

Abstract
Background
Mytilisepta virgata is a marine mussel commonly found along the coasts of Japan. Although this species has been the subject of occasional studies concerning its ecological role, growth and reproduction, it has been so far almost completely neglected from a genetic and molecular point of view. In the present study we present a high quality de novo assembled transcriptome of the Japanese purplish mussel, which represents the first publicly available collection of expressed sequences for this species.
Results
The assembled transcriptome comprises almost 50,000 contigs, with a N50 statistics of ~1 kilobase and a high estimated completeness based on the rate of BUSCOs identified, standing as one of the most exhaustive sequence resources available for mytiloid bivalves to date. Overall this data, accompanied by gene expression profiles from gills, digestive gland, mantle rim, foot and posterior adductor muscle, presents an accurate snapshot of the great functional specialization of these five tissues in adult mussels.
Conclusions
We highlight that one of the most striking features of the M. virgata transcriptome is the high abundance and diversification of lectin-like transcripts, which pertain to different gene families and appear to be expressed in particular in the digestive gland and in the gills. Therefore, these two tissues might be selected as preferential targets for the isolation of molecules with interesting carbohydrate-binding properties.
In addition, by molecular phylogenomics, we provide solid evidence in support of the classification of M. virgata within the Brachidontinae subfamily. This result is in agreement with the previously proposed hypothesis that the morphological features traditionally used to group Mytilisepta spp. and Septifer spp. within the same clade are inappropriate due to homoplasy.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4012-z) contains supplementary material, which is available to authorized users.
Keywords: Mussel, Transcriptome, RNA-seq, Lectins, Phylogenomics, Bivalve, Mollusk
Background
The purplish bifurcate mussel Mytilisepta virgata (Wiegmann, 1837), also known as Septifer virgatus, is a small bivalve mollusk species commonly found in the middle/upper intertidal zone of moderately wave-exposed shores along the coasts of Japan, Taiwan, and South Eastern China [1], between −10 and +70 cm above the mean tide level [2]. M. virgata is usually found in dense mussel beds, whose lower limit of vertical distribution is often determined by the association with the lower-intertidal mussel Hormomya mutabilis [3]. This mussel species developed remarkable morphological adaptations to cope with a wave-exposed environment, including a ventrally flattened, particularly resistant shell and stronger byssal attachment to the substrate compared to other subtidal and lower-intertidal sympatric mussel species [3–5]. Furthermore, M. virgata appears to be much more resistant to high temperature stress and air exposure than M. edulis, another invasive mussel species which preferentially occupies the lower part of the intertidal zone in the same geographical region [2]. Mature individuals are usually 45 mm long and live for approximately 4–5 years, although exceptional cases of 65 mm long specimen surviving up to 12 years have been recorded. Sexual maturity is reached after 12 months and, although fertility is maintained throughout the entire year, spawning events follow a bimodal pattern (the first one occurring in February–March, the second one in September–December) [6].
From a taxonomical point of view, M. virgata has long been considered as a member of the Septifer genus (and therefore named S. virgatus) and placed within the subfamily Septiferinae [7]. However, this clade was later found to be polyphyletic and, based on the revised hierarchical classification of NCBI Taxonomy, M. virgata, along with and most of the other species previously paced within this subfamily, has been moved to Mytilinae (Rafinesque, 1815). However, the current taxonomical classification still does not appear to accurately reflect the evolutionary history of these mussels. Indeed, more than a decade ago, molecular studies based on Cytochrome c oxidase subunit I (COI) first pointed out that M. virgata and the morphologically similar Septifer excisus (Wiegmann, 1837) pertained to two different clades within the order Mytiloida [8]. This observation is strongly supported by a recent study by Trovant and colleagues, which reported that COI and 18S/28S–based phylogeny identified both M. virgata and Mytilisepta bifurcata (Conrad, 1837) as members of the same clade, together with Perumytilus purpuratus (Lamarck, 1819) and Brachidontes rostratus (Dunker, 1857) (both pertaining to the Brachidontinae family), but distantly related to Septifer bilocularis [9]. Based on these results, the authors further suggested that Mytilisepta should be retained as a separate genus within Brachidontinae and that the septum structure involved in the insertion of the adductor muscle and used for the current morphological classification of different species within the Septifer genus is the product of homoplasy.
Apart from its disputed taxonomical placement, very limited scientific attention has been so far focused on M. virgata, with only a handful of studies dealing with its morphological adaptations [5], embryonic development [10], reproductive cycle [6] and population dynamics [2]. To date, only 31 nucleotide and 11 amino acid sequences have been deposited in public repositories for this species (data retrieved from NCBI, June 2017). These include several partial sequences used for phylogenetic analyses [9, 11, 12], microsatellites [13], a complete mitochondrial sequence (KX094521.1) and a few unrelated sequences referring to still unpublished manuscripts, highlighting the still nearly non-existing molecular knowledge of M. virgata, in stark contrast with the extensive investigations carried out in Mytilus spp. since the early 2000’s [14–18]. The only reliable data available about the genome of M. virgata is an assessment of its size made by flow cytometry. The calculated genome c-value, 1.08, further confirmed by a very similar estimate for the closely related Mytilisepta keenae (1.06) [19], reveals that the purplish Japanese mussel genome is about 2/3 the size of those of Mytilus spp. and among the smallest known mytiloid genomes, but quite in line with the average genome size for all bivalves (Animal Genome Size Database, http://www.genomesize.com/).
Due the narrow scientific interest posed so far on mussel species other than Mytilus spp., deep sea vent mussels (Bathymodiolus spp.) and Perna viridis, M. virgata represents an interesting alternative subject for the study of the evolution of Mytiloida and for the investigation of some peculiar gene family expansion events which specifically occurred in this lineage [20].
The main aim of the present work was to provide the first curated and publicly accessible genomic resource for this species. The de novo assembled and functionally annotated transcriptome, together with gene expression data from five different adult tissues, will serve as a sequence and gene expression database for future genetic and molecular studies. The data we present will provide a substantial contribution in the improvement of scientific studies in this marine mussel. While discussing the potential function of tissue-specific genes, we put a particular emphasis on expanded families encoding lectin-like proteins involved in carbohydrate recognition, a class of molecules with a great biotechnological potential which could find a practical application in many areas of biomedical and biological research [21, 22].
Methods
Collection of samples
The selected sampling site was a natural mussel bed found in a tide pool in Oshima, Saikai city (Nagasaki prefecture, Japan). Based on national and local fishing regulations, no permits were required for the collection of shellfish. Five adult mussels, whose shell size approximately ranged between 4 and 5 cm, were collected, dissected using razor blades and scissors, and tissues were immediately placed in RNAlater (Thermo Fisher Scientific, Waltham, USA). Namely, the following tissues were dissected and stored for subsequent RNA extraction: hemocytes (collected with a syringe), posterior adductor muscle, inner mantle, mantle rim, digestive gland, gills and foot (Fig. 1). The collected tissues were subsequently chopped into smaller parts weighting approximately 20 mg; for each of the sampled tissues, one of these slices of tissue was selected for each of the five specimens, placed in a vial containing TRIzol (Thermo Fisher Scientific, Waltham, USA) and homogenized. The total RNA, thereby representing a pool of five individual mussels, was extracted following the manufacturer’s instructions. The quality and quantity of the extracted RNAs were assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, USA). Only samples whose RNA Integrity Number was > = 9 were selected for RNA sequencing. Unfortunately the quality of the RNA extracted from hemocytes and inner mantle was not sufficient to proceed with the preparation of Illumina sequencing libraries.
Fig. 1.
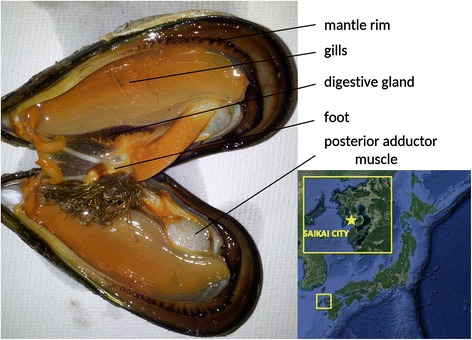
Internal anatomy of an adult Mytilisepta virgata specimen with indication of the five main tissues selected for this study and geographical location of the selected sampling site in Saikai city, Nagasaki prefecture (Japan)
Library preparation and sequencing of the samples
The preparation of cDNA libraries compatible with Illumina sequencing was carried out using the Lexogen SENSE mRNA-seq library prep kit v2 (Lexogen, Wien, Austria), aiming at an average fragment size of 400 nt. RNA-sequencing was performed at the DNA Sequencing Center of the Brigham Young University using a 2 × 125 bp paired-end strategy on a single lane of an Illumina HiSeq 2500 in high output mode with v4 reagents.
De novo assembly, quality assessment and annotation
Raw reads were demultiplexed, imported in the CLC Genomics Workbench v.9.5 environment (Qiagen, Hilden, Germany) and trimmed according to base-calling quality scores and presence of residual sequencing adaptors. Resulting reads shorter than 75 bp were discarded. Trimmed reads were used as an input for Trinity v.2.3.2 [23] on the Galaxy platform [24] with default parameters. The minimum contig length was set at 200 bp. As suggested by previous studies [25, 26], in order to remove unreliable sequences likely originated from excessive fragmentation of longer mRNA molecules or residual contamination from exogenous RNA, contigs displaying a very low sequencing coverage were removed; this procedure was based on a threshold of TPM = 1, calculated with the back-mapping of all trimmed reads on the full assembled transcriptome using the software Kallisto v0.43.0 [27]. All the contigs that did not reach this threshold were discarded. Contigs originated from the assembly of mitochondrial and ribosomal RNAs were identified by BLASTn [28] using the sequences KX094521.1 (mitochondrial) and KJ453817.1 (ribosomal) as queries (e-value threshold 1E−10). The quality of the transcriptome was assessed with BUSCO v.2 [29] based on the set of conserved orthologous genes of the Metazoa lineage according to OrthoDB v.9 [30].
For annotation purposes, only the longest transcript per gene was selected and subjected to virtual protein translation with TransDecoder v.3.0.1 (http://transdecoder.github.io), using a minimum predicted protein length of 100 amino acids. All the sequences were annotated with the Trinotate pipeline v.3.0 (https://trinotate.github.io) and assigned Gene Ontology categories [31] based on positive BLASTp and BLASTx [28] matches against the UniProtKB/Swiss-Prot database (e-value threshold, 1E−5). The annotation of Pfam conserved protein domains [32] was based on the identification by Hmmer v.3.1b2 [33]. In specific cases where no annotation could be assigned based on these criteria, remote structural similarities with templates deposited in the Protein DataBank were inspected with HHpred [34].
Gene expression analysis
Trimmed reads for each of the five tissues were mapped on the annotated transcriptome with the RNA-seq mapping tool of the CLC Genomics Workbench v.9.5 (Qiagen, Hilden, Germany) using the following parameters: length fraction: 0.75; similarity fraction: 0.98; maximum number of matching contigs: 10; paired-end reads distance was automatically estimated. The number of matched reads were converted into Transcripts Per Million (TPM) expression values [35], a measure which ensures efficient data normalization and comparability both within and between samples. The obtained TPM values, representing the average expression levels of a pool of five biological replicates, were subjected to a statistical analysis of gene expression with a Kal’s Z-test [36]. In detail, the expression profile of a given tissue was compared with the other four to identify differentially expressed genes (DEGs). The thresholds of fold change and False Discovery Rate-corrected p-value were set at 2 and 0.05, respectively. All DEGs exceeding these thresholds in all the four comparisons were marked as “tissue-specific”. TPM expression were processed by log2 transformation for visualization purpose to generate scatter and volcano plots. A gene expression heat map was created by Euclidean distance-based hierarchical clustering (with average linkage) of a representative set of genes whose expression exceeded 3000 TPM in any of the five tissues taken into account in this study. Heat maps were also similarly generated for genes encoding lectins.
Tissue-specific genes were subjected to a functional evaluation, assessed through hypergeometric tests on Gene Ontology and Pfam annotations [37], implemented in the CLC Genomics Workbench v.9.5. Significantly over-represented annotations were detected based on a p-value <0.01 and observed – expected value > = 5.
Validation by real-time PCR
The expression data obtained by in silico analyses as detailed in the section above was validated by Real-Time PCR on three individual adult mussel, collected and dissected as previously described. Extracted RNAs were used to synthetize cDNA with a qScript™ Flex cDNA Synthesis Kit, QuantaBio (Quanta BioSciences Inc., Gaithersburg, MD) following manufacturer’s instructions. Ten target genes (two for each tissue) were selected for validation among those displaying high tissue specificity in RNA-seq experiments. The primer sequences, designed with Primer3Plus (https://primer3plus.com/) to obtain amplicons of 100–150 nt size, are reported in Table 1. In addition, we selected two stable housekeeping genes, the elongation factor 1 alpha and the ribosomal protein S2 (the former from literature data [38, 39], the latter due to its constant TPM values), to normalize gene expression data among samples.
Table 1.
Primers used for real-time PCR validation
| target gene | forward primer (5′ - > 3′) | reverse primer(5′ - > 3′) |
|---|---|---|
| Chitotriosidase | TACTGCGATTGGCCATACAA | TGCCTGTGTAGTTCGAGGTG |
| Meprin A | GCTTGGGTACATCGACCACT | CTGCTTTGGTTCCAGTGTCA |
| Paramyosin | CCGAACTCGCAGAAAAGAAC | CGTAGAGCTTCCTCGGTGTC |
| Myosin, striated muscle | CTCTTGTTGCCCCAGGATTA | CTGGTAGCTCCACCAGCTTC |
| Acetylcholine receptor | GTCAAAGTCGGCCACTCACT | GTCTGACCGTCGTTGGTTCT |
| Glycine-rich secreted protein | CACACGGTCTTACTGGAGCA | GTTCGCCTTGTTCACCTTGT |
| Valine-rich secreted protein | AAAGTGCCATTCGAGACACC | GGGCTGGGAACTCTGAATTT |
| SCP domain containign protein | TCAAACACTGCGTCAAGACC | GGATCCACGTTTTCTCTTGC |
| Serine protease inhibitor | AGGCCAACTGCAAAAACG | CCGTCAACTCCACACACG |
| Tyrosinase-like protein 2 | CAGAGCCCTACCTCCAGATG | TGACTGCTCGCTTTGTATGG |
| EF1alpha | CTCTTCGTCTCCCACTCCAG | ACCAGGGAGAGCTTCAGTCA |
| Ribosomal protein S2 | GCCATTGCCAATACCTATGC | GCCTGGTTGACGAGTATGGT |
In detail, PCR reactions were prepared as follows: 5 μL of SsoAdvanced SYBR Green Supermix (Bio-Rad, Hercules, CA), 0.2 μL of the 10 μM forward and reverse primers, 1 μL of 1:20 diluted cDNA and water to reach a final reaction volume of 10 μL. Polymerase chain reaction (PCR) amplifications were carried out on a Real-Time C1000-CFX96 platform (Bio-rad), using the following thermal profile: 95° for 30 s, by 40 cycles at 95° (10 s) and 60° (20 s). The absence of non-specific amplification products was assessed with a melting curve analysis (65° to 95°). Gene expression values for the target genes were calculated using the delta Ct method and normalized on the average expression level of the two housekeeping genes. Results are shown as the average plus standard deviation of three technical replicates.
