

Abstract
The orexin 1 receptor (OX1R) and the kappa opioid receptor (KOR) are two G protein-coupled receptors (GPCRs) previously demonstrated to play important roles in modulating the rewarding effects of drugs of abuse such as cocaine. Using cells heterologously expressing both receptors, we investigated whether OX1R can regulate the function of KOR and vice versa. Activation of OX1R was found to attenuate agonist-activated KOR-mediated inhibition of cAMP production. In contrast, agonist-activated KOR-mediated β-arrestin recruitment and p38 activation were enhanced in the presence of activated OX1R. These effects are independent of OX1R internalization but are blocked in the presence of the JNK inhibitor SP-600125. OX1R signaling does not affect ligand binding by KOR. Taken together, these data suggest that OX1R signaling can modulate KOR function in a JNK-dependent manner, promoting preferential signaling of KOR via β-arrestin/p38 rather than Gαi. Conversely, Gαq coupling of OX1R is unaffected by activation of KOR, suggesting that this crosstalk is unidirectional. Given that KOR Gαi-mediated signaling events and β-arrestin-mediated signaling events are thought to promote distinct cellular responses and physiological outcomes downstream of KOR activation, this mechanism may have important implications on the behavioral effects of KOR activity.
Keywords: GPCR, orexin, dynorphin, crosstalk, JNK
1. Introduction
The neuropeptides orexin A and orexin B (also known as hypocretin-1 and hypocretin-2) are produced from the precursor pre-pro-orexin, which is expressed primarily by a restricted group of cells within the dorsolateral hypothalamus [1-3]. This limited population of orexin neurons projects to many different brain areas and regulates various behaviors including arousal [4], stress/anxiety [5], learning [6] and reward seeking [7]. These effects are mediated via activation of two widely-expressed Gαq-coupled G protein-coupled receptors (GPCRs)1, namely the orexin 1 and orexin 2 receptors (OX1R and OX2R [8]). Selective antagonism of OX1R reduces the rewarding effects of drugs of abuse such as cocaine [9], establishing OX1R as an important therapeutic target for the treatment of addiction. In contrast, activation of the kappa opioid receptor (KOR), also a GPCR, by its endogenous ligand dynorphin mediates depressive-like behaviors, and stimulation rather than blockade of KOR has been shown to reduce the rewarding effects of drugs [10]. Despite their contrasting functions, orexins and dynorphin can be packaged in the same synaptic vesicles [11], suggesting that they may function as co-neurotransmitters.
Most dopamine neurons in the ventral tegmental area (VTA), an important target for orexin neurons, respond to both orexins and dynorphin [11]. Co-stimulation with both neuropeptides can, however, result in opposing effects on the firing rates of different individual target neurons [11] and it is unclear how these responses are regulated [12]. Intra-VTA infusion of OX1R antagonist SB-334867 reduces cocaine self-administration in rats while intra-VTA infusion of the KOR antagonist norBNI has no effect on drug intake [11]. Interestingly, however, norBNI blocks the ability of SB-334867 to reduce drug intake [11]. Thus, it is conceivable that modulation of KOR function may occur downstream of OX1R activation. Such a relationship between these two GPCRs could explain the observed loss of SB-334867 efficacy in the presence of norBNI and would enhance our understanding of how target cells respond to orexin/dynorphin inputs.
Here we use cell lines heterologously expressing both receptors and show that OX1R signaling via Gαq can attenuate KOR-mediated Gαi signaling. This effect is lost in the presence of SP-600125, an inhibitor of c-Jun N-terminal kinase (JNK), activation of which has previously been associated with negative regulation of KOR [13]. In contrast, dynorphin-induced β-arrestin recruitment to KOR is increased in the presence of OX1R, in an SP-600125-dependent manner. Furthermore, OX1R signaling increases KOR-induced p38 activation, previously shown to require β-arrestin recruitment to KOR [14]. Taken together, our data suggest that OX1R signaling can modulate KOR function in a JNK-dependent manner such that, in response to its ligand dynorphin, KOR now preferentially signals via β-arrestin rather than via Gαi. This could be an important mechanism for determining how a target neuron responds to co-stimulation with orexins and dynorphin. Furthermore, this type of cross-talk may provide a general mechanism by which signaling specificity is regulated downstream of GPCRs.
2. Methods
2.1 Materials
All GPCR constructs were purchased from Missouri S&T cDNA Resource Center. The HA-dDRY-OX1R construct was generated using wildtype HA-OX1R and mutating Arg-144 (CGC) of the DRY motif to Ala (GCC) using the QuikChange II Site-Directed Mutagenesis Kit from Agilent Technologies. (Forward primer: 5′-GCTTCATCG CCCTGGACGCCTGGTATGCCATCTGC-3′, Reverse primer: 5′-GCAGATGGCATACCAGGCGTCCAGGGCGATGA AGC-3′). Note that mutation of the ‘DRY motif’ to abolish G protein-coupling has been described previously [15]. Dynorphin A (1-17) was purchased from American Peptide and orexin A from Tocris Bioscience. Rabbit phospho-p38, mouse p38 and rabbit phospho-JNK antibodies were purchased from Cell Signaling Technology, rat anti-HA from Roche and mouse anti-actin from Santa Cruz Biotechnology. Secondary antibodies were purchased from LI-COR Biosciences. Dynasore, H89 and norBNI were purchased from Tocris Bioscience, SP-600125 from Fisher Scientific, PKI from Santa Cruz Biotechnology and UBO-QIC was purchased from The University of Bonn.
2.2 Cell culture and transfection
CHO cells (ATCC) were grown in F12 media (Gibco) and Drx-KOR cells (DiscoveRx Corporation) in McCoy's media (ATCC) both supplemented with 10% fetal bovine serum (FBS, Gibco). All cells were maintained at 37°C, 5% CO2 and passaged every 3 days so that they remained <80% confluent. Transfections were performed using Fugene HD (Roche) as per manufacturer's recommendations. Stable expression of KOR in CHO-KOR cells was maintained using 250 μg/ml G418.
