

Abstract
Aim:
We sought to define age-associated DNA methylation changes in naive CD4+ T cells.
Materials & methods:
Naive CD4+ T cells were collected from 74 healthy individuals (age 19–66 years), and age-related DNA methylation changes were characterized.
Results:
We identified 11,431 age-associated CpG sites, 57% of which were hypermethylated with age. Hypermethylated sites were enriched in CpG islands and repressive transcription factor binding sites, while hypomethylated sites showed T cell specific enrichment in active enhancers marked by H3K27ac and H3K4me1. Our data emphasize cancer-related DNA methylation changes with age, and also reveal age-associated hypomethylation in immune-related pathways, such as T cell receptor signaling, FCγR-mediated phagocytosis, apoptosis and the mammalian target of rapamycin signaling pathway. The MAPK signaling pathway was hypermethylated with age, consistent with a defective MAPK signaling in aging T cells.
Conclusion:
Age-associated DNA methylation changes may alter regulatory mechanisms and signaling pathways that predispose to autoimmunity.
Keywords: : aging, autoimmunity, CD4+ T cells, epigenetics, EZH2, lupus, methylation, mTOR, PRC2
Understanding human aging only within genomic constrains is impossible, as genetics appears to explain only a small proportion of the observed variation in lifespan and health [1–3]. DNA methylation is arguably a promising complement to the genetic component of aging [4,5]. A growing number of human diseases and normal processes are linked to epigenetic alterations, emphasizing the importance of DNA methylation changes. Methylation changes have been identified as a hallmark of cancer, immunologic and neurologic disorders, as well as aging [6–10]. For example, it has been demonstrated that, using ‘epigenetic clock’, tumor tissues appear to age 40% faster than the corresponding normal tissue [11,12].
The connection between age-associated methylation sites and immunologic functions has been noted, but remains relatively less explored than age–cancer epigenetic similarity. The majority of age-associated DNA methylation studies use whole blood, or unfractionated peripheral blood mononuclear cells (PBMCs) [11,13–16]. Although age-associated DNA methylation changes have been suggested as a multitissue phenomenon, and several studies indeed show high similarity among each other, concerns remain that cell type and composition may significantly affect interpretation of the age-associated DNA methylation changes [10–13,16–22]. Thus, studying purified immune cell subsets may be the most feasible way to investigate age-associated changes and the corresponding immune function alterations.
This study focuses on age-associated changes in DNA methylation of naive CD4+ T cells, a lymphocyte population that differentiates into effector CD4+ T cells after activation by complexes of MHC and antigen [23]. Defects in T cell DNA methylation have been previously described in lupus, an autoimmune disease characterized by autoantibody production against a number of nuclear antigens [24,25]. The prevalence of these autoantibodies increases with age in healthy individuals [25], and T cells from aging individuals have been shown to demethylate and overexpress some of the same genes demethylated and overexpressed in T cells from lupus patients [26]. Furthermore, naive CD4+ T cells have previously been shown to have distinct hypomethylation and epigenetic poising of interferon-regulated genes in lupus [27]. In this study, we focused on characterizing age-sensitive DNA methylation sites in human naive CD4+ T cells, and explore the potential relationship between these age-dependent DNA methylation changes and autoimmunity.
Methods
Study demographics
Seventy four healthy female individuals were included in this study. A subset of these individuals was recruited for a prior study comparing the DNA methylation changes in naive CD4+ T cells of healthy European–Americans and African–Americans (GEO accession: GSE79237) [28]. The age of the participants ranged from 19 to 66 years, with a mean age of 40.8 and a median age of 40. The racial distribution of study participants was as follows: 47 European–Americans, 22 African–Americans, three Asians, one Hispanic and one Indian/Arab. Healthy participants were recruited from the Oklahoma Medical Research Foundation, University of Michigan Health System and Henry Ford Health System. The institutional review boards of the participating institutions approved this study and all participants provided written, informed consent prior to enrollment.
Naive CD4+ T cell isolation & DNA extraction
Naive CD4+ T cells were isolated from peripheral blood as previously described [27,28]. Briefly, PBMCs were isolated from peripheral blood using Ficoll-Paque™ PLUS (GE Healthcare, PA, USA). Naive CD4+ T cells were isolated from PBMCs by indirect labeling using the naive CD4+ T cell Isolation Kit II, human (Miltenyi Biotec, CA, USA). Other cells were bound by biotinylated antibodies and using antibiotin-labeled magnetic beads, bound to a column allowing untouched naive CD4+ T cells to flow through. DNA was isolated from untouched naive CD4+ T cells using the DNeasy Blood and Tissue Kit (Qiagen, MD, USA) according to manufacturer's instructions. Concentration and 260/280 absorbance ratio were measured using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, MA, USA).
Naive CD4+ T cell DNA methylation profiling
Five hundred nanogram of naive CD4+ T-cell DNA was bisulfite-converted using the EZ DNA Methylation™ Kit (Zymo Research, CA, USA) according to manufacturer's instructions. The bisulfite converted DNA was hybridized to Illumina Infinium HumanMethylation450 BeadChip arrays (Illumina, CA, USA) according to manufacturer's instructions. The BeadChip arrays were scanned with the iScan reader (Illumina). All samples passed Illumina array quality control measures.
