

Abstract
Phenylketonuria (PKU) and less severe hyperphenylalaninemia (HPA) constitute the most common inborn error of amino acid metabolism, and is most often caused by defects in phenylalanine hydroxylase (PAH) function resulting in accumulation of Phe to neurotoxic levels. Despite the success of dietary intervention in preventing permanent neurological damage, individuals living with PKU clamor for additional non-dietary therapies. The bulk of disease-associated mutations are PAH missense variants, which occur throughout the entire 452 amino acid human PAH protein. While some disease-associated mutations affect protein structure (e.g. truncations) and others encode catalytically dead variants, most have been viewed as defective in protein folding/stability. Here we refine this view to address how PKU-associated missense variants can perturb the equilibrium among alternate native PAH structures (resting-state PAH and activated PAH), thus shifting the tipping point of this equilibrium to a neurotoxic Phe concentration. This refined view of PKU introduces opportunities for the design or discovery of therapeutic pharmacological chaperones that can help restore the tipping point to healthy Phe levels and how such a therapeutic might work with or without the inhibitory pharmacological chaperone BH4. Dysregulation of an equilibrium of architecturally distinct native PAH structures departs from the concept of “misfolding”, provides an updated understanding of PKU, and presents an enhanced foundation for understanding genotype/phenotype relationships.
Keywords: Phenylketonuria, Phenylalanine hydroxylase, allostery, conformational selection, pharmacological chaperones
Phenylketonuria – a brief overview
Phenylalanine (Phe), one of the twenty common amino acids that are the building blocks of all protein, cannot be synthesized by humans and must be obtained through diet and/or protein catabolism. Regardless of intake, humans generally maintain blood (plasma) Phe in a narrow range, and nearly always below 120 μM. Individuals who fail to do so have disorders ranging from hyperphenylalaninemia (HPA, 120–360 μM Phe) to the most severe forms of phenylketonuria (PKU, >1200 μM Phe) (OMIM 261600). Current treatment strategies focus on lowering Phe levels, though guidelines continue to evolve (e.g. (1–5)). Untreated PKU during brain development (infancy, childhood, adolescence) can result in profound and irreversible neurocognitive damage. Poor control of Phe levels, at any life stage, can result in failures in executive function or a need for psychological or psychiatric care (6). Symptoms generally improve with reduction in blood Phe. Hence, lifelong control of blood Phe, through diet and/or pharmacological intervention, is the consensus recommendation for individuals living with HPA and PKU (hereafter together called PKU) (7).
PKU is the most common inborn error of amino acid metabolism, though frequency varies widely by country (~1:10,000 on average; 1:2,600 in Turkey; 1:100,000 in Japan). Newborn screening, early diagnosis, and a synthetic (Phe-controlled) diet during infancy and childhood has alleviated the most severe outcomes in countries where testing and intervention is available. However, neonatal testing, first introduced mid-20th Century (8), is not yet universal; treatment is expensive and insurance coverage for medical foods remains problematic (9, 10). One outcome of successful dietary interventions is an increased population of socially integrated individuals living with PKU who are advocating for expanded non-dietary therapies. Many affected individuals struggle to obtain sufficient overall nutrition containing enough protein to satisfy essential metabolic needs while keeping Phe levels below the neurotoxic range. This problem is acute during pregnancy when high maternal Phe can cause irreversible fetal brain damage, regardless of whether the child will have PKU. Compounded by perpetual hunger, the high cost and inferior taste of most medical foods, and the basic human need for social inclusion, dietary compliance is problematic. Treatments allowing increased natural protein intake are desirable. An improved understanding of the molecular bases for PKU can help reach this goal.
The industry to address the unmet medical need of PKU includes about eight companies providing specialized foods/formulas, and one marketed therapeutic (Kuvan®), which is effective for some patients (11). In addition, an injectable enzyme substitution therapy, Pegvaliase, is in clinical trials (12). However, a recent survey of patients indicates strong desire for new, preferably oral, interventions that will reduce the need for dietary restrictions (13). In short, there is a growing medical need, and a significant societal cost to not meeting this need. The consequences of untreated PKU has been estimated at a lifetime societal cost of >$1,000,000 per individual (12).
There is consensus that restoring the body’s ability to maintain Phe in the normal range is key to effective therapy, although it is unresolved whether the best indicator of disease is blood Phe, brain Phe, or downstream neurotransmitter concentrations (e.g. (14, 15)). Nevertheless, restoration of Phe regulation begins with understanding how unaffected individuals control Phe levels, and why there is extensive variation in phenotype among those who cannot regulate Phe. This article presents an updated framework for understanding human regulation of Phe, how this can go amiss, and ways of restoring affected individuals to a more normal Phe concentration range. The presented perspective is different from the common view; it is driven by, and focuses on, advances in knowledge of the structural biology of the enzyme phenylalanine hydroxylase (PAH; EC 1.14.16.1; OMIM 612349).
Regulation of Phe in humans
Phe in humans is regulated by PAH, which functions largely in the liver, and whose dysfunction is the cause of most PKU. An understanding of normal PAH structure/function relationships provides context for repairing abnormal PAH function. Dysfunctional PAH may have one or more incapacitated characteristics of normal PAH, while the other characteristics remain intact. PAH is able to convert Phe to another common amino acid, tyrosine, using molecular oxygen, tetrahydrobiopterin (BH4) and a non-heme iron; this chemistry occurs at the PAH active site. PAH accomplishes the regulation of Phe through a structural flexibility that allows the protein to exist as a mixture of native structures that represent “off” and “on”. In the “off” state (called resting-state PAH, (RS-PAH)), Phe cannot easily get into the active site. In the “on” state (called activated PAH (A-PAH)), the active site is fully accessible. RS-PAH and A-PAH exist as an equilibrating mixture of architecturally distinct assemblies, whose structures are used as symbols in Fig 1. At low Phe (< ~50 μM in normal individuals), the preferred PAH assembly is RS-PAH. RS-PAH is the molecular guard, watching and waiting, having a low affinity for Phe, and allowing a basal level of Phe to remain available for essential functions such as protein biosynthesis. As Phe levels rise, the PAH structural equilibrium shifts toward the activated assembly, which is A-PAH. A-PAH avidly binds Phe at the active site, converting it to tyrosine; this returns Phe levels to a concentration where RS-PAH again predominates. RS-PAH and A-PAH differ in the ability to convert Phe to tyrosine because they differ in their ability to productively bind Phe at the active site. However, the equilibrium between RS-PAH and A-PAH depends upon Phe binding to another site on the protein, called the allosteric site, which is formed by a structural change repositioning various parts of the PAH protein in the transition from RS-PAH to A-PAH. The precise structural location of this allosteric site, which Fig 1 shows as present only in the A-PAH structure, was first hypothesized in 2013 (16); it is now strongly supported by newly published structural studies (17–20). In contrast, some earlier studies had generally suggested that allosteric Phe binding involved an intermolecular interaction involving the regulatory domain; these studies were interpreted solely on the basis of an RS-PAH structure model and did not foresee the conformational change required for formation of A-PAH (e.g. (21)).