2.3 GloSensor (Promega) cAMP assay
This assay was performed according to manufacturer's instructions. Briefly, CHO-KOR cells were transiently transfected with pGlosensor-22F and the indicated receptor construct and 24 h later 10,000 cells/well were plated in a white 384-well plate in a volume of 20 μl CO2-independent media-10% FBS. After 24 h at 37°C, 20 μl GloSensor substrate/ equilibration buffer was added before incubation for 2 h at room temperature. Wells were then pretreated with 5 μl control or 500 nM orexin (Tocris Bioscience) for 10 min prior to treatment with 5 μl of 2 μM forskolin (MP Biomedical) or 2 μM forskolin/1 μM dynorphin (American Peptide) for 15 min. Luminescence was then measured using an EnVision plate reader. Data is normalized to the forskolin signal in the absence of orexin pretreatment for each transfection condition and error bars represent standard deviation from the mean of 3 separate experiments.
2.4 Calcium/FLIPR assay
10,000 cells/well were plated in a 384-well black clear-bottomed microtitre plate and incubated for 18 h at 37°C, 5% CO2. Growth media was then replaced with 25 μl assay buffer (HBSS, 20mM HEPES, 0.77 mg/ml probenecid, pH7.4). After a further 30 min at 37°C, 25 μl of ‘Calcium 5’ dye (Molecular Devices) was added to each well and plates were incubated at room temperature for 2 h in the dark. Following addition of orexin (10 μl, various concentrations), fluorescence was measured using a fluorescence imaging plate reader (FLIPR; Molecular Devices) and maximum response reads were divided by the baseline read taken before peptide addition for each well. Data is plotted as a % of the maximum read on the plate and error bars represent standard error of the mean of 3 replicates. Data shown is representative of 3 separate experiments.
2.5 Radioligand binding assay
To prepare membranes, cells were trypsinized and harvested in cold PBS. After 1 wash in cold PBS, cells were incubated in cold assay buffer (25 mM HEPES, 5 mM MgCl2, pH7.4) + 10 mM EDTA for 45 min. Then, after centrifugation at 45,000g at 4°C for 30 min, the supernatant was removed and the pellet was washed with cold assay buffer and resuspended in cold assay buffer + protease inhibitors (1 ml per T75 starting material). The suspension was then passed through a 27-gauge needle and protein concentration determined. 10 μg of membrane/ well of a 96-well plate was incubated with various concentrations of [3H]-U69 (Perkin Elmer) for 30 min at 37°C in a total volume of 100 μl assay buffer + 0.6% BSA. This was performed in the presence or absence of norBNI to allow for the subtraction of non-specific binding. Samples were then filtered using filters presoaked with 0.3% polyethyleneimine. Filters were then washed 5 times with assay buffer and allowed to dry before scintillation counting. Error bars represent standard error of the mean of 3 replicates and curves shown are representative of at least 3 separate experiments.
2.6 β-arrestin recruitment assay (PathHunter ™; DiscoveRx Corporation)
Direct interaction between a GPCR and β-arrestin can be monitored using enzyme fragment complementation between two inactive deletion mutants of β-galactosidase; one of the inactive β-gal fragments is fused to the carboxyl-terminus of the receptor and the other to β-arrestin. Recruitment of β-arrestin to the GPCR promotes restoration of functional β-gal enzyme, which can be detected using a luminescent substrate. This was performed according to manufacturer's instructions. Briefly, 5000 cells/well were plated in a white 384-well plate in a volume of 20 μl optimem supplemented with 2% charcoal-treated FBS. Following overnight incubation at 37°C, 5 μl dynorphin (various concentrations) was added to the plate and cells were incubated for a further 90 min at 37°C. 12 μl of the chemiluminescent PathHunter™ detection reagent was added and plates were incubated at room temperature in the dark for 1 h before luminescence was measured using an EnVision plate reader. Error bars represent standard error of the mean of 3 replicates and curves shown are representative of at least 3 separate experiments.
3. Results
3.1 OX1R signaling attenuates KOR-mediated inhibition of forskolin-stimulated cAMP production
KOR couples primarily to Gαi, activation of which inhibits adenylate cyclases, resulting in reduced intracellular levels of cyclic adenosine monophosphate (cAMP) [16]. To monitor KOR-mediated Gαi activation, CHO cells stably expressing KOR (CHO-KOR) were transiently transfected with ‘pGloSensor-22F’ cAMP biosensor (Promega). GloSensor is a form of firefly luciferase genetically modified by insertion of a cAMP-binding moiety such that its luciferase activity/light output is dependent on it binding to cAMP [17]. An EC90 (2 μM) of forskolin, a direct activator of adenylate cyclases, was used to increase the cellular cAMP/luciferase signal and provide an assay window for detecting a dynorphin-induced reduction of cellular cAMP levels.
Treatment of pGloSensor-transfected forskolin-treated CHO-KOR cells with 1 μM dynorphin for 15 min reduced the luciferase signal by 77 ± 4.8 % compared to that observed in cells treated with 2 μM forskolin alone, demonstrating a dynorphin-mediated reduction in forskolin-stimulated cAMP production (Figure 1A). Transient transfection of CHO-KOR cells with HA-OX1R and stimulation with 500 nM orexin A for 10 min prior to dynorphin treatment, resulted in an attenuation of the dynorphin-mediated reduction of cAMP (Figure 1A). Thus, after orexin A pre-treatment, dynorphin only reduced the GloSensor luciferase signal by 51 ± 7.2 % compared to that observed in response to forskolin alone (Figure 1A). Orexin A treatment does not itself affect the basal or forskolin-stimulated luciferase signals, indicating that any direct effects of OX1R on cAMP are not detectable in this assay. Futhermore, in the absence of OX1R, orexin A does not affect KOR responding to dynorphin, thus discounting any direct effect of orexin peptide on KOR function (Figure 1A). Finally, transfection and activation of an alternative Gαq-coupled receptor such as the 5-HT receptor 5HT2aR does not affect the KOR response to dynorphin (Figure 1A). Taken together, these results demonstrate that activation of OX1R can attenuate KOR-mediated inhibition of cellular cAMP production, suggesting that OX1R signaling negatively regulates KOR-induced Gαi activation.
Figure 1. OX1R signaling attenuates KOR-induced reduction in cellular cAMP levels.