Preprocessing of methylation data
The DNA methylation data were imported into a MethyLumiSet object using R software with the methylumi v. 2.16.0 package. Probes confounded with array batch (using BeadChip ID number) were removed (n = 1164). Nonspecific (n = 29,155), polymorphic (n = 62,344) and chromosome Y (n = 294) probes were also removed based on best practice recommendations [29]. Background correction and quantile normalization was performed using the dasen method from the wateRmelon v. 1.10.0 package. The batch effect of the Infinium I and II chemistries was adjusted using the BIMQ method [20]. Several visualization strategies provided by the wateRmelon and lumi v. 2.22.1 packages were utilized to ensure the quality of background correction/normalization. The batch effect was confirmed by principal component analysis (PCA) and removed using the ComBat function in the sva v. 3.18.0 R package. Three individuals (2 European–Americans and 1 African–American; mean age 38.6 years) were removed from further analysis at this stage, as ComBat cannot adjust for batch effect in a batch consisting of one sample. The background corrected, normalized and batch effect removed dataset was used for further analysis.
Regression analysis
To evaluate the association of methylation differences with age, a beta regression model was calculated using the betareg v. 3.0.5 R package, along with linear regression and Pearson correlation coefficient approaches. The beta regression model has been shown to be particularly well suited to test associations based on the distribution of methylation β values [30,31]. The model included race, BeadChip ID and sample chip placement as covariates. A Benjamini–Hochberg adjusted p-value threshold of 0.05 was selected as the threshold of statistical significance prior to performing the regression analysis. All subsequent genomic and epigenomic enrichment analyses, epigenomic similarity analysis, and functional enrichment analyses, as described below, were performed using age-dependent DNA methylation changes identified using this regression analysis.
Selection of CpG sites showing substantial age differences
To detect CpG sites showing large change in DNA methylation during aging, the β values were transformed to M values using the equation 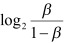 . The median M values between individuals in the higher 75th (n = 18; 53–66 years) and lower 25th (n = 17; 19–32 years) percentile of the age range were compared. CpG sites with |ΔM| >1 were selected [32]. The rank sums of the M values of the two groups were further compared using Wilcoxon test.
. The median M values between individuals in the higher 75th (n = 18; 53–66 years) and lower 25th (n = 17; 19–32 years) percentile of the age range were compared. CpG sites with |ΔM| >1 were selected [32]. The rank sums of the M values of the two groups were further compared using Wilcoxon test.
Genomic & epigenomic enrichment analysis
Positional and epigenomic enrichment analyses were performed as described previously [33]. Briefly, the enrichment analysis evaluates whether a set of age-associated CpG sites colocalizes with genome annotation datasets in a statistically significant manner, utilizing genomic coordinates of the CpG sites and genomic annotations in the hg19/GRCh37 human genome coordinate system. All CpG sites included on the Illumina Infinium 450K array were used as a ‘background’. Genomic coordinates of chromosome bands and transcription factor/regulator binding sites obtained by ChIP-seq from ENCODE (wgEncodeRegTfbsClusteredV3 data table) were obtained from the UCSC genome browser database [34]. Coordinates of gene/transcript types [35] were obtained from annotables R package. Genomic Evolutionary Rate Profiling (GERP) elements [36], CpG islands [37] and Functional Annotation of the Mammalian Genome enhancers [38], as well as chromatin states, 15-mark model, experimentally obtained histone modifications and gapped peaks from the Roadmap Epigenomics project [39] were obtained from the accompanying web sites. The two-tailed chi-square test was used to calculate enriched and depleted associations. While enriched associations imply significant concentration of CpG sites in the tested regions, depleted associations indicate that age-associated CpG sites are devoid of tested regions compared with background. All reported p-values were corrected using Benjamini–Hochberg procedure.
Epigenomic similarity analysis
To compare epigenomic signatures of age-associated CpG sites from different studies, we performed epigenomic similarity meta-analysis as described previously [33]. Briefly, age-associated CpG sites were tested for enrichment in multiple epigenomic annotations, and the corresponding epigenomic enrichment profiles of –log10- transformed p-values were compared using Pearson correlation coefficient and PCA. Experimentally obtained histone modification marks detected with gapped peak algorithm were used. These included H3K4me1, H3K4me3, H3K4me3, H3K9me3, H3K27me3, H3K27ac and H3K36me3. This analysis allows comparing epigenomic enrichments of age-associated CpG sites in the context of reference genome annotations.
Gene set functional enrichment analysis
Genes annotated with CpG sites hypermethylated or hypomethylated with age were tested for enrichment using ‘molecular function’, ‘biological process’ and ‘cellular component’ gene ontology annotations, and Kyoto Encyclopedia of Genes and Genomes canonical pathways [40]. Enrichment p-values were calculated using chi-square test in the GOStats v2.36.0 R package. The p-values were corrected for multiple testing using the Benjamini–Hochberg procedure.
Results
Naive CD4+ T-cell-specific age-associated methylation changes
DNA methylation profiles generated from naive CD4+ T cells of 71 healthy women (age range: 19–66 years) were included in our analysis. Figure 1 shows our analysis pipeline to characterize age-associated DNA methylation changes. PCA of the DNA methylation profiles identified the first principle component to be correlated with batch ID (PC1 accounting for 10.48% variability, p = 3.10 × 10-27; Supplementary Figure 1), suggesting technical batch effect. Although other parameters, such as race and sample chip placement, did not significantly contribute to variability of the data (not shown), all three variables including bath ID, sample chip placement and race, were included in the regression model as covariates in all analyses.