Figure 1. The equilibrium of architecturally distinct tetrameric PAH assemblies, shown in terms of a double-pan balance.

The transition between RS-PAH and A-PAH structures requires relocation of the PAH regulatory domain (shown in magenta); this relocation forms a multimer-specific allosteric Phe (black dot) binding site, which is distant from the active site, which is where Phe is converted to tyrosine. RS-PAH predominates at low Phe. Phe-stabilized A-PAH predominates at high Phe.
The presented Phe-centric view of PAH function stresses the regulation of Phe concentration, not the production of tyrosine. The need for tyrosine does not drive PAH activity; the availability of Phe does. To regulate Phe, PAH must be able to interconvert between the RS-PAH and A-PAH assemblies in response to fluctuations in Phe. PAH variants that cannot do this will likely be disease-associated. Thus, PAH variants defective in the ability to properly modulate the PAH structure equilibrium (Fig 1), expand our understanding of the repertoire of disease-associated PAH. The updated view of PKU does not dismiss the long-appreciated fact that a small number of disease-associated PAH variants 1) are truncated proteins that cannot fold and assemble properly or 2) are missing key active site components and cannot transform Phe to tyrosine, regardless of whether or not they fold, assemble, or convert between the RS-PAH and A-PAH structures. But the updated view departs from an oft-cited dogma that all other disease-associated PAH variants are defective in folding and/or stability. A portion of the >600 different disease-associated PAH variants have been shown to be prone to aggregation and/or degradation. These variants likely cannot properly form either the RS-PAH or the A-PAH structures. But an equally significant portion do not fall into this category. Unfortunately the concept of protein misfolding is often broadly applied in the absence of an appreciation for a metastable equilibrium of alternate protein structures, each of which is native, properly folded, and similar in energy (stability) to one another (22, 23). Here we abandon the concept of “misfolding with loss-of-function” and put a structural framework to prior proposals that disease-associated PAH variants can be defective in the allosteric response to elevated Phe in a manner that does not impair protein stability and/or promote aggregation (e.g. (21, 24)). On the basis of newly available structural information that defines the allosteric Phe binding site, we propose that there is a class of common disease-associated variants that are not defective in the ability to fold, but are defective in the ability to stabilize A-PAH. Other variants are likely defective in the ability to transition from the “off” state to the “on” state.
Structure changes required for PAH to respond to allosteric Phe binding
This section describes what is known about the structures of RS-PAH and A-PAH and how this knowledge reshapes our understanding of the breadth of PAH variants that can contribute to PKU.
PAH is encoded by a single gene (location 12q23.2), which normally yields a 452 amino-acid long protein that folds and assembles predominantly into a homotetramer (an assembly containing four copies of the same protein chain). PAH can exist as a dimer, but the factors controlling the dimer ⇔ tetramer equilibrium remain poorly understood and are not discussed herein. The PAH protein contains various structural/functional domains, which are described in Fig 2a. One or more domains are missing in two common truncation variants. The IVS12DS,G-A,+1 variant lacks the multimerization domain; the R111* variant lacks both catalytic and multimerization domains. There are also >600 disease-associated missense variants that have single amino acid changes (11, 25). These occur throughout the protein and are generally not at the enzyme active site. However, enzymatically defective active site variants include E280K, which perturbs the active site structure, and H285Y, which perturbs the iron binding site (26). In order to decipher how all the other, non-active site variants, may be impaired, one needs to know details about the structures of both RS-PAH and A-PAH. Structural understanding of full-length A-PAH remains limited to well-supported models (e.g. (16, 18, 20)), however 2016 saw publication of the first crystal structures of full-length PAH in the resting state (18, 20).
Figure 2. PAH structure and model.

a) The annotated domain structure of PAH illustrates the regulatory, catalytic, and multimerization domains, including subdomains. Numbered residues correspond to the termini and hinges. Coloring is reflected in the darker subunits of the molecular illustrations. b) The first crystal structure for full length PAH (PDB id 5DEN, 2.9 Å resolution (18)) is illustrated using orthogonal views (top and bottom), and represents RS-PAH. The bulk of the protein is shown in balls, while the C-terminal helix is in cartoon. Two of the four subunits are transparent. One opaque subunit is illustrated using lighter shades of the colors in part a. The subunits are labeled in cyan near the catalytic domains (top); they are labeled in red near the regulatory domain (bottom). The dotted white circle illustrates the autoregulatory region partially occluding the enzyme active site (the active site iron is obscured by the auto-inhibitory interaction). Blocked active-site access (partial or total) results in low activity. c) A composite homology model of the A-PAH tetramer (18), depicted as in part b. A-PAH is active because the active sites are accessible (white circle in top image, active site iron is visible). A-PAH contains a close association between the ACT domains of subunits located along the diagonal of the tetramer. Formation of this subunit-subunit interface was predicted in 2013 (16) along with the resultant two allosteric Phe binding sites (location indicated by green “Phe” label on top image). The oval in the bottom image shows the proposed multimer-specific interfaces that might form the binding site for a new pharmacological chaperones that could potentiate, rather than interfere with, allosteric Phe binding. d) An overlay of one subunit of the RS-PAH structure (darker shades) with one subunit of the A-PAH model (lighter shades) illustrates the relocation of the regulatory domain relative to the rest of the subunit. The regulatory domain is in cartoon, the catalytic domain is in balls, and the multimerization domain is in coil representation. Regulatory domain relocation results from a ~90° rotation relative to the catalytic domain; displacement of the regulatory domain in this way disallows active site occlusion by the autoregulatory region.