Phylogenomic analysis
The phylogenomic analysis was carried out based on a set of 445 single-copy orthologous genes present in the transcriptomes of M. virgata and 8 other mytiloid bivalve mollusk species, using the same strategy previously used by Biscotti and colleagues [25]. Namely, the species selected for this purpose were: Bathymodiolus azoricus, Bathymodiolus manusensis, Bathymodiolus platifrons, Geukensia demissa, Lithophaga lithophaga, Modiolus kurilensis, Modiolus modiolus, Modiolus philippinarum, Mytilus californianus, Mytilus coruscus, Mytilus galloprovincialis (as a representative of the M. edulis species complex), Perna viridis and Perumytilus purpuratus. The full set of the proteins encoded by the recently published genomes of B. platifrons and M. philippinarum were recovered. For all the other species, publicly available raw sequencing data was downloaded from the NCBI SRA database, imported in the CLC Genomics Workbench 9, trimmed based on quality as described above and assembled with the de novo assembly tool, setting both the word size and bubble size parameters to “automatic”. Coding sequences (CDSs) were then predicted with TransDecoder v.3.0.1 (http://transdecoder.github.io), as described above for M. virgata. Sequence data from the genome of the Pacific oyster Crassostrea gigas v.9 [40] were also included to provide a reliable outgroup species for the analysis, based on the recent identification of Ostreoida as a sister group to Mytiloida [41].
Following this procedure, reciprocal BLASTp searches were performed, between the target species, M. virgata and C. gigas (the outgroup), using an e-value threshold of 1 × 10−10; taking into account only the best BLAST hit and discarding all the sequences lacking significant homology in any of the comparisons (either because of an e-value lower than the threshold or because of sequence identity <50%). This finally allowed the identification of a set of conserved orthologous sequences, which were aligned with MUSCLE [42] and further processed with Gblocks v.0.91b to remove highly divergent regions or fragments missing in one or more species due to the incompleteness of transcriptome data [43]. The resulting trimmed alignments were then concatenated and used as an input for a ProtTest v.3.4 analysis [44] in order to identify the best-fitting model of molecular evolution based on the corrected Akaike Information Criterion [45]. The concatenated multiple sequence alignment file, consisting of 247 orthologous proteins unambiguously detected in all the species taken into account, was subjected to Bayesian phylogenetic inference analysis with MrBayes v.3.2 [46], based on the LG model of molecular evolution, with a Gamma-shaped distribution of rates across sites, a proportion of invariable sites and fixed (empirical) priors on state frequencies (LG + G + I + F), identified by ProtTest as the best-fitting model. Phylogenetic inference was carried out with two independent analyses run in parallel. The convergence of the parameters generated by the two MCMC analyses was evaluated with Tracer v.1.6 (http://beast.bio.ed.ac.uk/Tracer). The analysis was stopped when the Effective Sample Size of each estimated parameter reached a value higher than 200, without considering the initial 25% of the generated trees (removed due to the burnin process). Sampled trees were used to calculate a consensus phylogenetic tree, where poorly supported nodes (those displaying posterior probability <50%) were collapsed.
Results and discussion
The high quality transcriptome of Mytilisepta virgata
Following the application of quality filters, the de novo transcriptome assembly of M. virgata comprised 49,501 contigs with an average length of 679 nucleotides (Table 2). This number, apparently rather high if compared to the number of annotated genes in bivalve genomes (e.g. ~26,000 in C. gigas and ~33,000 in P. fucata) [40, 47], is possibly justified by at least three factors: (i) the poor knowledge of bivalve non-coding RNAs, that are abundant in mussels [14], including in M. virgata, where they account for 71% of the assembled contigs; (ii) the partial fragmentation of mRNAs in smaller contigs; (iii) the high genomic complexity and heterozygosity of the genome of mytiloids, previously pointed out in Mytilus spp. [16].
Table 2.
Sequencing, de novo assembly and annotation statistics
| Digestive gland trimmed reads | 48,106,544 |
|---|---|
| Posterior adductor muscle trimmed reads | 46,456,019 |
| Gills trimmed reads | 49,760,348 |
| Mantle rim trimmed reads | 53,492,730 |
| Foot trimmed reads | 86,035,069 |
| Number of contigs | 49,501 |
| Longest contigs | 9778 nt |
| Average contig length | 679 nt |
| N50 | 1046 |
| Contigs longer than 5Kb | 68 |
| Complete BUSCOs | 66% |
| Fragmented BUSCOs | 10% |
| Absent BUSCOs | 24% |
| Predicted protein-coding contigs (>100 aa) | 29% |
| GO cellular component annotation rate | 19% |
| GO molecular function annotation rate | 17% |
| GO biological process annotation rate | 17% |
| PFAM annotation rate | 23% |
| Mytiloid innovations (protein coding) | 2% |
N50, the metric most commonly used to assess the quality of a de novo assembly [48] was 1046, in line with those obtained in the de novo assembly of other species of the order Mytiloida using Illumina sequencing technologies [14, 15]. While mean contig length (679 nt) and N50 metrics can only provide a measure of the effectiveness of the de novo assembly pipeline, the completeness of a transcriptome can be only theoretically estimated through the comparison with its reference genome. However, a similar estimate can be performed also when a reference genome is not available, using a set of highly conserved single-copy orthologous sequences. These genes, expected to be found across all the genomes within a target taxa, can be defined as “Benchmarking Universal Single-Copy Orthologs” (BUSCOs) [29]. This analysis revealed that ~66% of the conserved metazoan orthologous genes was present and complete in the assembled transcriptome of M. virgata, whereas only ~10% were present but fragmented, and ~24% were absent (Fig. 2). This result highlights a slightly higher level of completeness of the M. virgata transcriptome compared to other mussel transcriptomes obtained with Illumina sequencing technologies from single tissues [14, 49], much higher than those obtained with more error-prone or lower throughput technologies (454 Life Sciences and Sanger sequencing) [18, 50, 51] and just slightly lower than bivalve genome-scale protein prediction in oysters [40, 47]. This supports our choice of combining RNA-seq from multiple tissues and different specimens to obtain a representative nearly-complete transcriptome. The residual incompleteness of the de novo assembled transcriptome, evidenced by the missing BUSCOs, can be explained by the fact that some genes were not expressed at all in the five tissues taken into account. Specifically, as no transcriptome from larval stages was sequenced in this study, genes related to embryonic development might have been entirely missing. At the same time, since inner mantle (invaded by gonads during the spawning season) and hemocytes could not be analyzed due to insufficient RNA quality, a number of genes displaying high specificity of expression in these two highly specialized tissues are likely to be absent in the assembled transcriptome. Finally, tightly regulated genes whose expression is turned on in response to particular stimuli are also expected to be absent due to naïve condition of the mussel specimens collected.
Fig. 2.
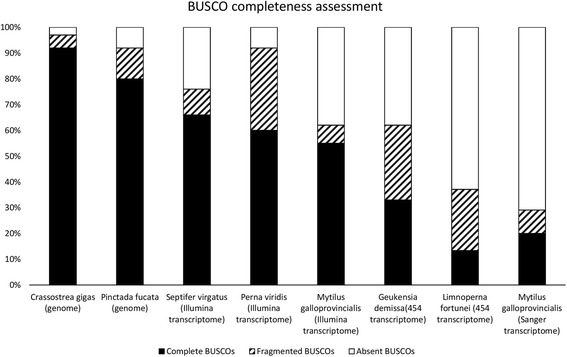
Comparison of transcriptome completeness, estimated with Busco v.2, among publicly available de novo assembled transcriptomes from Mytiloida and gene model predictions from the fully sequenced genomes of Crassostrea gigas and Pinctada fucata
Overall, poor expression can also lead to contig fragmentation, together with other factors such as transcriptome complexity, heterozygosity and inter-individual variability. Taking into account that mussels pertaining to the genus Mytilus display an astounding level of heterozygosity and are subject to significant genetic introgression [52], the relatively low rate of fragmented/complete BUSCOs (0.15) was somewhat unexpected. Since no information is currently available concerning the genetic diversity of M. virgata populations, these results possibly suggest that the mean level of heterozygosity in this species is significantly lower than that found in Mytilus spp., which would be consistent with a smaller effective population size.
The annotation rate by Trinotate was quite low, as only about 25% of the assembled contigs could be associated to a Gene Ontology term or to a Pfam domain. This observation, which is in line with previous results obtained from transcriptome studies in other mytiloids [14], is linked to at least three different factors: (i) partial fragmentation of contigs, as evidenced by BUSCO; (ii) high prevalence of taxonomically restricted genes; (iii) high prevalence of non-coding transcripts.
In detail, the presence of gene families restricted to Mytiloida has been previously described [53, 54] and, while this topic should be better investigated once the first complete mussel genome will become available, a brief analysis revealed that ~2% (1078) of the assembled sequences pertained to gene families exclusively found in mytiloids (as evidenced by the lack of BLAST homology with non-mytiloid bivalve genomes and transcriptomes, as opposed to significant homology against the publicly available genomes and transcriptomes of other mytiloids).
At the same time, the universe of non-coding RNAs in invertebrates remains to be fully investigated. While genome annotation pipelines, in most cases, currently disregard non-coding genes in newly assembled invertebrate genomes due to lack of homology and experimental evidence, the number of non-coding genes annotated in the genome of the model organism Caenorhabditis elegans has recently surpassed that of coding genes, starting to unveil the important role these RNAs cover in the biology of protostomes. Consistently with this observation, only 29% of the M. virgata contigs were found to contain an ORF longer than 100 codons, suggesting that a high fraction of non-coding sequences was indeed the primary reason of the low annotation rate.
Complete annotation data and gene expression levels are provided in Additional file 1.
Phylogenetic relationship with other mytiloids
The phylogenetic position of M. virgata and the classification of this species within the Septifer (Récluz, 1848) or the Mytilisepta (Habe, 1951) genus have been a matter of debate. Currently, although Septifer virgatus and Mytilisepta virgata are considered as synonyms by the World Register of Marine Species and by MolluscaBase [55], only the latter is marked as an accepted species name. Based on the same taxonomy reference databases, the genus Mytilisepta currently includes two other species, M. bifurcata (Conrad, 1837) and M. keenae (Nomura, 1936). On the other hand, nine different species are currently recognized as pertaining to the Septifer genus by this taxonomical authority. However, the Mytilisepta genus is currently not accepted by the NCBI taxonomy authority and M. virgata is therefore still listed as S. virgatus and further placed within Mytilinae subfamily.
The traditional classification of both Mytilisepta and Septifer species as members of the same genus is strictly based on morphological features, namely the presence of a septum which is involved in the attachment of the adductor muscle to the shell. However, recent molecular phylogenetic studies based on the analysis of 18S/28S nucleotide sequence have strongly suggested that the presence of this structure in the two mussel genera is the result of convergent evolution. Indeed, M. virgata and S. bilocularis pertain to highly divergent clades, with the former being related to mussel species pertaining to the Brachidontinae subfamily [9].
Taking advantage from the large amount of sequence data generated in the present study and transcriptomic dataset publicly available for other mytiloid species, we inferred the phylogenetic placement of M. virgata within the order Mytiloida by the use of Bayesian methods. Our phylogenomics approach, which took into account a set of 247 single-copy orthologous genes detected in all the studied species, is expected to provide a highly reliable estimate of the evolutionary relationship of M. virgata with other mussel species, as recently demonstrated by a series of phylogenomics studies which have helped to discern the phylogenetic relationship of Mollusca and Bivalvia [41, 56–58].
Before proceeding to the discussion of the placement of M. virgata, it is worth to briefly remind that the classical classification of mytiloids, currently used by NCBI Taxonomy, comprises seven subfamilies: (i) Bathymodiolinae, giant deep see mussels associated with hydrothermal vents; (ii) Brachidontinae (Nordsieck, 1969), small-sized “scorched mussels” adapted to life in the mid-intertidal zone; (iii) Crenellinae (Gray 1840), small-size byssal nets-creating benthic species commonly found in sandy and muddy seabeds; (iv) Lithophaginae (H. Adams & A. Adams, 1857), rock- or coral-boring mussels with a cylindrical, elongated shape; (v) Modiolinae (G. Termier & H. Termier, 1950), presenting a rounded outline and subterminal umbones, which usually live buried in the soft sediments of the subtidal zone; (vi) Mytilinae (Rafinesque, 1815), usually presenting a triangular shape and terminal umbones and commonly creating dense mussel beds in the intertidal zone of rocky shores; (vii) Septiferinae, previously including also M. virgata and other morphologically similar mussel species with am arcuate and convex anterior margin of the shell.
The phylogenetic tree obtained was highly supported in all its nodes by posterior probability values higher than 99%, indicating the nearly certain and unambiguous inference of the most likely evolutionary scenario which led to the radiation of mussels (Fig. 3). Remarkably, M. virgata was found to be closely related to P. purpuratus, supporting the results previously published by Trovant and colleagues based on ribosomal RNA sequence data [9], and clearly placed within the Brachidontinae (Nordsieck, 1969) subfamily, together with the ribbed horsemussel G. demissa. While no –omic scale sequence data is available for other species currently classified in the Septifer genus, molecular phylogeny produced by other authors suggest that S. excisus, S. bilocularis and other congeneric species are distantly related to M. virgata and do not belong to Brachidontinae, being more closely related to Mytilus (Linnaeus, 1758) and other species of the subfamily Mytilinae.
Fig. 3.