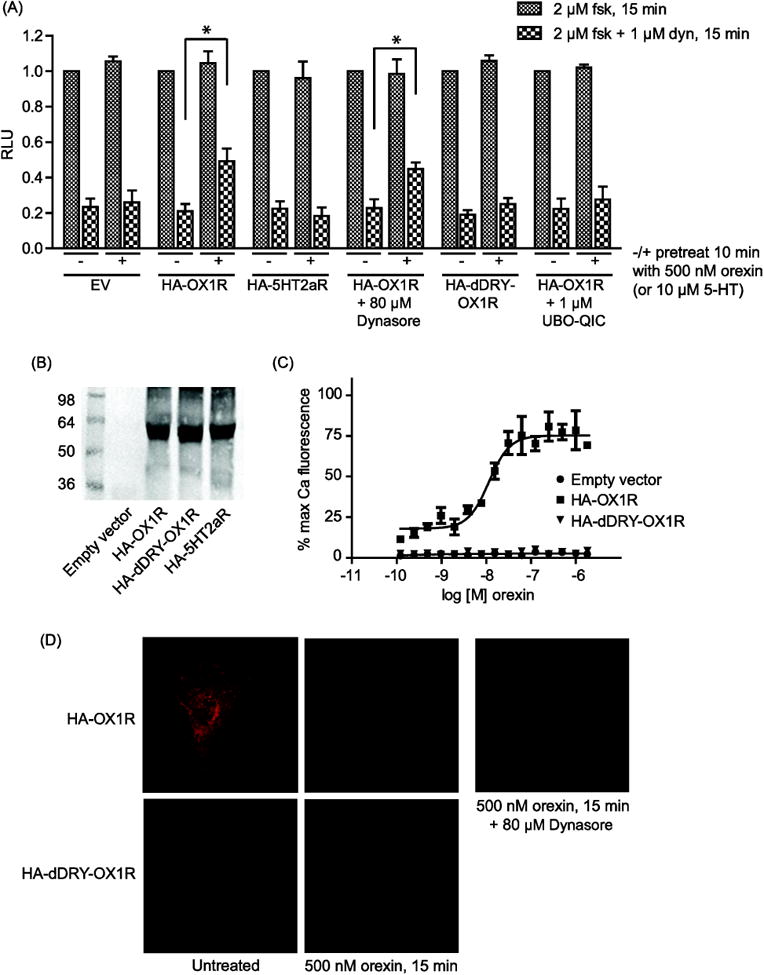
(A): CHO-KOR cells expressing ‘pGloSensor-22F’ cAMP sensor together with the indicated receptor construct or empty vector (EV) were incubated with or without 80 μM Dynasore or 1 μM Gq-inhibitor UBO-QIC for 2 h. Cells were pretreated or not with 500 nM orexin for 10 min prior to treatment with 2 μM forskolin (fsk) or 2 μM forskolin + 1 μM dynorphin (dyn) for 15 min, as indicated. Receptor expression was validated by western blot using a rat anti-HA antibody (Roche) (B). Data is normalized to the forskolin signal in the absence of orexin pretreatment for each transfection condition and error bars represent standard deviation from the mean of 3 separate experiments (* indicates p < 0.01). (C): HA-dDRY-OX1R fails to activate calcium signaling, consistent with a failure to activate Gαq. CHO cells were transfected as indicated and, after 48 h, treated with the indicated concentrations of orexin. Error bars represent standard error of the mean of 3 replicates and data shown is representative of 3 separate experiments. (D): CHO cells were transfected as indicated. After 48 h, cells were incubated with or without 80 μM Dynasore for 2 h then treated or not with 500 nM orexin for 15 min and fixed using PFA. Samples were analyzed by confocal microscopy using a rat anti-HA antibody (Roche) and an anti-rat Alexafluor-594 secondary antibody (Invitrogen).
OX1R has been shown to heterodimerize with the cannabinoid 1 receptor (CB1R) and orexin A treatment of cells co-expressing these receptors was found to promote their co-internalization [18]. We speculated that a similar relationship may exist between OX1R and KOR and that orexin A-induced co-internalization of OX1R with KOR could account for the reduced response of KOR to dynorphin following orexin A treatment. However, blocking endocytosis with a 2 hour incubation with 80 μM ‘Dynasore’, a cell-permeable inhibitor of dynamin GTPase activity, significantly reduced orexin A-induced OX1R internalization (Figure 1D) but did not affect the orexin A-induced attenuation of KOR-mediated reduction of cAMP production (Figure 1A), suggesting that OX1R internalization is not required for its ability to modulate the function of KOR.
To investigate the role that OX1R Gαq signaling may play in attenuating KOR activity, we introduced a single point mutation into the ‘DRY motif’ of wild type HA-OX1R such that the DRY motif arginine residue is changed to alanine (HA-dDRY-OX1R, see methods). Most Family A GPCRs possess the highly conserved DRY motif at the beginning of the second intracellular loop and substitution of the central arginine residue has been shown to abolish G protein coupling of many GPCRs [15, 19]. Like wildtype OX1R, HA-dDRY-OX1R expresses normally at the cell surface and internalizes in response to orexin A treatment (Figure 1B and 1D) but it is completely devoid of Gαq coupling as evidenced by its inability to promote intracellular calcium release in response to orexin (Figure 1C). We found that activation of HA-dDRY-OX1R fails to attenuate KOR-mediated inhibition of cAMP production (Figure 1A). Consistent with this, a 2 hour incubation with the Gαq inhibitor UBO-QIC [20] also prevents wildtype OX1R from attenuating KOR-mediated reduction of cellular cAMP levels (Figure 1A). Taken together, these results suggest that Gαq signaling downstream of OX1R rather than OX1R/KOR co-internalization is required for OX1R-mediated attenuation of KOR-mediated Gαi activation.
3.2 OX1R attenuation of KOR Gai signaling is lost in the presence of the JNK inhibitor SP-600125
Almost all family A GPCRs undergo agonist-induced homologous desensitization via a well-characterized mechanism involving GPCR kinases (GRKs) and β-arrestin. β-arrestin recruitment to agonist-occupied receptors sterically hinders G protein signaling and promotes clathrin-mediated receptor endocytosis. In contrast, mechanisms of heterologous desensitization, that is the desensitization of one GPCR in response to activation of another, are more diverse and less well understood. One report has shown that heterologous desensitization of KOR can occur in response to activation of the chemokine receptor CXCR4 [21]. Like OX1R modulation of KOR, this mechanism is also independent of receptor internalization [21] but further mechanistic details remain to be elucidated. Other studies have associated activation of the MAP kinase JNK with disruption of KOR signaling [13] although it is not clear whether or not JNK mediates heterologous desensitization of KOR.
