

Summary
Set8 is the only mammalian monomethyltransferase responsible for H4K20me1, a methyl mark critical for genomic integrity of eukaryotic cells. We present here a structural model for how Set8 uses multivalent interactions to bind and methylate the nucleosome based on crystallographic and solution studies of the Set8/nucleosome complex. Our studies indicate that Set8 employs its i-SET and c-SET domains to engage nucleosomal DNA 1 to 1.5 turns from the nucleosomal dyad and in so doing, positions the SET domain for catalysis with H4 Lys20. Surprisingly, we find that a basic N-terminal extension to the SET domain plays an even more prominent role in nucleosome binding, possibly by making an arginine anchor interaction with the nucleosome H2A/H2B acidic patch. We further show that PCNA and the nucleosome compete for binding to Set8 through this basic extension, suggesting a mechanism for how nucleosome binding protects Set8 from PCNA-dependent degradation during the cell cycle.
Graphical Abstract
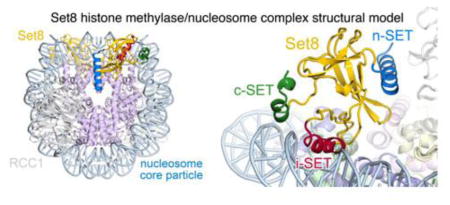
Introduction
The post-translational methylation of histone H4 on lysine 20 (H4K20me) is critical for the genomic integrity of eukaryotic cells. This modification plays key roles in DNA replication, DNA damage repair and silenced hetereochromatin [1–4]. Consequently, there is significant interest in the precise mechanistic role of H4K20 methylation and the enzymes responsible for installing the methyl marks.
Like other lysine residues, H4K20 can be mono-, di- or tri-methylated. Set8 (Pr-Set7, KMT5A) is the only known mammalian monomethyltransferase catalyzing the formation of H4K20me1. H4K20me1 is the preferred substrate for SUV4-20H1 and SUV4-20H2 histone methyltransferases to produce H4K20me2 and H4K20me3 [1–3]. These methyl marks may directly modulate chromatin compaction [5], or they may also recruit transacting replication factors in the ORC (replication origin complex) or chromatin regulators such as 53BP1 and L3MBTL1 [6–8]. For example, recent studies confirm the role of Set8’s enzymatic activity to recruit 53BP1 to site of double-strand DNA breaks [9]. The importance of the Set8 enzyme is also highlighted by studies showing that Set8 depletion results in severe changes to the cell including increased double stranded DNA breaks and defective cell cycle regulation [10–14]. Furthermore, Set8 is an essential gene in Drosophila and mice, with early embryonic lethality resulting from loss of Set8 [10,11,15,16].
Consistent with Set8’s role in cell cycle progression, the enzymatic activity of Set8 is controlled in a cell cycle dependent manner by regulating the levels of the Set8 protein [4,7,17]. Set8 protein is degraded upon ubiquitylation by the CRL4CDT2 ubiquitin ligase complex during S-phase and following DNA damage in a PCNA-dependent manner [18–22]. Like many other proteins that directly bind the PCNA (Proliferating Cell Nuclear Antigen) DNA replication clamp protein, Set8 contains a PCNA-interacting motif or PIP box which mediates this interaction.
Set8 is a member of the SET domain family of lysine methyltransferase [15,16]. Like other mammalian members of this family, Set8 contains additional n-SET and c-SET helical regions that flank the central SET domain (Fig. 1a) [23,24]. The SET domain itself is structurally conserved, but the two halves that constitute the domain are separated by an insertion, i-SET, that is variable in length and structure among SET family members [25]. This i-SET region contributes to substrate histone H4 peptide binding by cradling the peptide against the SET domain itself (Fig. 1b).
Figure 1.
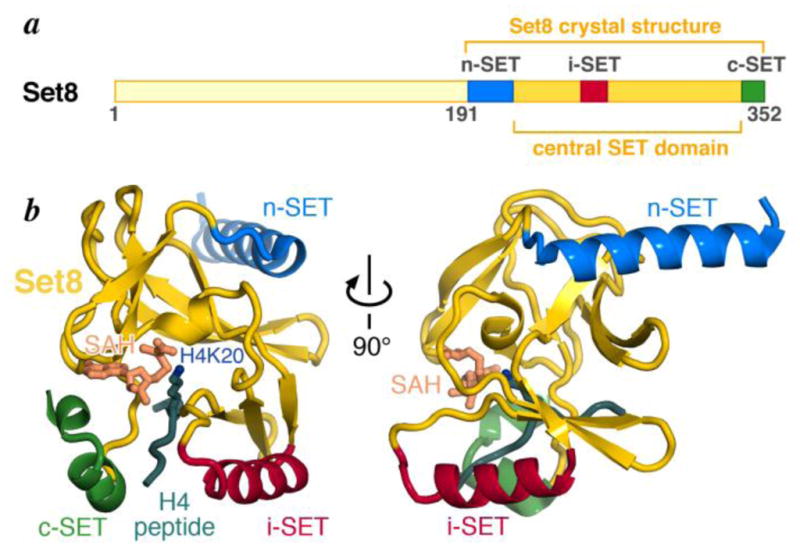
Set8 protein catalytic SET domain. (a) Cartoon showing the C-terminal SET domain and the locations of the n-SET, i-SET and c-SET regions, (b) Crystal structure of the Set8 SET domain (yellow) showing bound H4 peptide and substrate residue Lys20 (dark green), n-SET (blue), i-SET (red), c-SET (green) regions and product cofactor S-adenosyl homocysteine (SAH, sandy-brown). Coordinates from PDB id 1ZKK, chain A. All molecular structures presented were prepared using PyMOL molecular graphics software [48].
While crystallographic studies of Set8 bound to substrate peptides have provided valuable structural insights into substrate specificity [8,26,27], including the basis for Set8 being restricted to monomethylation of H4K20, we do not understand the structural basis for Set8’s preference for a nucleosome substrate. Set8 exhibits remarkably greater enzymatic activity on nucleosomes than on free histone substrates [15,16]. This suggests that Set8 must interact with other surfaces of the nucleosome in addition to the H4 N-terminal region surrounding the targeted H4K20 residue, but we lack information regarding these pertinent regions of Set8 or the nucleosome.
We have tackled the question of how Set8 interacts with the nucleosome through structural and biochemical studies. Our results show that Set8 binds to the nucleosome via multivalent interaction using both the SET domain and a basic region N-terminal to the SET domain. Intriguingly, the basic N-terminal region that we identify includes the PIP box that binds to PCNA. Our results thus explain Set8’s nucleosome specific activity, and provide insights into how Set8’s binding to the nucleosome protects it from degradation during the cell cycle.