The PAH tetramer is comprised of four identical protein chains, yet the crystal structure of RS-PAH (Fig 2b, PDBid 5DEN) is asymmetric, with the largest conformational variation in the C-terminal helices (18). An independent solution structure analysis using small angle X-ray scattering (SAXS) suggests an even more profound asymmetry in solution relative to the crystal structure (20). Comparison of crystal structures of RS-PAH suggests significant rocking motion in the entire resting-state structure that derives from movement of the C-terminal helices relative to each other (18). This is notable because the assembly of these helices is largely what holds the RS-PAH tetramer together. There are few other tetramer-specific inter-chain interactions. The kinetic characteristics of mammalian PAH suggest that RS-PAH may need to dissociate into dimers or monomers as part of its normal function (16, 27), but this remains hypothetical.
In contrast with positional variations seen in the C-terminal helices, the remainder of each subunit of the RS-PAH tetramer adopt the same conformation. This conformation is secured by interactions within each subunit that involve the regulatory domain contacting other parts of the subunit (18). Significant biophysical data supports the hypothesis that the regulatory domain undergoes dramatic repositioning in the transition to A-PAH (e.g. (16, 17, 19, 20, 28)). Therefore, the intrachain interactions that stabilize the RS-PAH conformation are likely forfeited in the transition to A-PAH. New intermolecular interactions, specific to A-PAH, are predicted to compensate for this loss and allow the overall stability of RS-PAH and A-PAH to be close enough that the position of the equilibrium can be shifted by ligand binding (or single amino acid substitutions). The key perturbing ligands will be those that have interactions specific to either RS-PAH or A-PAH, rather than those that bind to a site that is common to both structures.
Development of a model of A-PAH (Fig 2c) (16), began with the realization that PAH may not be an exception to the rule that ACT domains in proteins generally form homodimers (29, 30). The PAH ACT domain is a major component of the PAH regulatory domain (see Fig 2a). The Protein Data Bank (PDB) contains many protein structures that show ACT domain dimers to have amino acid ligands at the dimer interface. There are now examples in the PDB where ACT domain dimerization is stabilized by allosteric amino acid ligand binding and this serves as a structural foundation for gating active site access and regulating metabolism (31). In the RS-PAH tetramer (Fig 2b), where the ACT domains are not interacting with each other, the N-terminal portion of the regulatory domain (i.e. the autoregulatory region) impedes active site access; non-interacting ACT domains correspond to the autoinhibited conformation of PAH. An approximate 90° rotation of the regulatory domains relative to the catalytic domains of PAH was proposed to form the allosteric Phe binding site at the ACT domain dimer interface (16): this domain rotation (illustrated in Fig 2d) would drag along the autoregulatory region and remove (disallow) active site occlusion. Thus, for PAH, interacting ACT domains correspond to the activated conformation. With the understanding that all protein structures can sample a variety of conformations, in the current usage, RS-PAH refers to all conformations containing monomeric ACT domains while A-PAH refers to all conformations containing the ACT domain dimer. Although our prediction of the specific location of allosteric Phe binding at an ACT dimer interface was novel in 2013, it was preceded by biophysical studies that suggested that isolated PAH regulatory domain dimerization occurs in the absence of Phe and that Phe binding to the dimer causes a conformational change (32); earlier non-structural studies investigating the roles of PAH residues 46–48 and 65–69 in Phe binding to a PAH regulatory domain dimer (21). Several 2016 publications support the hypothetical location of Phe bound to A-PAH. These are: 1) an NMR study that positions the allosteric Phe ligand as bound to residues predicted to be at the PAH ACT-dimer interface (17), 2) two independent SAXS studies consistent with formation of the PAH ACT-domain dimer in the presence of Phe (18, 20), and 3) the X-ray crystal structure of a truncated PAH ACT domain (Fig 3a), assembled as a dimer, with Phe bound as predicted in the 2013 model (16, 19). The crystal structure (PDB id 5FII) precisely defines the amino acids that interact with allosterically bound Phe.
Figure 3. Phe-bound PAH ACT domain dimer.
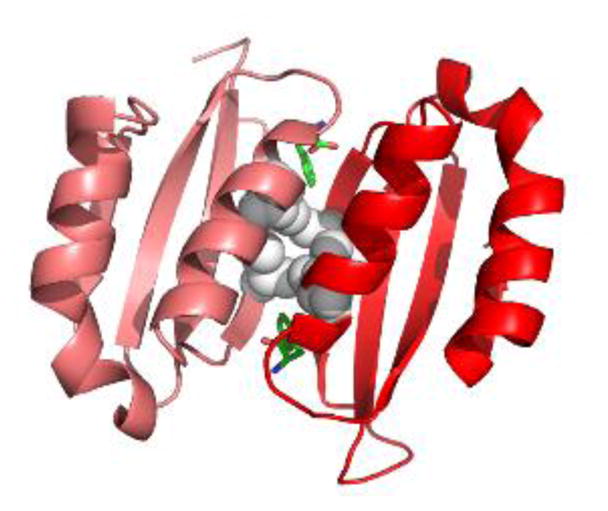
The crystal structure of a truncated human PAH ACT domain (PDB id 5FII, containing residues 34–111 (19)) forms a dimer and shows Phe (green) binding as predicted (18). The α-helices are predicted to be solvent exposed while the 8-stranded β-sheet is predicted to face into the tetramer. Grey balls are used for the side chains of Leu48 and Ile65. These four interacting residues contribute to the hydrophobic core of the ACT-domain dimer. L48S and I65T are each predicted to disrupt this important stabilizing interaction. Leu48 and Ile65 each have van der Waals interactions with the allosteric Phe.