We found that treatment of CHO-OX1R cells with orexin A resulted in phosphorylation of JNK at residues Thr183/Tyr185 (Figure 2A), indicating that JNK is activated downstream of OX1R. To the best of our knowledge this is the first time OX1R signaling to JNK has been demonstrated. This effect is lost in the presence of the Gαq inhibitor UBO-QIC (Figure 2A), suggesting that OX1R-mediated JNK activation is Gαq-dependent. When CHO-KOR cells, co-transfected with HA-OX1R and pGloSensor, are incubated for 2 hours with 20 μM of the JNK inhibitor SP-600125, activation of OX1R no longer attenuates KOR-mediated reduction of cAMP levels (Figure 2B). Thus, as shown previously, in the absence of SP-600125, dynorphin treatment reduces the forskolin-stimulated luciferase signal by 80 ± 4.8 % when OX1R is not activated but only by 54 ± 6.1 % when OX1R is activated. However, in the presence of SP-600125, the dynorphin-mediated reduction of forskolin-stimulated cAMP production does not significantly differ whether or not OX1R is activated (87 ± 8.2 % when OX1R is not activated and 88 ± 8.1 % when OX1R is activated; Figure 2B). These data suggest that JNK activation downstream of OX1R is required for attenuation of KOR Gαi signaling. Two different inhibitors (PKI and H89) of protein kinase A (PKA), which is involved in heterologous desensitization of several GPCRs including the delta opioid receptor (DOR) [22] and chemokine receptors [23], did not affect OX1R attenuation of KOR Gαi signaling (Figure 2B).
Figure 2. OX1R-mediated attenuation of KOR Gαi signaling is lost in the presence of the JNK inhibitor SP-600125.
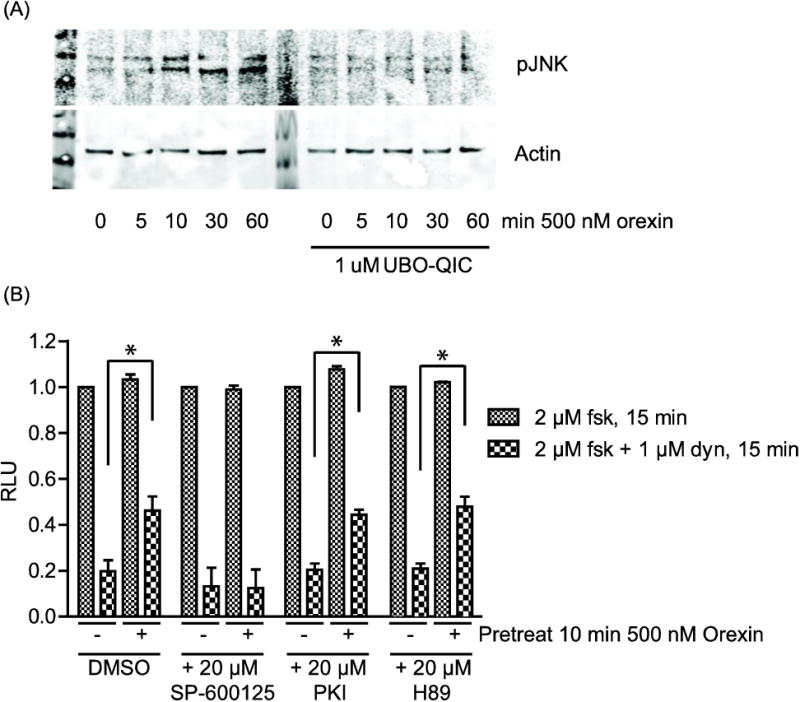
(A): CHO-OX1R cells were serum starved in optimem for 4 h in the presence or absence of Gαq inhibitor UBO-QIC prior to treatment with the indicated concentrations of orexin. Cell lysates were collected and equal amounts of protein were run for western blotting. Phospho-JNK (pJNK) levels were analyzed using a rabbit anti-pJNK antibody (Cell Signaling) and loading was controlled using a mouse anti-actin antibody (Santa Cruz). (B): CHO-KOR cells were co-transfected with ‘GloSensor-22F’ cAMP sensor and HA-OX1R. After 48 h, cells were incubated with or without 20 μM SP-600125, PKI or H89 for 2 h. Cells were then pretreated or not with 500 nM orexin for 10 min prior to treatment with 2 μM forskolin (fsk) or 2 μM forskolin + 1 μM dynorphin (dyn) for 15 min, as indicated. Data is normalized to the forskolin signal in the absence of orexin pretreatment for each condition and error bars represent standard deviation from the mean of 3 separate experiments (* indicates p < 0.01).
3.3 KOR activation does not affect OX1R calcium signaling
We next investigated whether OX1R modulation of KOR is reciprocal. CHO cells were transiently co-transfected with HA-OX1R and either KOR or empty vector and then treated with 1 μM dynorphin for 10 min prior to treatment with a concentration range of orexin A (Figure 3). Specific binding of the KOR-selective radiolabelled agonist [3H]-U69 indicates that KOR expresses well under these co-transfection conditions (Figure 4B). Orexin A-induced intracellular calcium release was monitored as a measure of Gαq activation downstream of OX1R. The potency and efficacy of orexin A did not significantly differ in the absence or presence of activated KOR (Figure 3), demonstrating that KOR activation does not modulate OX1R Gαq signaling. The EC50 of orexin A in the absence of activated KOR was 14.7 ± 4.6 nM compared with 13.3 ± 4.9 nM in the presence of activated KOR. This suggests that the crosstalk between these two GPCRs is unidirectional.
Figure 3. KOR activation does not affect OX1R calcium signaling.