Figure 1. . Analysis pipeline used to identify and characterize age-associated DNA methylation changes in naive CD4+ T cells.

Age-associated CpG sites are predominantly hypermethylated in naive CD4+ T cells
Our approach was two sided in that we aimed to identify CpG sites significantly hypermethylated or hypomethylated with age. A total of 11,431 CpG sites were identified as having methylation values with a significant association with age (Supplementary Table 1). Fifty six of these CpG sites were previously identified as a part of the 353 CpG sites defining an ‘age–epigenetic clock’ signature [12], which was statistically significant (p = 1.09 × 10-24, Fisher's exact test). Furthermore, 300 CpG sites identified in our study were a part of the 6366 CpG sites signature of increased variability with age, as identified in the recent study by Slieker et al. [41] (p = 2.06 × 10-15, Fisher's exact test).
A total of 6533 (57%) CpG sites were positively correlated (hypermethylated) with age compared with 4898 (43%) negatively correlated (hypomethylated) sites (Supplementary Table 1). Table 1 lists the top 20 most robust CpG sites associated with age (as measured by r2 value) observed in this study. In order to identify CpG sites showing the largest methylation differences at the extremes of age range, we compared M values between two groups of individuals in the lower 25th and upper 75th percentiles of the total age range, 56 CpG sites had a ΔM >1 and Benjamini–Hochberg corrected Wilcoxon p < 0.05 (Supplementary Table 2).
Table 1. . Top 20 CpG sites with DNA methylation levels that correlate with age in naive CD4+ T cells using beta regression analysis.
| CpG site | Age coefficient | Regression p-value | r2 | Pearson correlation | Pearson correlation p-value | Gene symbol |
|---|---|---|---|---|---|---|
| cg16867657 |
0.01470 |
2.42 × 10-25 |
0.65402 |
0.80879 |
0.00 × 1000 |
ELOVL2 |
| cg18898125 |
0.00870 |
8.75 × 10-18 |
0.57144 |
0.76013 |
1.51 × 10-14 |
NEFM |
| cg04212239 |
0.01685 |
9.29 × 10-15 |
0.54095 |
0.74173 |
1.37 × 10-13 |
SMC4 |
| cg19283806 |
-0.01833 |
1.64 × 10-14 |
0.53900 |
-0.72976 |
5.23 × 10-13 |
CCDC102B |
| cg00474657 |
-0.01392 |
7.36 × 10-14 |
0.52064 |
-0.72892 |
5.73 × 10-13 |
SPNS1 |
| cg01697487 |
-0.02314 |
7.36 × 10-14 |
0.52550 |
-0.72819 |
6.20 × 10-13 |
MYCBPAP |
| cg15353603 |
-0.02453 |
7.36 × 10-14 |
0.52392 |
-0.73051 |
4.82 × 10-13 |
DPP4 |
| cg24724428 |
0.01238 |
7.36 × 10-14 |
0.52689 |
0.72268 |
1.12 × 10-12 |
ELOVL2 |
| cg01409343 |
-0.02258 |
7.76 × 10-14 |
0.51800 |
-0.72437 |
9.35 × 10-13 |
TMEM49 |
| cg05762671 |
-0.02160 |
1.56 × 10-13 |
0.51405 |
-0.71969 |
1.53 × 10-12 |
KCTD19 |
| cg02202664 |
-0.02398 |
5.05 × 10-13 |
0.51321 |
-0.71588 |
2.27 × 10-12 |
TIAM2 |
| cg10701110 |
-0.01065 |
5.21 × 10-13 |
0.50237 |
-0.71352 |
2.89 × 10-12 |
ARHGAP30, USF1 |
| cg07642566 |
0.01136 |
5.96 × 10-13 |
0.50347 |
0.70857 |
4.75 × 10-12 |
C19orf30, MIR7–3 |
| cg14377791 |
-0.02071 |
7.60 × 10-13 |
0.51007 |
-0.70851 |
4.78 × 10-12 |
LOC400696 |
| cg17851795 |
-0.01366 |
9.67 × 10-13 |
0.50167 |
-0.71006 |
4.10 × 10-12 |
GPSM3, PBX2 |
| cg13029847 |
0.01120 |
1.07 × 10-12 |
0.50039 |
0.70446 |
7.13 × 10-12 |
SEZ6 |
| cg23003085 |
-0.01710 |
1.21 × 10-12 |
0.50237 |
-0.70558 |
6.39 × 10-12 |
PRDX5, TRMT112 |
| cg03917666 |
-0.02410 |
1.79 × 10-12 |
0.49840 |
-0.70761 |
5.23 × 10-12 |
|
| cg21915449 |
-0.01651 |
4.46 × 10-12 |
0.48747 |
-0.70049 |
1.05 × 10-11 |
HAVCR1 |
| cg15953461 | -0.01524 | 6.91 × 10-12 | 0.48626 | -0.69800 | 1.33 × 10-11 |
CpG site: Unique CpG locus identifier; Age coefficient: Age correlation coefficient, 1 year of age change corresponds to coefficient value change in methylation; Regression p-value: significance of the regression coefficient; r2: Percent of variance explained by the regression fit; Pearson correlation: Pearson correlation coefficient between age and methylation level; Pearson correlation p-value: Significance of the correlation.