The full-length A-PAH structure model (Fig 2c) remains an approximation. Overwhelming evidence supports Phe-responsive regulatory domain dimerization as the mechanism for allosteric control of PAH activity by Phe (17–20), but many structural details about A-PAH remain unknown. It remains unknown whether A-PAH has a distinct conformation for the C-terminal helices among monomers. Evidence for alternate conformations for the helices was originally suggested by the crystal structure of a truncated human PAH tetramer that lacked the regulatory domain (PDB id 2PAH) (33). The fold and/or location of the autoregulatory region in A-PAH is also unknown. In keeping with published biophysical studies (34), a compact conformation for the autoregulatory region is modeled with possible stabilizing interactions involving both the regulatory and catalytic domains (Fig 2c). Regardless of the details of the A-PAH structure, ACT domain dimerization dictates that stabilizing interactions in RS-PAH that involve the regulatory domain will be forfeited upon formation of A-PAH. The stabilizing interactions specific to A-PAH, which are predicted to compensate for these losses, are 1) at the ACT domain dimer interface, 2) interactions between the dimerized ACT domain and the catalytic and/or multimerization domains, and 3) yet unidentified interactions involving the repositioned/refolded autoregulatory region. Based on the structure of RS-PAH and the model of A-PAH, we predict that there will be an equilibrium between the two structures that do not significantly differ in intrinsic stability (see Fig 1). Phe can bind allosterically only to A-PAH, and allosteric Phe binding is predicted to stabilize the ACT-domain dimer interface. Thus, in the presence of Phe, A-PAH will be selectively stabilized and the equilibrium of PAH structures will be shifted towards the activated tetramer. This provides a structural foundation to the conformational selection model for PAH allostery (e.g. (35)), as distinct from an induced fit model, which requires an allosteric Phe binding site on RS-PAH. Despite computational studies assuming such a site (e.g. (36, 37)), it does not appear to exist (20). The apparent absence of allosteric Phe binding to the RS-PAH structure illustrated in Fig 2b constitutes a paradigm shift in understanding PAH structure/function relationships. This paradigm shift is away form an induced fit allosteric mechanism toward a conformational selection allosteric mechanism.
The PAH structure equilibrium and small molecule stabilization
Much prior and ongoing research on disease-associated PAH function has focused on protein stability (e.g. (38–43)). Unfortunately, such work has often used the terms “misfolded” or “misfolding” (in place of stability) when there is no measure of the folding process nor the final folded state. Within the context of an equilibrium of alternately assembled multimers, Fig 4 presents an updated view of PAH structure dynamics introducing an expanded context to analyze and understand normal PAH function and dysfunction within the context of small molecules that can alter the position of the equilibrium of PAH structures. Considering PKU in terms of the existence of an equilibrium of alternatively folded and/or assembled native structures is an important refinement to the imprecise generalization that “PAH misfolding” is a the most common basis for PKU. Some PKU-associated PAH variants exemplify a disequilibrium among functionally distinct native structures.
Figure 4. The equilibrium of PAH tetramers in the context of small molecule ligands (shown in red).

Newly synthesized PAH can be drawn into the equilibrium of folded native structures by BH4, which reversibly binds to RS-PAH. Inhibitory BH4 binding involves and secures the auto-inhibitory interaction (see Fig 5). Allosteric Phe binding to A-PAH stabilizes that structure and effectively competes with the inhibitory effect of BH4. Note that there is no direct interconversion between BH4-stabilized RS-PAH and Phe-stabilized A-PAH. By targeting multimer-specific small molecule binding sites, we foresee a new pharmacological chaperone (red asterisk) that can stabilize A-PAH without interfering with allosteric Phe binding (see Fig 2c). This is predicted to potentiate the sensitivity of A-PAH to allosteric Phe binding and restore the Phe response in a subset of disease-associated PAH variants.
Figure 4 shows newly synthesized PAH protein as folding and assembling into the equilibrium of native structures (RS-PAH and A-PAH tetramers). This process is in competition with protein misfolding/degradation. Any entity that stabilizes one or more of the native structures may help draw newly synthesized wild-type PAH or disease-associated PAH variants into the physiologically relevant equilibrium of native assemblies. The co-substrate BH4 can stabilize RS-PAH (as illustrated in Fig 5), shift the equilibrium in that direction, and act as an inhibitor (see below). Elevated Phe can bind allosterically to stabilize A-PAH (see Fig 2c, 3a), shift the equilibrium in that direction, and act as an activator. This is consistent with the well-established phenomenon that exposing PAH to BH4 causes inhibition of in vitro enzyme activity, while exposing PAH to Phe (> ~100 μM) causes activation (e.g. (44)). Inhibitory BH4 binding and activating Phe binding are both multimer-specific, each stabilizing their respective multimer, and helping to draw newly synthesized (or partially folded) PAH into the equilibrium of native structures. Inhibitory BH4 binding is specific for RS-PAH because this binding site includes hydrogen bonding to Ser23, an auto-inhibitory interaction that is disallowed in A-PAH (Fig 2b, Fig 5). Allosteric Phe binding is specific for A-PAH because the binding site is at the ACT domain dimer interface (Fig 2c, 3a), which is only present in the A-PAH structure. Furthermore, BH4 and Phe compete with each other to control the position of the equilibrium (Fig 4). This correlates with the observation that some individuals living with PKU are found to achieve lower Phe levels when treated with a lesser dose of BH4 (45). The equilibrium presented in Fig 4 also suggests that it may be possible to design or discover a pharmacological chaperone that will specifically bind to and stabilize A-PAH regardless of whether or not Phe is bound in the allosteric site. Unlike chaperones, which assist in protein folding, pharmacological chaperones are small molecules that stabilize a folded protein structure by binding to a specific ligand binding site (46). In order to sensitize the protein’s response to allosteric activation by Phe, the desired pharmacological chaperone will stabilize A-PAH by binding to a multimer-specific site that is different from the allosteric Phe binding site. This is distinct from an effector that binds to the allosteric site and activates PAH regardless of the Phe level. The latter runs the risk of constitutive activation (like enzyme replacement therapy), which can deplete Phe levels below those required for normal cellular functions. There have been a variety of approaches to identifying new pharmacological chaperones for PAH, most of which target the enzyme active site (47–50); it is not yet established if these agents selectively bind to and stabilize either RS-PAH or A-PAH.
Figure 5. BH4, bound two different ways at the PAH active site provides the molecular basis for stabilization of RS-PAH.

(a) An overlay of crystal structures (full length RS-PAH (white); PDB id 5DEN and PAH catalytic domain containing BH4 (cyan/green); PDB id 1J8U) illustrates that BH4 is positioned to make two stabilizing H-bonds to Ser23 which secure the auto-inhibitory interaction. (b) An alternate overlay (full length PAH (white); PDB id 5DEN and PAH catalytic domain containing BH4 and norleucine (cyan/green); PDB id 1MMT) shows BH4 sitting deeper in the active site and too far away to make the stabilizing H-bonds.
BH4 acting as a pharmacological chaperone
BH4 acting as a pharmacological chaperone was discovered in the process of discriminating between PKU caused by PAH dysfunction and PKU caused by rare defects in BH4 biosynthesis (51). Newly diagnosed patients, challenged by administration of BH4, were monitored for reduction in blood Phe as a diagnostic for defects in BH4 biosynthesis. However, decreased Phe levels in response to BH4 were also observed in a subset of patients whose PKU was caused by PAH dysfunction. In these cases, BH4 appeared to be working as a pharmacological chaperone to restore dysfunctional PAH to more normal function (52). Kuvan®, marketed by BioMarin Pharmaceuticals in the USA, is the name of the pharmacological form of BH4; it is reported as effective in lowering Phe levels for ~30% of PKU affected individuals for whom it has been tried (53). Much research has been directed at determining which genotypes are likely to respond to BH4, with varying results (e.g. (11, 39, 52, 54–58)).