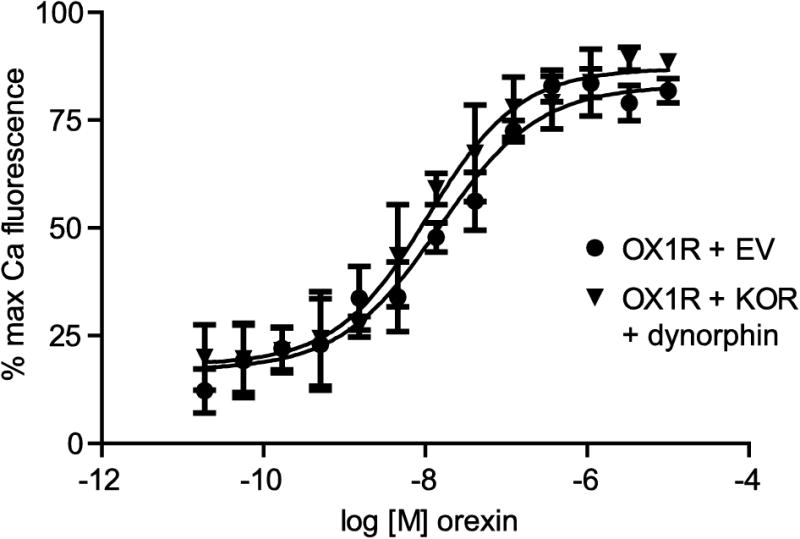
CHO cells were transfected as indicated. After 48 h, cells were loaded with calcium-sensitive dye for 2 h. Cells were then treated with 1 μM dynorphin for 10 min prior to treatment with the indicated concentrations of orexin. Error bars represent standard error of the mean of three replicates and data shown is representative of 3 separate experiments. The mean EC50s from the 3 separate experiments are quoted in the text.
Figure 4. OX1R activation does not affect KOR ligand binding.
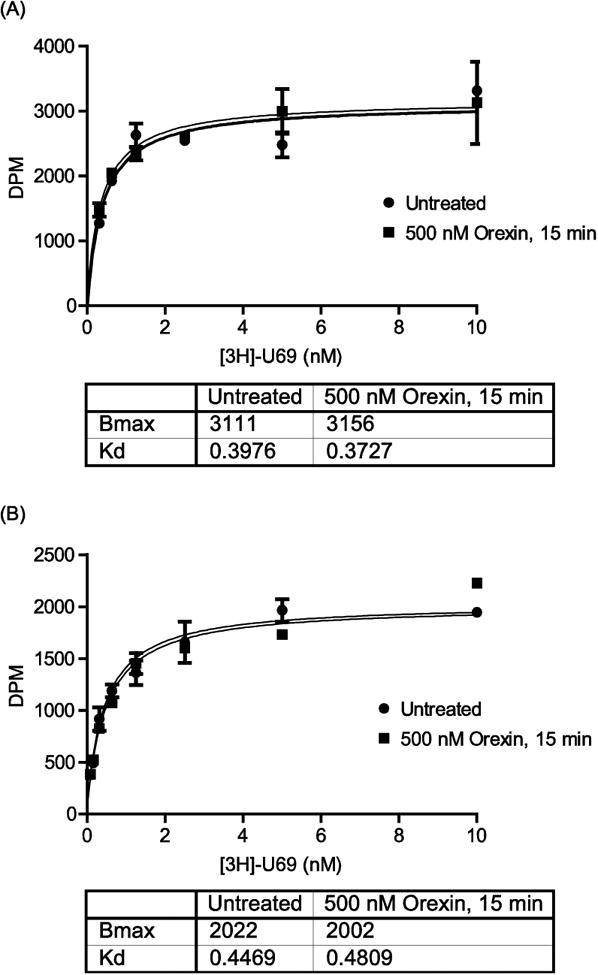
(A): CHO-KOR cells were transfected with HA-OX1R. (B): CHO cells were co-transfected with KOR + HA-OX1R. (A) and (B): Cells were treated or not with 500 nM orexin for 15 min prior to harvesting. Cell membranes were purified and 10 μg membranes were incubated per well of a 96-well plate in the presence or absence of 20 μM norBNI to allow subtraction of non-specific binding. [3H]-U69 was added at the indicated concentrations and incubated for 30 min at 37°C. Samples were filtered and washed before scintillation counting. Error bars represent the standard error of the mean of 3 replicates and data shown is representative of 3 separate experiments.
3.4 OX1R activation does not affect KOR ligand binding
We performed saturation binding assays with radiolabelled KOR agonist [3H]-U69 using membranes prepared from our CHO-KOR cells transiently transfected with HA-OX1R (Figure 4A). The Bmax and Kd of [3H]-U69 binding to KOR was unaffected when cells were treated with 500 nM orexin for 15 min prior to membrane harvesting (Figure 4A), indicating that OX1R signaling does not affect KOR ligand binding. To ensure that this observed lack of effect on ligand binding in the CHO-KOR cells was not due to low transfection efficiency of HA-OX1R, we performed the same experiment using membranes prepared from parental CHO cells that were transiently co-transfected with HA-OX1R and KOR, a condition in which we know OX1R expresses and functions normally (Figure 3). Experiments performed using membranes prepared from these cells also showed that OX1R activation did not affect the Bmx or Kd of [3H]-U69 binding to KOR (Figure 4B). These results suggest that OX1R signaling acts to modulate how KOR responds to agonist activation rather than the ability of KOR to bind to its ligand.
3.5 OX1R enhances KOR-mediated β-arrestin recruitment and P38 activation
KOR, like most GPCRs, engages both G protein-dependent and G protein-independent signaling pathways in response to agonist binding [16, 24]. Our data suggests that OX1R signaling can reduce KOR-mediated Gαi activation. We next asked whether OX1R can also modulate G protein-independent signaling by KOR. To do this we monitored KOR β-arrestin recruitment using the PathHunter™ assay (DiscoveRx Corporation) as described in section 2.6.