CpG sites associated with age differ in their genomic location & regulatory context
We sought to investigate previously reported observations that CpG sites hypermethylated or hypomethylated with age are located in distinct genomic compartments and enriched in different epigenomic elements [13,14,42–44]. To identify chromosomal regions with unusually high concentration of age-associated CpG sites, we performed enrichment analysis of CpG sites on chromosomes and chromosomal bands (Figure 2). CpG sites hypermethylated with age were enriched on chromosome 2q36.2 (p = 1.55 × 10-30), 2q24.1 (p = 8.69 × 10-19), 2q31.1 (p = 5.19 × 10-14), 6q16.3 (p = 2.51 × 10-12) and several other bands. They were also depleted on chromosome 6p21.33 (p = 3.40 × 10-09), 1p36.32 (p = 4.62 × 10-04) and others (Supplementary Table 3). On the contrary, CpG sites hypomethylated with age were enriched on chromosome 6p21.32 (p = 1.52 × 10-14), 6p21.33 (p = 2.81 × 10-10), 17q23.1 (p = 1.52 × 10-13), 3p21.2 (p = 4.81 × 10-13), and depleted on chromosome 12q24.33 (p = 9.66 × 10-04) (Supplementary Table 3). Expectedly, some enrichments were mutually exclusive, for example, CpG sites hypermethylated with age were strongly depleted in the 6p21.33 chromosomal band (p = 3.40 × 10-09), while CpG sites hypomethylated with age were enriched in this band (p = 2.81 × 10-10). These results confirm our hypothesis that age-associated hypermethylated and hypomethylated events may involve different genomic compartments.
Figure 2. . Genomic colocalization of naive CD4+ age-associated CpG methylation sites in chromosomal bands.

Significant enrichments (red bars) and depletions (green bars) for CpG sites hypermethylated (A & B) and hypomethylated (C & D) with age are shown.
Y-axis represents -log10-transformed p-values obtained using chi-squared test.
The differential localization of CpG sites hypermethylated or hypomethylated with age has been observed in the meta-analysis of multiple independent studies [17]. To compare genomic location and the corresponding epigenomic signatures of the age-associated CpG sites detected in our study with other studies, we performed epigenomic similarity analysis of our hypermethylated or hypomethylated age-associated sites and those identified in six other studies [13,14,19,42,43,45]. Expectedly, CpG sites hypermethylated or hypomethylated with age are grouped into distinct regulatory clusters (Figure 3). PCA further confirmed this observation, with the age-correlation accounting for 86.06% variability explained by the first principle component (p = 6.78 × 10-05) (Supplementary Figure 2). Our list of CpG sites hypomethylated with age expectedly showed high regulatory similarity with those identified in CD4+ T cells by Reynolds et al. [42] (Pearson coefficient = 0.82, p < 1.00 × 10-16), indicating that the location and regulatory impact of age-associated CpG sites identified in our study in naive CD4+ T cells are most similar to those previously identified in total CD4+ T cells (Figure 3).
Figure 3. . Epigenomic similarities among sets of CpG sites hypermethylated and hypomethylated with age identified in previous studies, compared with one another and to this study.

Set-specific epigenomic enrichment profiles were obtained using Roadmap Epigenomics histone modification data, and pair-wise correlated using Pearson correlation coefficient. The resulting correlation matrix was clustered using Euclidean distance/ward.D clustering metrics. A color gradient from blue to red indicates Pearson correlation coefficient values, with blue being -1 and red being 1. Groups of hypermethylated and hypomethylated sets of sites are marked with purple and green color of the dendrogram and the side bars, respectively. The following studies were compared (Study name, number of hypermethylated sites, number of hypomethylated sites, tissue types used): Alisch et al. (2012), 479 hypermethylated sites, 1599 hypomethylated sites, peripheral blood cells; Marttila et al. (2015), 3925 hypermethylated sites, 4615 hypomethylated sites, peripheral blood mononuclear cells; Weidner et al. (2014), 58 hypermethylated sites, 44 hypomethylated sites, whole blood; Reynolds et al. (2014), 2049 hypermethylated sites, 546 hypomethylated sites, CD4+ T cells; Heyn et al. (2012), 1219 hypermethylated sites, 1986 hypomethylated sites, cord blood of newborns and CD4+ T cells of centenarians; and Florath et al. (2012), 119 hypermethylated sites, 43 hypomethylated sites, whole blood. Data taken from [13,14,19,42,43,45].
Hypermethylated, but not hypomethylated age-associated CpG sites are enriched in CpG islands & conserved regions
Regulatory enrichment analysis identified CpG sites hypermethylated with age as enriched in known CpG islands (p = 6.15 × 10-04) and evolutionary conserved GERP elements (p = 2.38 × 10-36). CpG sites hypomethylated with age were significantly depleted in CpG islands (p = 1.89 × 10-287) and GERP elements (p = 5.16 × 10-13).
Cell type specific enrichment of CpG sites hypomethylated with age
Enrichment analysis of age-associated CpG sites for overlap with known classes of genes and transcripts [35], identified hypermethylated CpG sites to be enriched in protein-coding genes (p = 6.26 × 10-04). CpG sites hypomethylated with age were enriched in cell type specific genes, ‘T-cell J receptor gene’ (p = 8.50 × 10-23) and ‘all T-cell receptor genes combined’ (p = 6.12 × 10-08). Both CpG sites hypermethylated and hypomethylated with age were depleted in ‘pseudogene’ transcript types (p = 8.13 × 10-11 and p = 1.83 × 10-12, respectively). These results suggest that CpG sites hypermethylated with age may regulate noncell type specific expression of protein-coding genes, while CpG sites hypomethylated with age regulate T cell specific gene expression activity.
