mutant serine tRNA is recognized and aminoacylated by both leucyl- and seryl-tRNA synthetases (LeuRS and SerRS, respectively) (15), yielding a charged tRNA that can incorporate either Leu or Ser at CUG codons. This leads to proteome-wide plasticity, such that each protein in the cell is represented by a mixture of polypeptides containing Ser or Leu at such sites. The basal level of Ser and Leu incorporated at CUG sites (3% leucine and 97% serine in rich medium) differs in different ecological niches and under different environmental conditions, contributing to proteomic and phenotypic diversification (16). In a previous study, we increased Leu misincorporation at CUG sites to 22% by inserting a copy of a yeast Leu tDNACAGLeu gene into the genome of C. albicans strain SN148 and could show that this strain was slightly more tolerant to fluconazole than the wild-type (WT) strain when grown on solid agar plates containing this antifungal (16). This result suggested that CUG mistranslation might have a role in antifungal drug tolerance. However, the underlying molecular mechanism by which proteomic plasticity results in fluconazole resistance is unknown. As mistranslation is associated with antibiotic resistance in bacteria (17–19), we hypothesized that it may also be relevant to the evolution of fluconazole resistance in C. albicans.
In the present study, we investigated how CUG mistranslation contributes to the acquisition of fluconazole resistance by evolving WT and hypermistranslating strains of C. albicans in parallel in the absence and presence of fluconazole. These strains were then characterized by genome resequencing, array-based comparative genome hybridization (CGH), and gene expression analysis. We found that mistranslation accelerated the acquisition of fluconazole resistance in C. albicans and revealed a number of genomic features and key genes that provide insights into how CUG mistranslation promotes resistance to fluconazole.
RESULTS
To clarify the role of mistranslation in fluconazole resistance, we used C. albicans strains that incorporate Leu at CUG sites at different levels; a control strain (T0) incorporates the WT levels of 3% Leu and 97% Ser, and a hypermistranslating strain (T1) incorporates 22 and 78% Leu and Ser at the same sites, respectively (16). We asked initially if mistranslation alters the frequency of acquisition of fluconazole resistance during evolution. For this, strains T0 and T1 were evolved by serial passage in increasing concentrations of fluconazole (Fig. 1; see Fig. S1 in the supplemental material) and genetically tested at three different time points, i.e., at the beginning of the experiment (no fluconazole, no suffix), at medium fluconazole concentrations (16 to 32 µg/ml, suffix FM), and at a high fluconazole concentration (256 µg/ml, suffix FH) (Fig. 1; Fig. S1). Both strains were also evolved in the absence of fluconazole to test for effects due to mistranslation alone (suffix NF).
FIG 1 .

Fluconazole resistance profiles during evolution. (A) During evolution with fluconazole (FLZ), control strain TO (3.2% natural mistranslation; blue line) and hypermistranslating strain T1 (22.5% leucine misincorporation; red line) showed increased resistance to the drug over time yet revealed marked differences in the speed of resistance acquisition. Both strains evolved without (w/o) the drug (green and gray lines) did not show increased resistance. (B) E tests after 10 days of evolution. Hypermistranslating strain T1 rapidly adapted to the drug and showed complete resistance to fluconazole, while WT strain T0 was still sensitive.
Experimental design used in this study. RNA for gene expression analysis was extracted from growth duplicates of the same strains (Tx, TxFM, and TxFH) of which the whole genome was sequenced (WGS; genomic DNA extraction). Additionally, genomic DNA of three T1FH clones was extracted for array-based CGH analysis. The intensity of the red color of arrows indicates increasing fluconazole concentrations. Download FIG S1, EPS file, 0.4 MB (448.3KB, eps) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
When evolved without fluconazole, the T0NF and T1NF strains did not exhibit a visible difference in resistance (Fig. 1, green and gray lines). In contrast, the evolution of either strain T0 or T1 in the presence of fluconazole resulted in the acquisition of high-level resistance (MIC of 256 μg/ml) during evolution with no difference in the mutation rate, albeit with a marked difference in the speed of acquisition. The hypermistranslating T1 strain became resistant (T1FH, MIC of 256 μg/ml) within 10 days of passaging (~2 days per passage), while control strain T0 began to show increased resistance after 1 week of evolution (T0FM) and reached a MIC of 256 µg/ml (T0FH) after 1 month of passaging (Fig. 1). Thus, increased levels of mistranslation appeared to speed up the emergence of fluconazole resistance. To address the generality of this phenomenon, we asked if mistranslation also promotes resistance to caspofungin, an antifungal drug of clinical relevance belonging to the echinocandin class. While the MICs for both strains T0 and T1 were in the same range (0.094 to 0.125 µg/ml), only the T1 hypermistranslating strain produced microcolonies within the entire inhibition ellipse of the E test. These microcolonies also grew in liquid medium containing caspofungin concentrations of up to 32 µg/ml (Fig. S2).
Caspofungin susceptibility test. E tests of strains T0 and T1 revealed similar MICs (0.094 to 0.125 µg/ml), yet only hypermistranslating strain T1 produced microcolonies within the entire inhibition ellipse. These microcolonies also grew in liquid medium containing caspofungin concentrations of up to 32 µg/ml. Download FIG S2, EPS file, 0.8 MB (871.8KB, eps) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differences in resistance to fluconazole.
To clarify how resistance to fluconazole evolved in both strains T0 and T1, we resequenced the genomes of the evolved strains by using the Illumina sequencing platform and analyzed gene expression with the Agilent DNA microarray platform. The T0 strain acquired a known gain-of-function mutation (V877F) in transcription factor MRR1 (20), a positive transcriptional regulator of the multidrug efflux pump gene MDR1; consistent with this, MDR1 expression was upregulated 5.3-fold (Table S1; Fig. S3). The hypermistranslating T1 strain was remarkably different; it acquired a previously described A736V gain-of-function mutation in the transcriptional activator TAC1, which drives high levels of expression of the CDR1 and CDR2 genes, which encode the ABC transporters (21) (CDR1, 4.8-fold; CDR2, 3.9-fold; Table S1; Fig. S3). Analysis of the genomic DNA revealed LOH of ERG11, the target of fluconazole, in T1FM and T1FH; no such LOH event was observed in T0FM and T0FH. In addition, a previously unknown mutation (S35C) in the oxidosqualene cyclase gene ERG7 was also detected in T0FM, T0FH, and T1FM (Table S1). Moreover, the expression of genes encoding components of the ergosterol biosynthesis pathway, the molecular target of fluconazole, were upregulated in both T0 and T1 fluconazole-resistant strains (Fig. S3). Taken together, these results suggest that both strains activated the common mechanisms of azole resistance, yet they did so via different drug efflux pathways and through different types of genomic alterations.
The left panel is a schema depicting major events that allow strain T1 to acquire fluconazole resistance rapidly. Drug efflux in strain T1 seems to be mediated mainly via the ABC superfamily transporters CDR1 and CDR2, while in strain T0, drug efflux is likely due to the multidrug pump encoded by MDR1. The right panel shows GO BP heatmaps that depict log-fold changes in gene expression. Green letters represent DEGs in T1FH/T1NF, i.e., those involved in steroid biosynthesis, glucose transport, and the chitin biosynthetic process. Download FIG S3, EPS file, 1.4 MB (1.4MB, eps) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Nonsynonymous codon changes in TAC1, MRR1, and ERG7. N.ofAA, position of amino acid; T0.Codon, codon in control strain T0; T0.AA, amino acid in control strain T0; S.Codon, codon in the strain tested; S.AA, amino acid in the strain tested. Download TABLE S1, DOCX file, 0.02 MB (19.2KB, docx) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genomic alterations induced by mistranslation and fluconazole.
A combination of whole-genome sequencing (WGS) and high-density array-based CGH data analysis provided deeper insight into the role of mistranslation in genome evolution.