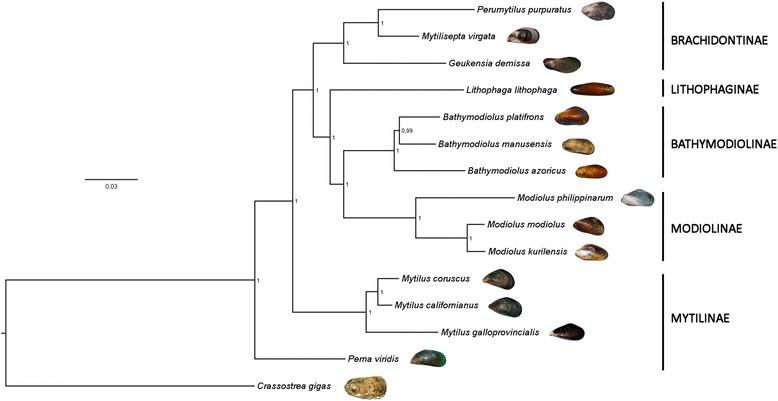
Bayesian phylogeny of Mytiloida. The classical taxonomical classification of mytiloids, according to NCBI Taxonomy, is shown on the right side of the figure. No species pertaining to the subfamilies Crenellinae and Septiferinae could be included due to the absence of available sequence data. Note the placement of M. virgata within the Brachidontinae subfamily. The number indicated on the nodes of the tree indicate posterior probability support values
Based on these results, the current official naming of M. virgata as S. virgatus at the NCBI taxonomy database does not appear to be appropriate, and it should be updated following the official nomenclature already adopted by WoRMS and MolluscaBase. Moreover, Mytilisepta appears to be clearly pertaining to the Brachidontinae subfamily, which at the present time only lists the Brachidontes (Swainson, 1840), Geukensia (Van der Poel, 1956), Ischadium (jules-Brown, 1905) and Perumytilus (Olsson, 1961) genera in the NCBI taxonomy database.
Overview of gene expression profiles
As an overall trend, the five analyzed tissues displayed widely diverse gene expression profiles, characterized by a broad prevalence of tissue-specific genes (Fig. 4). Cumulative expression plots revealed that one of the major differences among tissues can be attributed to the high energetic investment in the synthesis of a very few mRNA species by some tissues (e.g. foot) compared to a more even distribution of the transcriptional efforts by other tissues (e.g. gills). This factor can be efficiently estimated by the relative contribution to global transcriptional activity of the 100 most highly expressed genes in any given sample. We define the reciprocal of this value as Transcriptomic Diversity Index (TDI). Therefore, a high TDI indicates the expression of a broad range of transcripts, whereas a low TDI points out a low diversity in the mRNA population. The TDI values calculated for the Mytilisepta tissues were, from the highest to the lowest: 3.57 (gills), 3.03 (mantle rim), 2.63 (digestive gland), 2.08 (posterior adductor muscle) and 1.59 (foot) (Fig. 4, panel a). As a further confirmation of the remarkable functional specialization of bivalve tissues, just a relatively low number of genes (235), mostly encoding fundamental housekeeping genes, were found to be expressed at relatively high levels (TPM > 100) in all the five tissues analyzed (Fig. 4, panel b).
Fig. 4.
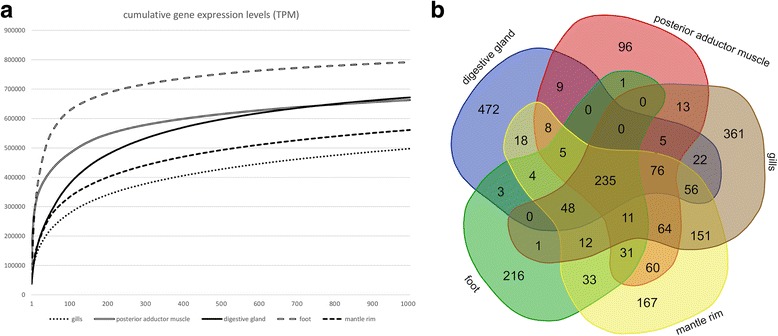
Panel (a) cumulative gene expression of the 1000 most expressed genes per tissue. The gene expression level on the Y axis is measured as TPM. Panel (b) Venn diagram displaying the overlap between highly expressed transcripts (TPM > 100) among the five tissues subjected to RNA-seq
The functional differentiation of the five mussel tissues appears to be evident from the hierarchical clustering of genes based on their pattern of expression (Fig. 5, panel a). The digestive gland and the foot, in particular, are characterized by two clusters of genes with high tissue specificity, nearly undetectable outside the main site of production. At a first sight, the hierarchical clustering analysis does not permit to clearly identify equally important differences among mantle rim, gills and posterior adductor muscle. However, while these differences are not as evident as those observed for foot and digestive gland, they might influence in a highly significant manner the function of a tissue (see the following sections).
Fig. 5.
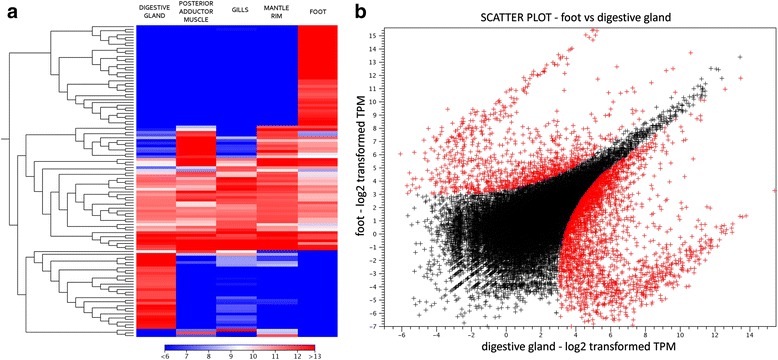
Panel (a) Euclidean distance-based hierarchical clustering, calculated based on average linkage, of the Mytilisepta virgata transcripts reaching an expression level higher than 3000 TPM in at least on out of the five tissues analyzed. Panel (b) Gene expression scatter plot comparing the expression profiles of digestive and foot in Mytilisepta virgata. The log2 normalized TPM gene expression levels are reported on the X and Y axes. Differentially expressed genes are marked in red
Digestive gland transcriptional profile
The digestive gland is the main tissue involved in the digestion of food particles, in the conjugation of nutrients to carriers for their transportation through circulation, in the metabolism of xenobiotics and in the excretion process. In addition, it also covers an important role in the long-term storage of nutrient reserves to be used during gametogenesis or long-lasting stress periods. Two main cell types are responsible for the fundamental functions of this organ, i.e. digestive cells and basophil secretory cells. These cells are located in the blind end of digestive tubules branching from the main digestive ducts. Here, highly abundant columnar digestive cells adsorb food particles by pinocytosis and proceed to their digestion upon the fusion of endocytotic phagosomes with lysosomes. On the other hand, pyramid-shaped secretory cells are thought to be mainly involved in the release of enzymes involved in the extracellular breakdown of major food particles [59].
Mussels feed on a wide variety of food particles found in the water column, including phytoplankton, micro-zooplankton, bacteria and detritus, whose abundance largely varies both seasonally and geographically. For this reason, mussels display limited food preference, mostly based on size selection [60], and they actively produce of a broad range of digestive enzymes required for the assimilation of diverse nutrients, i.e. fat, carbohydrates and proteins found in different food sources.
Coherently with these observations, the expression profile of the M. virgata digestive gland was typical of a highly specialized tissue, displaying tissue-specificity for 2043 genes, as evidenced by the expression scatter plots (Fig. 5, panel b) and a low TDI (2.63). The transcriptional profile was dominated, as expected, by digestive enzymes. The gene set enrichment analysis indeed clearly highlighted that a major energetic effort is spent in the synthesis of enzymes involved in the processing of the two most abundant biomasses in nature, cellulose and chitin [61], i.e. cellulases (identified as members of the glycosyl hydrolase families 9 and 10) and chitinases (pertaining to the glycosyl hydrolases family 16) (Table 3). The overwhelming presence of these classes of enzymes among the most highly expressed genes is probably ascribable to the wide availability of micro-zooplankton and microalgae as potential food sources in the oceans.
Table 3.
The 25 most highly expressed genes in Mytilisepta virgata digestive gland and representative significantly over-represented annotations by gene set enrichment analysis
| Most expressed genes | Over-represented annotations | |||
|---|---|---|---|---|
| Annotation | TPM | Term | Class | p-value |
| Vdg3 | 43,813.51 | Extracellular space | CC | 0.00 |
| Probable apolipoprotein | 14,067.11 | ShK domain-like | PFAM | 0.00 |
| Probable apolipoprotein | 12,230.17 | Ependymin | PFAM | 0.00 |
| Alpha tubulin | 11,375.80 | Lectin C-type domain | PFAM | 1.11E-16 |
| Elongation factor 1 alpha | 11,074.06 | C1q domain | PFAM | 2.22E-16 |
| Ependymin-related protein | 11,054.83 | von Willebrand factor type C domain | PFAM | 1.07E-14 |
| Ependymin-related protein | 8275.80 | Cellulose catabolic process | BP | 3.83E-12 |
| Vdg3 | 8084.66 | Lysosome | CC | 7.27E-12 |
| Chitotriosidase | 7926.27 | Cellulase activity | MF | 4.21E-11 |
| Unknown | 6439.28 | Glycosyl hydrolase family 9 | PFAM | 8.40E-11 |
| Probable apolipoprotein | 6230.41 | alpha/beta hydrolase fold | PFAM | 2.00E-10 |
| Cytoplasmic actin | 6103.04 | Carboxylesterase family | PFAM | 3.77E-10 |
| Calcium binding protein | 5887.19 | Trypsin | PFAM | 6.07E-10 |
| Meprin A | 5695.11 | Glycosyl hydrolases family 16 | PFAM | 8.76E-9 |
| 60S ribosomal protein P0 | 5364.73 | Serine-type endopeptidase activity | MF | 1.34E-8 |
| Ependymin-related protein | 5342.49 | MAM domain, meprin/A5/mu | PFAM | 2.76E-8 |
| Urokinase-like protein | 5283.92 | Carbohydrate metabolic process | BP | 5.91E-8 |
| Ganglioside GM2 activator | 5163.45 | Lipid transporter activity | MF | 8.20E-8 |
| Putative metalloprotease | 4892.49 | Antistasin family | PFAM | 1.36E-7 |
| Alpha tubulin | 4698.32 | Sulfatase | PFAM | 1.17E-6 |
| Spondin-like protein | 4503.80 | Glycosyl hydrolase family 10 | PFAM | 5.83E-6 |
| 40S ribosomal protein SA | 4288.17 | Peptide metabolic process | BP | 4.26E-6 |
| C1q domain-containing protein | 4279.26 | Lipase | PFAM | 8.19E-6 |
| Ependymin-related protein | 3725.69 | Prolyl oligopeptidase family | PFAM | 8.39E-5 |
| Calcium binding protein | 3528.24 | Xenobiotic metabolic process | BP | 4.56E-4 |
See the complete list in Additional file 4
TPM Transcript Per Million, PFAM Pfam conserved domains, BP Gene Ontology Biological Process, MF Gene Ontology Molecular Function, CC Gene Ontology Cellular Component
At the same time, fat-digesting enzymes (lipases, in particular) and proteases were also found to be expressed at highly significant levels. The latter include trypsin-, meprin- and papain-like proteinases, cathepsins, several serine-type endopeptidases and a large family of putative metalloproteases characterized by the ShK domain [62]. The high expression of protease inhibitors, such as antistasin, targeting trypsin [63], and Kazal-type serine-protease inhibitors [64], is also in line with the strong production of the aforementioned proteolytic enzymes, whose functional dysregulation could lead to serious cellular and tissue damage. Altogether, while some of these annotations are likely linked to extracellular digestive processes, others clearly point towards lysosomes, which are found in high numbers in vacuolated digestive cells. Among these, sulfatases and of ganglioside GM2 activator, highly expressed in the digestive gland, are probably the most relevant. The former is involved in the lysosomal cleavage of sulfated carbohydrates; the latter is a cofactor for the lysosomal digestion of gangliosides.
As a result of its pronounced endocytotic activity, the digestive gland is also a main site of bioaccumulation for pollutants, heavy metals and biotoxins [65, 66], which are processed by detoxifying enzymes. The specialization of the digestive gland in this activity is indeed evidenced by the overrepresentation of oxidoreductases and carboxylesterases, phase I detoxifying enzymes which introduce polar groups in xenobiotics to increase their solubility and promote their excretion.
Furthermore, consistently with the function homologous to that covered by liver in vertebrates, a few highly expressed genes in the digestive gland of Mytilisepta encode apolipoproteins, devoted to the binding and transportation of lipids in the circulatory system. In particular, the second and the third most highly expressed genes (Table 3), despite lacking detectable primary sequence similarity, display a remarkable predicted structural similarity with apolipoproteins, identified by HHpred as detailed in the materials and methods section.
Similarly to what has been previously reported in M. galloprovincialis [14, 67], the most highly expressed gene in the digestive gland of Japanese mussels was vdg3, a developmental marker of the maturation of this organ in juveniles [68], whose precise biological function still remains to be fully elucidated. Vdg3, which is probably encoded by a few highly similar paralogous genes (like in the Mediterranean mussel [69]), accounts for more than 4% of the total transcriptional activity of the M. virgata digestive gland.
In addition, it is worth of note that the digestive gland, besides being involved in the secretion of enzymes for extracellular digestion, also releases in the extracellular environment a large number of proteins with potential carbohydrate binding properties, most notably C-type lectins and C1q domain-containing (C1qDC) proteins (Table 3; see the following sections for a detailed overview on these gene families).
Foot transcriptional profile
The foot is an important tissue both in the larval and in the adult phases of mussel life. Although the foot has progressively lost its ancestral function as the main locomotory organ along bivalve evolution, it still retains a limited role in the movement of pediveliger larvae and juvenile mussels. In adult specimens, the foot is the main responsible of the attachment to suitable substrates through the secretion of byssal threads, robust and elastic fibers produced by a specialized secretory glands, which have attracted a considerable interest due to their potential biotechnological applications [70].
The gene expression profile (TDI = 1.59, see Fig. 4 panel a) pointed out that the foot is by far the most specialized tissue among those subjected to RNA-seq and the one producing the lowest variety of transcripts. As evidenced by gene set enrichment analyses, most of the 769 foot-specific transcripts identified in M. virgata were indeed clearly associated with the production of byssus. In particular, ~15% of the total transcriptional activity was used for the production of collagens and ~13.5% for the production of byssal cuticle proteins (Table 4). Precollagens are the major constituents of the fibrous core of byssal threads, contribute to their self-assembly and account for more than 70% of their mass [71, 72]. On the other hand, the astounding resistance of these threads is provided by a stiff coating by proteins rich in modified amino acids, such as trans-4-hydroxyproline, trans-2,3-cis-3,4-dihydroxyproline, and L-3,4-dihydroxyphenylalanine (L-DOPA), which form a 2–5 μm thick highly resistant cuticle [73]. We found a significant sequence similarity between the byssal cuticle proteins of M. virgata, those of the deep sea vent mussel Bathymodiolus thermophilus, and an adhesive polyphenolic foot protein of Septifer bifurcatus. These low-complexity proteins in fact consist of a high number of consecutive repeats of conserved 17-mers, which present, in all the three species, a well detectable Y-X-X-X-Y-K motif required for adhesion [74] (Additional file 2). Besides these two classes of sequences, the gene expression profile of the foot was dominated by other low-complexity proteins, including YGH-rich proteins similar to those previously identified in Mytilus coruscus [75], and proteins containing EGF-like domains, which characterize cross-linking mussel adhesive plaque proteins [76].