Hypermethylated CpGs are associated with PRC2, while hypomethylated CpGs are enriched in enhancers
The different types of enhancer regions enriched in CpG sites hypermethylated or hypomethylated with age were also observed in the histone mark enrichment analysis focused on previously obtained histone mark ChIP-seq data in primary naive CD4+ T cells [39]. CpG sites hypermethylated with age were enriched in H3K27me3 (p = 1.06 × 10-305), a mark of polycomb repressive complex II (PRC2) binding, and H3K4me1 (p = 1.06 × 10-305), a classical activating mark, that together with H3K27me3, marks bivalent promoters [46]. These bivalent promoters, repressed in the absence of differentiation signals, are considered to regulate temporal expression of developmental genes. CpG sites hypomethylated with age in naive CD4+ T cells were enriched in H3K4me1 (p = 2.44 × 10-228) and H3K27ac (p = 6.96 × 10-207), a mark of active enhancers (Supplementary Table 3).
EZH2 & SUZ12 binding sites are enriched in CpGs hypermethylated with age
We investigated whether age-associated CpG sites are enriched in binding sites of transcription factors and chromatin remodeling complexes. Binding sites of two core proteins of PRC2 complex, EZH2 (p = 1.61 × 10-304) and SUZ12 (p = 5.73 × 10-181) were over-represented in hypermethylated CpG sites, but the opposite trend was observed in CpG sites hypomethylated with age (under-represented EZH2 (p = 4.37 × 10-51) and SUZ12 (p = 6.13 × 10-20). Intriguingly, enrichment analysis of the recently released EED binding sites, another core protein of PRC2 complex, identified EED as being enriched in both hypermethylated and hypomethylated CpG sites (p < 3.00 × 10-39, Supplementary Table 3).
Among transcription factor binding sites, CpG sites hypomethylated with age were also enriched in BATF (p = 4.1 × 10-159), a negative regulator of AP-1 mediated signaling [47], and IKZF1 (p = 1.09 × 10-101) and BCL11A (p = 1.81 × 10-67), transcription factors important in lymphocyte development [48–50]. Also enriched was IRF4 (p = 1.37 × 10-45), a lymphocyte-specific transcription factor that negatively regulates Toll-like receptor signaling. CpG sites hypermethylated or hypomethylated with age were depleted in PHF8 and KDM5B transcription factor binding sites (PHF8 p = 5.54 × 10-84 and 1.47 × 10-25, KDM5B p = 1.06 × 10-52 and 1.03 × 10-16 for hypermethylated and hypomethylated age-associated CpG sites, respectively). Both transcription factors are members of the Jumonji family of proteins that play a role in chromatin remodeling and histone demethylation [51,52] (Figure 4 & Supplementary Table 3).
Figure 4. . Genomic colocalization of naive CD4+ T cell age-associated CpG methylation sites in transcription factor binding sites.

Significant enrichments (red bars) and depletions (green bars) for CpG sites hypermethylated (A & B) and hypomethylated (C & D) with age are shown.
Y-axis represents -log10-transformed p-values obtained using chi-squared test.
Functional enrichments of genes annotated with age-associated CpG sites
A total of 3183 and 3022 unique genes were annotated with CpG sites hypermethylated and hypomethylated with age, respectively (Table 2 & Supplementary Table 4). Genes associated with CpG sites hypermethylated with age were enriched in ‘Hedgehog signaling pathway’ (p 1.10 × 10-05) (Table 3), downregulation of which has been associated with age-related diseases [53]. Other pathways included ‘pathways in cancer’ (p = 1.10 × 10-05), ‘focal adhesion’ (p = 8.35 × 10-04), ‘Wnt signaling pathway’ (p = 8.35 × 10-04) and ‘MAPK signaling pathway’ (p = 4.35 × 10-03) (Table 3 & Supplementary Table 5). Genes associated with CpG sites hypomethylated with age were enriched in two metabolism-related pathways, ‘insulin signaling pathway’ (p = 4.94 × 10-04) and ‘mammalian target of rapamycin (mTOR) signaling pathway’ (p = 2.95 × 10-03), (Table 4). Further reflecting immune links of age-associated methylation profiles, ‘Fc gamma R-mediated phagocytosis’ (p = 6.37 × 10-04) and ‘T cell receptor signaling pathway’ (p = 8.41 × 10-04) were also enriched in genes associated with hypomethylated CpGs. Cancer-related pathways, such as ‘small cell lung cancer’ (p = 8.41 × 10-04), ‘acute myeloid leukemia’ (p = 8.41 × 10-04) and ‘apoptosis’ (p = 8.41 × 10-04) were also among the top most significant hypomethylated pathways (Table 4 & Supplementary Table 5).