Treatment of CHO cells co-expressing the engineered KOR and β-arrestin constructs (Drx-KOR cells) with dynorphin results in a concentration-dependent increase in luminescence, indicative of β-arrestin recruitment to KOR (Figure 5A). Transient transfection of HA-OX1R into Drx-KOR cells enhances dynorphin-induced luminescence, as shown by an increase in the span between the ECmax signal and the baseline luminescence relative to empty vector-transfected control cells (Figure 5A and 5C). This suggests that OX1R can enhance dynorphin-mediated β-arrestin recruitment to KOR. We observed that transfection of unrelated GPCRs into Drx-KOR cells increases the basal luminescence signal, presumably due to a proximity effect in which there is generally more β-arrestin in the vicinity of the plasma membrane when another GPCR is over-expressed. However, an unrelated GPCR such as the Galanin receptor GalR1 does not affect the dynorphin-induced β-arrestin recruitment to KOR as the span between the ECmax signal and the baseline luminescence is not affected (Figure 5A and 5C). Strikingly, in the presence of the JNK inhibitor SP-600125 the effect of OX1R on dynorphin-mediated KOR β-arrestin recruitment is completely blocked (Figure 5B and 5C). SP-600125 itself does not affect KOR β-arrestin recruitment in the absence of OX1R (Figure 5C). These data suggest that OX1R enhances dynorphin-mediated β-arrestin recruitment to KOR and, like OX1R attenuation of KOR Gαi signaling (Figure 2B), this enhancement is also JNK-dependent.
Figure 5. OX1R promotes β-arrestin recruitment and p38 activation by KOR.
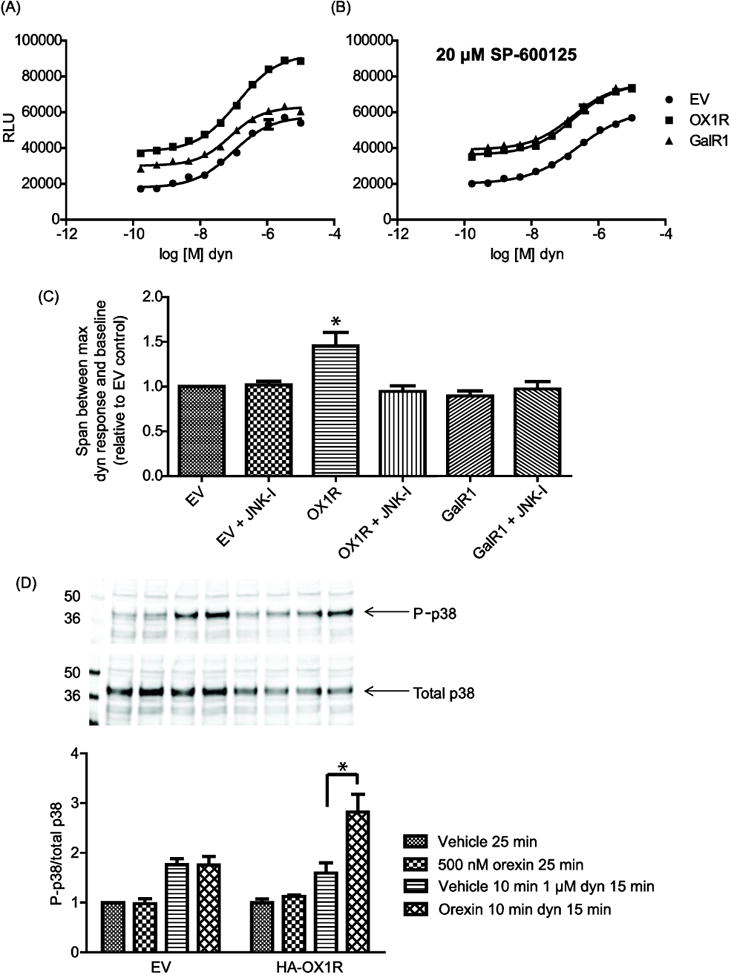
(A) and (B): Drx-KOR cells were transfected as indicated. 24 h later, cells were plated in the presence or absence of 20 μM SP-600125. After a further 18 h, cells were incubated with the indicated concentrations of dynorphin (dyn) for 90 min before addition of the 3-component PathHunter™ reagent mix. Error bars represent standard error of the mean of 3 replicates and data shown is representative of 3 separate experiments. In (C), the maximum dynorphin response minus baseline span is averaged from the 3 separate experiments and plotted relative to that seen in empty vector (EV) transfected cells. Error bars represent the standard deviation from the mean of the 3 separate experiments. JNK-I = JNK inhibitor SP-600125. (D): CHO-KOR cells were transfected with empty vector (EV) or HA-OX1R as indicated. After serum starving in optimem for 4 h, cells were treated as indicated and harvested. Lysates were analyzed by western blotting using rabbit anti-phospho-p38 (P-p38) and mouse anti-total p38 (Cell Signaling). Bands were quantified from 3 separate experiments and P-p38 values were divided by total p38 in each condition. The average P-p38/Total p38 values are plotted relative to the untreated EV condition with error bars representing the standard deviation from the mean of the 3 separate experiments (* indicates p < 0.01).
Note that orexin A-mediated stimulation of OX1R is not required to see an effect of OX1R on dynorphin-mediated KOR β-arrestin recruitment in Drx-KOR cells. This is in contrast to OX1R attenuation of KOR-mediated inhibition of cellular cAMP production in CHO-KOR cells, which requires OX1R activation (Figure 1A). To explain this observation we hypothesize that some degree of constitutive signaling from OX1R to JNK is occurring in the Drx-KOR cells. This might be due to differing expression levels of OX1R when it is transiently transfected into the two different cell lines. It is also possible that the high levels of β-arrestin that are expressed in the Drx-KOR cells could mediate constitutive signaling from OX1R to JNK. This would not be evident in the GloSensor cAMP assay performed in CHO-KOR cells as they express endogenous levels of β-arrestin. If we activate OX1R (or any GPCR) prior to stimulating KOR we find that, due to the non-specific proximity effect discussed above, the basal signal is increased to such an extent that we can no longer see an effect of KOR stimulation in this assay.
KOR activation of the MAPK p38 has been reported to be a β-arrestin-dependent signaling event [14]. Treatment of CHO-KOR cells with 1 μM dynorphin for 15 min resulted in a 1.76 ± 0.12 fold increase in phospho-p38/total p38, indicative of KOR-mediated p38 activation (Figure 5D) and in good agreement with literature values [14]. Transfection of HA-OX1R and treatment with 500 nM orexin for 10 min prior to dynorphin treatment resulted in a 2.82 ± 0.36 fold increase in phospho-p38/total p38 relative to untreated cells (Figure 5D). Orexin treatment in the presence or absence of transfected HA-OX1R does not itself affect p38 activation after 25 min (Figure 5D). Taken together, these results demonstrate that OX1R enhances both dynorphin-induced β-arrestin recruitment and p38 activation by KOR.