Results
A minimal domain of Set8 sufficient to methylate and to bind nucleosomes
Crystal structures and biochemical studies of the Set8 SET domain revealed a structured region from residues 192-352 sufficient to monomethylate a histone H4 peptide at Lys20 [23,24]. However, we failed in our attempts to form a complex of Set8(191-352) with the nucleosome. We therefore performed a deletion analysis to determine the minimal domain of Set8 that would methylate and bind a nucleosome substrate. Histone methyltransferase activity was determined using a filter-binding assay. To measure nucleosome binding, we monitored fluorescence quenching of Oregon Green-488 conjugated to recombinant nucleosomes at histone H4 Q27C. We find that full length Set8 binds to nucleosomes with an apparent dissociation constant of 5.3 nM. In contrast, Set8(191-352) has 13% of nucleosome methyltransferase activity and we are unable to measure the very weak binding of Set8(191-352) to nucleosomes (Fig. 2). Examination of other Set8 truncations shows that Set8(153-352) has similar methyltransferase activity (113%) and nucleosome binding (Kd = 7.9 nM) to full length Set8, while Set8(175-352) retains significant methyltransferase activity (102%) but binds nucleosomes much more weakly (Kd = 1.8 μM). A further deletion of 6 residues in Set8(181-352) reduces methyltransferase activity to 79% and binding to 4.8 μM. We also examined the Set8(153-352) H347F mutant that was shown to increase binding to H4 peptide approximately 20 fold [23], but we find this mutant has similar nucleosome methyltransferase activity and a modest increase in binding affinity to nucleosomes (Kd = 4.5 nM vs 7.9 nM for the unmutated protein). These results indicate that Set8(153-352) constitutes a minimal domain that methylates and binds nucleosomes with significant contributions from residues 153-174 and 181-190 located N-terminal to the structured SET domain.
Figure 2.

N-terminal deletion analysis of Set8 shows residues which precede SET domain are involved in nucleosomal histone methyltransferase activity and nucleosome binding. (a) Summary of results with extent of individual truncations shown in respective cartoons. Histone methyltransferase activity on nucleosome substrates are provided relative to full length Set8, while the dissociation constants for Set8 variants binding to fluorescently labeled nucleosomes is shown on the right. (b) Nucleosome binding data for Set8 truncations. Nucleosome fluorescently labeled with Oregon Green 488 on H4Q27C were titrated with Set8 variants in triplicate, and the normalized fluorescence change plotted as a function of Set8 concentration.
Use of RCC1 to crystallize the Set8/nucleosome complex
Based on this analysis of Set8 truncations, we selected Set8(153-352) containing the H347F mutation [Set8Δ1(H347F)] for structural studies. While we were able to reconstitute, purify and concentrate monodisperse Set8Δ1/nucleosome complex, we were not able to grow crystals of the complex. As part of our efforts to explore alternate strategies for crystallizing the Set8/nucleosome complex, we considered using the RCC1 chromatin factor as a co-crystallization agent. This approach was inspired by the packing of individual RCC1/nucleosome complexes in the crystals used to determine the RCC1/nucleosome X-ray structure [28,29]. The crystal packing leaves the histone H4 tail and regions adjacent to this tail accessible, suggesting the possibility that Set8 could bind to the nucleosome in the context of the crystalline RCC1/nucleosome complex. We first examined if Set8 could bind to RCC1/nucleosome in solution by comparing the elution of Set8Δ1/nucleosome, RCC1/nucleosome and Set8Δ1/RCC1/nucleosome complexes by Superdex 200 size exclusion chromatography. We observe slightly faster elution of RCC1/nucleosome and nucleosome upon Set8Δ1 addition, and SDS-PAGE analysis of the peak fractions confirms Set8, RCC1 and the nucleosome coelute as a complex (Supplementary Fig. 2).
In contrast to our unsuccessful Set8Δ1/nucleosome crystallization trials, our Set8Δ1/RCC1/nucleosome crystallization trials produced single crystals. To confirm the presence of Set8Δ1 in such crystals, we trace labeled Set8Δ1 with carboxyrhodamine [30–32], purified reconstituted Set8Δ1/RCC1/nucleosome complex by size exclusion chromatography, set up crystallization trials and examined resulting crystals with a fluorescence microscope (Supplementary Fig. 3a, b). The observed fluorescence indicated Set8Δ1 is present in the crystals. To further validate this conclusion, we washed individual crystals and examined the contents by SDS-PAGE. Bands for each of the RCC1, Set8Δ1, and individual histone proteins were detected by Coomassie Blue staining, and the carboxyrhodamine labeled the Set8 band was additionally detected by fluorescence (Supplementary Fig. 3c). These results indicated that we had crystallized a ternary complex of Set8Δ1, RCC1 and the nucleosome core particle. We note that we obtained crystals of Set8Δ1/RCC1/nucleosome using yeast RCC1 (Srm1), but not the Drosophila RCC1 that we had used in our RCC1/nucleosome crystal structure [28,29].
Initial diffraction studies of these crystals produced diffraction to only 8–10 Å, but we were able to improve order in the crystals through post-crystallization dehydration soaks [29,33]. We collected and processed a 4.5 Å resolution data set, and performed molecular replacement using a polyalanine model of a yeast RCC1/nucleosome structure (Girish, Huang, Makde and Tan, unpublished) and a polyalanine model of the Set8 SET domain (PDB coordinates 1ZKK, chain A). Two potential molecular replacement (MR) solutions were obtained with logarithm likelihood gain (LLG) of 446 and 380 for MR models 1 and 2, respectively. Other molecular replacement solutions were not considered further since they included substantial steric clashes. Both models contain a pseudosymmetric half of the nucleosome and one molecule each of RCC1 and Set8 in the asymmetric unit. We manually corrected close contacts in COOT and performed rigid body refinement followed by restrained refinement of the models using REFMAC5 [34,35] to Rwork/Rfree of 34.2% and 40.5% for model 1 and Rwork/Rfree of 45.0%/47.0% for model 2. The electron density for RCC1 and the histone cores is continuous and well defined in model 1 but significantly less so in model 2, while the electron density for Set8 is discontinuous and poorly defined in both models 1 and 2 (Supplementary Fig. 4). Attempts to further refine these crystallographic models were not successful.
The structure of yeast RCC1/nucleosome in Set8/RCC1/nucleosome MR models 1 and 2 are similar and resemble the Drosophila RCC1/nucleosome in RCC1 switchback loop/histone dimer acidic patch interactions (Fig. 3) [28]. While Set8 binds directly to the nucleosome in both models, the orientation of Set8 in the MR models 1 and 2 are very different, with different regions of Set8 in the two models interacting with the nucleosome. In both cases, no interactions are observed between Set8 and RCC1.
Figure 3.