Table 4.
The 25 most highly expressed genes in Mytilisepta virgata foot and representative significantly over-represented annotations by gene set enrichment analysis
| Most expressed genes | Over-represented annotations | |||
|---|---|---|---|---|
| Annotation | TPM | Term | Class | p-value |
| Byssal cuticle protein | 45,262.97 | Collagen triple helix repeat (20 copies) | PFAM | 0.00 |
| Byssal cuticle protein | 43,379.14 | Serine-type endopeptidase inhibitor activity | MF | 0.00 |
| Collagen-like protein | 42,699.96 | Negative regulation of coagulation | BP | 1.11E-16 |
| Byssal cuticle protein | 34,293.81 | Common central domain of tyrosinase | PFAM | 3.33E-16 |
| Collagen-like protein | 33,886.43 | Extracellular region | CC | 3.33E-16 |
| Serine protease inhibitor | 30,056.14 | Negative regulation of endopeptidase activity | BP | 6.77E-15 |
| Low complexity protein | 26,250.15 | Animal haem peroxidase | PFAM | 6.77E-14 |
| Collagen-like protein | 17,017.56 | Kazal-type serine protease inhibitor domain | PFAM | 4.35E-12 |
| YGH-rich protein-2 | 16,065.78 | Peroxidase activity | MF | 5.69E-12 |
| Collagen-like protein | 15,351.63 | Proteinaceous extracellular matrix | CC | 6.47E-12 |
| Collagen-like protein | 13,389.57 | Tissue inhibitor of metalloproteinase | PFAM | 6.83E-10 |
| Byssal cuticle protein | 12,178.71 | Collagen trimer | CC | 2.14E-8 |
| Collagen-like protein | 10,965.75 | Response to oxidative stress | BP | 5.45E-8 |
| Elongation factor 1 alpha | 10,722.32 | Regulation of keratinocyte differentiation | BP | 8.93E-8 |
| Collagen-like protein | 10,461.88 | Hydrogen peroxide catabolic process | BP | 1.05E-7 |
| YGH-rich protein-1 | 10,019.89 | Transforming growth factor beta binding | MF | 1.63E-7 |
| Precollagen NG | 9810.75 | Metal ion binding | MF | 8.77E-7 |
| YGH-rich protein-1 | 8119.26 | Extracellular space | CC | 9.20E-7 |
| Serine protease inhibitor | 7406.30 | Heme binding | MF | 2.55E-6 |
| Serine protease inhibitor | 7362.32 | Oxidoreductase activity | MF | 6.45E-6 |
| Collagen-like protein | 7349.68 | WAP-type ‘four-disulfide core’ | PFAM | 1.01E-5 |
| Collagen-like protein | 7307.02 | Quinone binding | MF | 1.26E-5 |
| Unknown | 7104.95 | Human growth factor-like EGF | PFAM | 1.78E-5 |
| Kazal-type serine proteinase inhibitor | 7095.67 | EGF-like domain | PFAM | 6.84E-5 |
| Low complexity protein | 6445.09 | Kunitz/Bovine pancreatic trypsin inhibitor domain | PFAM | 7.62E-5 |
See the complete list in Additional file 4
TPM Transcript Per Million, PFAM Pfam conserved domains, BP Gene Ontology Biological Process, MF Gene Ontology Molecular Function, CC Gene Ontology Cellular Component
Interestingly, tyrosinases were the most important class of enzymes expressed in this tissue: 13 out of the 18 tyrosinase sequences found in the purplish mussel transcriptome were indeed specifically expressed in the foot. This data is in agreement with the importance of this enzymatic activity in the conversion of tyrosine residues to L-DOPA and, consequently, to dopaquinone in Tyr-rich byssal cuticle proteins [77, 78]. At the same time, the high observed activity of antioxidant enzymes (peroxidases in particular) is explained by the need of maintaining a redox balance to allow the modification of L-DOPA-rich adhesion proteins [79, 80].
Consistently with previous reports, many proteinase inhibitors were selectively expressed at high levels in the foot. Among these, the strong expression of serine-type endopeptidases and alpha-2 macroglobulins is certainly worth of a note. Moreover, the foot-specificity of six WAP-like proteins suggest that they may act as suppressors of elastase-type serine proteases, preventing laminin degradation [81]. These enzyme inhibitors may altogether constitute an efficient protecting system that prevents the degradation of byssal threads by extracellular proteases [75].
Gills transcriptional profile
In all non-protobranch bivalves, gills are large organs whose structure follows the curvature of the shell, dividing the mantle cavity between inhalant and exhalant chambers and providing a large surface area exposed to the incoming water flow. Their lamellar structure, strengthened by collagen, makes them well suited for both gas exchange and filter-feeding. Gills are highly vascularized by vessels which bring deoxygenated hemolymph to the filaments, where the gas exchange takes place, and subsequently recirculate oxygenated fluid to the whole body through an efferent vein. The feeding function on the other hand is carried out by specialized cilia that cover the entire branchial surface and convey food particles carried by inflowing water towards the labial palps. Overall, gills are very complex organs that combine functionally and structurally diverse cell types, including neuronal cells that depart from a visceral ganglion.
Dissimilarly from other highly specialized tissues (i.e. foot and digestive gland), gills do not show the expression of a well-evident group of highly tissue-specific genes in the scatter plots (Additional file 3). As mentioned above, this is possibly linked to the diversity of cell types present in this complex organ, resulting in a partial overlap with the transcriptomes of the other four tissues analyzed. As a matter of fact, gills represent the richest out of the five tissues in terms of transcriptional diversity (TDI = 3.57) (Fig. 4, panel a).
In any case, the number of genes whose expression was statistically significantly higher in this tissue compared to the others was rather high (2877). These genes were, for the most part, linked to the movement of motile cilia, the main players in the filter-feeding process, present at very high densities on the whole surface of the gills. In particular the expression of axonemal dyneins, that drive ciliary movement by regulating the relative sliding of microtubule doublets [82], was very prominent, as well as that of tektins, fundamental for ciliar assembly and functionality [83] (Table 5). As the function of dyneins require ATP, mitochondria-eating proteins, significantly overexpressed, may be used to improve mitochondrial function, enhancing energy production [84]. The strong production of several genes encoding neuronal acetylcholine receptor subunits [85] was also likely related to the coordinated movement of cilia. In this context, acetylcholine could lead to a strong enhancement of ciliary beat frequency and Shisa-like proteins, also over-represented, may act as regulators of its trafficking [86].
Table 5.
The 25 most highly expressed genes in Mytilisepta virgata gills and representative significantly over-represented annotations by gene set enrichment analysis
| Most expressed genes | Over-represented annotations | |||
|---|---|---|---|---|
| Annotation | TPM | Term | Class | p-value |
| Alpha tubulin | 55,938.25 | Dynein heavy chain and region D6 of dynein motor | PFAM | 1.43E-13 |
| 40S ribosomal protein SA | 16,641.12 | Microtubule | CC | 0.00 |
| Elongation factor 1 alpha | 11,612.37 | Microtubule motor activity | MF | 6.20E-14 |
| Alpha tubulin | 8853.84 | Microtubule-based movement | BP | 2.52E-10 |
| Beta tubulin | 8710.17 | Motile cilium | CC | 3.06E-10 |
| Actin | 8589.14 | Axoneme assembly | BP | 1.57E-9 |
| Alpha tubulin | 8158.42 | Cilium movement | BP | 2.22E-9 |
| Low complexity glycine-rich protein | 7335.45 | Acetylcholine-activated cation-selective channel activity | MF | 5.94E-8 |
| Beta tubulin | 5932.28 | C1q domain | PFAM | 3.23E-7 |
| Actin | 5614.26 | ATP-binding dynein motor region D5 | PFAM | 1.32E-6 |
| Beta tubulin | 5144.94 | Tektin family | PFAM | 2.15E-6 |
| Beta tubulin | 5004.75 | Axonemal dynein complex | CC | 2.63E-6 |
| Actin | 4296.64 | Neurotransmitter-gated ion-channel ligand binding domain | PFAM | 6.18E-6 |
| 60s ribosomal protein P0 | 4296.41 | Dynein complex | CC | 1.06E-5 |
| non coding RNA | 4137.29 | ATPase activity | MF | 1.56E-5 |
| Ferritin | 3614.02 | Shisa/Wnt and FGF inhibitory regulator | PFAM | 3.71E-5 |
| Ribosomial Protein S10 | 3367.63 | Microtubule-binding stalk of dynein motor | PFAM | 4.65E-5 |
| Cyclophilin B | 3364.42 | Collagen trimer | CC | 5.40E-5 |
| Transcription factor ATF4 | 3351.91 | Immunoglobulin C1-set domain | PFAM | 7.45E-5 |
| Beta tubulin | 3296.13 | Fibrinogen beta and gamma chains, C-terminal globular domain | PFAM | 1.44E-4 |
| non coding RNA | 3201.67 | LITAF-like zinc ribbon domain | PFAM | 1.60E-4 |
| Cytochrome c oxidase polypeptide | 3059.53 | TIR domain | PFAM | 2.28E-4 |
| Outer dense fiber protein 3 | 3038.49 | Mitochondria-eating protein | PFAM | 2.42E-4 |
| Ribosomal Protein L15 | 2921.58 | Neurotransmitter-gated ion-channel transmembrane region | PFAM | 6.71E-4 |
| Ribosomal Protein S4 | 2769.43 | Cadherin domain | PFAM | 7.75E-4 |
See the complete list in Additional file 4
TPM Transcript Per Million, PFAM Pfam conserved domains, BP Gene Ontology Biological Process, MF Gene Ontology Molecular Function, CC Gene Ontology Cellular Component
The high expression of a number of genes encoding extracellular matrix components is certainly connected with the lamellar organization of branchial filaments, which facilitates gaseous exchanges for the respiratory function. Collagens, cadherins and the over-represented “secreted acidic and rich in cysteine Ca binding region proteins”, corresponding to osteonectin-like glycoproteins, appear to be the most important players in the establishment of these complex morphological structures. No specific domain or annotation related to oxygen binding could be identified as unequivocally linked to gills, probably due to the fact that this function is carried out by circulating cells which are also found in other tissues.
Immunity markers certainly represent one of the most noticeable characterizing gene expression signatures of gills. This may be correlated with the large surface of contact with the external environment and, consequently, with potentially pathogenic microorganisms. Both the gene families of C1qDC and FREPs (discussed in detail in the following sections), lectin-like proteins potentially involved in Pathogen Associated Molecular Patterns (PAMPs) recognition [87, 88], were over-represented in the gills transcriptome of the Japanese purplish mussel. At the same time, some TIR-domain containing proteins were also preferentially expressed in the gills. The TIR domain is found in a number of proteins linked to the detection of foreign ligands and to the ransduction of immune signals inside the cell. In detail, the expression of the cytosolic gene products MyD88, STING and ecTIR-DC families 6, 11 and 13 was particularly elevated in this tissue [89]. Moreover, a number of LITAF-like transcription factors, which may positively regulate the production of pro-inflammatory cytokines [90], were also selectively expressed in the gills.
While these signatures could be, at least in part, linked to the presence of hemocytes carried by hemolymph vessels, these data suggest that the gills of M. virgata might play an important role as a first line of defense towards pathogens, specifically by releasing soluble factors in the mucus that covers most of the branchial surface.
Mantle rim transcriptional profile
In mussels, the mantle is a two-lobed tissue that covers the entire inner face of the shells and hosts gonadal development. During the non-reproductive season however, the mantle becomes a very thin and nearly-transparent tissue, whose main functions are the storage of nutrients and the deposition of the shell. The region close to the edges, external to the pallial line of attachment to the shell, is known as mantle rim; this modified part of the mantle tissue is darkly pigmented and possesses tentacles with sensorial function which can be extruded from the shell when the valves are open. Furthermore, the mantle rim is rich in muscle fibers and nerves that permit the retraction of tentacles upon chemical, physical or light stimuli. The portion of tissue sampled in the present experiment corresponds to the large inhalant syphon that regulates the flux of water to the mantle cavity. In mussels, this modified region of mantle is less specialized than other bivalves, as it does not form an exhalant tube like in burrowing bivalve species.
The gene expression profile of the mantle rim only displayed little specialization, as evidenced by the expression scatter plots (Additional file 3). It was indeed characterized by a remarkable overlap with those of the other tissues and a relatively low number of genes (694) were specifically expressed in this tissue and just a very few out of these were expressed at very high levels (Table 6). Possibly owing to the combination of multiple cell types with widely diverse functions, the mantle rim produced a very broad range of transcripts and was only second to the gills for transcriptional diversity (TDI = 3.03, Fig. 4, panel a).
Table 6.
The 25 most highly expressed genes in Mytilisepta virgata mantle rim and representative significantly over-represented annotations by gene set enrichment analysis
| Most expressed genes | Over-represented annotations | |||
|---|---|---|---|---|
| Annotation | TPM | Term | Class | p-value |
| Actin | 40,034.02 | Proteinaceous extracellular matrix | CC | 1.84E-7 |
| Elongation factor 1 alpha | 17,159.08 | Extracellular region | CC | 1.87E-7 |
| alpha tubulin | 16,259.80 | Chitin binding Peritrophin-A domain | PFAM | 4.58E-7 |
| 60s ribosomal protein L23 | 15,108.07 | Matrixin | PFAM | 1.14E-6 |
| Actin | 15,102.97 | Calcium ion binding | MF | 2.65E-6 |
| 40S ribosomal protein SA | 11,849.66 | WAP-type (Whey Acidic Protein) ‘four-disulfide core’ | PFAM | 3.65E-6 |
| Actin | 11,356.99 | Extracellular matrix structural constituent | MF | 3.66E-6 |
| Alpha tubulin | 8547.04 | Immunoglobulin C1-set domain | PFAM | 1.51E-5 |
| Ferritin | 7899.68 | Serine-type endopeptidase inhibitor activity | MF | 3.27E-5 |
| Myosin | 7382.76 | Collagen catabolic process | BP | 1.09E-4 |
| low complexity protein - collagen-like | 6579.48 | Homeobox domain | PFAM | 1.24E-4 |
| 60s ribosomal protein P0 | 5142.34 | CD80-like C2-set immunoglobulin domain | PFAM | 1.42E-4 |
| Beta tubulin | 4972.09 | Metalloendopeptidase activity | MF | 1.58E-4 |
| Beta tubulin | 4461.74 | Cell junction | CC | 8.89E-4 |
| Superoxide Dismutase | 4220.54 | Cadherin domain | PFAM | 9.51E-4 |
| Actin | 4066.75 | Extracellular space | CC | 1.77E-3 |
| small heat shock protein 24.1 | 3933.60 | Immunoglobulin V-set domain | PFAM | 3.06E-3 |
| Paramyosin | 3898.32 | Basement membrane | CC | 3.57E-3 |
| non coding RNA? | 3796.60 | Immunoglobulin I-set domain | PFAM | 7.51E-3 |
| non coding RNA? | 3710.19 | Integral component of membrane | CC | 7.74E-3 |
| Myosin | 3690.69 | / | / | / |
| non coding RNA? | 3538.31 | / | / | / |
| Cyclophilin | 3464.80 | / | / | / |
| Ribosomal protein S4 | 3459.95 | / | / | / |
| Adenine nucleotide translocator | 3431.98 | / | / | / |
See the complete list in Additional file 4
TPM Transcript Per Million, PFAM Pfam conserved domains, BP Gene Ontology Biological Process, MF Gene Ontology Molecular Function, CC Gene Ontology Cellular Component
The most enriched annotations were linked to the extracellular matrix building and remodeling. The included matrixins, Zinc-dependent extracellular proteases involved in the degradation of components of the extracellular matrix [91], metalloendopeptidases and several proteinase inhibitors (e.g. WAP-like proteins, serine-type endopeptidase inhibitors, etc.), which might regulate their enzymatic activity. Consistently with the high prevalence of connective tissue-related genes, annotations linked to cell-cell and cell-extracellular matrix (e.g. immunoglobulin and cadherin domains) were also over-represented. Another class of enriched proteins was that encoded by homeobox genes; while most of these only displayed limited sequence similarity to homeobox genes whose function has been previously characterized, one of these was clearly homologous to Mohawk, a critical regulator of tendon differentiation [92].