By comparing genotype changes that were heterozygous in the nonevolved control strain and homozygous in the other samples analyzed, we found that strain T0 accumulated a moderate number of such events (<500) during evolution, regardless of whether fluconazole was present (T0FM and T0FH) or absent (T0NF). In contrast, the hypermistranslator T1 accumulated 2,193 LOH events during evolution in the absence of fluconazole (T1NF) and >5,100 LOH events in the presence of fluconazole (T1FM and T1FH). Thus, mistranslation alone resulted in the accumulation of a higher number of LOH events during evolution and the combination of mistranslation and fluconazole (strain T1) more than doubled the number of LOH events. Interestingly, in the resistant T1FH strain, a small group of open reading frames were especially rich in such LOH events, with orf19.2850 (yeast ortholog involved in telomeric silencing), MEC1 (encodes a cell cycle checkpoint protein with a role in genome integrity), HAL9 (yeast ortholog involved in salt tolerance), and INT1 (has a role in morphogenesis and adhesion) having more than 51 LOH events (median, 5 LOH events).
Further, the evolution of the hypermistranslating T1 strain resulted in the appearance of two large LOH regions that include most of the above-mentioned events. A large LOH region on chromosome 5 (Chr5) appeared exclusively in the presence of fluconazole (T1FM and T1FH), while an LOH tract in the left arm of ChrR appeared regardless of drug treatment (T1NF, T1FM, and T1FH) (Fig. S4), suggesting that it was already present in the parental T1 strain or that it occurs at a recombination hotspot.
LOH events in quickly evolving strain T1 (WGS data). While LOH on Chr5 appeared only under drug pressure (T1FM and T1FH), the LOH on the left arm of ChrR appeared regardless of fluconazole treatment (T1NF, T1FM, and T1FH). Download FIG S4, EPS file, 0.4 MB (376.7KB, eps) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Notably, in both LOH regions, several loci encoding aminoacyl-tRNA synthetases and tRNA modification enzymes lost heterozygosity in strain T1 during evolution. We observed homozygosis of the cytosolic leucyl-tRNA synthetase gene locus (LeuRS; CDC60, ChrR) regardless of drug treatment. LeuRS charges the atypical tRNACAGSer with leucine and is central to Leu/Ser ambiguity in mistranslation in C. albicans (22). Similar LOH events during evolution of strain T1 involved loci encoding the mitochondrial (MSM1) and cytosolic (MES1) methionyl-tRNA synthetases; the mitochondrial tryptophanyl-tRNA synthetase (MSW1); and the cytosolic phenylalanyl (FRS1)-, putative prolyl (PRS)-, putative aspartyl (DPS1-1)-, and tryptophanyl (WRS1)-tRNA synthetases; as well as orf19.3956 (orthologs have glutaminyl-tRNA synthase activity), orf19.2387 (a putative tRNA-Pro synthetase), and orf19.2382 (protein similar to isoleucyl-tRNA synthetase). Apart from WRS1 (Chr1), these genes are all located within the LOH regions of ChrR and Chr5, suggesting that the tRNA aminoacylation system might be a driving force for the LOH on those chromosomes (Fig. 2; Fig. S4).
FIG 2 .

YMAP visualization of SNP-CGH data for three T1FH clones. The copy numbers and SNP-allele ratios of all three T1FH samples were visualized with YMAP (49). Changes in the copy number estimate for regions relative to the parental strain are illustrated by dark bars drawn up- or downward, depending on the direction and magnitude of the change. These strains appear to be tetraploid on the basis of the copy number estimates for Chr7 (and most of ChrR in clone T1FHb), which are 3/4 of the other chromosomes. Color illustrates SNP status across regions. Heterozygous regions are gray, white regions do not have SNPs in the SC5314 reference sequence, and cyan is homozygous “a” alleles (e.g., aaaa on Chr5 in all three clones and ChrR in T1FHa and T1FHc and aaa in T1FHb). Intermediate ratios are indicated by intermediate colors. Blue shade represents more copies of “a” alleles (e.g., aab on the central trisomic region of ChrR in T1FHb), and purple shade represents more copies of the “b” alleles (e.g., abb on Chr7 in all three clones). The portion of ChrR in clone T1FHb to the right of the rDNA region (blue dot) is present in five copies. Major repeat sequences are represented by black dots.
Since LOH events on Chr5 appear frequently in fluconazole-resistant C. albicans isolates (10), these data support the idea that mistranslation, LOH of Chr5, and fluconazole resistance are linked. To determine if genes affected by LOH events during evolution were selected because of their serine codon usage, we computed the relative synonymous codon usage (RSCU) of the seven serine codons of strains T1 and T1FH in all genes (with Anaconda [23]) and compared the two sets codon by codon. We found statistically significant differences in UCA, UCC, and UCG usage, which was decreased in the LOH-affected genes of the nonevolved T1 strain, and in UCG usage, which was decreased in the LOH-affected genes of T1FH (Table S2A).
CUG codon usage. (A) Statistical comparison of the altered set of genes for each evolutionary event and strain (T1 and T1FH genes affected by LOH and T1FH genes affected by copy number variations) and the rest of the genome. Significant results show that T1 genes affected by LOH are depleted of UCA, UCC, and UCG codons; T1FH genes affected by LOH are depleted of UCG codons; and T1FH genes affected by copy number variations are enriched in UCA codons and depleted of UCC and UCG codons. RSCU values for serine codons of each gene were calculated with Anaconda (23) and compared through Student t tests corrected by the Bonferroni method. (B) Numbers of CUG codons that differ in both alleles (a and b) in genes that lost heterozygosity or changed copy number in T1FH (data from CGD). Notably, the large LOH regions on Chr5 and ChrR are aaaa (see Fig. 2), which caused the highlighted changes in CTG codon usage. All of the genes that have a positive difference led to a reduction in CTG codons (blue) by changing CTG to other codons, while those having a negative difference increased the number of CTG codons (green) by changing other codons to CTG. Further, all of the genes with an increased copy number (red) had a positive difference, while those with a reduced copy number (orange) had a negative difference. Download TABLE S2, PDF file, 0.1 MB (141.6KB, pdf) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Interestingly, the number of CUG codons differs between alleles (a and b) of genes that lost heterozygosity in T1FH (Table S2B); genes with a positive difference (a > b) underwent a reduction in the number of CTG codons, and genes with a negative difference (a < b) had an increase in the number of CTG codons. This suggests that the loss of one allele (Table S2B) had a balancing effect on the number of CTG codons.
As mentioned above, the LOH in the left arm of ChrR was present in all evolved T1 clones, regardless of whether cells were untreated (T1NF) or treated (T1FM and T1FH) with fluconazole (Fig. S4). The results imply that homozygosis in this region was already present as a standing variation in the starting population and was likely selected because of the presence of genes that are important in overcoming the potentially lethal effect of increased mistranslation. Because of their immediate availability and higher initial frequency, this standing variation (beneficial alleles) may speed up adaptation to new environments if compared to the time required for new beneficial mutations to arise (24). This LOH region on ChrR is enriched in genes with gene ontology (GO) terms related to peptide transport, cellular and nucleic acid metabolic processes, chromatin silencing at telomeres, and tRNA aminoacylation (Table S3). Of note, genes involved in transcriptional silencing by heterochromatin and aminoacylation were enriched among genes with sequence changes, and they represent two critical steps required for the accurate transfer of genetic information, a feature of particular importance under increased mistranslation.
GO BP categories of genes located within the region of ChrR that lost heterozygosity in all evolved T1 strains regardless of whether cells were untreated (T1NF) or treated with the drug (T1FM and T1FH). For genes with >10 LOH events, the corrected P value is <0.005. Download TABLE S3, DOCX file, 0.02 MB (19.6KB, docx) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Copy number variation upon fluconazole exposure.
WGS revealed that the evolution of strain T1 in the presence of fluconazole also led to loss of chromosomal repeat regions in Chr6, Chr4, and Chr2 and of a few telomere-proximal genes (e.g., CTA24, RRN3, TLO5). Furthermore, it showed that evolution of strain T1 in the presence of fluconazole was accompanied by the apparent gain and loss of regions of Chr1, Chr4, Chr5, and ChrR. Specifically, T1FM had increased copies of Chr1 (164001 to 168200), Chr4 (46800 to 47200 and 518001 to 530000), Chr5 (744001 to 752000 and 860001 to 876000), and ChrR (1884001 to 1898000). Within these amplified chromosomal regions were, apart from the genes of the rDNA locus and repeat regions of Chr1, genes functioning in the regulation of transcription. Many of the amplified genome regions in strain T1FM were not detectable at the end of evolution in the presence of fluconazole. Rather, T1FH showed gains almost exclusively on Chr7 (150001 to 170000, 262001 to 328000, 318001 to 324000, 360001 to 380000, and 482001 to 576000), mainly located in the region between WHI3 and MRS7b. The RSCU for serine codons in genes affected by copy number variation in T1FH showed increased usage of UCA and lower usage of UCC and UCG than the rest of the genome. Notably, the balancing effect of CUG codon usage observed for LOH was also detectable for copy number variation (Table S2).