Crystallographic molecular replacement models for the Set8/RCC1/nucleosome complex. (a) and (b) Overview of molecular replacement models 1 and 2, respectively, looking down on the histone surface. Set8 is shown in the same colors used in Figure 1, while RCC1 is shown in white, and histones H2A, H2B, H3, H4 and nucleosomal DNA are shown in pale yellow, pink, cornflower blue, light green and light blue, respectively. (c) and (d) Molecular replacement models 1 and 2 viewed looking from the nucleosome dyad. This view highlights that model 1 Set8 employs its i-SET and c-SET regions to interact with nucleosomal DNA 1 to 1.5 superhelical turns from the nucleosome dyad, whereas model 2 Set8’s n-SET region interacts with DNA 2 superhelical turns from the nucleosomal dyad. (e) and (f) Molecular replacement models 1 and 2 oriented to highlight H4 Asp24 in each molecular replacement model, and the position of H4 Arg23 in the H4 peptide and S-adenosyl homocysteine entity both modeled based on PDB id 1ZKK chain A. The RCC1 molecules are omitted for clarity.
Set8/RCC1/nucleosome MR model 1
In MR model 1, the Set8 i-SET helix, which contains a cluster of three basic residues (K256, K257 and R258), is positioned in the DNA major groove of the nucleosome at SHL±1 (Fig. 3a, c). Resting above the adjacent DNA minor groove is the c-SET domain containing the basic residue K341 in position to interact with the DNA backbone. In contrast, the n-SET helix projects from the face of the SET domain opposite the nucleosome interface and makes no contact with histones or nucleosomal DNA. Complementing the Set8 i-SET and c-SET interactions with the nucleosome DNA are Set8-histone protein-protein interactions by the Set8 loop between strands β6 and β7 located above histone H4 α2 helix. Potential contacts include charged interactions between Set8 Lys280 and histone H4 Glu52, and additional hydrogen bonding interactions.
Although electron density for the histone H4 tail around target Lys20 is not visible, we can model the H4 tail based on crystal structures of the Set8 catalytic domain complexed with an H4 peptide spanning residues 16-23 [23,24]. The modeled H4 peptide is sandwiched between the Set8 SET domain on top and the nucleosome DNA at SHL±1.5 below, and bordered on the sides by the i-SET and c-SET domains, which interact with nucleosomal DNA (Fig. 3e). The C-alpha atom of H4 Arg23 is 13 Å from the C-alpha atom of H4 Asp24 in this model, suggesting that if this model is correct, at least four residues starting from H4 D24 would need to be unraveled from the short helix extension to the H4 histone fold core. Aside from this, there do not appear to be any other constraints limiting the H4 tail to bind in the Set8 active site.
Set8/RCC1/nucleosome MR model 2
The predominant interaction of Set8 with the nucleosome in MR model 2 is Set8’s n-SET domain, which lies across the nucleosome DNA major groove at SHL±2 (Fig. 3b, d). In addition, the loop between Set8 strands β9 and β10 may interact with the adjacent minor groove, and the C-terminal end of the n-SET domain is positioned over the histone H3 α1 helix. Neither the Set8 i-SET nor the c-SET domains are close to the nucleosome, and no interactions between Set8 with the histone H4 α2 helix are evident.
The H4(16-23) peptide modeled in MR model 2 is located on a Set8 surface that faces away from the nucleosome and is solvent exposed (Fig. 3f), in contrast to the H4 peptide modeled in MR model 1. The H4 Arg23 C-alpha atom is 23 Å from the H4 Asp24 C-alpha atom, but even more problematic than this distance are the relative locations of these two residues. The Set8 SET domain is positioned between the H4 histone fold and the modeled H4 peptide, blocking a path for the H4 tail to bind to Set8, thus increasing the effective distance between the nucleosome surface and the Set8 active site, and raising the question of how the H4 peptide around Lys20 could reach into the Set8 active site in MR model 2.
Basic residues in i-SET and c-SET helices are important for Set8 methyltransferase activity and nucleosome binding
Our crystallography studies provided two distinct structural models for how Set8 binds to the nucleosome, but these studies did not unambiguously distinguish between the two models. While MR model 1 appears more plausible because the Set8 position is consistent with previous structural information for how Set8 binds to its histone H4 substrate and because of the better crystallographic statistics and electron density, we sought additional evidence to distinguish between the two models. Since Set8 employs the n-SET, the i-SET and the c-SET regions differently to bind to the nucleosome in the two structural models, we engineered mutations into these regions (Fig. 4a, b) and tested the activity of the mutant Set8 proteins in nucleosome methyltransferase and nucleosome binding (Fig. 5). We used the same Set8(153-352) H347F construct used for our crystallographic studies for this analysis. We focused on basic residues because each of these three regions interacts with nucleosomal DNA in the structural models. We find that mutating basic residues in the n-SET, i-Set and c-SET region slightly decrease nucleosome binding affinity by a factor of only 3.5 to 5 fold (Fig. 5). Mutations in the n-SET and c-SET domains did not significantly affect Set8’s nucleosome methyltransferase activity, but mutations in the i-SET domain decrease methyltransferase activity by a factor of two. Combining the i-SET and c-SET mutations further decreased methyltransferase activity (32% of wild-type) and significantly weakened nucleosome binding (Kd = 330 nM). We also prepared hSet8Δ1(K280A, H347F) but aggregation of this mutant protein prevented us from testing the predicted contact between Set8 Lys280 and histone H4 Glu52. Since model 1 is more consistent with how Set8 binds the histone H4 tail substrate (model 2 does not provide a plausible means for Set8 to bind the H4 tail) and since mutations designed to disrupt model 1 interfaces have a larger effect on Set8 enzymatic and substrate binding activity, we believe that model 1 better describes how Set8 binds to the nucleosome. Model 1’s direct involvement of the Set8 i-SET region in nucleosome binding is also consistent with the variable i-SET region contributing substrate specificity amongst the SET domain proteins.
Figure 4.

Sites of interactions between Set8 and the nucleosome. (a) and (b) Location of residues targeted for site-directed mutagenesis studies in the n-SET, i-SET and c-SET regions of molecular replacement models 1 and 2. The C-alpha positions of each mutated residue is shown as a sphere, and the corresponding residues listed. Same colors as in Figure 3. (c) and (d) The nucleosomal acidic patch created by histone H2A and its position relative to the N-terminal end of the structured Set8 SET domain in molecular replacement models 1 and 2. Whereas model 1 Set8’s n-SET helix points in the direction of the nucleosome acidic patch facilitating a possible interaction between Set8 residues R188, R189 with the acidic patch, model 2 Set8’s n-SET helix points away from the nucleosome acidic patch. The RCC1 molecules are omitted for clarity.
Figure 5.

Site-directed mutagenesis of Set8 n-SET, i-SET and c-SET regions to distinguish between molecular replacement models 1 and 2. The combinations of mutations targeting the i-SET and c-SET regions predicted to bind to nucleosomal DNA in model 1 significantly reduced histone methyltransferase and nucleosome binding activities, whereas mutations in the n-SET region implicated in model 2 had only minor effects.