Posterior adductor muscle transcriptional profile
In bivalves, valve closure is controlled by the contraction of the anterior and posterior adductor muscles. However, in all byssus-producing bivalves the anterior muscle is generally much smaller than the posterior one, to the point that in some mussel species (i.e. Perna spp.) the anterior muscle scar is not visible anymore. The large posterior adductor muscle is also closely associated to a sinus (the pericardial sac), which is part of the mussel open circulatory system and is commonly used in laboratory practice for the withdrawal of hemolymph with a syringe [93].
The transcriptional profile of the M. virgata posterior adductor muscle was particularly poor in terms of transcriptional diversity (TDI = 2.08), being only second to the foot (Fig. 4, panel a). Although the muscle can be certainly considered as a highly specialized tissue, it was characterized by a relatively small number (1089) of tissue-specific genes, probably due to the presence of muscular tissue also in the foot and mantle rim (Fig. 4, panel b; Fig. 5, panel a). As expected, the most highly expressed genes encoded structural components of muscle fibers, including actin and myosin, approximately accounting for 2 and 0.7% of total transcriptional activity, respectively. The enriched annotations contained conserved protein domains commonly associated with muscle adhesion proteins, such as immunoglobulins and fibronectin type III domains, and typical muscle components, such as myofibrils, sarcomere, Z disc, A, I and M bands, and functions, such as telethonin and actinin binding, contraction, response to calcium and sarcomere morphogenesis (Table 7). Curiously, “condensed nuclear chromosome” and “mitotic chromosome condensation” were also listed among the most significantly enriched GO terms. This is linked to the high expression of many contigs corresponding to titin, one of the longest known proteins in metazoans, with over 30,000 amino acids, which mRNA was fragmented in the Mytilisepta de novo assembly. Besides its main function as a structural component of the sarcomere, titin has been indeed linked to chromosome stabilization [94].
Table 7.
The 25 most highly expressed genes in Septifer virgatus posterior adductor muscle and representative significantly over-represented annotations by gene set enrichment analysis
| Most expressed genes | Over-represented annotations | |||
|---|---|---|---|---|
| Annotation | TPM | Term | Class | p-value |
| Actin | 150,779.49 | Condensed nuclear chromosome | CC | 4.11E-15 |
| 60s ribosomal protein L23 | 69,416.79 | Skeletal muscle myosin thick filament assembly | BP | 1.09E-14 |
| Actin | 44,602.27 | Immunoglobulin I-set domain | PFAM | 1.89E-14 |
| Paramyosin | 39,709.12 | Sarcomere organization | BP | 1.42E-13 |
| Myosin | 28,949.91 | M band | CC | 2.14E-13 |
| RS-rich protein 1 | 27,745.06 | I band | CC | 4.35E-13 |
| Myosin | 16,064.48 | Structural constituent of muscle | MF | 2.31E-12 |
| 40S ribosomal protein SA | 15,155.63 | Muscle alpha-actinin binding | MF | 2.64E-12 |
| Myosin | 12,525.17 | Z disc | CC | 3.45E-12 |
| Actin | 11,992.63 | A band | CC | 7.25E-12 |
| Myosin | 9275.62 | Mitotic chromosome condensation | BP | 3.29E-11 |
| Alpha tubulin | 8768.82 | Myofibril | CC | 3.65E-10 |
| Ferritin | 8517.40 | Myosin filament | CC | 5.51E-10 |
| Elongation factor 1 alpha | 8411.27 | Structural molecule activity conferring elasticity | MF | 5.85E-10 |
| PDZ domain containign protein | 7804.25 | Telethonin binding | MF | 5.85E-10 |
| Non coding RNA | 5844.46 | Sarcomere | CC | 5.99E-10 |
| Ribosomal Protein S10 | 5212.90 | Striated muscle thin filament | CC | 1.73E-9 |
| Actin | 4986.83 | Muscle myosin complex | CC | 1.73E-9 |
| Non coding RNA | 4561.28 | Actinin binding | MF | 1.73E-9 |
| Alpha tubulin | 4344.93 | Myosin tail | PFAM | 5.07E-7 |
| Unknown | 4209.22 | Fibronectin type III domain | PFAM | 5.33E-6 |
| 60s ribosomal protein P0 | 4157.43 | Skeletal muscle thin filament assembly | BP | 4.27E-9 |
| Small heat shock protein 24.1 | 3846.08 | Sarcomerogenesis | BP | 4.27E-9 |
| Beta tubulin | 3725.55 | Response to calcium ion | BP | 4.86E-7 |
| Superoxide dismutase | 3712.29 | Muscle contraction | BP | 6.84E-7 |
See the complete list in Additional file 4
TPM Transcript Per Million, PFAM Pfam conserved domains, BP Gene Ontology Biological Process, MF Gene Ontology Molecular Function, CC Gene Ontology Cellular Component
Validation of gene expression patterns
Taking into account previous reports of high heterozygosity [69] and high inter-individual variability of expression [95] in marine mussels, and considering the fact that RNA sequencing was carried out on a pool of five specimens, we proceeded to a validation of gene expression levels on individual mussels. This was achieved by Real-Time PCR experiments, performed on three biological replicates, which targeted ten tissue-specific genes (Table 1). Overall, the results obtained fully confirmed the observations collected by the in silico analysis of RNA-seq data, supporting our experimental approach for the detection of significant differences of expression among tissues (Fig. 6). In particular, chitotriosidase and meprin A, typical digestive enzymes, displayed high specificity to the digestive gland. Paramyosin and striated muscle myosin, encoding structural components of muscles, were highly expressed only in the posterior adductor muscle. A gills-specific acetylcholine receptor possibly involved in coordinating the movement of cilia and a glycine-rich secreted protein of unknown function were confirmed to be specifically expressed in this multifunctional tissue. A serine protease inhibitor and a tyrosinase-like protein, both important in byssogenesis, were expressed as high levels in the foot, as expected. Finally, an SCP domain-containing protein and a valine-rich protein of unknown function were confirmed as mantle rim-specific gene products.
Fig. 6.
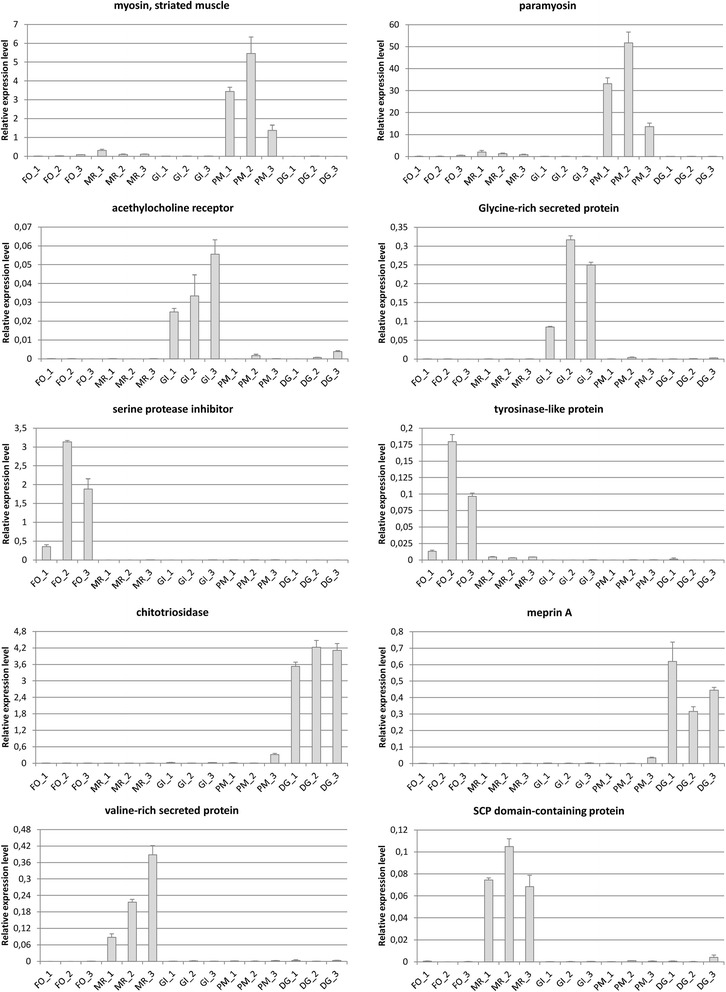
Gene expression levels of 10 selected genes in M. virgata. FO: foot; MR: mantle rim; GI: gills; PM: posterior adductor muscle; DG: digestive gland. “1”, “2” and “3” indicate the three biological replicates. Histogram bars represent the expression relative to housekeeping genes elongation factor 1 alpha and ribosomal protein S2. Error bars represent the standard deviation of three technical replicates
It is worth noticing that, despite the maintenance of tissue specificity, some of the target genes displayed relevant inter-individual differences in their expression levels. While these variations are not surprising in the light of previous reports [95], they reveal that a number of unpredictable factors of difficult monitoring might influence gene expression of M. virgata in the marine environment. As an example, the two foot-specific genes displayed an expression level more than 10 times lower in specimen 1 compared to the two other biological replicates (Fig. 6). This difference might indicate a lower byssogenic activity, a process which is well known to be influenced by multiple environmental parameters [96], including the intensity of water currents and exposure to waves, factors which could not be controlled in the sites of sampling.
In summary, the results obtained by RT-PCR confirmed that the pooling approach we used was appropriate to detect major differences in gene expression among tissues, permitting to pinpoint gene products which cover an important physiological function in the different body districts of mussels. At the same time however, the non-negligible inter-individual differences observed point out that extra care should be taken into account in future experiments aimed at assessing the individual response of M. virgata to abiotic and biotic stress.
The high prevalence of lectins in the Mytilisepta virgata transcriptome
Bivalve mollusks are known to produce a very broad range of secreted proteins containing carbohydrate-binding domains (CRDs). It has been suggested that these lectin-like molecules might function as Pattern Recognition Receptors (PRRs), acting as a first line of defense towards pathogens. Indeed, due to their high sequence diversity, bivalve lectins could potentially broaden the spectrum of recognition for Microbe Associated Molecular Patterns (MAMPs) or PAMPs, depending on whether the whole microbiota or just its pathogenic fraction are taken into account. However, alternative functions have been suggested. Specifically, the lectins found in the mucus that covers digestive organs have been linked to the feeding process, thanks to their ability to recognize carbohydrates exposed on the surface of food particles [97]. Others might be involved in establishing genetic barriers between species by regulating the species-specific recognition of congenetic gametes [98–100].
Many different types of bivalve lectins have been isolated since the early ‘90 through classical biochemical methods [101–103] and many more have been discovered in recent years in different bivalve species. Although most of these pertain to four distinct families, i.e. C-type lectins, fibrinogen-related proteins (FREPs), galectins and C1qDC proteins, others have been also found, including SUEL-type lectins, [104], lipopolysaccharide and β-1,3-glucan binding proteins [105], mytilectins [106] and F-type lectins [107].
Besides covering fundamental roles in bivalve physiology, some bivalve lectins also possess highly interesting binding or cytotoxic properties which might warrant future studies aimed at developing potential biotechnological applications. For example, the recently identified MytiLec is cytotoxic against some cancer cell types [108], and it has been suggested that a hemolytic C-type lectin from the freshwater clam Villorita cyprinoides could be potentially used as a clot lysing molecule [109].
Some bivalve lectin-encoding gene families are extremely expanded and comprise several hundred members, as suggested by proteomic [110] and transcriptome studies [20, 111, 112] and later confirmed on a whole genome scale in oyster [113]. The purplish Japanese mussel is no exception, as some important lectin-like gene families were also found among the most abundant ones in the de novo assembled transcriptome (Table 8).
Table 8.
The 25 most highly represented PFAM conserved domains in the Mytilisepta virgata transcriptome
| Domain name | PFAM code | domain count |
|---|---|---|
| Protein kinase domain | PF00069 | 200 |
| Immunoglobulin domain | PF00047 | 190 |
| Protein tyrosine kinase | PF07714 | 183 |
| Ankyrin repeats (3 copies) | PF12796 | 182 |
| Ankyrin repeats (many copies) | PF13857 | 175 |
| EF-hand domain pair | PF13833 | 161 |
| C1q domain | PF00386 | 161 |
| Immunoglobulin I-set domain | PF07679 | 161 |
| Ankyrin repeat | PF00023 | 160 |
| EF hand | PF00036 | 157 |
| Lectin C-type domain | PF00059 | 154 |
| EGF-like domain | PF00008 | 149 |
| EF-hand domain | PF13405 | 147 |
| AAA domain | PF00004 | 139 |
| Zinc finger, C3HC4 type (RING finger) | PF00097 | 129 |
| 50S ribosome-binding GTPase | PF01926 | 118 |
| Leucine rich repeat | PF13855 | 117 |
| 7 transmembrane receptor (rhodopsin family) | PF00001 | 117 |
| WD domain, G-beta repeat | PF00400 | 110 |
| Immunoglobulin V-set domain | PF07686 | 109 |
| B-box zinc finger | PF00643 | 108 |
| von Willebrand factor type A domain | PF00092 | 106 |
| Leucine Rich repeats (2 copies) | PF12799 | 103 |
| Tetratricopeptide repeat | PF00515 | 103 |
| Zinc finger, C2H2 type | PF00096 | 102 |
| F5/8 type C domain | PF00754 | 42 |
| Fibrinogen C-terminal globular domain | PF00147 | 37 |
| Galactose binding lectin domain | PF02140 | 23 |
| Ricin-type beta-trefoil lectin domain-like | PF14200 | 6 |
| H-type lectin domain | PF09458 | 6 |
| Galactoside-binding lectin | PF00337 | 2 |
Domains with carbohydrate-binding properties are marked in bold. A few additional less abundant lectin-like domains are listed in the bottom part of the table
Similarly to other mussel species [38], C1qDC proteins were found in very high number in M. virgata (161), suggesting the presence of several hundred genes in the genome of this species, in line with the previously demonstrated massive lineage-specific C1q gene family expansion event which occurred in the class Bivalvia [87]. C1qDC proteins have been implicated on multiple occasions in the innate immune response of different mollusks, where they have been often designed as sialic acid-binding lectins [114–116]. The expression profile of C1qDC genes in Mytilisepta closely resembles that of the Pacific oyster [87], since the most of such genes are selectively expressed in the digestive gland or in the gills. However, ~25% of mussel C1qDC proteins are produced ubiquitously by all tissues (Fig. 7).