To gain deeper insight into the segmental aneuploidies, we performed single nucleotide polymorphism (SNP)-CGH analysis of three T1FH clones (Fig. 2). Our CGH analysis suggested that all of our clones were nearly tetraploid, with Chr7 being trisomic. It appears that either the b homolog reduplicated or one copy of the a homolog was lost, leading to the Chr7 abb trisomy in T1FH (Fig. 2). One of the T1FH clones (T1FHb) was also trisomic (aaa) for the common LOH region of ChrR for which the other two clones were tetraploid (aaaa). In the T1FHb clone, the LOH region (aaa) of ChrR was followed by an additional trisomic LOH region (abb) (Fig. 2). Another feature of this clone was the copy number losses that appeared to the right of the rDNA locus of ChrR. WGS showed that during evolution in the drug, T1FM increased the copy number of the rDNA locus, albeit transiently, as the chromosomal amplifications disappeared in the highly resistant T1FH strain. Thus, our results support the observations that the rDNA locus is a fragile site that can cause copy number variation on chromosome ends (25, 26) and additionally might be relevant for a rapid adaptation to fluconazole.
Gene expression deregulation induced by mistranslation.
Comparing the gene expression patterns of all of the strains and conditions used in this study revealed that during evolution in the drug, strain T0 showed a fluconazole-induced stress response, while strain T1 upregulated several mating-related genes, as well as genes encoding sugar transporters (Fig. 3). To distinguish the effects of adaptation to fluconazole from the effects of mistranslation, we compared the mRNA expression patterns of the hypermistranslating T1 strain that evolved in the presence (T1FH) and absence of the drug (T1NF) (Fig. S5). Gene set enrichment analysis (GSEA) (27) of T1FN detected deregulation of expression of translation genes, including those involved in tRNA and rRNA processing, tRNA methylation and modification, mRNA splicing, cytosolic and mitochondrial ribosomal subunit biosynthesis, and ribosomal biogenesis and assembly (Fig. S5). In T1FH, treatment with fluconazole resulted in downregulation of genes related to ribosome biogenesis, e.g., those required for maturation of large-subunit rRNA genes (Fig. S5). These GSEA results suggest that the hypermistranslating strain (T1) evolved in the absence of fluconazole had remodeled tRNA and rRNA processing and that addition of fluconazole repressed protein synthesis (Fig. S5). Mistranslation also upregulated genes involved in organic acid and amino acid metabolic processes and downregulated genes involved in glycoside and carbohydrate metabolic processes, cellular iron homeostasis, oxidation-reduction processes, and glycolysis (Fig. S6). The FMP45 gene, which is involved in sensitivity to toxic ergosterol analogs and is induced during mating, was downregulated during evolution of the hypermistranslating T1 strain in the absence of fluconazole (T1NF) but upregulated in the presence of the drug (T1FH) (Fig. S6), suggesting that FMP45 may also play a role in fluconazole resistance.
FIG 3 .
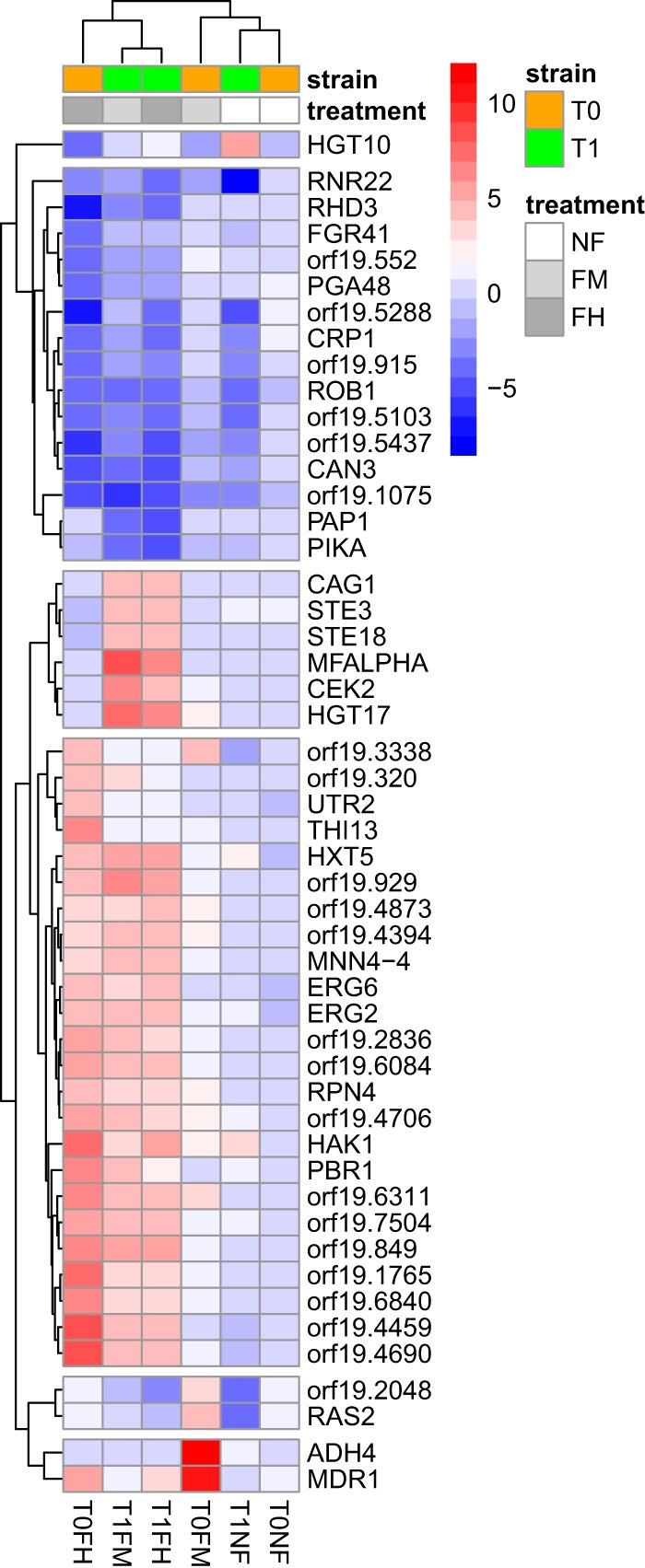
Gene expression heatmap. Depicted are DEGs with a 4-log-fold change for at least one comparison and a false discovery rate-adjusted P value of <1e−18.
Gene set enrichment networks. (A) Network displaying gene sets enriched in hypermistranslating strain T1FH relative to strain T0FH evolved in the presence of fluconazole. (B) Network displaying gene sets enriched in strain T1 evolved in the presence of a high fluconazole concentration (T1FH) relative to strain T1 evolved without the drug (T1NF). (C) Network displaying gene sets enriched in hypermistranslating strain T1 evolved without the drug (T1NF) versus nonevolved strain T1. Edge width represents the number of shared genes, node size represents the number of annotated genes, and node color represents the enrichment based on the number of upregulated (red) or downregulated (blue) genes in the set. The 10 genes ranked highest in each gene set are shown. Download FIG S5, PDF file, 0.9 MB (979.1KB, pdf) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes expressed differentially in T1FH and T1NF and functional analysis. (Top) Venn diagram in which each colored area represents a set of DEGs (differential expression threshold of ≥3.0, adjusted P value of <0.01). Only one gene, FMP45, is significantly upregulated in T1FH and downregulated in T1NF. The top two GO terms for each condition are shown. (Bottom) GO BP categories enriched below a P value of 0.01 in all of the expression sets shown in the Venn diagram. Download FIG S6, PDF file, 1 MB (1MB, pdf) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Mating and cell surface alterations.