Role of Set8 basic region preceding SET domain in nucleosome substrate binding
Although the Set8 construct we used for our crystallization studies, Set8(153-352) H347F was extended by 40 residues N-terminal to the SET domain (residues 193-352), we did not observe electron density for this region. Nevertheless, our deletion analysis suggests this region is important for interaction with the nucleosome since Set8(175-352) and Set(181-352) were approximately 200 to 600-fold weaker in nucleosome binding than Set8(153-352) and Set8(191-352) fails to bind the nucleosome altogether (Fig. 2). The major feature of Set8 between residues 153 and 192 is its basic nature, with 14 Arg or Lys residues (35% of 40 residues) and only 2 acidic residues (5% of 40 residues) (Fig. 6a). Among the basic residues in the Set8(175-191) region critical for nucleosome binding are R179, K180, R188 and R189. We find that the Set8(153-352) R188A, R189A mutant has reduced nucleosome methyltransferase and much lower nucleosome binding activity (56% of full length, wild-type Set8 enzymatic activity, and 2.5 μM nucleosome binding dissociation constant or over 300 fold lower binding affinity compared to the equivalent wild-type protein) (Fig. 6b). In contrast, the effect of the R179A, K180A mutations is much more muted with similar enzymatic activity as the wild-type equivalent and about 2.5x decrease in nucleosome binding affinity. The combination of R179A, K180A with R188A, R189A produced similar enzymatic activity and even weaker nucleosome binding as compared to the R188A, R189A mutant. We note that the effect of the R188A, R189A mutations on nucleosome binding was larger than the effect of the combined i-SET and c-SET basic mutations in the structured SET domain. Thus, our results suggest a major role for R188, R189 residues and a minor role for R179, K180 residues in nucleosome substrate recognition by Set8. Binding experiments using the fluorescent probe installed on a different position (H4 G56C within the histone fold) produced similar trends in binding for the subset of Set8 variants examined, suggesting our conclusions are not dependent on the location of the probe at H4 Q27C near the H4 N-terminal tail (Supplementary Table 2).
Figure 6.

Critical role of Set8 basic regions N-terminal to the SET domain. (a) Amino acid sequence of Set8(153-200). The PIP (PCNA Interacting Peptide) box is show as a red box and conserved PIP residues are shown underlined in red. Basic residues R179, K180, R188, R189 within the PIP box are shown in blue while other basic residues N-terminal to the n-SET helix are show in black. The location of the n-SET helix observed in crystal structures is shown as a blue box. (b) Mutations in the Set8 basic region highlight the critical role of Set8 R188 and R189 and the nucleosome acidic patch in Set8/nucleosome interactions. Acidic patch mutant nucleosomes contain an N-terminal hexahistidine tag. Similarly tagged wild-type nucleosomes have no effect on nucleosome binding or methylation (data not shown).
We next asked what aspect of the nucleosome might be targeted by the R188, R189 residues. The short distance of R189 from K195, the first residue of the n-SET helix, constrains the nucleosome surfaces with which R188 and R189 could interact. We also note that the histone dimer contains an acidic patch that is targeted by what we have termed the arginine anchor. All crystal structures to date of a chromatin factor or enzyme in complex with the nucleosome except for the chromatosome employ an arginine residue from the chromatin protein to interact with the histone dimer acidic patch [28,36–41]. In model 1, the N-terminus of the Set8 n-SET domain is poised above the acidic patch, possibly positioning Set8 R188 and/or R189 to interact with the acidic patch (Fig. 4c) At first glance, it would appear that Set8 K193 is almost 30 Å away from the acidic patch and therefore too far away for R188 or R189 to bind to the acidic patch.
However, since Set8 residues 195-202 appear to be structured as a helix in the Set8/peptides structures due to crystal contact interactions and not due to interactions within the SET domain [23,24], Set8 residues 190-202 could bridge the distance to the acidic patch as an extended polypeptide chain. In contrast, in model 2 the N-terminus of the Set8 n-SET domain is pointing away from the histone disc towards nucleosomal DNA (Fig. 4d). We therefore examined the effect of removing this histone dimer acidic patch on the Set8’s activity. We find that full length Set8 has only 3.8% of its histone methyltransferase activity on nucleosomes containing the quadruple H2A E61A, E64A, D90, E92A mutation compared to wild-type nucleosomes (Fig. 6b). This is a larger effect on methyltransferase activity than any of the Set8 truncations or mutations that we analyzed. Similarly, Set8Δ1 lacking the N-terminal 152 residues has 5.4% histone methyltransferase activity on nucleosomes with the same acidic patch mutation. The acidic patch mutations had a significantly greater effect on nucleosome binding for the Set8Δ1 truncation (Kd = 1.3 μM or 160 fold weaker binding compared to wild-type nucleosomes) than it did for full length Set8 (Kd = 60 nM or 11 fold weaker binding compared to wild-type nucleosomes). We find that combining the Set8Δ1(R188A, R189A) basic mutations with the nucleosome acidic patch mutations results in only slightly weaker binding to the nucleosome compared to the R188A, R189A mutations or the nucleosome acidic patch mutations on their own. The finding that the Set8 R188A, R189A basic mutations and the nucleosome acidic patch mutations each severely affect nucleosome binding but the combination of the mutations is not additive suggests the possibility that the Set8 R188A, R189A basic region interacts with the nucleosome acidic patch.
The Set8 R188A, R189A basic region mediates competitive binding of Set8 to PCNA and to the nucleosome
Set8 protein levels are regulated by ubiquitylation-mediated proteolysis during the cell cycle by binding to PCNA [18–22]. Set8 contains two PIP or PCNA interaction boxes, but only the second PIP box is required for PCNA-dependent ubiquitylation of Set8 [18,21,22]. This second PIP box is located in the region which includes Set8 residues 178 to 190 (Fig. 6a). This directly overlaps the same Set8 basic region that we identified as playing a critical role in the Set8 enzyme binding to its nucleosome substrate. The crystal structure of the homologous PIP box peptide from the p21 protein bound to PCNA shows extensive interaction between the PIP box and PCNA [42]. Specifically, PCNA interacts with the p21 peptide residues which correspond to Set8(178-190), including the p21 R155 equivalent of Set8 R189. Prompted by this observation that the same Set8 region apparently interacts with both PCNA and the nucleosome, we asked if Set8 can bind simultaneously with both PCNA and the nucleosome.
We used the same nucleosome binding assay which monitors quenching of Oregon Green-488 conjugated to H4 Q27C nucleosomes and examined the ability of PCNA to compete Set8 from the nucleosome. In this assay, quenching is observed upon Set8 binding to the nucleosome, If Set8 is competed off the nucleosome by the addition of PCNA, a dequenching of this quenched fluorescence should be observed, leading to fluorescence levels comparable to the nucleosome alone in the absence of Set8. As expected, adding PCNA to the nucleosomes did not change fluorescence of the labeled nucleosomes since PCNA is not anticipated to interact with the nucleosome. When a saturating amount of Set8 is added to nucleosomes, binding of Set8 is detected as quenching of the nucleosome fluorescence signal (normalized fluorescence change of 1.0 in Figure 7). Titrating PCNA into this pre-formed Set8/nucleosome complex results in an increase in the fluorescence (returning the relative fluorescence change to 0 in Figure 7). We confirmed that this fluorescence dequenching was due to direct binding of PCNA to Set8’s PIP domain using a Set8 PIP domain F184A, Y185A mutant shown to be defective for binding to PCNA [18,21]. In contrast to wild type Set8, PCNA was severely compromised in its ability to compete this PIP mutant Set8 off the nucleosome, while the Set8 mutant’s nucleosome binding was only minimally affected (data not shown). These results suggest that the Set8 PIP domain mediates interactions with both PCNA and the nucleosome. While it is possible that Set8, PCNA and the nucleosome can form a ternary complex, such an interpretation would require Set8 to not quench the labeled nucleosome only in the context of the ternary complex. While we were unable to rule this out, we believe such a scenario is unlikely given that the labeled position on the nucleosome is proximal to the nucleosomal binding site of Set8’s SET domain near the H4 tail and distant from the acidic patch where the PIP domain is expected to bind.