Fig. 7.
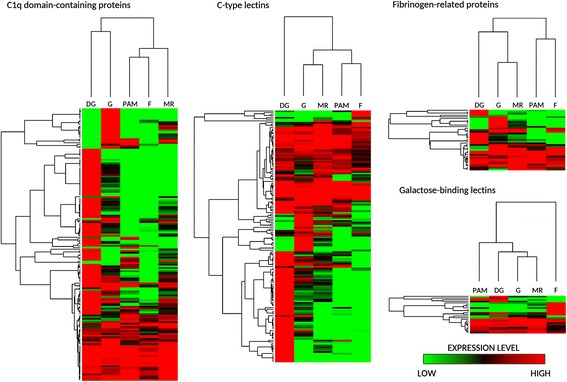
Heat maps summarizing gene expression patterns of C1q domain-containing proteins, C-type lectins, Fibrinogen-related proteins and Galactose-binding lectins in Mytilisepta virgata. Heat maps were built with log2 transformed gene expression levels; genes and tissues were hierarchically clusterized based on Euclidean distances, estimated with average linkage method. DG: digestive gland; G: gills; PAM: posterior adductor muscle; F: foot; MR: mantle rim
C-type lectins represent another large family of carbohydrate-binding proteins, comprising 154 members in M. virgata. These proteins, which structurally resemble the mannose-binding lectin (MBL) involved in the lectin pathway of the vertebrate complement system, have been often linked to bivalve immunity as PRRs [117–119]. However, coherently with their remarkable structural diversification, they could also be involved in other functions, such as particle capture [120] and gamete recognition [98]. Overall, the pattern of expression of C-type lectins was similar to that of C1qDC proteins, with two well-distinct groups of digestive gland- and gills- specific genes and others found at similar levels in all tissues (Fig. 7).
The role of FREPs, first described in gastropod mollusks [88], has later been demonstrated also in bivalves [121, 122]. Despite inconclusive phylogeny, molluscan FREPs structurally resemble vertebrate ficolins which, like MBL, can activate the complement system through the lectin pathway. FREPs possess an astounding sequence diversity in Mytilus spp., where they are though to provide a broad array of PRRs [123]. A total of 37 different FREPs could be identified in the purplish mussel, where they appear to be expressed in a broad range of tissues, but in particular in the gills (Fig. 7).
A remarkable number of proteins bearing a galactose-binding domain (23) was also found to be encoded by Mytilisepta mRNAs. This domain, first identified in SUEL lectins from sea urchin eggs, allows the preferential binding of L-rhamnose, in addition to D-galactose [124]. Although most of them are expressed in all the main tissues of mussels, some are clearly foot-specific. On the other hand, the galactoside-binding domain, which characterizes galectins, was only identified in two sequences encoding two structurally different galectins with two and four consecutive CRDs, respectively.
Consistently with previous reports in M. galloprovincialis [20], the F5/8 type C domain, typical of fucose-binding (F-type) lectins, was also detected in high abundance (42) in the Mytilisepta transcriptome. However, the function of F-type lectins has been only marginally investigated in bivalves so far and, taking into account that the F5/8 type C domain is also found in many non-lectin proteins (e.g. coagulation factors and bindins), the data currently available is insufficient to classify the M. virgata sequences as bona fide F-type lectins.
Six different proteins bearing a ricin-type beta-trefoil lectin-like domain are present in the de novo assembled transcriptome. However, none of these can be classified as mytilectins, despite the fact these novel carbohydrate binding proteins have been first characterized and described as a multigenic family in M. galloprovincialis [106, 108]. This unexpected absence may either reflect a lack of expression in physiological conditions or a highly specific expression to one of the tissues which could not be sampled in the current experiment (e.g. hemocytes or inner mantle).
The H-type lectin domain is capable of binding N-acetylgalactosamine and it has been described in the agglutinin of land snails, proteins which are part of the innate immune system, protecting fertilized eggs from bacteria [125, 126]. Although to date no H-type lectin has ever been characterized in bivalve mollusks, six assembled transcripts pertain to this family in M. virgata.
Altogether these data confirm the high prevalence of transcripts encoding lectins in bivalve transcriptomes. Although the large majority of the putative carbohydrate-binding proteins encoded by the transcripts identified in M. virgata either pertaining to the C1qDC or to the C-type lectin families, many others, with distinct binding properties and potential biotechnological applications, are also present. Overall, gene expression data point out the digestive gland as the most lectin-rich tissue, followed by the gills, while on the other hand the mantle rim, foot and posterior adductor muscle only produce a limited set of lectin-like molecules. While this remarkable expression might be linked to food particle selection, it is also consistent with the emerging role of peripheral tissues in the bivalve innate immune system, coherently with the extensive contact of gills and digestive tract with the external environments and microbes associated with sea water, sediment and food particles.
Conclusions
As more and more bivalve species become the subject of –omic studies thanks to the development of cost-effective sequencing technologies, most transcriptomic studies are usually either focused on single tissues or on samples obtained from whole body, and just a very few have been so far dedicated to the investigation of the highly specialized function of tissues in a comparative way. With the present study, we tried to fill a gap in the genetic knowledge of M. virgata, providing a detailed snapshot of the gene expression profile of most of the main tissues of this marine mussel species. Besides revealing the high tissue-specificity of genes fundamental for mussel immune defense, feeding and attachment to the substrate, we also provide compelling evidence bimolecular phylogeny that the Japanese purplish mussel is not closely related to Septifer (Récluz, 1848) and that is should be instead considered as part of the Brachidontinae subfamily.
Additional files
Annotations and gene expression levels of Mytilisepta virgata contigs. (XLSX 5900 kb)
Multiple sequence alignment of the consensus of the 17-mer repeated units present in the byssal cuticle proteins of Mytilisepta virgata, Septifer biforcatus and Bathymodiolus thermophilus. (PDF 122 kb)
Scatter plots representing paired comparisons of gene expression profiles between tissues. (PDF 1316 kb)
Complete results of gene set enrichment analyses. (XLSX 29 kb)
Acknowledgements
We would like to thank Dr. Giulia Moro and Dr. Martina Ianniello for technical help. We thank Dr. Jose Liétor Gallego for providing shell photographs.
Funding
This project was performed within the frame of the project “Molecular characterization of Myticalins: a new family of linearcationic AMPs from Mytilus galloprovincialis”, supported by the program Finanziamento di Ateneo per la Ricerca Scientifica 2015 (FRA 2015) of the University of Trieste. Yuki Fujii and Hideaki Fujita received research funds from Nagasaki International University and grants-in-aid from the Ministry of Education, Science, Culture, and Sport (25,840,119 to YF, and 460,086 to HF). Yasuhiro Ozeki received research funds from the Yokohama City University (20160951101). This study was supported in part by Grants-In-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS). The funding agencies played no role in the design of the study, sample collection, analysis or interpretation of the data nor in the writing of the manuscript.
Availability of data and materials
Raw sequencing reads have been deposited in the NCBI SRA database (Accession ID: SRP096873) under the umbrella Bioproject PRJNA361571, linked to Biosamples SAMN06233921–5.
The final set of high quality assembled contigs was deposited in the NCBI Transcriptome Shotgun Assembly database under the accession ID GFKS00000000.
Contig annotation and expression data are available in Additional file 1.
The multiple sequence alignment of the 17-mer repeated units of byssal cuticle proteins is available in Additional file 2.
Gene expression scatter plots are available in Additional file 3.
The complete results of hypergeometric tests on annotations are available in Additional file 4.
Abbreviations
- BUSCO
Benchmarking Universal Single-Copy Ortholog
- C1qDC
C1q domain-containing
- CDS
Coding sequence
- COI
Cytochrome c oxidase subunit I
- CRD
Carbohydrate recognition domain
- DEG
Differentially expressed gene
- FREP
Fibrinogen-related protein
- L-DOPA
L-3,4-dihydroxyphenylalanine
- MAPM
Microbe Associated Molecular Pattern
- PAMP
Pathogen Associated Molecular Pattern
- PCR
Polymerase chain reaction
- PRR
Pattern Recognition Receptor
- TDI
Transcriptomic Diversity Index
- TPM
Transcripts Per Million
Authors’ contributions
MG performed the assembly, annotation and bioinformatics analyses, designed validation experiments and wrote the paper. MG and YF conceived the project and experimental plan. YF, TK, SS and KY collected the biological material, dissected the animals and prepared tissues for RNA extraction. FS extracted RNA from samples, evaluated its quality, prepared sequencing libraries and performed validation experiments by RT-PCR. AP contributed to bioinformatics analyses and supervised the pipeline of analysis. YF, IH, YO and HF provided inputs for the biological interpretation of results and performed the analysis of lectin gene families. All the authors provided a critical contribution for the improvement of the first draft of the manuscript and approved its final version.
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-017-4012-z) contains supplementary material, which is available to authorized users.
Contributor Information
Marco Gerdol, Email: mgerdol@units.it.
Yuki Fujii, Email: yfujii@niu.ac.jp.
Imtiaj Hasan, Email: hasanimtiaj@yahoo.co.uk.
Toru Koike, Email: to_ru4321@yahoo.co.jp.
Shunsuke Shimojo, Email: syunsyunsyunborder@yahoo.co.jp.
Francesca Spazzali, Email: francesca.spazzali@studenti.units.it.
Kaname Yamamoto, Email: Kaname0225you@gmail.com.
Yasuhiro Ozeki, Email: ozeki@yokohama-cu.ac.jp.
Alberto Pallavicini, Email: pallavic@units.it.
Hideaki Fujita, Email: fujita@niu.ac.jp.
References
- 1.Lee SY, Morton B. The Hong Kong Mytilidae. Proc. second Int. workshop Malacofauna Hong Kong south. China Hong Kong. 1983;1985:49–76. [Google Scholar]
- 2.Huq KA. Ecological studies on the population of Septifer virgatus (Bivalvia:Mytilidae) on a rocky shore in Mutsu Bay, northern Japan [Internet]. Tohoku University; 1997 [cited 2017 Jan 11] Available from: http://ci.nii.ac.jp/naid/500000142556?l=en
- 3.Iwasaki K. Distribution and Bed Structure of the Two Intertidal Mussels, Septifer virgatus (Wiegmann) and Hormomya mutabilis (Gould). Publ Seto Mar Biol Lab. 1994;36:223–47.
- 4.Fuji A, Nomura H. Community structure of the rocky shore macrobenthos in southern Hokkaido. Japan Mar Biol. 1990;107:471–477. doi: 10.1007/BF01313431. [DOI] [Google Scholar]
- 5.Seed R, Richardson CA. Evolutionary traits in Perna Viridis (Linnaeus) and Septifer Virgatus (Wiegmann) (Bivalvia: Mytilidae) J Exp Mar Biol Ecol. 1999;239:273–287. doi: 10.1016/S0022-0981(99)00043-X. [DOI] [Google Scholar]
- 6.Morton B. The population dynamics and reproductive cycle of Septifer Virgatus (Bivalvia: Mytilidae) on an exposed rocky shore in Hong Kong. J Zool. 1995;235:485–500. doi: 10.1111/j.1469-7998.1995.tb01764.x. [DOI] [Google Scholar]
- 7.Scarlato O, Starobogatov Y. O sisteme podotriada Mytileina (Bivalvia). Molliuski Oznovnye Rezul’taty Ikh Izycheniiya [Internet]. Akademiia Nauk SSSR, Zoologicheskii Institut. Nauka. Leningrad: Likharev, IM; 1979 [cited 2017 Jan 9]. p. 22–5. Available from: http://bionames.org/references/8012d29920104d11d9edbcbc42fcf325
- 8.Matsumoto M. Phylogenetic analysis of the subclass Pteriomorphia (Bivalvia) from mtDNA COI sequences. Mol Phylogenet Evol. 2003;27:429–440. doi: 10.1016/S1055-7903(03)00013-7. [DOI] [PubMed] [Google Scholar]
- 9.Trovant B, Orensanz JM (Lobo), Ruzzante DE, Stotz W, Basso NG. Scorched mussels (BIVALVIA: MYTILIDAE: BRACHIDONTINAE) from the temperate coasts of South America: Phylogenetic relationships, trans-Pacific connections and the footprints of Quaternary glaciations Mol. Phylogenet. Evol. 2015;82, Part A:60–74. [DOI] [PubMed]
- 10.Kurita Y, Deguchi R, Wada H. Early development and cleavage pattern of the Japanese purple mussel. Septifer virgatus Zoolog Sci. 2009;26:814–820. doi: 10.2108/zsj.26.814. [DOI] [PubMed] [Google Scholar]
- 11.Combosch DJ, Collins TM, Glover EA, Graf DL, Harper EM, Healy JM, et al. A family-level tree of life for bivalves based on a sanger-sequencing approach. Mol Phylogenet Evol. 2017;107:191–208. doi: 10.1016/j.ympev.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Lee T, Foighil D. Placing the Floridian marine genetic disjunction into a regional evolutionary context using the scorched mussel, Brachidontes Exustus. Species Complex Evolution. 2005;59:2139–2158. [PubMed] [Google Scholar]
- 13.Martínez-Lage A, Rodríguez-Fariña F, González-Tizón A, Méndez J. Origin and evolution of Mytilus mussel satellite DNAs. Genome. 2005;48:247–256. doi: 10.1139/g04-115. [DOI] [PubMed] [Google Scholar]
- 14.Gerdol M, Moro GD, Manfrin C, Milandri A, Riccardi E, Beran A, et al. RNA sequencing and de novo assembly of the digestive gland transcriptome in Mytilus Galloprovincialis fed with toxinogenic and non-toxic strains of Alexandrium Minutum. BMC Res Notes. 2014;7:722. doi: 10.1186/1756-0500-7-722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moreira R, Pereiro P, Canchaya C, Posada D, Figueras A, Novoa B. RNA-Seq in Mytilus Galloprovincialis: comparative transcriptomics and expression profiles among different tissues. BMC Genomics. 2015;16:728. doi: 10.1186/s12864-015-1817-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murgarella M, Puiu D, Novoa B, Figueras A, Posada D, Canchaya C. Correction: a first insight into the genome of the filter-feeder mussel Mytilus Galloprovincialis. PLoS One. 2016;11 doi: 10.1371/journal.pone.0160081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Philipp EER, Kraemer L, Melzner F, Poustka AJ, Thieme S, Findeisen U, et al. Massively parallel RNA sequencing identifies a complex immune gene repertoire in the lophotrochozoan Mytilus Edulis. PLoS One. 2012;7 doi: 10.1371/journal.pone.0033091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venier P, Pittà CD, Bernante F, Varotto L, Nardi BD, Bovo G, et al. MytiBase: a knowledgebase of mussel (M. galloprovincialis) transcribed sequences. BMC Genomics. 2009;10:–72. [DOI] [PMC free article] [PubMed]
- 19.Ieyama H, Kameoka O, Tan T, Yamasaki J. Chromosomes and nuclear DNA contents of some species of Mytilidae. Venus. 53:327–31.