WGS and CGH also showed that T1 lost the MTLa locus (including the nonsex genes PAP1, PIKa, and OBPa) during evolution in fluconazole (Fig. 2 and 4; Fig. S7) and increased the copy number of several genes on Chr7 involved in mating, transport, and translation, including orf19.6583, orf19.7038, FLU1, WHI3, TOM70, NBP2, YCF1, and GCN2 (Table 1).
FIG 4 .

Highlighted features of quickly evolving and highly resistant strain T1. Mistranslation appears to affect the phosphatidylinositol signal transduction pathways that control several cell cycle events, cell membrane and cell wall remodeling, as well as protein synthesis and turnover, via GCN2, most of which are known to enhance drug resistance. The main features of highly drug-resistant strain T1FH are depicted, i.e., loss of MTLa and the nonsex gene PIKa; phosphatidylinositol signaling and downstream targets such as DNA repair, cell wall remodeling, and mating; GCN2-mediated modulation of protein synthesis; drug efflux pumps; and bud neck formation.
TABLE 1 .
Copy number variation in T1FHa
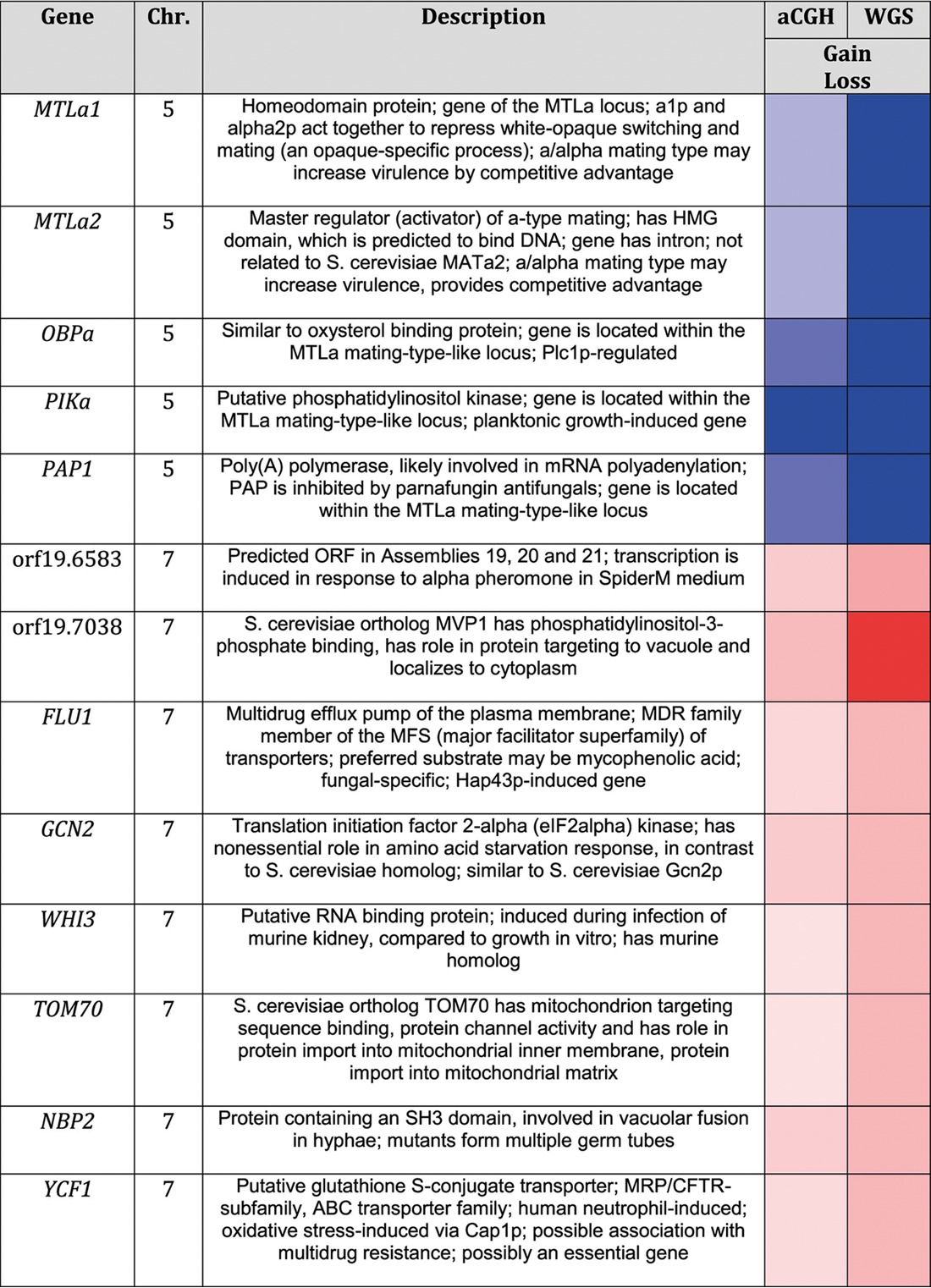
Genes that revealed high concordance in copy number changes in both SNP-CGH and WGS analysis are shown. Apart from the MTL genes that were also lost in T1FM, all of the gene alterations listed appeared exclusively in strain T1FH. Colors: red, copy number gain; blue, copy number loss. Increasing color intensity reflects an increase in gain or loss, respectively.
(A) Flow cytometry profiles of two T1FH clones. Red, diploid control; blue, sample. The T1FH profile indicates a mixed population of diploids, tetraploids, and some cells with greater ploidy. (B) Confirmation of loss of the MTLa locus in T1FH by targeted PCR. Colony PCR products of the MTLa1 (red) and MTLalpha1 (blue) genes visualized on an agarose gel. MTLa1 is absent from all tested clones of the evolved and highly resistant strain T1FH. For the sequences of the MTLa primers used, see I. Miranda, R. Rocha, M. C. Santos, D. D. Mateus, G. R. Moura, L. Carreto, and M. A. Santos, PLoS One 2:e996, 2007. Download FIG S7, EPS file, 0.4 MB (452.4KB, eps) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In addition to those genome alterations, in T1FH, we observed a 6.5-fold increase in the expression of MFalpha, the alpha factor mating pheromone gene, and a gene set enrichment of mating-specific terms like bud neck and ascospore formation, pheromone-dependent transcription involved in conjugation with cellular fusion, and terms related to virulence, like adhesion to a host and immune response (Fig. 3; Fig. S6). These gene expression alterations in T1FH are possibly related to loss of the MTLa locus and/or the LOH of Chr5 (28–30).
DISCUSSION
Mistranslation is linked to elevated mutagenesis and the appearance of drug resistance in bacteria (17, 31), but its role in eukaryotes is still poorly understood. In this study, we used experimental evolution to examine the ability of a C. albicans strain that mistranslates at high levels to acquire resistance to fluconazole. By comparing changes in a hypermistranslating strain with those in a WT strain subjected to or excluded from fluconazole treatment, we could detect contributions of mistranslation to drug resistance at both the genome and transcription levels. During evolution in the absence of fluconazole, the resistance to fluconazole of both the WT T0 and hypermistranslating T1 strains was similar. In contrast, strain T1 became resistant to fluconazole at a much earlier time point during evolution in the drug. Our data suggest that different ways of upregulating drug efflux pumps, together with genomic changes in drug target genes, may speed up the acquisition of resistance in mistranslating strains. Increased drug efflux in the T0FM strain was likely mediated via the multidrug efflux pump encoded by MDR1, while T1FM overexpressed the ABC superfamily transporter genes CDR1 and CDR2. Additionally, in T1FM, ERG11, the gene encoding the fluconazole target protein, became homozygous. An elevated copy number of the FLU1 gene and the open reading frame for a putative multidrug resistance protein (orf19.3218) may also have contributed to fluconazole resistance in T1.
Recent literature suggests that a common mechanism for the acquisition of drug resistance involves LOH, which is often accompanied by aneuploidy (11). In T1FH, the two major LOH regions of ChrR and Chr5 include a large proportion of genes involved in both translation and drug resistance. This suggests that allele-specific gene functions may contribute to rapid adaptation to azoles and/or to stabilization of the mistranslation state and that CTG codon usage seems to have a balancing effect on such genomic events (Table S2). Further, the tRNA aminoacylation system might be a driving force for the LOH in hypermistranslators.