Figure 7.

PCNA competes with the nucleosome for binding to Set8 via the Set8 PIP box. Nucleosome core particles (NCP) fluorescently labeled on H4 Q27C with Oregon Green 488 were incubated with buffer (black), Set8Δ1 (red) and Set8Δ1 with the PIP F184A, Y185A mutations shown to disrupt binding to PCNA (blue) were titrated with PCNA. A normalized fluorescence change value of 1.0 represents full binding of nucleosomes by Set8, and a value of 0 signifies no binding. The decrease in fluorescence signal of Set8Δ1/NCP upon incubation with PCNA demonstrates that PCNA sequesters wild-type Set8 from the nucleosome, while Set8 defective for binding to PCNA continues to bind the nucleosome in the presence of PCNA.
Discussion
We have combined crystal diffraction data and biochemical characterization to determine a structural model for how the Set8 histone methyltransferase enzyme interacts with its nucleosome substrate. Our model indicates that the relatively compact Set8 protein uses at least three distinct regions to interact with the nucleosome. A basic N-terminal extension is the primary determinant of nucleosome binding and interacts with the nucleosomal H2A/H2B histone dimer acidic patch and likely nucleosomal DNA to anchor Set8 to the nucleosome. This positions the SET domain for interaction with the nucleosome face near the targeted methylation site. Finally, the Set8 catalytic site engages the H4 tail and methylates lysine 20. The multivalent interactions with the nucleosome and particularly nucleosomal DNA observed or predicted by our studies provides a molecular basis for why the Set8 enzyme greatly prefers a nucleosome substrate over histone proteins or peptides.
We have used a novel approach employing the chromatin factor RCC1 as a cocrystallization protein for crystallizing the Set8/nucleosome complex. Using diffraction data collected from these crystals, we obtained two crystallographic molecular replacement solutions using Set8 and RCC1/nucleosome structures as search models. We then challenged these models by examining the consequences of mutating Set8 residues in key nucleosome interfaces. Our results are consistent with Set8 using its i-SET and c-SET regions to interact with nucleosomal DNA 1 to 1.5 DNA helical turns from the nucleosomal dyad. Notably, mutation of the i-SET region alone or in combination with the c-SET impairs enzymatic activity to a greater degree than nucleosome binding when compared to truncations or mutations of Set8 regions adjacent to the SET domain. One possible explanation is that the SET domain nucleosome interactions do not function in Set8 recruitment, but rather orient Set8 on the nucleosome surface for optimal catalysis. Our preferred model is broadly consistent with previous crystal structures of the Set8 SET domain bound to its substrate histone H4 peptide given correct orientation of the Set8 catalytic site relative to the H4 histone fold core. However, the depth of the Set8 catalytic cleft requires at least four residues of the H4 tail that are typically structured in nucleosome crystal structures (residues 24-27) to unfold and extend to reach the Set8 active site. We note that unraveling of the H4 tail is not a special requirement of our particular structural model. Given how embedded the H4 tail is within the Set8 catalytic cleft, it is difficult to explain how Set8, in any model, could bind to the H4 tail without unfolding some of the structured H4.
Although the Set8 catalytic SET domain interacts directly with the nucleosome, these interactions are not sufficient for detectable nucleosome binding. We have identified a positively charged N-terminal extension to the SET domain spanning residues 153-190 that is required to restore nucleosome binding and methylation by the SET domain to levels comparable to the full-length protein. Further dissection of this region implicates sequences between 181-190 and 153-174. Within the Set8(181-190) sequence, the Set8 R188, R189 basic cluster plays a significant role in nucleosome binding and activity. The nearby R179, K180 basic cluster contributes only minimally, suggesting that the interactions mediated by Set8 R188, R189 residues with the nucleosome are more than simply nonspecific electrostatic interactions. We provide evidence that Set8 R188, R189 likely interacts with the histone H2A/H2B dimer acidic patch on the nucleosome: mutation of the nucleosome acidic patch has a similar deleterious effect on Set8 binding as the Set8 R188A, R189A mutation, while combining these mutations (Set8 R188A, R189A binding to nucleosomes mutated in the acidic patch) weakens binding only slightly further. We would have expected an additive effect on nucleosome binding mutations if the Set8 and histone mutations affected separate binding surfaces. Interestingly, mutation of the acidic patch results in a larger impairment of enzymatic activity than mutation of the Set8 R188, R189 cluster. This suggests that elimination of the acidic patch may result in an additional unanticipated effect on nucleosome methylation possibly through re-orienting Set8 on the nucleosome surface. Previous reports that Set8 does not rely on the acidic patch for nucleosomal methylation may have missed this effect due to use of significantly higher Set8 concentrations or single point mutation of the acidic patch [43]. Binding of the nucleosome acidic patch by a chromatin protein arginine residue (the “arginine anchor”) has been observed in every chromatin protein/nucleosome crystal structure determined to date (complexes of the nucleosome with RCC1, Sir3 and the PRC1 ubiquitylation module proteins, and with the LANA and CENP-C peptides). We propose here that Set8 also employs an arginine anchor to bind to the nucleosome.
The Set8 arginine residues that may act as an arginine anchor to bind to the nucleosome acidic patch are included within the PCNA interacting peptide sequence (PIP box) of Set8. We show that PCNA and the nucleosome compete for binding to Set8, consistent with the extensive interactions made by the homologous PIP box of p21 with PCNA including direct interactions between arginine residues equivalent to Set8 R188 and R189. Given that PCNA binding to Set8 is required for Set8 ubiquitylation and subsequent proteolytic degradation, this finding suggests an eloquent mechanism for ensuring that Set8 is protected from proteolytic degradation when it is engaged on the nucleosome. As long as Set8 is bound to the nucleosome, the signal to recruit PNCA is obscured and proteolysis is prevented. Our results also exemplify how a PIP box can serve multiple functions: it is utilized to bind to PCNA but it also contributes to nucleosome binding in the case of Set8.