Figure 5a illustrates the molecular basis for BH4 inhibition through stabilization of RS-PAH, as previously deduced from structures of truncated PAH (59–61). Shown is an overlay of the crystal structures of full length PAH and the isolated PAH catalytic domain bound to BH4. Figure 5b illustrates a similar overlay using a catalytic domain structure that also has norleucine (a Phe analog) at the enzyme active site. In the absence of a Phe analog, BH4 is positioned to form two hydrogen bonds to Ser23, securing the auto-inhibitory interaction (Fig 5a). Kinetic evidence for the importance of this molecular interaction to BH4 inhibition comes from stereospecificity for the 6R (not the 6S) isomer of BH4, and lack of inhibition by 6-methyltetrahydropterin (6MPH4), a BH4 analog that lacks the corresponding hydroxyl group (44); each of these pterins supports tyrosine formation. Figure 5b shows that, in the presence of norleucine, BH4 moves deeper into the active site and is no longer positioned to H-bond with the auto-regulatory region (stabilize the RS-PAH structure). This structural basis for loss of autoinhibition when Phe itself is bound at the active site was previously described (62). Thus, BH4 serves as a pharmacological chaperone by binding near the edge of the active site and securing the auto-inhibitory interaction through a reversible interaction that impedes active site access. Molecular motions available to RS-PAH include a hinge at the boundary between the autoregulatory region and the ACT domain (see Fig 2a); motion at this hinge likely accounts for the transient nature of auto-inhibition. It is important to note that there are conformational variants of RS-PAH in which autoinhibition is more or less transient. For example, PAH phosphorylation at Ser-16 appears to destabilize the autoinhibited conformation of RS-PAH without rotating the entire regulatory domain, as would be necessary to form A-PAH (63, 64).
The vast array of disease-associated PAH variants
Normal PAH function, seen as the regulation of Phe, requires accommodation of two architecturally distinct tetramer structures (RS-PAH and A-PAH) as well as the transient molecular interactions that may be required to transition between these structures. Although normal PAH can adroitly accomplish these reversible structural changes and maintain Phe in a healthy concentration range, the PAH of many individuals living with PKU cannot. This failure of normal function may result from the disease-associated amino acid change stabilizing RS-PAH, destabilizing A-PAH, or destabilizing a transient structure that PAH must sample in order to transition from RS-PAH to the A-PAH structure. In each of these cases, higher Phe levels would be required to shift the equilibrium toward A-PAH. With this in mind, it is perhaps not all that surprising that there are myriad disease-associated PAH missense mutations and these occur throughout the entire protein.
Any of the >600 amino acid substitution that may stabilize (or destabilize) one or more of the folded/assembled structures has the potential to contribute to disease by shifting the tipping-point for the equilibrium shown in Fig 1. Identifying these is the subject of ongoing investigation. However, based on the available crystal structures, two of the more common disease-associated alleles encode missense PAH variants, L48S and I65T, which are predicted to perturb intermolecular interactions important to the stability of A-PAH. Leu48 and Ile65 contribute to the hydrophobic core of the ACT domain dimer (see Fig 3b) and both make van der Waals contact with the allosteric Phe in the crystal structure of the Phe-bound ACT domain (19). Consistent with this prediction, the I65T variant has been reported to abolish Phe binding to the PAH regulatory domain (21). In contrast, kinetic analysis using a tetrameric fusion protein shows the I65T variant to have a relatively normal affinity for Phe (39), though the activity suggests that the fusion itself favors release of autoinhibition. Although sophisticated kinetic analysis are reported for L48S (e.g. (45)), direct Phe binding to the regulatory domain has not. Unfortunately published kinetic analysis on many disease associated PAH variants use intact fusion proteins and/or determine the allosteric response only at 1 mM Phe. These data do not address more subtle changes that would become apparent at lower Phe concentrations (e.g. 100 – 200 μM). It is also interesting that patients expressing the L48S and/or I65T variants are reported as having highly variable responses to BH4 therapy (54, 58, 65).
Further contribution to heterogeneity in genotype/phenotype correlation stems from the fact that most patients are compound heterozygotes whose genes encode two different disease-associated PAH proteins. In many instances, these patients will have PAH tetramers that are heteromeric, though not necessarily in a 50:50 mixture. There are in vitro studies supporting interallelic complementation in which one gene product dominates the phenotype (56, 65–67).
The precedent for the updated view of PAH and PKU is porphobilinogen synthase and the inborn error ALAD porphyria
Perturbation of a highly responsive equilibrium between two architecturally distinct protein multimers has been established as the molecular basis for ALAD porphyria, a very rare inborn error of metabolism caused by dysfunction of the enzyme porphobilinogen synthase (68). In this disease, the position of a multimeric equilibrium is altered in each of the eight disease-associated missense variants (69). Similar to PAH variants, the amino acid substitutions associated with ALAD porphyria occur throughout the protein. In vitro screening established that a few dozen compounds, some of which are drugs (70) and some of which are environmental contaminants (71), inhibit this protein by shifting the multimeric equilibrium toward the low-activity multimer. One might expect that the sensitive equilibrium proposed for PAH could also respond to small molecules, such as drugs and environmental contaminants, for which individual exposure would vary considerably, providing an environmental contribution to variations in genotype/phenotype correlations.
CONCLUSION
PKU is a complex disease usually caused by PAH dysfunction. PAH is described in terms of a sensitive equilibrium between RS-PAH and A-PAH structures, for which there is new structural information (17–20). The position of this equilibrium is governed by available Phe, which selectively binds to and stabilizes A-PAH. The equilibrium position will also respond to anything that perturbs the intrinsic stability of either structure (e.g. single amino acid variants of PAH, molecules that selectively bind to either RS-PAH or A-PAH, such as drugs, foods, environmental contaminants, or metabolic byproducts). BH4 acts as an effective pharmacological chaperone for some disease-associated PAH by selectively binding to and stabilizing RS-PAH. This helps draw newly synthesized PAH into the equilibrium of native structures, but requires higher Phe to shift the equilibrium toward A-PAH. The envisaged pharmacological chaperone would bind to A-PAH in a manner that does not interfere with allosteric Phe binding. Although significant new structural data helps define this updated view of PKU, an important remaining “unknown” is the detailed structure of A-PAH.