- 20.Gerdol M, Venier P. An updated molecular basis for mussel immunity. Fish Shellfish Immunol. 2015;46:17–38. doi: 10.1016/j.fsi.2015.02.013. [DOI] [PubMed] [Google Scholar]
- 21.Oliveira C, Teixeira JA, Domingues L. Recombinant production of plant lectins in microbial systems for biomedical application – the frutalin case study. Front Plant Sci. 2014;5:390. [DOI] [PMC free article] [PubMed]
- 22.Dan X, Liu W, Ng TB. Development and applications of Lectins as biological tools in biomedical research. Med Res Rev. 2016;36:221–247. doi: 10.1002/med.21363. [DOI] [PubMed] [Google Scholar]
- 23.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, et al. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44:W3–10. doi: 10.1093/nar/gkw343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biscotti MA, Gerdol M, Canapa A, Forconi M, Olmo E, Pallavicini A, et al. The Lungfish Transcriptome: A Glimpse into Molecular Evolution Events at the Transition from Water to Land. Sci Rep. 2016;6:21517. [DOI] [PMC free article] [PubMed]
- 26.Carniel FC, Gerdol M, Montagner A, Banchi E, De Moro G, Manfrin C, et al. New features of desiccation tolerance in the lichen photobiont Trebouxia Gelatinosa are revealed by a transcriptomic approach. Plant Mol Biol. 2016;91:319–339. doi: 10.1007/s11103-016-0468-5. [DOI] [PubMed] [Google Scholar]
- 27.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 28.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 29.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinforma Oxf Engl. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 30.Kriventseva EV, Rahman N, Espinosa O, Zdobnov EM. OrthoDB: the hierarchical catalog of eukaryotic orthologs. Nucleic Acids Res. 2008;36:D271–D275. doi: 10.1093/nar/gkm845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci Theor Den Biowissenschaften. 2012;131:281–285. doi: 10.1007/s12064-012-0162-3. [DOI] [PubMed] [Google Scholar]
- 36.Kal AJ, van Zonneveld AJ, Benes V, van den Berg M, Koerkamp MG, Albermann K, et al. Dynamics of gene expression revealed by comparison of serial analysis of gene expression transcript profiles from yeast grown on two different carbon sources. Mol Biol Cell. 1999;10:1859–1872. doi: 10.1091/mbc.10.6.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falcon S, Gentleman R. Hypergeometric Testing Used for Gene Set Enrichment Analysis. Bioconductor Case Stud. [Internet]. Springer New York; 2008 [cited 2017 Jan 11]. p. 207–20. Available from: http://link.springer.com/chapter/10.1007/978-0-387-77240-0_14
- 38.Gerdol M, Manfrin C, De Moro G, Figueras A, Novoa B, Venier P, et al. The C1q domain containing proteins of the Mediterranean mussel Mytilus Galloprovincialis: a widespread and diverse family of immune-related molecules. Dev Comp Immunol. 2011;35:635–643. doi: 10.1016/j.dci.2011.01.018. [DOI] [PubMed] [Google Scholar]
- 39.Gerdol M, De Moro G, Manfrin C, Venier P, Pallavicini A. Big defensins and mytimacins, new AMP families of the Mediterranean mussel Mytilus Galloprovincialis. Dev Comp Immunol. 2012;36:390–399. doi: 10.1016/j.dci.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Zhang G, Fang X, Guo X, Li L, Luo R, Xu F, et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature. 2012;490:49–54. doi: 10.1038/nature11413. [DOI] [PubMed] [Google Scholar]
- 41.Lemer S, González VL, Bieler R, Giribet G. Cementing mussels to oysters in the pteriomorphian tree: a phylogenomic approach. Proc R Soc B. 2016;283:20160857. doi: 10.1098/rspb.2016.0857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 44.Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;btr088. [DOI] [PMC free article] [PubMed]
- 45.Hurvich CM, Tsai C-L. Regression and time series model selection in small samples. Biometrika. 1989;76:297–307. doi: 10.1093/biomet/76.2.297. [DOI] [Google Scholar]
- 46.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinforma. Oxf. Engl. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 47.Takeuchi T, Kawashima T, Koyanagi R, Gyoja F, Tanaka M, Ikuta T, et al. Draft genome of the pearl oyster Pinctada Fucata: a platform for understanding bivalve biology. DNA res. Int. J. Rapid Publ. Rep. Genes Genomes. 2012;19:117–130. doi: 10.1093/dnares/dss005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Earl D, Bradnam K, John JS, Darling A, Lin D, Fass J, et al. Assemblathon 1: a competitive assessment of de novo short read assembly methods. Genome Res. 2011;21:2224–2241. doi: 10.1101/gr.126599.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guerette PA, Hoon S, Seow Y, Raida M, Masic A, Wong FT, et al. Accelerating the design of biomimetic materials by integrating RNA-seq with proteomics and materials science. Nat Biotechnol. 2013;31:908–915. doi: 10.1038/nbt.2671. [DOI] [PubMed] [Google Scholar]
- 50.Fields PA, Eurich C, Gao WL, Cela B. Changes in protein expression in the salt marsh mussel Geukensia Demissa: evidence for a shift from anaerobic to aerobic metabolism during prolonged aerial exposure. J Exp Biol. 2014;217:1601–1612. doi: 10.1242/jeb.101758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uliano-Silva M, Americo JA, Brindeiro R, Dondero F, Prosdocimi F. Rebelo M de F. Gene Discovery through Transcriptome Sequencing for the Invasive Mussel Limnoperna fortunei PLOS ONE. 2014;9 doi: 10.1371/journal.pone.0102973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fraïsse C, Belkhir K, Welch JJ, Bierne N. Local interspecies introgression is the main cause of extreme levels of intraspecific differentiation in mussels. Mol. Ecol. 2015; [DOI] [PubMed]
- 53.Gerdol M, Puillandre N, Moro GD, Guarnaccia C, Lucafò M, Benincasa M, et al. Identification and characterization of a novel family of cysteine-rich peptides (MgCRP-I) from Mytilus Galloprovincialis. Genome Biol Evol. 2015;7:2203–2219. doi: 10.1093/gbe/evv133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sonthi M, Toubiana M, Pallavicini A, Venier P, Roch P. Diversity of coding sequences and gene structures of the antifungal peptide Mytimycin (MytM) from the Mediterranean mussel. Mar Biotechnol. 2011;13:857–867. doi: 10.1007/s10126-010-9345-4. [DOI] [PubMed] [Google Scholar]
- 55.Sartori A, Bouchet P, Huber M. Mytilisepta virgata (Wiegmann, 1837) [Internet]. MolluscaBase. 2015; Available from: http://molluscabase.org/aphia.php?p=taxdetails&id=506157
- 56.González VL, Andrade SCS, Bieler R, Collins TM, Dunn CW, Mikkelsen PM, et al. A phylogenetic backbone for Bivalvia: an RNA-seq approach. Proc R Soc B Biol Sci. 2015;282:20142332. [DOI] [PMC free article] [PubMed]
- 57.Kocot KM, Cannon JT, Todt C, Citarella MR, Kohn AB, Meyer A, et al. Phylogenomics reveals deep molluscan relationships. Nature. 2011;477:452–456. doi: 10.1038/nature10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith SA, Wilson NG, Goetz FE, Feehery C, Andrade SCS, Rouse GW, et al. Resolving the evolutionary relationships of molluscs with phylogenomic tools. Nature. 2011;480:364–367. doi: 10.1038/nature10526. [DOI] [PubMed] [Google Scholar]
- 59.Weinstein J. Fine structure of the digestive tubule of the eastern oyster, Crassostrea Virginica (Gmelin 1971) J Shellfish Res. 1995;14:97–103. [Google Scholar]
- 60.Picoche C, Le Gendre R, Flye-Sainte-Marie J, Françoise S, Maheux F, Simon B, et al. Towards the determination of Mytilus Edulis food preferences using the dynamic energy budget (DEB) theory. PLoS One. 2014;9 doi: 10.1371/journal.pone.0109796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tharanathan RN, Kittur FS. Chitin--the undisputed biomolecule of great potential. Crit Rev Food Sci Nutr. 2003;43:61–87. doi: 10.1080/10408690390826455. [DOI] [PubMed] [Google Scholar]
- 62.Rangaraju S, Khoo KK, Feng Z-P, Crossley G, Nugent D, Khaytin I, et al. Potassium channel modulation by a toxin domain in matrix metalloprotease 23. J Biol Chem. 2010;285:9124–9136. doi: 10.1074/jbc.M109.071266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nikapitiya C, De Zoysa M, Oh C, Lee Y, Ekanayake PM, Whang I, et al. Disk abalone (Haliotis Discus Discus) expresses a novel antistasin-like serine protease inhibitor: molecular cloning and immune response against bacterial infection. Fish Shellfish Immunol. 2010;28:661–671. doi: 10.1016/j.fsi.2009.12.027. [DOI] [PubMed] [Google Scholar]
- 64.M Laskowski J, Kato I. Protein Inhibitors of Proteinases. Ann Rev Biochem. 1980;45:593–626. [DOI] [PubMed]
- 65.Manfrin C, De Moro G, Torboli V, Venier P, Pallavicini A, Gerdol M. Physiological and molecular responses of bivalves to toxic dinoflagellates. Invertebr Surviv J. 2012;9:184–199. [Google Scholar]
- 66.Sakellari A, Karavoltsos S, Theodorou D, Dassenakis M, Scoullos M. Bioaccumulation of metals (cd, cu, Zn) by the marine bivalves M. Galloprovincialis, P. Radiata, V. Verrucosa and C. Chione in Mediterranean coastal microenvironments: association with metal bioavailability. Environ. Monit. Assessment 2013;185:3383–3395. [DOI] [PubMed]
- 67.Craft JA, Gilbert JA, Temperton B, Dempsey KE, Ashelford K, Tiwari B, et al. Pyrosequencing of Mytilus Galloprovincialis cDNAs: tissue-specific expression patterns. PLoS One. 2010;5 doi: 10.1371/journal.pone.0008875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jackson DJ, Ellemor N, Degnan BM. Correlating gene expression with larval competence, and the effect of age and parentage on metamorphosis in the tropical abalone Haliotis Asinina. Mar Biol. 2005;147:681–697. doi: 10.1007/s00227-005-1603-z. [DOI] [Google Scholar]
- 69.Murgarella M, Puiu D, Novoa B, Figueras A, Posada D, Canchaya C. A first insight into the genome of the filter-feeder mussel Mytilus Galloprovincialis. PLoS One. 2016;11 doi: 10.1371/journal.pone.0151561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Waite JH. The Formation of Mussel Byssus: Anatomy of a Natural Manufacturing Process. In: Case PDST, editor. Struct. Cell. Synth. Assem. Biopolym. [Internet]. Springer Berlin Heidelberg; 1992 [cited 2017 Jan 25]. p. 27–54. Available from: http://link.springer.com/chapter/10.1007/978-3-540-47207-0_2 [DOI] [PubMed]
- 71.Harrington MJ, Waite JH. Holdfast heroics: comparing the molecular and mechanical properties of Mytilus Californianus byssal threads. J Exp Biol. 2007;210:4307–4318. doi: 10.1242/jeb.009753. [DOI] [PubMed] [Google Scholar]
- 72.Qin X-X, Coyne KJ, Waite JH. Tough tendons MUSSEL BYSSUS HAS COLLAGEN WITH SILK-LIKE DOMAINS. J Biol Chem. 1997;272:32623–32627. doi: 10.1074/jbc.272.51.32623. [DOI] [PubMed] [Google Scholar]
- 73.Holten-Andersen N, Zhao H, Waite JH. Stiff coatings on compliant biofibers: the cuticle of Mytilus Californianus byssal threads. Biochemistry (Mosc) 2009;48:2752–2759. doi: 10.1021/bi900018m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rzepecki LM, Chin SS, Waite JH, Lavin MF. Molecular diversity of marine glues: polyphenolic proteins from five mussel species. Mol Mar Biol Biotechnol. 1991;1:78–88. [PubMed] [Google Scholar]
- 75.Qin C, Pan Q, Qi Q, Fan M, Sun J, Li N, et al. In-depth proteomic analysis of the byssus from marine mussel Mytilus Coruscus. J Proteome. 2016;144:87–98. doi: 10.1016/j.jprot.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 76.Inoue K, Takeuchi Y, Miki D, Odo S. Mussel adhesive plaque protein gene is a novel member of epidermal growth factor-like gene family. J Biol Chem. 1995;270:6698–6701. doi: 10.1074/jbc.270.12.6698. [DOI] [PubMed] [Google Scholar]
- 77.Waite JH. The phylogeny and chemical diversity of quinone-tanned glues and varnishes. Comp Biochem Physiol -- Part B Biochem. 1990;97:19–29. doi: 10.1016/0305-0491(90)90172-P. [DOI] [PubMed] [Google Scholar]
- 78.Waite JH. Catechol oxidase in the byssus of the common mussel, Mytilus Edulis L. J Mar Biol Assoc U K. 1985;65:359–371. doi: 10.1017/S0025315400050487. [DOI] [Google Scholar]
- 79.Anderson TH, Yu J, Estrada A, Hammer MU, Waite JH, Israelachvili JN. The contribution of DOPA to substrate–peptide adhesion and internal cohesion of mussel-inspired synthetic peptide films. Adv Funct Mater. 2010;20:4196–4205. doi: 10.1002/adfm.201000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu J, Wei W, Danner E, Ashley RK, Israelachvili JN, Waite JH. Mussel protein adhesion depends on interprotein thiol-mediated redox modulation. Nat Chem Biol. 2011;7:588–590. doi: 10.1038/nchembio.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nukumi N, Iwamori T, Kano K, Naito K, Tojo H. Whey acidic protein (WAP) regulates the proliferation of mammary epithelial cells by preventing serine protease from degrading laminin. J Cell Physiol. 2007;213:793–800. doi: 10.1002/jcp.21155. [DOI] [PubMed] [Google Scholar]