Our deletion analysis suggests that Set8(153-174) also contributes to nucleosome binding as we detect a decrease in binding affinity from ~8 nM to 1800 nM when this region is deleted. This effect is comparable to that observed for the R188A, R189A basic cluster mutation. As shown in Fig. 6a, Set8(153-174) contains additional basic amino acids (8 lysine and 1 arginine residues). Since we believe the PIP box that immediately follows this region binds to the histone dimer acidic patch situated next to nucleosomal DNA, it seems likely that basic residues within Set8(153-174) may interact with nucleosomal DNA near the acidic patch (approximately 5 helical turns from the nucleosome dyad). Interestingly, unlike mutation of the PIP box basic cluster, truncation of Set8(153-174) did not appreciably affect histone methyltransferase activity on nucleosome substrates. Thus, this region may similarly enhance Set8-nucleosome affinity, but unlike the proposed PIP box-acidic patch interaction plays a minimal role in orienting the SET domain for catalysis.
Set8’s multivalent interactions with the nucleosome may also explain why the electron density for Set8 in our Set8/RCC1/nucleosome crystallographic electron density maps was so poor as well as why we did not observe electron density for the Set8 basic residues interacting with the nucleosome acidic patch. As noted in the Results section, electron density for RCC1 as well as for the histone and DNA components of the nucleosome was far better than the electron density for Set8 particularly in molecular replacement model 1. Based on our studies which suggest that Set8 and RCC1 both bind to the nucleosome acidic patch, we suspect that the occupancy of Set8 in the crystal may be less than the expected two Set8 molecules per nucleosome due to competition with RCC1 for binding to the nucleosome. Since Set8 is able to make multivalent interactions with the nucleosome via the Set8 basic N-terminal extension and its SET domain, Set8 could presumably form a triple complex with RCC1 and the nucleosome even if interactions via the acidic patch were blocked by RCC1. Another possibility is that Set8 is present in stoichiometric amounts in the crystal, but the competition between RCC1 and Set8 for the acidic patch leaves Set8 less structurally constrained when bound via to the SET domain to the nucleosome. We have tried without success to substantiate these speculations. For example, comparison of SDS-PAGE band intensities of purified Set8/RCC1/nucleosome complex with washed Set8/RCC1/nucleosome did not provide definitive results for the stoichiometry of Set8 to nucleosome. Competition binding experiments show that RCC1 can compete for nucleosome binding with Set8 (Supplementary Fig. 1c). However, unlike PCNA which neutralizes the PIP box, RCC1 is unable to completely remove Set8 from the nucleosome, likely due to the multivalent nature of the Set8-nucleosome interaction.
The Set8/nucleosome model adds to a small but growing number of structural descriptions of how chromatin factors and enzymes interact with the nucleosome. A common feature is that the chromatin factors and enzymes recognize not just individual elements of the nucleosome, but rather the architecture of the nucleosome. In the case of RCC1, the PRC1 ubiquitylation enzyme and now Set8, interactions are made to both histone and DNA components of the nucleosome ensuring binding occurs in a chromatin context. Despite these broad similarities and the apparent common use of an arginine anchor, the details of the interactions are different reflecting the different functions including, in the case of PRC1 and Set8, the different histone tails (H2A C-terminal tail for PRC1 and the H4 N-terminal tail for Set8) targeted by the chromatin enzymes. Thus, although paradigms such as the arginine anchor may guide our understanding of how chromatin enzymes bind to the nucleosome, many more unique features are likely to govern how each of these enzymes targets catalysis to unique nucleosomal surfaces.
Material and Methods
Recombinant proteins and nucleosomes
The human Set8 gene (UniProtKB #Q9NQR1) and truncation or mutated variants were expressed as a combination Strept tag-decahistidine tagged protein using the pST50Tr expression vector [44] in BL21(DE3)pLysS cells at 18°C. Each fusion protein was purified by Talon metal affinity chromatography (Clontech), the fusion affinity tag removed by tobacco etch virus protease and the untagged protein further purified by SourceS cation-exchange chromatography (GE Healthcare). The yeast RCC1 gene, Srm1, was amplified from genomic DNA and similarly expressed using the pST50Tr expression vector in BL21(DE3)pLysS cells at 18°C. Tagged yeast RCC1 was purified using Talon metal affinity chromatography, followed by TEV protease digestion to remove the affinity tag and the untagged protein further purified by SourceQ anion-exchange chromatography (GE Healthcare). Human PCNA was expressed in pST50Tr with an N-terminal (Gly-Ser)4 linker between the Strept-decahistidine combination tag and PCNA to facilitate TEV protease cleavage. After Talon metal affinity purification and TEV treatment, the GS4hPCNA protein was purified by SourceQ anion-exchange chromatography. Recombinant Xenopus histones and nucleosome core particles were prepared essentially as described previously, including SourceQ HPLC of the reconstituted nucleosome core particles. [45]. All proteins were analyzed by dynamic light scattering and determined to be non-aggregated. Selected Set8 proteins used for crystallization were N-terminally labeled with carboxyrhodamine as previously described [30–32]. Briefly, Set8 was reacted with two molar equivalents of 5-(and 6-)Carboxyrhodamine succinimidyl ester (Life Technologies) at pH 7.5 followed by purification using a Superdex 200 column. Typically labeling percentages of approximately 3–5% were achieved.
Reconstitution and purification of Set8/RCC1/nucleosome complex
To generate the Set8/RCC1/nucleosome complex, yeast RCC1(2-482) protein was mixed in 10 mM Tris-Cl pH 7.6, 50 mM sodium chloride, 1 mM dithiothreitol (DTT) with recombinant nucleosome core particles containing Xenopus core histones and 149 bp Widom 601 DNA in a 2.2:1 RCC1:nucleosome molar ratio followed by the addition of human Set8 protein (153-352) H347F in a 2.2:1 Set8:nucleosome molar ratio. The reconstituted complex was then purified on a Superdex 200 HR size-exclusion chromatography column (GE Healthcare) in 10 mM Tris-HCl pH 7.5, 50 mM sodium acetate, 1 mM dithiothreitol (DTT) and 0.1 mM phenyl methyl sulfonyl fluoride (PMSF) and concentrated by filtration centrifugation in the same buffer. For complexes containing trace labeled Set8, N-terminal carboxyrhodamine labeled Set8 was doped into reconstitutions to give a 0.5% final labeling percentage.