Acknowledgments
The author acknowledges the contributions of past coauthors (16, 18) in the evolution of the updated view of PKU as described herein. Particular insight derived from my colleague Roland L. Dunbrack, Jr. and my student Emilia C. Arturo, who also provided assistance in preparation of the illustrations. Grant support for our work on PAH has been provided by the Developmental Therapeutics Program at the Fox Chase Cancer Center, the Pennsylvania Tobacco Settlement Fund (CURE), BioMarin Pharmaceuticals, the National PKU Alliance, and the National Institutes of Health grants R01 NS100081 and P01 CA006927.
The author declares no conflict of interest.
Abbreviations
- PAH
Phenylalanine hydroxylase
- PKU
phenylketonuria
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Singh RH, et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet Med. 2014;16(2):121–131. doi: 10.1038/gim.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jahja R, Huijbregts SC, de Sonneville LM, van der Meere JJ, van Spronsen FJ. Neurocognitive evidence for revision of treatment targets and guidelines for phenylketonuria. J Pediatr. 2014;164(4):895–899. e892. doi: 10.1016/j.jpeds.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 3.Greene CL, Longo N. National Institutes of Health (NIH) review of evidence in phenylalanine hydroxylase deficiency (phenylketonuria) and recommendations/guidelines for therapy from the American College of Medical Genetics (ACMG) and Genetics Metabolic Dietitians International (GMDI) Mol Genet Metab. 2014;112(2):85–86. doi: 10.1016/j.ymgme.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Camp KM, et al. Phenylketonuria Scientific Review Conference: state of the science and future research needs. Mol Genet Metab. 2014;112(2):87–122. doi: 10.1016/j.ymgme.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 5.van Spronsen FJ. Phenylketonuria management from an European perspective: a commentary. Mol Genet Metab. 2010;100(2):107–110. doi: 10.1016/j.ymgme.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 6.Gentile JK, Ten Hoedt AE, Bosch AM. Psychosocial aspects of PKU: hidden disabilities--a review. Mol Genet Metab. 2010;99(Suppl 1):S64–67. doi: 10.1016/j.ymgme.2009.10.183. [DOI] [PubMed] [Google Scholar]
- 7.Vockley J, et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med. 2014;16(2):188–200. doi: 10.1038/gim.2013.157. [DOI] [PubMed] [Google Scholar]
- 8.Guthrie R, Susi A. A Simple Phenylalanine Method for Detecting Phenylketonuria in Large Populations of Newborn Infants. Pediatrics. 1963;32:338–343. [PubMed] [Google Scholar]
- 9.Huntington K, Buist NRM. Medical Food for Treatment of Inborn Errors of Metabolism and State Legislative Mandates. Top Clin Nutr. 2009;24(4):289–306. [Google Scholar]
- 10.Buist NRM, Huntington K. Scene from the USA: The illogic of mandating screening without also providing for treatment. Journal of Inherited Metabolic Disease. 2007;30(4):445–446. doi: 10.1007/s10545-007-9988-0. [DOI] [PubMed] [Google Scholar]
- 11.Wettstein S, et al. Linking genotypes database with locus-specific database and genotype-phenotype correlation in phenylketonuria. Eur J Hum Genet. 2015;23(3):302–309. doi: 10.1038/ejhg.2014.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blau N, Longo N. Alternative therapies to address the unmet medical needs of patients with phenylketonuria. Expert opinion on pharmacotherapy. 2015;16(6):791–800. doi: 10.1517/14656566.2015.1013030. [DOI] [PubMed] [Google Scholar]
- 13.Brown CS, Lichter-Konecki U. Phenylketonuria (PKU): A problem solved? Molecular genetics and metabolism reports. 2016;6:8–12. doi: 10.1016/j.ymgmr.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waisbren SE, et al. Improved Measurement of Brain Phenylalanine and Tyrosine Related to Neuropsychological Functioning in Phenylketonuria. JIMD Rep. 2016 doi: 10.1007/8904_2016_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yano S, Moseley K, Fu X, Azen C. Evaluation of Tetrahydrobiopterin Therapy with Large Neutral Amino Acid Supplementation in Phenylketonuria: Effects on Potential Peripheral Biomarkers, Melatonin and Dopamine, for Brain Monoamine Neurotransmitters. PLoS One. 2016;11(8):e0160892. doi: 10.1371/journal.pone.0160892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaffe EK, Stith L, Lawrence SH, Andrake M, Dunbrack RL., Jr A new model for allosteric regulation of phenylalanine hydroxylase: implications for disease and therapeutics. Arch Biochem Biophys. 2013;530(2):73–82. doi: 10.1016/j.abb.2012.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang S, Fitzpatrick PF. Identification of the Allosteric Site for Phenylalanine in Rat Phenylalanine Hydroxylase. J Biol Chem. 2016;291(14):7418–7425. doi: 10.1074/jbc.M115.709998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arturo EC, et al. First structure of full-length mammalian phenylalanine hydroxylase reveals the architecture of an autoinhibited tetramer. Proc Natl Acad Sci U S A. 2016;113(9):2394–2399. doi: 10.1073/pnas.1516967113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel D, Kopec J, Fitzpatrick F, McCorvie TJ, Yue WW. Structural basis for ligand-dependent dimerization of phenylalanine hydroxylase regulatory domain. Scientific reports. 2016;6:23748. doi: 10.1038/srep23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meisburger SP, et al. Domain Movements upon Activation of Phenylalanine Hydroxylase Characterized by Crystallography and Chromatography-Coupled Small-Angle X-ray Scattering. J Am Chem Soc. 2016;138(20):6506–6516. doi: 10.1021/jacs.6b01563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gjetting T, Petersen M, Guldberg P, Guttler F. Missense mutations in the N-terminal domain of human phenylalanine hydroxylase interfere with binding of regulatory phenylalanine. Am J Hum Genet. 2001;68(6):1353–1360. doi: 10.1086/320604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaffe EK. Morpheeins - a new structural paradigm for allosteric regulation. Trends in Biochemical Sciences. 2005;30(9):490–497. doi: 10.1016/j.tibs.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Selwood T, Jaffe EK. Dynamic dissociating homo-oligomers and the control of protein function. Arch Biochem Biophys. 2012;519(2):131–143. doi: 10.1016/j.abb.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gersting SW, et al. Activation of phenylalanine hydroxylase induces positive cooperativity toward the natural cofactor. J Biol Chem. 2010;285(40):30686–30697. doi: 10.1074/jbc.M110.124016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scriver CR, et al. PAHdb 2003: what a locus-specific knowledgebase can do. Hum Mutat. 2003;21(4):333–344. doi: 10.1002/humu.10200. [DOI] [PubMed] [Google Scholar]