Interestingly, mistranslation led to both upregulation of amino acid metabolism and mutations in tRNA synthetases (T1NF), with the latter being increased under drug pressure (T1FH). For example, PIKa encodes a phosphatidylinositol kinase and is located within the MTLa locus that was lost on Chr5. While ΔMTLa1 ΔMTLa2 allows growth even in the highest levels of fluconazole (32), ΔPIKa may play a role in translation initiation. In Saccharomyces cerevisiae conditional mutants of the PIKa ortholog, Pik1 controls translation initiation independently of TOR (33). S. cerevisiae Pik1 mutation leads to a 4-fold increase in the phosphorylation of translation initiation factor 2 alpha (elf2α). Further, the general amino acid control (GCN) response induces the amino acid biosynthesis pathways upon the accumulation of uncharged tRNAs (34, 35). The increase in the copy number of elf2α kinase GCN2 observed in the T1FH strain suggests that high levels of elf2α phosphorylation could be a specific translational modification required for adaptation to fluconazole.
Another feature that appeared in T1 during drug resistance evolution was the accumulation of a large number of SNPs and codon changes in the PGA, IFF, and ALS genes, which encode glycosyl-phosphatidylinositol (GPI)-anchored proteins. Additionally, the AGM1 gene, which is involved in the chitin biosynthetic pathway (synthesis of UDP-N-acetylglucosamine), was specifically upregulated in evolved T1 strains (Fig. S3). N-Acetylglucosamine is a moiety of the synthesis of GPI anchors and a building block of chitin (36). Sequences encoding GPI-anchored adhesins are rich in CUG codons, and a recent study demonstrated that CUG mistranslation enhances the ability of C. albicans to adhere to different surfaces, especially under stress conditions (37). Remodeling of the cell wall and modification of adhesion properties are important features of drug resistance and virulence (38, 39). Consistent with this, T1FH flocculated and showed strong adhesion to surfaces in liquid medium (data not shown). Hence, by expanding the variability of cell wall proteins, CUG codon ambiguity could allow C. albicans to accelerate the acquisition of antifungal resistance.
Several serine/threonine kinases and serine/threonine phosphatases lost heterozygosity and/or changed gene expression levels in T1FH. Here, overexpression of the SHA3 gene, encoding a Ser/Thr kinase, which regulates glucose transport, is likely responsible for the upregulation of sugar import genes in the resistant T1FH strain. Members of the transporter-encoding HGT and HXT gene families were also overexpressed. As the genes regulating the first steps of glycolysis (PGI1, PFK1, and PFK2) were downregulated and INO1, which encodes the key enzyme of myo-inositol biosynthesis, was upregulated, we assume that the imported glucose is channeled into myo-inositol biosynthesis in T1FH, which is essential for growth and virulence in C. albicans (40). Additionally, the Mec1 gene, which is located on the right arm of Chr5, was one of the few genes that were highly enriched in LOH events (Mec1, 60 [median, 5] LOH events) exclusively in T1 strains exposed to fluconazole. Mec1 is a serine threonine- and leucine-rich putative phosphatidylinositol kinase that, upon DNA damage, phosphorylates a large set of proteins, and together with its yeast ortholog, Mec1 was repeatedly linked to genome instability, telomere maintenance, and drug response (3, 41, 42). Flow cytometry (Fig. S7) suggested that T1FH cells have multiple ploidy states, including diploids, tetraploids, and a smaller population of cells with >4c ploidy. Alterations in ploidy were recently shown to occur soon after C. albicans cells are exposed to fluconazole, and it is thought that they provide the first step in the formation of aneuploid cells with a wide range of phenotypic variability (43).
Taken together, our data suggest that mistranslation mediated more rapid evolution of fluconazole resistance via a range of mechanisms, including the classical effects on efflux and ergosterol biosynthesis, among others. Dissection of the genes necessary and sufficient for this evolution will require additional studies. In particular, parallel evolution of multiple clones of both hypermistranslating and WT strains is necessary to clarify the adaptive and mechanistic relevance of the multiple and important mutations identified in this study. Our data leave no doubt that mistranslation accelerates the evolution of resistance to fluconazole in C. albicans; whether mistranslation is a major driver of mutagenesis in drug stress will be fascinating to study in future work.
MATERIALS AND METHODS
Strains and growth conditions.
The C. albicans strains used in this study were engineered to exhibit different levels of leucine misincorporation at serine CUG codons. The T0 control strain incorporates 3.2% Leu and 96.8% Ser at CUG codons (normal level of mistranslation in C. albicans); T1 incorporates 22.5% Leu and 77.5% Ser at CUG codons (16). All strains were constructed with the SN148 parental strain (arg4Δ/arg4Δ leu2Δ/leu2Δ his1Δ/his1Δ ura3Δ::imm43/ura3Δ::imm43 iro1Δ::imm43/iro1Δ::imm43) (44) by homologous recombination as described in reference 16.
Incorporation of Leu and Ser at CUG codons was determined with a previously developed gain-of-function reporter (16). Fluorescence was quantified with a Zeiss MC80 Axioplan 2 light microscope equipped for epifluorescence microscopy with the HE38 filter set (Carl Zeiss AG). Images were taken with an AxioCam HRc camera and analyzed with ImageJ software (https://imagej.nih.gov/ij/) as previously described (16).
Strains were grown in synthetic defined (SD) medium without uracil containing 0.67% yeast nitrogen base, 2% glucose, and 0.2% dropout mix (with 2% agar for solid medium only) at 30°C.
Experimental evolution.
Resistance to fluconazole was monitored by the E-test method (AB Biodisk, BioMérieux). Cells were grown to mid-log phase, washed in 1× phosphate-buffered saline, and diluted to an optical density at 600 nm of 0.015. A 150-μl volume of cells was plated on SD minus uracil agar plates (pH 7) with glass beads and allowed to dry for 15 to 30 min before a fluconazole E-test strip (0.016 to 256 μg/ml; AB Biodisk) was applied. Plates were incubated at 30°C for 48 h, and the MIC was determined as the concentration at which the first growth inhibition ellipse occurred. To perform the evolution and E-test experiments in parallel, cells were incubated in liquid (SD minus uracil) medium adjusted to 1× and 2× the last measured fluconazole (Sigma-Aldrich) MIC. Depending on growth in the respective concentrations of fluconazole, passages were pursued until strains were able to grow in liquid medium containing a fluconazole concentration of 256 μg/ml (Fig. S1). Additionally, control clones were evolved for an equal number of passages without fluconazole to study the effects of mistranslation and the antifungal (T0NF and T1NF).
Nucleic acid extraction.
RNA and DNA were extracted from strains at the beginning (T0 and T1), middle (T0FM and T1FM), and end of evolution with fluconazole (T0FH and T1FH) and from the evolved control strains (T0NF and T1NF) (Fig. S1). Additionally, to test for clonal diversity, resistant strains were grown in the highest concentration plated on SD minus uracil agar plates and three single colonies (T1FHa to T1FHc) were picked for DNA extraction.
RNA was extracted from mid-exponential-phase cells by the hot phenol method. DNase I (Invitrogen)-treated RNA was resuspended in RNase-free water, and RNA quantity and integrity were assessed by UV/Vis spectrometry (NanoDrop; Thermo Scientific) and with the Agilent 2100 Bioanalyzer, respectively. Only RNAs with an integrity number of >9 were selected for further analysis.
DNA was extracted with the Genomic-tip 100/G kit (Qiagen) in accordance with the manufacturer’s instructions. DNA was resuspended in EB buffer, and DNA quantity and integrity were assessed with the Quant-iT PicoGreen double-stranded DNA quantitation assay and by agarose gel electrophoresis.
Transcriptome analysis.
Custom gene expression SurePrint G3 microarrays (Agilent-065138) were hybridized with CY3-labeled cRNA of growth duplicates in accordance with the manufacturer’s instructions (Agilent Low Input Quick Amp Labeling kit). Arrays were scanned on an Agilent G2505C microarray scanner, and microarray scan data were extracted with Agilent feature extraction software.