Crystallization and structure solution
Crystals of the Set8/RCC1/nucleosome complex were grown in microbatch by mixing 1 μl of 10 to 15 mg/ml complex in 10 mM Tris-HCl pH 7.5, 50 mM sodium acetate, 1 mM DTT and 0.1 mM PMSF with 1 μl of 25 mM sodium acetate pH 5.5, 25 to 50 mM sodium citrate, 1 mM DTT, 2 to 8% PEG2000-MME and then overlaid with 75 μl of Al’s oil (an equal-volume mixture of silicon and paraffin oils) at 4°C in 96-well microtiter plates. Crystallization drops were visualized using a Nikon SMZ1000 stereoscope equipped with a CoolPix 990 camera and incorporation of trace fluorescently labeled Set8 was confirmed using a Nikon SMZ1500 stereoscope equipped with a Nikon Intensilight C-HGFI light source, a DsRed filter (Ex 545/30 nm, DM 570 nm, BA 620/60 nm), and a CoolSnapEZ Turbo 1394 camera. To further validate crystal contents, single crystals were washed in stabilizing solution prior to dissolution and separation by SDS-PAGE, fluorescence scanning using a Typhoon 9410 variable mode imager (λex= 532 nm and λem= 580 nm) from GE Healthcare, and Coomassie staining. Crystals were dehydrated following the soaking protocol previously described for Drosophila RCC1/nucleosome complex crystals [28,29]. The optimized soaking solution contained 25 mM sodium acetate pH 5.5, 35 mM sodium citrate, 1 mM DTT, 5% ethanol, 8% PEG2000-MME and a stepwise increase in PEG500-DME from 0–24% in 2% increments of 15 min before flash cooling in liquid nitrogen. Diffraction data were collected with an ADSC Quantum 315 CCD detector at Advanced Photon Source’s NE-CAT beamline 24-ID-E, and processed with the XDS program (Supplementary Table 1). Molecular replacement (MR) was performed in PHASER [46] using polyalanine models of the yeast RCC1/nucleosome complex (unpublished) and human Set8 (PDB 1ZKK, chain A). The MR models were evaluated for packing clashes using COOT. After several rounds of rigid body refinement MR models were restrain refined using REFMAC5 [34,35]. However, further attempts of model building and refinement were unsuccessful due to poor quality of the electron density map.
Histone Methyltransferase (HMTase) assay
Histone methyltransferase assays were carried out in triplicate in a final volume of 10 μl, containing 2 μl of 20 μM recombinant nucleosome core particles and 1.2 μl of 3H labeled S-adenosyl methionine (MP Biomedicals) in 60 mM Tris-HC1 pH 8.0, 50 mM NaCl, 20% glycerol, 1 mM DTT and 0.1 mM PMSF. The reaction was started by the addition of 2 μl of 10 nM Set8 and incubated at 30°C for 30 min before 8 μl of the reaction mixture was spotted onto P81 phosphocellulose filter paper (Whatman). Dried filter discs were washed 3 times in 50 mM sodium carbonate pH 9.0 and dried before scintillation counting. The wild type and mutant Set8 enzyme were diluted to ensure that the measured activity remained in the linear range of the assay.
High-throughput interactions by fluorescence intensity (HiFi) nucleosome binding assay
Clear bottom 384-well microplates were prepared as published previously [47]. Xenopus histone H4Q27C protein was labeled with Oregon Green 488 maleimide, and reconstituted into recombinant nucleosomes. 2 nM of these fluorescently labeled nucleosomes were titrated with 2 nM to 20 μM Set8 protein in 40 μl reaction volumes containing 20 mM Tris-HCl, pH 7.5, 125 mM NaCl, 5% glycerol, 5 mM DTT, 0.01% of both CHAPS (3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate) and NP40 (nonyl phenoxypolyethoxylethanol) detergents, and 0.1 mg/ml BSA for 15 min at room temperature in the dark before scanning at the appropriate wavelength (λex= 488 nm and λem= 526 nm) using a Typhoon 9410 variable mode fluoroimager. Fluorescence intensities were quantified using the program ImageQuant TL, and the data points were fitted with a non-linear regression curve using PROFIT software. All experiments were performed in triplicates. Due to the use of 2 nM labeled nucleosomes in binding experiments, Kd values below 10 nM may slightly underestimate affinity.
Accession Numbers
Crystallographic structure factors and coordinates for molecular replacement (MR) model 1 have been deposited in the Protein Data Bank with accession number 5HQ2.
Supplementary Material
Highlights.
How histone methylases interact with their nucleosome substrate is not understood.
We have crystallized the Set8 histone methylase in complex with the nucleosome.
We describe a structural model based on crystallographic and biochemical data.
Our model shows how Set8 makes multivalent interactions with the nucleosome
Our results explain how nucleosome binding protects Set8 from cell cycle proteolysis
Acknowledgments
We would like to thank the staff of APS NE-CAT beamlines 24ID-E and 24ID-C for their assistance during synchrotron data collection; Hemant Yennawar and Neela Yennawar at the Penn State Huck Institute X-ray core facility; Allen Minns and Mike Moore for reagent preparation; Jiehuan Huang, Matt Jennings and Sang-Ah Kim for assistance with crystallographic data collection; and members of the Tan laboratory and the Penn State Center for Eukaryotic Gene Regulation for helpful discussions. This work was supported by the National Institutes of Health grants GM088236 and GM111651 to S.T. and Damon Runyon Cancer Research Foundation grants DR-2107-12 and DFS-14-15 to R.K.M..
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jørgensen S, Schotta G, Sørensen CS. Histone H4 Lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013;41:2797–2806. doi: 10.1093/nar/gkt012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beck DB, Oda H, Shen SS, Reinberg D. PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. 2012;26:325–337. doi: 10.1101/gad.177444.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brustel J, Tardat M, Kirsh O, Grimaud C, Julien E. Coupling mitosis to DNA replication: the emerging role of the histone H4-lysine 20 methyltransferase PR-Set7. Trends Cell Biol. 2011;21:452–460. doi: 10.1016/j.tcb.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Wu S, Rice JC. A new regulator of the cell cycle: the PR-Set7 histone methyltransferase. Cell Cycle. 2011;10:68–72. doi: 10.4161/cc.10.1.14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu X, Simon MD, Chodaparambil JV, Hansen JC, Shokat KM, Luger K. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat Struct Mol Biol. 2008;15:1122–1124. doi: 10.1038/nsmb.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.West LE, Roy S, Lachmi-Weiner K, Hayashi R, Shi X, Appella E, et al. The MBT repeats of L3MBTL1 link SET8-mediated p53 methylation at lysine 382 to target gene repression. Journal of Biological Chemistry. 2010;285:37725–37732. doi: 10.1074/jbc.M110.139527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee J, Zhou P. SETting the clock for histone H4 monomethylation. Molecular Cell. 2010;40:345–346. doi: 10.1016/j.molcel.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang H, Mizzen CA. The multiple facets of histone H4-lysine 20 methylation. Biochem Cell Biol. 2009;87:151–161. doi: 10.1139/O08-131. [DOI] [PubMed] [Google Scholar]