- 26.Erlandsen H, Stevens RC. The structural basis of phenylketonuria. Mol Genet Metab. 1999;68(2):103–125. doi: 10.1006/mgme.1999.2922. [DOI] [PubMed] [Google Scholar]
- 27.Jaffe EK. Impact of quaternary structure dynamics on allosteric drug discovery. Curr Top Med Chem. 2013;13(1):55–63. doi: 10.2174/1568026611313010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang S, Hinck AP, Fitzpatrick PF. The Amino Acid Specificity for Activation of Phenylalanine Hydroxylase Matches the Specificity for Stabilization of Regulatory Domain Dimers. Biochemistry. 2015;54(33):5167–5174. doi: 10.1021/acs.biochem.5b00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chipman DM, Shaanan B. The ACT domain family. Curr Opin Struct Biol. 2001;11(6):694–700. doi: 10.1016/s0959-440x(01)00272-x. [DOI] [PubMed] [Google Scholar]
- 30.Grant GA. The ACT domain: a small molecule binding domain and its role as a common regulatory element. J Biol Chem. 2006;281(45):33825–33829. doi: 10.1074/jbc.R600024200. [DOI] [PubMed] [Google Scholar]
- 31.Lang EJM, Cross PJ, Mittelstadt G, Jameson GB, Parker EJ. Allosteric ACTion: the varied ACT domains regulating enzymes of amino-acid metabolism. Curr Opin Struct Biol. 2014;29:102–111. doi: 10.1016/j.sbi.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 32.Li J, Ilangovan U, Daubner SC, Hinck AP, Fitzpatrick PF. Direct evidence for a phenylalanine site in the regulatory domain of phenylalanine hydroxylase. Arch Biochem Biophys. 2011;505(2):250–255. doi: 10.1016/j.abb.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fusetti F, Erlandsen H, Flatmark T, Stevens RC. Structure of tetrameric human phenylalanine hydroxylase and its implications for phenylketonuria. J Biol Chem. 1998;273(27):16962–16967. doi: 10.1074/jbc.273.27.16962. [DOI] [PubMed] [Google Scholar]
- 34.Horne J, Jennings IG, Teh T, Gooley PR, Kobe B. Structural characterization of the N-terminal autoregulatory sequence of phenylalanine hydroxylase. Protein Sci. 2002;11(8):2041–2047. doi: 10.1110/ps.4560102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiman R, Jones SH, Gray DW. Mechanism of phenylalanine regulation of phenylalanine hydroxylase. J Biol Chem. 1990;265(20):11633–11642. [PubMed] [Google Scholar]
- 36.Carluccio C, Fraternali F, Salvatore F, Fornili A, Zagari A. Structural Features of the Regulatory ACT Domain of Phenylalanine Hydroxylase. PLoS One. 2013;8(11):e79482. doi: 10.1371/journal.pone.0079482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carluccio C, Fraternali F, Salvatore F, Fornili A, Zagari A. Towards the identification of the allosteric Phe-binding site in phenylalanine hydroxylase. Journal of biomolecular structure & dynamics. 2016;34(3):497–507. doi: 10.1080/07391102.2015.1052016. [DOI] [PubMed] [Google Scholar]
- 38.Gamez A, Perez B, Ugarte M, Desviat LR. Expression analysis of phenylketonuria mutations. Effect on folding and stability of the phenylalanine hydroxylase protein. J Biol Chem. 2000;275(38):29737–29742. doi: 10.1074/jbc.M003231200. [DOI] [PubMed] [Google Scholar]
- 39.Erlandsen H, et al. Correction of kinetic and stability defects by tetrahydrobiopterin in phenylketonuria patients with certain phenylalanine hydroxylase mutations. Proc Natl Acad Sci U S A. 2004;101(48):16903–16908. doi: 10.1073/pnas.0407256101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pey AL, Stricher F, Serrano L, Martinez A. Predicted effects of missense mutations on native-state stability account for phenotypic outcome in phenylketonuria, a paradigm of misfolding diseases. Am J Hum Genet. 2007;81(5):1006–1024. doi: 10.1086/521879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gersting SW, et al. Loss of function in phenylketonuria is caused by impaired molecular motions and conformational instability. Am J Hum Genet. 2008;83(1):5–17. doi: 10.1016/j.ajhg.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cerreto M, et al. Natural phenylalanine hydroxylase variants that confer a mild phenotype affect the enzyme’s conformational stability and oligomerization equilibrium. Biochim Biophys Acta. 2011;1812(11):1435–1445. doi: 10.1016/j.bbadis.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 43.Shi Z, Sellers J, Moult J. Protein stability and in vivo concentration of missense mutations in phenylalanine hydroxylase. Proteins. 2012;80(1):61–70. doi: 10.1002/prot.23159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaufman S. Regulation of the activity of hepatic phenylalanine hydroxylase. Advances in enzyme regulation. 1986;25:37–64. doi: 10.1016/0065-2571(86)90007-5. [DOI] [PubMed] [Google Scholar]
- 45.Danecka MK, et al. Mapping the functional landscape of frequent phenylalanine hydroxylase (PAH) genotypes promotes personalised medicine in phenylketonuria. J Med Genet. 2015;52(3):175–185. doi: 10.1136/jmedgenet-2014-102621. [DOI] [PubMed] [Google Scholar]
- 46.Ringe D, Petsko GA. What are pharmacological chaperones and why are they interesting? J Biol. 2009;8(9):80. doi: 10.1186/jbiol186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pey AL, et al. Identification of pharmacological chaperones as potential therapeutic agents to treat phenylketonuria. J Clin Invest. 2008;118(8):2858–2867. doi: 10.1172/JCI34355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Santos-Sierra S, et al. Novel pharmacological chaperones that correct phenylketonuria in mice. Hum Mol Genet. 2012;21(8):1877–1887. doi: 10.1093/hmg/dds001. [DOI] [PubMed] [Google Scholar]