Raw gene expression microarray data were normalized with limmaGUI software (R/Bioconductor, Boston, MA) (45). Differentially expressed genes (DEGs) were extracted with limma (45) by using a differential threshold of 1.5 and a false discovery rate-corrected P value of 0.05. Functional enrichment was performed with GSEA (27) based on GO terms. Strains evolved in the presence of fluconazole (TxFM, TxFH) were initially compared to the respective untreated and nonevolved strain (Tx). Additionally, TxFH strains were compared among themselves and against the respective control strain (TxNF). For each comparison, GO sets with >15 and <500 annotated genes were selected as enriched if the corrected P value was <0.01 (1,000 gene set permutations). Enriched sets were related upon shared genes with the Cytoscape plugin EnrichmentMap (46). Figure 3 was generated with the R package pheatmap (47). Voronto software (48) was used for visualization of gene expression on the basis of GO biological process (BP) categories (Fig. S3).
Whole-genome resequencing.
One paired-end library was prepared for each sample in accordance with Illumina DNA sample preparation protocols. Libraries were sequenced with the Illumina genome analyzer IIx.
Raw sequence data (101- and 146-bp paired-end reads) from eight samples (T0, T1, T0FM, T1FM, T0FH, T1FH, T0NF, and T1NF) were initially trimmed by removing consecutive bases on both the 5′ and 3′ flanks with base qualities of <20. Trimmed reads that did not pass the filtering criteria for ambiguity (N content of <5%), complexity (score of ≥10), length (50 bases or longer), and average base quality (≥20) were removed with PRINSEQ (49).
Remaining reads were mapped to the reference genome of C. albicans obtained from the Candida genome database (http://www.candidagenome.org/; assembly 21) with BWA version 0.6.2 (50). Processing and filtering of mapped reads were done with SAMtools version 0.1.17 (51). After removal of duplicates, read pairs where both reads mapped on different chromosomes were removed. Additionally, read pairs where one or both reads were not mapped or had low mapping quality (<37) were removed.
The control strain used in this experiment, T0, is genetically slightly different from SC5314, which was used to construct the reference genome. SAMtools was used to produce read pileups, detect single nucleotide variants, and call genotypes. Indels were not called. Bases with a read depth of <5, a low quality of REF/ALT polymorphism (<20), low genotype quality (<20), and low consensus quality (<|20|) were called as unknown genotype. Additionally, unknown genotypes were called if a base had more than three allele types and the read depths of all of the allele types were >5.
For the eight samples, we analyzed genomic features (coding, noncoding, or repeat region, etc.) and LOH of bases with different genotype compared with the control strain. Structural variations in coding regions were also analyzed for changes in codon usage.
Copy number analysis was performed with CNAnorm (52). Aligned reads of each sample were split into 2-kb windows, and reads were counted. CNAnorm was used to estimate copy numbers with normalization of paired control and test samples.
SNP-CGH.
SNP-CGH array analysis was performed with custom Agilent arrays (eArray Design ID 038464 [53]). Three T1FH clones (T1FHa to T1FHc) were compared with control strain T0. Genomic DNA was digested with AluI and RsaI, amplified, labeled, and hybridized in accordance with the manufacturer’s instructions with the Agilent Genomic DNA enzymatic labeling kit (Agilent). Arrays were scanned on an Agilent G2505C microarray scanner, and microarray scan data were extracted with Agilent feature extraction software. All of the samples analyzed in this study passed the quality control performed with the R/Bioconductor packages GLAD (positional biases) and arrayQualityMetrics (statistical distributions) without any relevant issue (54, 55). Genomic events for SNP-CGH data were predicted by computing hidden Markov model states via the R/Bioconductor SNP-CGH package (56) by using default parameters. Genome ratios were plotted with the R/Bioconductor SNP-CGH package and mapped against C. albicans chromosome annotations (http://www.candidagenome.org/; assembly 21). Allele fractions were calculated with a custom R script from the SNP probes in the SNP-CGH data.
Additionally, the data were visualized with YMAP (57). Therefore, the array data files produced during extraction were uploaded into YMAP for processing by using the SNP-CGH array processing option with the GC bias correction option selected to correct for artifacts in the array data caused by variations in percent GC across the genome. After YMAP processed the data, it produced cartoon figures displaying copy number and allelic ratio changes across the genome for each clone. See the legend to Fig. 2 for details of how the data are presented and for interpretation of the data for each of the three clones (T1FHa to T1FHc).
Accession number(s).
The raw microarray data obtained in this study have been submitted to the Gene Expression Omnibus and assigned GenBank accession numbers GSE60121 and GSE60122. The array CGH data are listed in the Gene Expression Omnibus under GenBank accession number GSE60120. Sequencing data were archived in ArrayExpress under accession number E-SYBR-13.
ACKNOWLEDGMENTS
We thank Geraldine Butler for granting access to the design of the C. albicans gene expression microarray.
We have no competing interests.
T.W., A.R.B., and L.C. performed experiments. I.G.G. and M.B. sequenced strains. R.S. and D.A. analyzed microarray data. W.L. and J.R. analyzed sequencing data. T.W., N.T., G.R.M., A.C.-N., D.C., J.B., and M.A.S.S. interpreted data. T.W., J.B., and M.A.S.S. wrote the paper.
This work was supported by European Union Framework Program 7 (EU-FP7) Sybaris Consortium Project 242220, the Portuguese Foundation for Science and Technology through project PTDC/IMIMIC/5350/2014, the Institute for Biomedicine—iBiMED (UID/BIM/4501/2013), the European Research Council under the European Community’s Seventh Framework Program (FP7/2007-2013/ERC grant 340087), and European structural funds through the Portugal2020 and FEDER programs. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Brown GD, Denning DW, Gow NAR, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization 2014. Antimicrobial resistance global report on surveillance 2014. WHO Press, ; Geneva, Switzerland. [Google Scholar]
- 3.Legrand M, Chan CL, Jauert PA, Kirkpatrick DT. 2007. Role of DNA mismatch repair and double-strand break repair in genome stability and antifungal drug resistance in Candida albicans. Eukaryot Cell 6:2194–2205. doi: 10.1128/EC.00299-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selmecki A, Forche A, Berman J. 2010. Genomic plasticity of the human fungal pathogen Candida albicans. Eukaryot Cell 9:991–1008. doi: 10.1128/EC.00060-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shapiro RS, Robbins N, Cowen LE. 2011. Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol Mol Biol Rev 75:213–267. doi: 10.1128/MMBR.00045-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selmecki AM, Dulmage K, Cowen LE, Anderson JB, Berman J. 2009. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet 5:e1000705. doi: 10.1371/journal.pgen.1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morio F, Pagniez F, Besse M, Gay-Andrieu F, Miegeville M, Le Pape P. 2013. Deciphering azole resistance mechanisms with a focus on transcription factor-encoding genes TAC1, MRR1 and UPC2 in a set of fluconazole-resistant clinical isolates of Candida albicans. Int J Antimicrob Agents 42:410–415. doi: 10.1016/j.ijantimicag.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 8.Coste AT, Karababa M, Ischer F, Bille J, Sanglard D. 2004. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of Candida albicans ABC transporters CDR1 and CDR2. Eukaryot Cell 3:1639–1652. doi: 10.1128/EC.3.6.1639-1652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schillig R, Morschhäuser J. 2013. Analysis of a fungus-specific transcription factor family, the Candida albicans zinc cluster proteins, by artificial activation. Mol Microbiol 89:1003–1017. doi: 10.1111/mmi.12327. [DOI] [PubMed] [Google Scholar]