Table 2. . Top 20 genes with the largest number of CpG sites hypermethylated or hypomethylated with age in naive CD4+ T cells.
|
Genes annotated with CpG sites hypermethylated with age | ||
|---|---|---|
| Gene symbol | Number of CpG probes | Description |
|
PRRT1 |
22 |
Proline-rich transmembrane protein 1 |
|
MAD1L1 |
21 |
MAD1 mitotic arrest deficient-like 1 (yeast) |
|
MSI2 |
14 |
Musashi RNA-binding protein 2 |
|
KCNQ1 |
13 |
Potassium channel, voltage gated KQT-like subfamily Q, member 1 |
|
SOX2OT |
12 |
SOX2 overlapping transcript |
|
PITX2 |
11 |
Paired-like homeodomain 2 |
|
REC8 |
11 |
REC8 meiotic recombination protein |
|
BLCAP |
10 |
Bladder cancer associated protein |
|
FAM124B |
10 |
Family with sequence similarity 124B |
|
GNA12 |
10 |
Guanine nucleotide binding protein (G protein) alpha 12 |
|
JARID2 |
10 |
Jumonji, AT rich interactive domain 2 |
|
NNAT |
10 |
Neuronatin |
|
NR4A2 |
10 |
Nuclear receptor subfamily 4, group A, member 2 |
|
GALNT2 |
9 |
Polypeptide N-acetylgalactosaminyltransferase 2 |
|
SYNGAP1 |
9 |
Synaptic Ras GTPase activating protein 1 |
|
ZIC1 |
9 |
Zic family member 1 |
|
C10orf26 |
8 |
WW domain binding protein 1-like |
|
FOXG1 |
8 |
Forkhead box G1 |
|
GIPR |
8 |
Gastric inhibitory polypeptide receptor |
|
H2AFY |
8 |
H2A histone family, member Y |
|
Genes annotated with CpG sites hypomethylated with age | ||
|
Gene symbol
|
Number of CpG probes
|
Description
|
|
IFT140 |
15 |
Intraflagellar transport 140 |
|
PRDM16 |
15 |
PR domain containing 16 |
|
KIAA1949 |
12 |
Protein phosphatase 1 regulatory subunit 18 |
|
RASA3 |
12 |
RAS p21 protein activator 3 |
|
RPTOR |
12 |
Regulatory associated protein of MTOR, complex 1 |
|
DAXX |
11 |
Death-domain associated protein |
|
ZC3H12D |
9 |
Zinc finger CCCH-type containing 12D |
|
B3GALT4 |
8 |
UDP-Gal:betaGlcNAc beta 1,3-galactosyltransferase, polypeptide 4 |
|
EHMT2 |
8 |
Euchromatic histone-lysine N-methyltransferase 2 |
|
KALRN |
8 |
Kalirin, RhoGEF kinase |
|
TAP1 |
8 |
Transporter 1, ATP-binding cassette, sub-family B (MDR/TAP) |
|
TMEM49 |
8 |
Vacuole membrane protein 1 |
|
PLEC1 |
7 |
Plectin |
|
SKI |
7 |
SKI proto-oncogene |
|
ABHD14A |
6 |
Abhydrolase domain containing 14A |
|
ABHD14B |
6 |
Abhydrolase domain containing 14B |
|
CISH |
6 |
Cytokine inducible SH2-containing protein |
|
CUTA |
6 |
cutA divalent cation tolerance homolog (Escherichia coli) |
|
DGKZ |
6 |
Diacylglycerol kinase, ζ |
| HCCA2 | 6 | MOB kinase activator 2 |
Table 3. . Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis of genes annotated with CpG sites hypermethylated with age in naive CD4+ T cells.
| Kyoto Encyclopedia of Genes and Genomes pathway | FDR-adjusted p-value | Gene symbol |
|---|---|---|
| Hedgehog signaling pathway |
1.10 × 10-05 |
BMP4, ZIC2, CSNK1E, WNT10A, WNT5B, GLI3, WNT6, BMP2, BMP7, BMP8B, BTRC, CSNK1A1, FBXW11, GAS1, GLI1, LRP2, PRKACA, PRKACG, WNT1, WNT2B, WNT3, WNT5A, WNT7A, WNT9A |
| Pathways in cancer |
1.10 × 10-05 |