4. Discussion
Many examples of signaling by one GPCR being influenced by activation of another GPCR have been described, however mechanistic information for such examples of crosstalk is often lacking [25, 26]. The ability of the KOR-selective antagonist norBNI to block the behavioral effects of OX1R-selective antagonists in rats [11] prompted us to investigate whether or not OX1R activation can modulate the function of KOR or vice versa. We found that KOR function can be regulated by OX1R signaling via a mechanism that is sensitive to JNK inhibition. This mechanism does not affect KOR ligand binding but rather modulates KOR responding to its peptide agonist dynorphin. Thus, OX1R signaling enhances KOR-mediated dynorphin-induced β-arrestin recruitment and p38 activation while attenuating dynorphin-induced inhibition of intracellular cAMP production (Figure 6C). In response to agonist binding, GPCRs can often activate both G protein-dependent and G protein-independent signaling pathways. Signaling specificity in different cell types can be controlled by the presence of different complements of regulatory proteins, including GPCR kinases, scaffolds and adaptor proteins, which modulate the function of a given GPCR to ensure that it activates the appropriate pathway(s) in an appropriate temporal and spatial manner [27-29]. Our work demonstrates that KOR can signal differently depending on whether or not OX1R is also activated in the same cell. Such mechanisms of crosstalk between GPCRs, as well as mechanisms of GPCR heterologous desensitization, though poorly characterized, likely play an important role in determining GPCR signaling specificity.
Figure 6. OX1R signaling modulates KOR function.
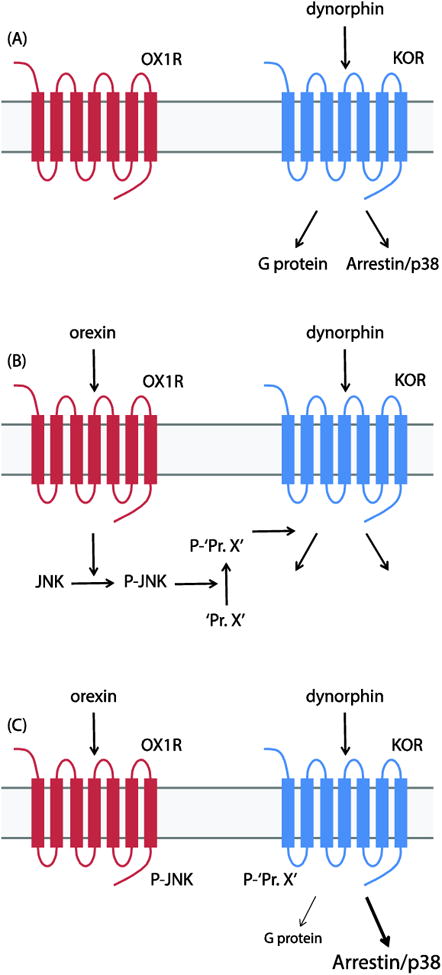
(A) KOR activates Gαi as well as β-arrestin-dependent pathways in response to dynorphin stimulation. (B) Stimulation of OX1R results in JNK activation. We hypothesize that an unknown KOR accessory protein (‘Pr. X’) is a JNK substrate. (C) In the presence of phospho-Pr. X (P-‘Pr. X’), dynorphin stimulation of KOR results in increased β-arrestin/p38 signaling and reduced Gαi signaling.
Our data also highlights the importance of considering the effects of co-transmitters on the functions of neuropeptides and neuropeptide receptors that are traditionally studied in isolation. In dopamine neurons in the VTA for example, which commonly respond to both orexins and dynorphin [11], KOR might signal preferentially to β-arrestin/p38 in response to an orexin/dynorphin input compared to stimulation with dynorphin alone. Furthermore, responding to orexin/dynorphin co-stimulation could differ in individual target neurons depending on the availability of certain JNK scaffolds or substrates. Interestingly, while Gαi signaling is thought to mediate the analgesic effects of KOR agonism, KOR p38 activation is thought to mediate dysphoria and stress-induced reinstatement of drug seeking [30-32]. Thus, the presence or absence of orexin co-stimulation and/or the availability of JNK scaffolds or substrates could have important implications for the behavioral effects of KOR signaling.
We hypothesize that an unknown KOR accessory protein (‘Pr. X’ in Figure 6) mediates the JNK-dependent modulation of KOR function by OX1R. We are currently working to identify JNK substrates and/or interacting proteins downstream of OX1R that could be responsible for these effects. It is possible that KOR itself is a direct substrate for JNK but, while it is known to phosphorylate several adaptor/scaffold proteins as well as other kinases [33], JNK has no previously-reported substrates that are GPCRs. The group of Chavkin et al have proposed the existence of a hypothetical ‘jammer’ protein that blocks KOR G protein signaling in a JNK-dependent manner in order to mediate the effects of certain long-acting KOR antagonists that are known to activate JNK [13, 34]. It will be interesting to see whether the mechanism of action of such long-acting KOR antagonists overlaps at all with the mechanism of OX1R-mediated KOR regulation.
Conclusions
OX1R signaling can attenuate KOR-mediated reduction of cellular cAMP levels while enhancing KOR-mediated β-arrestin recruitment and p38 activation.
These effects are unidirectional as OX1R signaling is not affected by KOR activation.
OX1R modulation of KOR does not require OX1R internalization but is blocked in the presence of SP-600125, an inhibitor of the MAPK JNK, which we have shown is activated downstream of OX1R.
JNK-dependent crosstalk between OX1R and KOR could influence responding of dopamine neurons in the VTA, which commonly express both receptors.
Highlights.
OX1R signaling can attenuate KOR-mediated modulation of cellular cAMP levels.
In contrast, OX1R enhances KOR-mediated β-arrestin recruitment and p38 activation.
KOR activation does not affect OX1R-mediated calcium signaling.
Modulation of KOR by OX1R is blocked by the JNK inhibitor SP-600125.
Acknowledgments
We thank Dr. Paul Kenny for helpful discussion and Dr. Laura Bohn and Dr. Cullen Schmid for their critical reviewing of the manuscript. This work was supported by NIH grant number P01-DA026838.