- 10.Selmecki A, Forche A, Berman J. 2006. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science 313:367–370. doi: 10.1126/science.1128242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ford CB, Funt JM, Abbey D, Issi L, Guiducci C, Martinez DA, Delorey T, Li B, White TC, Cuomo C, Rao RP, Berman J, Thompson DA, Regev A. 2015. The evolution of drug resistance in clinical isolates of Candida albicans. Elife 4:e00662. doi: 10.7554/eLife.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dunkel N, Morschhäuser J. 2011. Loss of heterozygosity at an unlinked genomic locus is responsible for the phenotype of a Candida albicans sap4Δ sap5Δ sap6Δ mutant. Eukaryot Cell 10:54–62. doi: 10.1128/EC.00281-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.White TC. 1997. The presence of an R467K amino acid substitution and loss of allelic variation correlate with an azole-resistant lanosterol 14α demethylase in Candida albicans. Antimicrob Agents Chemother 41:1488–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moura GR, Carreto LC, Santos MA. 2009. Genetic code ambiguity: an unexpected source of proteome innovation and phenotypic diversity. Curr Opin Microbiol 12:631–637. doi: 10.1016/j.mib.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 15.Butler G, Rasmussen MD, Lin MF, Santos MA, Sakthikumar S, Munro CA, Rheinbay E, Grabherr M, Forche A, Reedy JL, Agrafioti I, Arnaud MB, Bates S, Brown AJ, Brunke S, Costanzo MC, Fitzpatrick DA, de Groot PW, Harris D, Hoyer LL, Hube B, Klis FM, Kodira C, Lennard N, Logue ME, Martin R, Neiman AM, Nikolaou E, Quail MA, Quinn J, Santos MC, Schmitzberger FF, Sherlock G, Shah P, Silverstein KA, Skrzypek MS, Soll D, Staggs R, Stansfield I, Stumpf MP. 2009. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459:657–662. doi: 10.1038/nature08064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bezerra AR, Simões J, Lee W, Rung J, Weil T, Gut IG, Gut M, Bayés M, Rizzetto L, Cavalieri D, Giovannini G, Bozza S, Romani L, Kapushesky M, Moura GR, Santos MA. 2013. Reversion of a fungal genetic code alteration links proteome instability with genomic and phenotypic diversification. Proc Natl Acad Sci U S A 110:11079–11084. doi: 10.1073/pnas.1302094110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Javid B, Sorrentino F, Toosky M, Zheng W, Pinkham JT, Jain N, Pan M, Deighan P, Rubin EJ. 2014. Mycobacterial mistranslation is necessary and sufficient for rifampicin phenotypic resistance. Proc Natl Acad Sci U S A 111:1132–1137. doi: 10.1073/pnas.1317580111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balashov S, Humayun MZ. 2002. Mistranslation induced by streptomycin provokes a RecABC/RuvABC-dependent mutator phenotype in Escherichia coli cells. J Mol Biol 315:513–527. doi: 10.1006/jmbi.2001.5273. [DOI] [PubMed] [Google Scholar]
- 19.Al Mamun AAM, Humayun MZ. 2009. Spontaneous mutagenesis is elevated in protease-defective cells. Mol Microbiol 71:629–639. doi: 10.1111/j.1365-2958.2008.06551.x. [DOI] [PubMed] [Google Scholar]
- 20.Eddouzi J, Parker JE, Vale-Silva LA, Coste A, Ischer F, Kelly S, Manai M, Sanglard D. 2013. Molecular mechanisms of drug resistance in clinical Candida species isolated from Tunisian hospitals. Antimicrob Agents Chemother 57:3182–3193. doi: 10.1128/AAC.00555-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coste A, Selmecki A, Forche A, Diogo D, Bougnoux ME, d’Enfert C, Berman J, Sanglard D. 2007. Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryot Cell 6:1889–1904. doi: 10.1128/EC.00151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rocha R, Pereira PJB, Santos MA, Macedo-Ribeiro S. 2011. Unveiling the structural basis for translational ambiguity tolerance in a human fungal pathogen. Proc Natl Acad Sci U S A 108:14091–14096. doi: 10.1073/pnas.1102835108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moura G, Pinheiro M, Silva R, Miranda I, Afreixo V, Dias G, Freitas A, Oliveira JL, Santos MA. 2005. Comparative context analysis of codon pairs on an ORFeome scale. Genome Biol 6:R28. doi: 10.1186/gb-2005-6-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrett RDH, Schluter D. 2008. Adaptation from standing genetic variation. Trends Ecol Evol 23:38–44. doi: 10.1016/j.tree.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 25.Rustchenko EP, Curran TM, Sherman F. 1993. Variations in the number of ribosomal DNA units in morphological mutants and normal strains of Candida albicans and in normal strains of Saccharomyces cerevisiae. J Bacteriol 175:7189–7199. doi: 10.1128/jb.175.22.7189-7199.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perepnikhatka V, Fischer FJ, Niimi M, Baker RA, Cannon RD, Wang YK, Sherman F, Rustchenko E. 1999. Specific chromosome alterations in fluconazole-resistant mutants of Candida albicans. J Bacteriol 181:4041–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berman J, Sudbery PE. 2002. Candida albicans: a molecular revolution built on lessons from budding yeast. Nat Rev Genet 3:918–930. doi: 10.1038/nrg948. [DOI] [PubMed] [Google Scholar]
- 29.Hickman MA, Zeng G, Forche A, Hirakawa MP, Abbey D, Harrison BD, Wang YM, Su CH, Bennett RJ, Wang Y, Berman J. 2013. The “obligate diploid” Candida albicans forms mating-competent haploids. Nature 494:55–59. doi: 10.1038/nature11865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennett RJ, Johnson AD. 2005. Mating in Candida albicans and the search for a sexual cycle. Annu Rev Microbiol 59:233–255. doi: 10.1146/annurev.micro.59.030804.121310. [DOI] [PubMed] [Google Scholar]
- 31.Al Mamun AAM, Gautam S, Humayun MZ. 2006. Hypermutagenesis in mutA cells is mediated by mistranslational corruption of polymerase, and is accompanied by replication fork collapse. Mol Microbiol 62:1752–1763. doi: 10.1111/j.1365-2958.2006.05490.x. [DOI] [PubMed] [Google Scholar]
- 32.Rustad TR, Stevens DA, Pfaller MA, White TC. 2002. Homozygosity at the Candida albicans MTL locus associated with azole resistance. Microbiology 148:1061–1072. doi: 10.1099/00221287-148-4-1061. [DOI] [PubMed] [Google Scholar]
- 33.Cameroni E, De Virgilio C, Deloche O. 2006. Phosphatidylinositol 4-phosphate is required for translation initiation in Saccharomyces cerevisiae. J Biol Chem 281:38139–38149. doi: 10.1074/jbc.M601060200. [DOI] [PubMed] [Google Scholar]
- 34.Tournu H, Tripathi G, Bertram G, Macaskill S, Mavor A, Walker L, Odds FC, Gow NAR, Brown AJP. 2005. Global role of the protein kinase Gcn2 in the human pathogen Candida albicans global. Eukaryot Cell 4:1687–1696. doi: 10.1128/EC.4.10.1687-1696.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dever TE. 2002. Gene-specific regulation by general translation factors. Cell 108:545–556. doi: 10.1016/S0092-8674(02)00642-6. [DOI] [PubMed] [Google Scholar]
- 36.Wendland J, Schaub Y, Walther A. 2009. N-Acetylglucosamine utilization by Saccharomyces cerevisiae based on expression of Candida albicans NAG genes. Appl Environ Microbiol 75:5840–5845. doi: 10.1128/AEM.00053-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miranda I, Silva-Dias A, Rocha R, Teixeira-Santos R, Coelho C, Gonçalves T, Santos MAS, Pina-Vaz C, Solis NV, Filler SG, Rodrigues AG. 2013. Candida albicans CUG mistranslation is a mechanism to create cell surface variation. mBio 4:e00285-13. doi: 10.1128/mBio.00285-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gregori C, Glaser W, Frohner IE, Reinoso-Martín C, Rupp S, Schüller C, Kuchler K. 2011. Efg1 controls caspofungin-induced cell aggregation of Candida albicans through the adhesin Als1. Eukaryot Cell 10:1694–1704. doi: 10.1128/EC.05187-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rizzetto L, Weil T, Cavalieri D. 2015. Systems level dissection of Candida recognition by dectins: a matter of fungal morphology and site of infection. Pathogens 4:639–661. doi: 10.3390/pathogens4030639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reynolds TB. 2009. Strategies for acquiring the phospholipid metabolite inositol in pathogenic bacteria, fungi and protozoa: making it and taking it. Microbiology 155:1386–1396. doi: 10.1099/mic.0.025718-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kolodner RD, Putnam CD, Myung K. 2002. Maintenance of genome stability in Saccharomyces cerevisiae. Science 297:552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- 42.Tkach JM, Yimit A, Lee AY, Riffle M, Costanzo M, Jaschob D, Hendry JA, Ou J, Moffat J, Boone C, Davis TN, Nislow C, Brown GW. 2012. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat Cell Biol 14:966–976. doi: 10.1038/ncb2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harrison BD, Hashemi J, Bibi M, Pulver R, Bavli D, Nahmias Y, Wellington M, Sapiro G, Berman J. 2014. A tetraploid intermediate precedes aneuploid formation in yeasts exposed to fluconazole. PLoS Biol 12:e1001815. doi: 10.1371/journal.pbio.1001815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noble SM, Johnson AD. 2005. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot Cell 4:298–309. doi: 10.1128/EC.4.2.298-309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smyth GK. 2005. Limma: linear models for microarray data, p 397–420. In Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S. (ed), Bioinformatics and computational biology solutions using R and Bioconductor. Springer, New York, NY. [Google Scholar]