- 82.Porter ME, Sale WS. The 9 + 2 Axoneme anchors multiple inner arm Dyneins and a network of kinases and phosphatases that control motility. J Cell Biol. 2000;151:37–42. doi: 10.1083/jcb.151.5.F37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Amos LA. The tektin family of microtubule-stabilizing proteins. Genome Biol. 2008;9:229. doi: 10.1186/gb-2008-9-7-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miyamoto Y, Kitamura N, Nakamura Y, Futamura M, Miyamoto T, Yoshida M, et al. Possible existence of lysosome-like Organella within mitochondria and its role in mitochondrial quality control. PLoS One. 2011;6 doi: 10.1371/journal.pone.0016054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zagoory O, Braiman A, Priel Z. The mechanism of Ciliary stimulation by acetylcholine. J Gen Physiol. 2002;119:329–339. doi: 10.1085/jgp.20028519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pei J, Grishin NV. Unexpected diversity in Shisa-like proteins suggests the importance of their roles as transmembrane adaptors. Cell Signal. 2012;24:758–769. doi: 10.1016/j.cellsig.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gerdol M, Venier P, Pallavicini A. The genome of the Pacific oyster Crassostrea Gigas brings new insights on the massive expansion of the C1q gene family in Bivalvia. Dev Comp Immunol. 2015;49:59–71. doi: 10.1016/j.dci.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 88.Hanington PC, Zhang S-M. The primary role of fibrinogen-related proteins in invertebrates is defense, not coagulation. J Innate Immun. 2011;3:17–27. doi: 10.1159/000321882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gerdol M, Venier P, Edomi P, Pallavicini A. Diversity and evolution of TIR-domain-containing proteins in bivalves and Metazoa: new insights from comparative genomics. Dev Comp Immunol. 2017;70:145–164. doi: 10.1016/j.dci.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 90.Stucchi A, Reed K, O’Brien M, Cerda S, Andrews C, Gower A, et al. A new transcription factor that regulates TNF-alpha gene expression, LITAF, is increased in intestinal tissues from patients with CD and UC. Inflamm Bowel Dis. 2006;12:581–587. doi: 10.1097/01.MIB.0000225338.14356.d5. [DOI] [PubMed] [Google Scholar]
- 91.Lepage T, Gache C. Early expression of a collagenase-like hatching enzyme gene in the sea urchin embryo. EMBO J. 1990;9:3003–3012. doi: 10.1002/j.1460-2075.1990.tb07493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ito Y, Toriuchi N, Yoshitaka T, Ueno-Kudoh H, Sato T, Yokoyama S, et al. The Mohawk homeobox gene is a critical regulator of tendon differentiation. Proc Natl Acad Sci U S A. 2010;107:10538–10542. doi: 10.1073/pnas.1000525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Al-Subiai SN, Jha AN, Moody AJ. Contamination of bivalve haemolymph samples by adductor muscle components: implications for biomarker studies. Ecotoxicology. 2009;18:334. doi: 10.1007/s10646-008-0287-9. [DOI] [PubMed] [Google Scholar]
- 94.Machado C, Sunkel CE, Andrew DJ. Human autoantibodies reveal Titin as a chromosomal protein. J Cell Biol. 1998;141:321–333. doi: 10.1083/jcb.141.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li H, Venier P, Prado-Alvárez M, Gestal C, Toubiana M, Quartesan R, et al. Expression of Mytilus immune genes in response to experimental challenges varied according to the site of collection. Fish Shellfish Immunol. 2010;28:640–648. doi: 10.1016/j.fsi.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 96.Grutters BMC, Verhofstad MJJM, van der Velde G, Rajagopal S, Leuven RSEW. A comparative study of byssogenesis on zebra and quagga mussels: the effects of water temperature, salinity and light-dark cycle. Biofouling. 2012;28:121–129. doi: 10.1080/08927014.2012.654779. [DOI] [PubMed] [Google Scholar]
- 97.Emmanuelle PE, Mickael P, Evan W, Shumway SE, Bassem A. Lectins associated with the feeding organs of the oyster crassostrea virginica can mediate particle selection. Biol Bull. 2009;217:130–141. doi: 10.1086/BBLv217n2p130. [DOI] [PubMed] [Google Scholar]
- 98.Lima TG, McCartney MA. Adaptive evolution of M3 lysin--a candidate gamete recognition protein in the Mytilus Edulis species complex. Mol Biol Evol. 2013;30:2688–2698. doi: 10.1093/molbev/mst165. [DOI] [PubMed] [Google Scholar]
- 99.Springer SA, Crespi BJ. Adaptive gamete-recognition divergence in a hybridizing Mytilus population. Evolution. 2007;61:772–783. doi: 10.1111/j.1558-5646.2007.00073.x. [DOI] [PubMed] [Google Scholar]
- 100.Wu Q, Li L, Zhang G. Crassostrea Angulata bindin gene and the divergence of fucose-binding lectin repeats among three species of Crassostrea. Mar Biotechnol N Y N. 2011;13:327–335. doi: 10.1007/s10126-010-9304-0. [DOI] [PubMed] [Google Scholar]
- 101.Odo S, Kamino K, Kanai S, Maruyama T, Harayama S. Biochemical characterization of a Ca2+−dependent lectin from the hemolymph of a photosymbiotic marine bivalve, Tridacna derasa (röding) J Biochem (Tokyo) 1995;117:965–973. doi: 10.1093/oxfordjournals.jbchem.a124828. [DOI] [PubMed] [Google Scholar]
- 102.Suzuki T, Mori K. Hemolymph lectin of the pearl oyster, Pinctada Fucata Martensii: a possible non-self recognition system. Dev Comp Immunol. 1990;14:161–173. doi: 10.1016/0145-305X(90)90088-V. [DOI] [PubMed] [Google Scholar]
- 103.Tunkijjanukij S, Olafsen JA. Sialic acid-binding lectin with antibacterial activity from the horse mussel: further characterization and immunolocalization. Dev Comp Immunol. 1998;22:139–150. doi: 10.1016/S0145-305X(98)00017-2. [DOI] [PubMed] [Google Scholar]
- 104.Naganuma T, Ogawa T, Hirabayashi J, Kasai K, Kamiya H, Muramoto K. Isolation, characterization and molecular evolution of a novel pearl shell lectin from a marine bivalve. Pteria penguin Mol Divers. 2006;10:607–618. doi: 10.1007/s11030-006-9051-3. [DOI] [PubMed] [Google Scholar]
- 105.Liu SX, Qi ZH, Zhang JJ, He CB, Gao XG, Li HJ. Lipopolysaccharide and β-1,3-glucan binding protein in the hard clam (Meretrix meretrix): molecular characterization and expression analysis. Genet Mol Res GMR. 2014;13:4956–4966. doi: 10.4238/2014.July.4.10. [DOI] [PubMed] [Google Scholar]
- 106.Fujii Y, Dohmae N, Takio K, Kawsar SMA, Matsumoto R, Hasan I, et al. A lectin from the mussel Mytilus Galloprovincialis has a highly novel primary structure and induces glycan-mediated cytotoxicity of globotriaosylceramide-expressing lymphoma cells. J Biol Chem. 2012;287:44772–44783. doi: 10.1074/jbc.M112.418012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Anju A, Jeswin J, Thomas PC, Vijayan KK. Molecular cloning, characterization and expression analysis of F-type lectin from pearl oyster Pinctada Fucata. Fish Shellfish Immunol. 2013;35:170–174. doi: 10.1016/j.fsi.2013.03.359. [DOI] [PubMed] [Google Scholar]
- 108.Hasan I, Sugawara S, Fujii Y, Koide Y, Terada D, Iimura N, et al. MytiLec, a mussel R-type Lectin, interacts with surface glycan Gb3 on Burkitt’s lymphoma cells to trigger apoptosis through multiple pathways. Mar Drugs. 2015;13:7377–7389. doi: 10.3390/md13127071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sudhakar GRL, Vincent SGP. Purification and characterization of a novel C-type hemolytic lectin for clot lysis from the fresh water clam Villorita cyprinoides: a possible natural thrombolytic agent against myocardial infarction. Fish Shellfish Immunol. 2014;36:367–373. doi: 10.1016/j.fsi.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 110.Pales E, Koller A, Allam B. Proteomic characterization of mucosal secretions in the eastern oyster. J Proteomics. 2016;132:63–76. doi: 10.1016/j.jprot.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 111.Kang Y-S, Kim Y-M, Park K-I, Kim Cho S, Choi K-S, Cho M. Analysis of EST and lectin expressions in hemocytes of manila clams (Ruditapes philippinarum) (Bivalvia: Mollusca) infected with Perkinsus olseni. Dev Comp Immunol. 2006;30:1119–1131. doi: 10.1016/j.dci.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 112.McDowell IC, Modak TH, Lane CE, Gomez-Chiarri M. Multi-species protein similarity clustering reveals novel expanded immune gene families in the eastern oyster Crassostrea Virginica. Fish Shellfish Immunol. 2016;53:13–23. doi: 10.1016/j.fsi.2016.03.157. [DOI] [PubMed] [Google Scholar]
- 113.Zhang L, Li L, Guo X, Litman GW, Dishaw LJ, Zhang G. Massive expansion and functional divergence of innate immune genes in a protostome. Sci Rep. 2015;5:8693. [DOI] [PMC free article] [PubMed]
- 114.Jing X, Espinosa EP, Perrigault M, Allam B. Identification, molecular characterization and expression analysis of a mucosal C-type lectin in the eastern oyster. Fish Shellfish Immunol. 2011;30:851–858. doi: 10.1016/j.fsi.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 115.Li C, Yu S, Zhao J, Su X, Li T. Cloning and characterization of a sialic acid binding lectins (SABL) from manila clam Venerupis Philippinarum. Fish Shellfish Immunol. 2011;30:1202–1206. doi: 10.1016/j.fsi.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 116.Yang J, Wei X, Liu X, Xu J, Yang D, Yang J, et al. Cloning and transcriptional analysis of two sialic acid-binding lectins (SABLs) from razor clam Solen Grandis. Fish Shellfish Immunol. 2012;32:578–585. doi: 10.1016/j.fsi.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 117.Bao XB, He CB, Fu CD, Wang B, Zhao XM, Gao XG, et al. A C-type lectin fold gene from Japanese scallop Mizuhopecten yessoensis, involved with immunity and metamorphosis. Genet Mol Res. 2015;14:2253–2267. doi: 10.4238/2015.March.27.11. [DOI] [PubMed] [Google Scholar]
- 118.Kong P, Wang L, Zhang H, Song X, Zhou Z, Yang J, et al. A novel C-type lectin from bay scallop Argopecten Irradians (AiCTL-7) agglutinating fungi with mannose specificity. Fish Shellfish Immunol. 2011;30:836–844. doi: 10.1016/j.fsi.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 119.Zhang J, Qiu R, Hu Y-H. HdhCTL1 is a novel C-type lectin of abalone Haliotis Discus Hannai that agglutinates gram-negative bacterial pathogens. Fish Shellfish Immunol. 2014;41:466–472. doi: 10.1016/j.fsi.2014.09.032. [DOI] [PubMed] [Google Scholar]
- 120.Pales Espinosa E, Perrigault M, Allam B. Identification and molecular characterization of a mucosal lectin (MeML) from the blue mussel Mytilus Edulis and its potential role in particle capture. Comp Biochem Physiol A Mol Integr Physiol. 2010;156:495–501. doi: 10.1016/j.cbpa.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 121.Gorbushin AM, Iakovleva NV. A new gene family of single fibrinogen domain lectins in Mytilus. Fish Shellfish Immunol. 2011;30:434–438. doi: 10.1016/j.fsi.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 122.Zhang H, Wang L, Song L, Song X, Wang B, Mu C, et al. A fibrinogen-related protein from bay scallop Argopecten Irradians involved in innate immunity as pattern recognition receptor. Fish Shellfish Immunol. 2009;26:56–64. doi: 10.1016/j.fsi.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 123.Romero A, Dios S, Poisa-Beiro L, Costa MM, Posada D, Figueras A, et al. Individual sequence variability and functional activities of fibrinogen-related proteins (FREPs) in the Mediterranean mussel (Mytilus Galloprovincialis) suggest ancient and complex immune recognition models in invertebrates. Dev Comp Immunol. 2011;35:334–344. doi: 10.1016/j.dci.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 124.Ozeki Y, Matsui T, Suzuki M, Titani K. Amino acid sequence and molecular characterization of a D-galactoside-specific lectin purified from sea urchin (Anthocidaris crassispina) eggs. Biochemistry (Mosc.). 1991;30:2391–2394. [DOI] [PubMed]
- 125.Pietrzyk AJ, Bujacz A, Mak P, Potempa B, Niedziela T. Structural studies of Helix Aspersa agglutinin complexed with GalNAc: a lectin that serves as a diagnostic tool. Int J Biol Macromol. 2015;81:1059–1068. doi: 10.1016/j.ijbiomac.2015.09.044. [DOI] [PubMed] [Google Scholar]
- 126.Sanchez J-F, Lescar J, Chazalet V, Audfray A, Gagnon J, Alvarez R, et al. Biochemical and structural analysis of Helix Pomatia agglutinin. A hexameric lectin with a novel fold. J Biol Chem. 2006;281:20171–20180. doi: 10.1074/jbc.M603452200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Annotations and gene expression levels of Mytilisepta virgata contigs. (XLSX 5900 kb)
Multiple sequence alignment of the consensus of the 17-mer repeated units present in the byssal cuticle proteins of Mytilisepta virgata, Septifer biforcatus and Bathymodiolus thermophilus. (PDF 122 kb)
Scatter plots representing paired comparisons of gene expression profiles between tissues. (PDF 1316 kb)
Complete results of gene set enrichment analyses. (XLSX 29 kb)
Data Availability Statement
Raw sequencing reads have been deposited in the NCBI SRA database (Accession ID: SRP096873) under the umbrella Bioproject PRJNA361571, linked to Biosamples SAMN06233921–5.
The final set of high quality assembled contigs was deposited in the NCBI Transcriptome Shotgun Assembly database under the accession ID GFKS00000000.
Contig annotation and expression data are available in Additional file 1.
The multiple sequence alignment of the 17-mer repeated units of byssal cuticle proteins is available in Additional file 2.
Gene expression scatter plots are available in Additional file 3.
The complete results of hypergeometric tests on annotations are available in Additional file 4.