Footnotes
Abbreviations: GPCR – G protein-coupled receptor; OX1R – orexin 1 receptor; KOR – kappa opioid receptor; JNK – c-Jun N-terminal kinase; cAMP – cyclic adenosine monophosphate; VTA – ventral tegmental area; CHO – Chinese hamster ovary; FLIPR – fluorescence in situ plate reader; PKA – protein kinase A; MAPK – mitogen-activated protein kinase; FBS – fetal bovine serum; HBSS – Hank's buffered saline solution; DOR – delta opioid receptor; GalR1 – galanin receptor 1.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. Proc Natl Acad Sci U S A. 1998;95:322–327. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richarson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Cell. 1998;92 doi: 10.1016/s0092-8674(02)09256-5. 1 page following 696. [DOI] [PubMed] [Google Scholar]
- 4.Alexandre C, Andermann ML, Scammell TE. Current opinion in neurobiology. 2013;23:752–759. doi: 10.1016/j.conb.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson PL, Molosh A, Fitz SD, Truitt WA, Shekhar A. Progress in brain research. 2012;198:133–161. doi: 10.1016/B978-0-444-59489-1.00009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wheeler DS, Wan S, Miller A, Angeli N, Adileh B, Hu W, Holland PC. The European journal of neuroscience. 2014;40:2359–2377. doi: 10.1111/ejn.12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakurai T. Nature reviews. Neuroscience. 2014;15:719–731. doi: 10.1038/nrn3837. [DOI] [PubMed] [Google Scholar]
- 8.Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, Elmquist JK. The Journal of comparative neurology. 2001;435:6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- 9.Hollander JA, Pham D, Fowler CD, Kenny PJ. Frontiers in behavioral neuroscience. 2012;6:47. doi: 10.3389/fnbeh.2012.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wee S, Koob GF. Psychopharmacology. 2010;210:121–135. doi: 10.1007/s00213-010-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muschamp JW, Hollander JA, Thompson JL, Voren G, Hassinger LC, Onvani S, Kamenecka TM, Borgland SL, Kenny PJ, Carlezon WA., Jr Proc Natl Acad Sci U S A. 2014;111:E1648–1655. doi: 10.1073/pnas.1315542111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahler SV, Moorman DE, Smith RJ, James MH, Aston-Jones G. Nature neuroscience. 2014;17:1298–1303. doi: 10.1038/nn.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, Li S, Chavkin C. J Biol Chem. 2007;282:29803–29811. doi: 10.1074/jbc.M705540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruchas MR, Macey TA, Lowe JD, Chavkin C. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. Proc Natl Acad Sci U S A. 2003;100:10782–10787. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruchas MR, Chavkin C. Psychopharmacology. 2010;210:137–147. doi: 10.1007/s00213-010-1806-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vardy E, Mosier PD, Frankowski KJ, Wu H, Katritch V, Westkaemper RB, Aube J, Stevens RC, Roth BL. J Biol Chem. 2013;288:34470–34483. doi: 10.1074/jbc.M113.515668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ward RJ, Pediani JD, Milligan G. J Biol Chem. 2011;286:37414–37428. doi: 10.1074/jbc.M111.287649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rovati GE, Capra V, Neubig RR. Molecular pharmacology. 2007;71:959–964. doi: 10.1124/mol.106.029470. [DOI] [PubMed] [Google Scholar]
- 20.Karpinsky-Semper D, Volmar CH, Brothers SP, Slepak VZ. Molecular pharmacology. 2014;85:758–768. doi: 10.1124/mol.114.091843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finley MJ, Chen X, Bardi G, Davey P, Geller EB, Zhang L, Adler MW, Rogers TJ. Journal of neuroimmunology. 2008;197:114–123. doi: 10.1016/j.jneuroim.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu W, Chen C, Li JG, Dimattio K, Wang Y, Unterwald E, Liu-Chen LY. Life sciences. 2013;92:1101–1109. doi: 10.1016/j.lfs.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang N, Yang D, Dong H, Chen Q, Dimitrova DI, Rogers TJ, Sitkovsky M, Oppenheim JJ. Blood. 2006;108:38–44. doi: 10.1182/blood-2005-06-2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmid CL, Streicher JM, Groer CE, Munro TA, Zhou L, Bohn LM. J Biol Chem. 2013;288:22387–22398. doi: 10.1074/jbc.M113.476234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Werry TD, Wilkinson GF, Willars GB. The Biochemical journal. 2003;374:281–296. doi: 10.1042/BJ20030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prezeau L, Rives ML, Comps-Agrar L, Maurel D, Kniazeff J, Pin JP. Current opinion in pharmacology. 2010;10:6–13. doi: 10.1016/j.coph.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 27.Liggett SB. Science signaling. 2011;4:pe36. doi: 10.1126/scisignal.2002331. [DOI] [PubMed] [Google Scholar]
- 28.Zhou L, Bohn LM. Current opinion in cell biology. 2014;27:102–108. doi: 10.1016/j.ceb.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maurice P, Benleulmi-Chaachoua A, Jockers R. Sub-cellular biochemistry. 2012;63:225–240. doi: 10.1007/978-94-007-4765-4_12. [DOI] [PubMed] [Google Scholar]
- 30.Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C. Proc Natl Acad Sci U S A. 2009;106:19168–19173. doi: 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, Lemos JC, Hagan CE, Neumaier JF, Quintana A, Palmiter RD, Chavkin C. Neuron. 2011;71:498–511. doi: 10.1016/j.neuron.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.White KL, Scopton AP, Rives ML, Bikbulatov RV, Polepally PR, Brown PJ, Kenakin T, Javitch JA, Zjawiony JK, Roth BL. Molecular pharmacology. 2014;85:83–90. doi: 10.1124/mol.113.089649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY, Ng DC. Biochimica et biophysica acta. 2010;1804:463–475. doi: 10.1016/j.bbapap.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 34.Melief EJ, Miyatake M, Carroll FI, Beguin C, Carlezon WA, Jr, Cohen BM, Grimwood S, Mitch CH, Rorick-Kehn L, Chavkin C. Molecular pharmacology. 2011;80:920–929. doi: 10.1124/mol.111.074195. [DOI] [PMC free article] [PubMed] [Google Scholar]