- 9.Dulev S, Tkach J, Lin S, Batada NN. SET8 methyltransferase activity during the DNA double-strand break response is required for recruitment of 53BP1. EMBO Rep. 2014;15:1163–1174. doi: 10.15252/embr.201439434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oda H, Okamoto I, Murphy N, Chu J, Price SM, Shen MM, et al. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol Cell Biol. 2009;29:2278–2295. doi: 10.1128/MCB.01768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jørgensen S, Elvers I, Trelle MB, Menzel T, Eskildsen M, Jensen ON, et al. The histone methyltransferase SET8 is required for S-phase progression. J Cell Biol. 2007;179:1337–1345. doi: 10.1083/jcb.200706150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tardat M, Murr R, Herceg Z, Sardet C, Julien E. PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J Cell Biol. 2007;179:1413–1426. doi: 10.1083/jcb.200706179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakaguchi A, Steward R. Aberrant monomethylation of histone H4 lysine 20 activates the DNA damage checkpoint in Drosophila melanogaster. J Cell Biol. 2007;176:155–162. doi: 10.1083/jcb.200607178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houston SI, McManus KJ, Adams MM, Sims JK, Carpenter PB, Hendzel MJ, et al. Catalytic function of the PR-Set7 histone H4 lysine 20 monomethyltransferase is essential for mitotic entry and genomic stability. J Biol Chem. 2008;283:19478–19488. doi: 10.1074/jbc.M710579200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y, et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Molecular Cell. 2002;9:1201–1213. doi: 10.1016/s1097-2765(02)00548-8. [DOI] [PubMed] [Google Scholar]
- 16.Fang J, Feng Q, Ketel CS, Wang H, Cao R, Xia L, et al. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr Biol. 2002;12:1086–1099. doi: 10.1016/s0960-9822(02)00924-7. [DOI] [PubMed] [Google Scholar]
- 17.David R. Cell cycle: Disposing of SETD8. Nat Rev Mol Cell Biol. 2010;11:819. doi: 10.1038/nrm3020. [DOI] [PubMed] [Google Scholar]
- 18.Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Molecular Cell. 2010;40:9–21. doi: 10.1016/j.molcel.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, et al. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Molecular Cell. 2010;40:22–33. doi: 10.1016/j.molcel.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oda H, Hübner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, et al. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Molecular Cell. 2010;40:364–376. doi: 10.1016/j.molcel.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, et al. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol. 2010;12:1086–1093. doi: 10.1038/ncb2113. [DOI] [PubMed] [Google Scholar]
- 22.Jørgensen S, Eskildsen M, Fugger K, Hansen L, Larsen MSY, Kousholt AN, et al. SET8 is degraded via PCNA-coupled CRL4(CDT2) ubiquitylation in S phase and after UV irradiation. J Cell Biol. 2011;192:43–54. doi: 10.1083/jcb.201009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Couture JF, Collazo E, Brunzelle JS, Trievel RC. Structural and functional analysis of SET8, a histone H4 Lys-20 methyltransferase. 2005;19:1455–1465. doi: 10.1101/gad.1318405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao B, Jing C, Kelly G, Walker PA, Muskett FW, Frenkiel TA, et al. Specificity and mechanism of the histone methyltransferase Pr-Set7. 2005;19:1444–1454. doi: 10.1101/gad.1315905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian C, Zhou MM. SET domain protein lysine methyltransferases: Structure, specificity and catalysis. Cell Mol Life Sci. 2006;63:2755–2763. doi: 10.1007/s00018-006-6274-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marmorstein R, Trievel RC. Histone modifying enzymes: structures, mechanisms, and specificities. Biochim Biophys Acta. 2009;1789:58–68. doi: 10.1016/j.bbagrm.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Del Rizzo PA, Trievel RC. Substrate and product specificities of SET domain methyltransferases. Epigenetics. 2011;6:1059–1067. doi: 10.4161/epi.6.9.16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makde RD, England JR, Yennawar HP, Tan S. Structure of RCC1 chromatin factor bound to the nucleosome core particle. 2010;467:562–566. doi: 10.1038/nature09321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makde RD, Tan S. Strategies for crystallizing a chromatin protein in complex with the nucleosome core particle. Anal Biochem. 2013;442:138–145. doi: 10.1016/j.ab.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forsythe E, Achari A, Pusey ML. Trace fluorescent labeling for high-throughput crystallography. Acta Crystallogr D Biol Crystallogr. 2006;62:339–346. doi: 10.1107/S0907444906000813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pusey M, Barcena J, Morris M, Singhal A, Yuan Q, Ng J. Trace fluorescent labeling for protein crystallization. Acta Crystallogr F Struct Biol Commun. 2015;71:806–814. doi: 10.1107/S2053230X15008626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer A, Betzel C, Pusey M. Latest methods of fluorescence-based protein crystal identification. Acta Crystallogr F Struct Biol Commun. 2015;71:121–131. doi: 10.1107/S2053230X15000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heras B, Martin JL. Post-crystallization treatments for improving diffraction quality of protein crystals. Acta Crystallogr D Biol Crystallogr. 2005;61:1173–1180. doi: 10.1107/S0907444905019451. [DOI] [PubMed] [Google Scholar]
- 34.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 35.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McGinty RK, Tan S. Nucleosome Structure and Function. Chem Rev. 2015;115:2255–2273. doi: 10.1021/cr500373h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, et al. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science. 2006;311:856–861. doi: 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- 38.Armache KJ, Garlick JD, Canzio D, Narlikar GJ, Kingston RE. Structural basis of silencing: Sir3 BAH domain in complex with a nucleosome at 3.0 Å resolution. Science. 2011;334:977–982. doi: 10.1126/science.1210915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato H, Jiang J, Zhou BR, Rozendaal M, Feng H, Ghirlando R, et al. A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP-C. Science. 2013;340:1110–1113. doi: 10.1126/science.1235532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McGinty RK, Henrici RC, Tan S. Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. 2014;514:591–596. doi: 10.1038/nature13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou B-R, Jiang J, Feng H, Ghirlando R, Xiao TS, Bai Y. Structural Mechanisms of Nucleosome Recognition by Linker Histones. Molecular Cell. 2015 doi: 10.1016/j.molcel.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gulbis JM, Kelman Z, Hurwitz J, O’Donnell M, Kuriyan J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell. 1996;87:297–306. doi: 10.1016/s0092-8674(00)81347-1. [DOI] [PubMed] [Google Scholar]
- 43.Leung JW, Agarwal P, Canny MD, Gong F, Robison AD, Finkelstein IJ, et al. Nucleosome acidic patch promotes RNF168- and RING1B/BMI1-dependent H2AX and H2A ubiquitination and DNA damage signaling. PLoS Genet. 2014;10:e1004178. doi: 10.1371/journal.pgen.1004178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan S, Kern RC, Selleck W. The pST44 polycistronic expression system for producing protein complexes in Escherichia coli. Protein Expr Purif. 2005;40:385–395. doi: 10.1016/j.pep.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 45.Luger K, Rechsteiner TJ, Richmond TJ. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol Biol. 1999;119:1–16. doi: 10.1385/1-59259-681-9:1. [DOI] [PubMed] [Google Scholar]
- 46.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winkler DD, Luger K, Hieb AR. Quantifying Chromatin-Associated Interactions: The HI-FI System. Methods in Enzymology. 2012;512:243–274. doi: 10.1016/B978-0-12-391940-3.00011-1. [DOI] [PubMed] [Google Scholar]
- 48.Schrodinger,, LLC. The PyMOL Molecular Graphics System, Version 1.4. 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.