- 46.Merico D, Isserlin R, Stueker O, Emili A, Bader GD. 2010. Enrichment map: a network-based method for gene set enrichment visualization and interpretation. PLoS One 5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kolde R. 2015. pheat map: Pretty Heat maps. R package version 1.0.8. The R Foundation, Vienna, Austria. [Google Scholar]
- 48.Santamaría R, Pierre P. 2012. Voronto: mapper for expression data to ontologies. Bioinformatics 28:2281–2282. doi: 10.1093/bioinformatics/bts428. [DOI] [PubMed] [Google Scholar]
- 49.Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup . 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gusnanto A, Wood HM, Pawitan Y, Rabbitts P, Berri S. 2012. Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from next-generation sequence data. Bioinformatics 28:40–47. doi: 10.1093/bioinformatics/btr593. [DOI] [PubMed] [Google Scholar]
- 53.Abbey D, Hickman M, Gresham D, Berman J. 2011. High-resolution SNP/CGH microarrays reveal the accumulation of loss of heterozygosity in commonly used Candida albicans strains. G3 1:523–530. doi: 10.1534/g3.111.000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hupe P. 2011. GLAD: gain and loss analysis of DNA. R package version 2.28.1. The R Foundation, Vienna, Austria. [Google Scholar]
- 55.Kauffmann A, Huber W. 2014. arrayQualityMetrics: Quality metrics on microarray data sets. R package version 3.20.0. The R Foundation, Vienna, Austria. [Google Scholar]
- 56.Fridlyand J, Dimitrov P. 2013. aCGH: classes and functions for array comparative genomic hybridization data. R package version 1.36.0. The R Foundation, Vienna, Austria. [Google Scholar]
- 57.Abbey DA, Funt J, Lurie-Weinberger MN, Thompson DA, Regev A, Myers CL, Berman J. 2014. Y MAP: a pipeline for visualization of copy number variation and loss of heterozygosity in eukaryotic pathogens. Genome Med 6:100. doi: 10.1186/s13073-014-0100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental design used in this study. RNA for gene expression analysis was extracted from growth duplicates of the same strains (Tx, TxFM, and TxFH) of which the whole genome was sequenced (WGS; genomic DNA extraction). Additionally, genomic DNA of three T1FH clones was extracted for array-based CGH analysis. The intensity of the red color of arrows indicates increasing fluconazole concentrations. Download FIG S1, EPS file, 0.4 MB (448.3KB, eps) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Caspofungin susceptibility test. E tests of strains T0 and T1 revealed similar MICs (0.094 to 0.125 µg/ml), yet only hypermistranslating strain T1 produced microcolonies within the entire inhibition ellipse. These microcolonies also grew in liquid medium containing caspofungin concentrations of up to 32 µg/ml. Download FIG S2, EPS file, 0.8 MB (871.8KB, eps) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The left panel is a schema depicting major events that allow strain T1 to acquire fluconazole resistance rapidly. Drug efflux in strain T1 seems to be mediated mainly via the ABC superfamily transporters CDR1 and CDR2, while in strain T0, drug efflux is likely due to the multidrug pump encoded by MDR1. The right panel shows GO BP heatmaps that depict log-fold changes in gene expression. Green letters represent DEGs in T1FH/T1NF, i.e., those involved in steroid biosynthesis, glucose transport, and the chitin biosynthetic process. Download FIG S3, EPS file, 1.4 MB (1.4MB, eps) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Nonsynonymous codon changes in TAC1, MRR1, and ERG7. N.ofAA, position of amino acid; T0.Codon, codon in control strain T0; T0.AA, amino acid in control strain T0; S.Codon, codon in the strain tested; S.AA, amino acid in the strain tested. Download TABLE S1, DOCX file, 0.02 MB (19.2KB, docx) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
LOH events in quickly evolving strain T1 (WGS data). While LOH on Chr5 appeared only under drug pressure (T1FM and T1FH), the LOH on the left arm of ChrR appeared regardless of fluconazole treatment (T1NF, T1FM, and T1FH). Download FIG S4, EPS file, 0.4 MB (376.7KB, eps) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
CUG codon usage. (A) Statistical comparison of the altered set of genes for each evolutionary event and strain (T1 and T1FH genes affected by LOH and T1FH genes affected by copy number variations) and the rest of the genome. Significant results show that T1 genes affected by LOH are depleted of UCA, UCC, and UCG codons; T1FH genes affected by LOH are depleted of UCG codons; and T1FH genes affected by copy number variations are enriched in UCA codons and depleted of UCC and UCG codons. RSCU values for serine codons of each gene were calculated with Anaconda (23) and compared through Student t tests corrected by the Bonferroni method. (B) Numbers of CUG codons that differ in both alleles (a and b) in genes that lost heterozygosity or changed copy number in T1FH (data from CGD). Notably, the large LOH regions on Chr5 and ChrR are aaaa (see Fig. 2), which caused the highlighted changes in CTG codon usage. All of the genes that have a positive difference led to a reduction in CTG codons (blue) by changing CTG to other codons, while those having a negative difference increased the number of CTG codons (green) by changing other codons to CTG. Further, all of the genes with an increased copy number (red) had a positive difference, while those with a reduced copy number (orange) had a negative difference. Download TABLE S2, PDF file, 0.1 MB (141.6KB, pdf) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
GO BP categories of genes located within the region of ChrR that lost heterozygosity in all evolved T1 strains regardless of whether cells were untreated (T1NF) or treated with the drug (T1FM and T1FH). For genes with >10 LOH events, the corrected P value is <0.005. Download TABLE S3, DOCX file, 0.02 MB (19.6KB, docx) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Gene set enrichment networks. (A) Network displaying gene sets enriched in hypermistranslating strain T1FH relative to strain T0FH evolved in the presence of fluconazole. (B) Network displaying gene sets enriched in strain T1 evolved in the presence of a high fluconazole concentration (T1FH) relative to strain T1 evolved without the drug (T1NF). (C) Network displaying gene sets enriched in hypermistranslating strain T1 evolved without the drug (T1NF) versus nonevolved strain T1. Edge width represents the number of shared genes, node size represents the number of annotated genes, and node color represents the enrichment based on the number of upregulated (red) or downregulated (blue) genes in the set. The 10 genes ranked highest in each gene set are shown. Download FIG S5, PDF file, 0.9 MB (979.1KB, pdf) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Genes expressed differentially in T1FH and T1NF and functional analysis. (Top) Venn diagram in which each colored area represents a set of DEGs (differential expression threshold of ≥3.0, adjusted P value of <0.01). Only one gene, FMP45, is significantly upregulated in T1FH and downregulated in T1NF. The top two GO terms for each condition are shown. (Bottom) GO BP categories enriched below a P value of 0.01 in all of the expression sets shown in the Venn diagram. Download FIG S6, PDF file, 1 MB (1MB, pdf) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(A) Flow cytometry profiles of two T1FH clones. Red, diploid control; blue, sample. The T1FH profile indicates a mixed population of diploids, tetraploids, and some cells with greater ploidy. (B) Confirmation of loss of the MTLa locus in T1FH by targeted PCR. Colony PCR products of the MTLa1 (red) and MTLalpha1 (blue) genes visualized on an agarose gel. MTLa1 is absent from all tested clones of the evolved and highly resistant strain T1FH. For the sequences of the MTLa primers used, see I. Miranda, R. Rocha, M. C. Santos, D. D. Mateus, G. R. Moura, L. Carreto, and M. A. Santos, PLoS One 2:e996, 2007. Download FIG S7, EPS file, 0.4 MB (452.4KB, eps) .
Copyright © 2017 Weil et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.