- 49.Torreblanca R, Lira-Navarrete E, Sancho J, Hurtado-Guerrero R. Structural and mechanistic basis of the interaction between a pharmacological chaperone and human phenylalanine hydroxylase. Chembiochem. 2012;13(9):1266–1269. doi: 10.1002/cbic.201200188. [DOI] [PubMed] [Google Scholar]
- 50.Montalbano F, et al. Phenylalanine iminoboronates as new phenylalanine hydroxylase modulators. Rsc Adv. 2014;4(105):61022–61027. [Google Scholar]
- 51.Kure S, et al. Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J Pediatr. 1999;135(3):375–378. doi: 10.1016/s0022-3476(99)70138-1. [DOI] [PubMed] [Google Scholar]
- 52.Pey AL, et al. Mechanisms underlying responsiveness to tetrahydrobiopterin in mild phenylketonuria mutations. Hum Mutat. 2004;24(5):388–399. doi: 10.1002/humu.20097. [DOI] [PubMed] [Google Scholar]
- 53.Trefz F, et al. Tetrahydrobiopterin (BH4) responsiveness in neonates with hyperphenylalaninemia: a semi-mechanistically-based, nonlinear mixed-effect modeling. Mol Genet Metab. 2015;114(4):564–569. doi: 10.1016/j.ymgme.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 54.Daniele A, et al. Functional and structural characterization of novel mutations and genotype-phenotype correlation in 51 phenylalanine hydroxylase deficient families from Southern Italy. FEBS J. 2009;276:2048–2059. doi: 10.1111/j.1742-4658.2009.06940.x. (Copyright (C) 2014 American Chemical Society (ACS). All Rights Reserved.) [DOI] [PubMed] [Google Scholar]
- 55.Leuders S, et al. Influence of PAH Genotype on Sapropterin Response in PKU: Results of a Single-Center Cohort Study. JIMD Rep. 2014;13:101–109. doi: 10.1007/8904_2013_263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muntau AC, et al. Tetrahydrobiopterin as an alternative treatment for mild phenylketonuria. N Engl J Med. 2002;347(26):2122–2132. doi: 10.1056/NEJMoa021654. [DOI] [PubMed] [Google Scholar]
- 57.Pey AL, Desviat LR, Gamez A, Ugarte M, Perez B. Phenylketonuria: genotype-phenotype correlations based on expression analysis of structural and functional mutations in PAH. Hum Mutat. 2003;21(4):370–378. doi: 10.1002/humu.10198. [DOI] [PubMed] [Google Scholar]
- 58.Trefz FK, Scheible D, Goetz H, Frauendienst-Egger G. Significance of genotype in tetrahydrobiopterin-responsive phenylketonuria. J Inherited Metab Dis. 2009;32:22–26. doi: 10.1007/s10545-008-0940-8. [DOI] [PubMed] [Google Scholar]
- 59.Erlandsen H, Bjorgo E, Flatmark T, Stevens RC. Crystal structure and site-specific mutagenesis of pterin-bound human phenylalanine hydroxylase. Biochemistry. 2000;39(9):2208–2217. doi: 10.1021/bi992531+. [DOI] [PubMed] [Google Scholar]
- 60.Andersen OA, Flatmark T, Hough E. High resolution crystal structures of the catalytic domain of human phenylalanine hydroxylase in its catalytically active Fe(II) form and binary complex with tetrahydrobiopterin. J Mol Biol. 2001;314(2):279–291. doi: 10.1006/jmbi.2001.5061. [DOI] [PubMed] [Google Scholar]
- 61.Pey AL, Thorolfsson M, Teigen K, Ugarte M, Martinez A. Thermodynamic characterization of the binding of tetrahydropterins to phenylalanine hydroxylase. J Am Chem Soc. 2004;126(42):13670–13678. doi: 10.1021/ja047713s. [DOI] [PubMed] [Google Scholar]
- 62.Solstad T, Stokka AJ, Andersen OA, Flatmark T. Studies on the regulatory properties of the pterin cofactor and dopamine bound at the active site of human phenylalanine hydroxylase. Eur J Biochem. 2003;270(5):981–990. doi: 10.1046/j.1432-1033.2003.03471.x. [DOI] [PubMed] [Google Scholar]
- 63.Fitzpatrick PF. Allosteric regulation of phenylalanine hydroxylase. Arch Biochem Biophys. 2012;519(2):194–201. doi: 10.1016/j.abb.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li J, Fitzpatrick PF. Regulation of phenylalanine hydroxylase: Conformational changes upon phosphorylation detected by H/D exchange and mass spectrometry. Arch Biochem Biophys. 2013 doi: 10.1016/j.abb.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Djordjevic M, et al. Molecular Genetics and Genotype-Based Estimation of BH4-Responsiveness in Serbian PKU Patients: Spotlight on Phenotypic Implications of p.L48S. JIMD Rep. 2013;9:49–58. doi: 10.1007/8904_2012_178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leandro J, Nascimento C, de Almeida IT, Leandro P. Co-expression of different subunits of human phenylalanine hydroxylase: evidence of negative interallelic complementation. Biochim Biophys Acta. 2006;1762(5):544–550. doi: 10.1016/j.bbadis.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 67.Shen N, et al. Co-expression of phenylalanine hydroxylase variants and effects of interallelic complementation on in vitro enzyme activity and genotype-phenotype correlation. Mol Genet Metab. 2016;117(3):328–335. doi: 10.1016/j.ymgme.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 68.Maruno M, et al. Highly heterogeneous nature of delta-aminolevulinate dehydratase (ALAD) deficiencies in ALAD porphyria. Blood. 2001;97(10):2972–2978. doi: 10.1182/blood.v97.10.2972. [DOI] [PubMed] [Google Scholar]
- 69.Jaffe EK, Stith L. ALAD porphyria is a conformational disease. American Journal of Human Genetics. 2007;80(2):329–337. doi: 10.1086/511444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lawrence SH, Selwood T, Jaffe EK. Diverse clinical compounds alter the quaternary structure and inhibit the activity of an essential enzyme. ChemMedChem. 2011;6(6):1067–1073. doi: 10.1002/cmdc.201100009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lawrence SH, Selwood T, Jaffe EK. Environmental contaminants perturb fragile protein assemblies and inhibit normal protein function. Curr Chem Biol. 2013;7:196–206. doi: 10.2174/2212796811307020011. [DOI] [PMC free article] [PubMed] [Google Scholar]