SPI1, CDK6, FGF17, BMP4, MECOM, PDGFB, AXIN2, CTNNA2, IGF1R, TRAF3, WNT10A, WNT5B, ZBTB16, AKT3, BCL2, BRCA2, CDKN1A, CSF3R, EPAS1, FGF2, GLI3, GSTP1, JUP, LAMC1, RAC1, RARA, RXRG, SMAD3, WNT6, ABL1, APPL1, AXIN1, BCL2L1, BCR, BIRC3, BMP2, CDKN2A, EGLN3, FGF10, FGF19, FGF9, FN1, FOS, FZD6, GLI1, GRB2, HDAC1, ITGA6, JAK1, KIT, KRAS, LAMA1, LAMC2, MAP2K1, MITF, MLH1, MMP9, NKX3–1, NTRK1, PAX8, PIAS1, PIK3R3, PTEN, RALB, RALGDS, SMAD2, STAT1, STAT3, STAT5A, TCF7L1, TFG, TGFBR1, TGFBR2, TPM3, TPR, VHL, WNT1, WNT2B, WNT3, WNT5A, WNT7A, WNT9A |
| Adherens junction |
1.27 × 10-04 |
TJP1, CTNND1, CTNNA2, IGF1R, LMO7, RAC1, SMAD3, ACTN1, ACTN2, ACTN4, BAIAP2, CSNK2A1, FYN, IQGAP1, NLK, PTPRJ, PVRL1, SMAD2, SNAI1, SORBS1, SRC, TCF7L1, TGFBR1, TGFBR2, WAS, WASL |
| Maturity onset diabetes of the young |
8.35 × 10-04 |
NR5A2, NKX6–1, NEUROD1, PAX6, HES1, MAFA, ONECUT1, GCK, HHEX, HNF1B, NEUROG3, NKX2–2 |
| Focal adhesion |
8.35 × 10-04 |
PIP5K1C, PDGFB, IGF1R, PARVA, AKT3, BCL2, CCND3, FLNC, LAMC1, PAK6, PARVG, PPP1CC, RAC1, THBS2, VTN, ACTN1, ACTN2, ACTN4, BCAR1, BIRC3, CAPN2, CAV1, COL11A2, COL5A2, COL6A3, COL6A6, DOCK1, FLT1, FLT4, FN1, FYN, GRB2, ITGA10, ITGA11, ITGA6, ITGA8, ITGB5, LAMA1, LAMC2, MAP2K1, PAK2, PDPK1, PIK3R3, PPP1CA, PTEN, SRC, THBS1, THBS4, TNXB, VAV3 |
| Wnt signaling pathway |
8.35 × 10-04 |
LRP5, AXIN2, CSNK1E, DKK4, NKD1, PPP2CA, SFRP2, SOX17, WNT10A, WNT5B, CAMK2D, CCND3, CTNNBIP1, NFATC1, NFATC2, RAC1, SMAD3, WNT6, AXIN1, BTRC, CSNK1A1, CSNK2A1, DKK2, FBXW11, FRAT1, FZD6, NLK, PPP2R5A, PPP3R1, PRKACA, PRKACG, SMAD2, TBL1XR1, TCF7L1, WNT1, WNT2B, WNT3, WNT5A, WNT7A, WNT9A |
| Regulation of actin cytoskeleton |
3.28 × 10-03 |
GNA12, ARHGEF7, FGF17, PIP5K1C, PDGFB, CHRM2, EZR, FGF2, GSN, ITGAE, ITGAM, MSN, PAK6, PPP1CC, RAC1, ACTN1, ACTN2, ACTN4, BAIAP2, BCAR1, CD14, CHRM4, CHRM5, CSK, DIAPH3, DOCK1, FGF10, FGF19, FGF9, FN1, INSRR, IQGAP1, ITGA10, ITGA11, ITGA6, ITGA8, ITGB5, KRAS, MAP2K1, MYH10, PAK2, PFN3, PIK3R3, PPP1CA, SLC9A1, SSH1, TIAM1, VAV3, WAS, WASL |
| Arrhythmogenic right ventricular cardiomyopathy |
4.17 × 10-03 |
CACNG8, CTNNA2, DSP, JUP, ACTN1, ACTN2, ACTN4, CACNA2D1, CACNA2D2, CACNA2D4, CACNB2, CACNG2, CACNG3, CACNG4, ITGA10, ITGA11, ITGA6, ITGA8, ITGB5, LMNA, SGCD, TCF7L1 |
| Neuroactive ligand-receptor interaction |
4.17 × 10-03 |
GIPR, GRM2, CNR2, GRIK5, HTR6, P2RX1, GALR2, GRIK2, NR3C1, PTGDR, TSHR, ADORA3, CHRM2, CHRNB4, EDNRB, F2RL1, GLRA1, GRIN2D, GRM5, HTR1E, PRLHR, SSTR3, ADORA2B, ADRA1A, ADRB1, ADRB3, AVPR1A, CALCRL, CHRM4, CHRM5, CHRNA5, CNR1, DRD2, GABRA1, GABRA2, GABRB3, GABRG2, GABRQ, GABRR2, GLRA3, GRIA2, GRIN1, GRM6, GRM8, HTR2B, HTR4, HTR5A, LEP, LEPR, MCHR2, MLNR, NPY1R, NPY5R, NTSR2, OPRL1, P2RY1, SSTR2, TACR3, TBXA2R, VIPR2 |
| MAPK signaling pathway | 4.35 × 10-03 | GNA12, CACNA1I, RPS6KA2, FGF17, MAP3K8, CACNG8, MECOM, PDGFB, MAP4K4, AKT3, CACNA1G, DUSP14, DUSP6, FGF2, FLNC, NFATC2, RAC1, RASGRP2, CACNA1B, CACNA2D1, CACNA2D2, CACNA2D4, CACNB2, CACNG2, CACNG3, CACNG4, CD14, DUSP4, DUSP8, ECSIT, FGF10, FGF19, FGF9, FOS, GRB2, HSPA2, KRAS, MAP2K1, MAP2K4, MAP2K5, MAP2K6, MAP3K3, MAP3K5, MAP3K6, MAPK14, MAPK8IP1, MAPKAPK2, MAPKAPK3, NLK, NTRK1, PAK2, PPP3R1, PRKACA, PRKACG, RASGRP4, RPS6KA4, TGFBR1, TGFBR2, ZAK* |
FDR: False discovery rate.
Table 4. . Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis of genes annotated with CpG sites hypomethylated with age in naive CD4+ T cells.
| Kyoto Encyclopedia of Genes and Genomes pathway | FDR-adjusted p-value | Gene symbol |
|---|---|---|
| Insulin signaling pathway |
4.94 × 10-04 |
RPTOR, BAD, PDE3B, PIK3CD, MAPK10, SREBF1, AKT1, FLOT1, G6PC, PIK3R1, PIK3R2, PRKAG2, PYGM, AKT3, CBL, CRKL, EIF4E, EIF4E2, EIF4EBP1, FASN, FLOT2, GYS1, INPP5D, INS, IRS1, MKNK2, PHKG2, PIK3CA, PIK3R5, PPP1CA, PPP1R3B, PRKAR2B, PRKCI, PTPN1, PTPRF, RAPGEF1, RPS6KB1, RPS6KB2, SOCS1, SOCS2, SORBS1, TSC2 |
| Fc gamma R-mediated phagocytosis |
6.37 × 10-04 |
PRKCA, PIK3CD, AKT1, ARPC4, DNM1, LAT, PIK3R1, PIK3R2, AKT3, ASAP2, ASAP3, CRKL, DNM3, INPP5D, LYN, MARCKSL1, MYO10, PIK3CA, PIK3R5, PIP5K1A, PIP5K1B, PIP5K1C, PLA2G4D, PLCG2, PRKCB, RAC2, RPS6KB1, RPS6KB2, WAS, WASF2, WASF3 |
| Small cell lung cancer |
8.41 × 10-04 |
RXRB, PIK3CD, AKT1, COL4A1, PIK3R1, PIK3R2, TRAF4, TRAF5, AKT3, BCL2, BIRC2, CCND1, CDK2, E2F2, IKBKG, LAMA3, LAMA4, LAMA5, LAMB4, NFKB1, NFKBIA, PIAS3, PIK3CA, PIK3R5, PTK2, RXRA, TRAF1, TRAF2 |
| T cell receptor signaling pathway |
8.41 × 10-04 |
PTPN6, TNF, PIK3CD, MAP3K14, AKT1, CARD11, CD4, FYN, LAT, PIK3R1, PIK3R2, ZAP70, AKT3, CBL, CD28, CD40LG, CTLA4, GRAP2, IKBKG, IL5, ITK, MAP2K7, MAPK12, NFATC1, NFKB1, NFKBIA, PAK6, PDCD1, PIK3CA, PIK3R5, PRKCQ, RASGRP1, RHOA |
| Acute myeloid leukemia |
8.41 × 10-04 |
BAD, PIK3CD, STAT5A, AKT1, PIK3R1, PIK3R2, PIM2, PPARD, AKT3, CCND1, EIF4EBP1, IKBKG, NFKB1, PIK3CA, PIK3R5, PIM1, RPS6KB1, RPS6KB2, STAT3, STAT5B, TCF7 |
| Apoptosis |
8.41 × 10-04 |
TNF, BAD, PIK3CD, CAPN2, MAP3K14, AKT1, BID, IRAK4, PIK3R1, PIK3R2, AKT3, ATM, BCL2, BIRC2, CASP7, CASP8, CFLAR, ENDOG, IKBKG, NFKB1, NFKBIA, NGF, NTRK1, PIK3CA, PIK3R5, PRKAR2B, TRADD, TRAF2 |
| Toxoplasmosis |
1.43 × 10-03 |
TNF, BAD, PIK3CD, HLA-DOA, MAPK10, AKT1, HLA-DPB1, HSPA1L, IRAK4, MAP2K3, PIK3R1, PIK3R2, AKT3, BCL2, BIRC2, CASP8, CCR5, CD40LG, GNAI2, HLA-DMB, IKBKG, IL10RA, IL12A, JAK1, LAMA3, LAMA4, LAMA5, LAMB4, LDLR, MAPK12, NFKB1, NFKBIA, PIK3CA, PIK3R5, PLA2G12B, SOCS1, STAT3 |
| Phosphatidylinositol signaling system |
1.43 × 10-03 |
DGKZ, PRKCA, PIK3CD, INPP4B, PI4KB, PIK3R1, PIK3R2, CDS2, DGKI, INPP4A, INPP5A, INPP5D, INPP5E, ITPK1, ITPR2, PIK3C2G, PIK3CA, PIK3R5, PIP5K1A, PIP5K1B, PIP5K1C, PLCG2, PLCZ1, PRKCB, SYNJ2 |
| Endocytosis |
1.43 × 10-03 |
ARAP1, HLA-F, IGF1R, SMURF1, EHD1, ERBB3, HLA-E, AP2A2, DNM1, EHD2, HLA-B, HSPA1L, IL2RB, IQSEC3, SMAP1, ACAP1, ADRBK1, AGAP2, ARAP3, ASAP2, ASAP3, CBL, CCR5, CHMP6, DNM3, FGFR2, GRK4, GRK6, HGS, HLA-A, HLA-C, IL2RA, LDLR, MET, NEDD4L, NTRK1, PARD6A, PDCD6IP, PIP5K1A, PIP5K1B, PIP5K1C, PRKCI, PSD, PSD4, RAB7A, RABEP1, RHOA, SH3GL1, SMAD6, SRC, TFRC |
| mTOR signaling pathway | 2.95 × 10-03 | RPTOR, PIK3CD, AKT1, PIK3R1, PIK3R2, AKT3, CAB39, EIF4E, EIF4E2, EIF4EBP1, INS, PIK3CA, PIK3R5, RPS6KA1, RPS6KB1, RPS6KB2, TSC2, VEGFA* |
FDR: False discovery rate; mTOR: Mammalian target of rapamycin.
Discussion
The biological process of aging can be conceptualized as the interplay of genetics and intrinsic and extrinsic stress experienced by an individual beginning at conception and continuing throughout the lifespan [54]. These exposures accumulate across time and are reflected by the phenomenon of ‘epigenetic drift’ by which the human methylome can vary widely between individuals, even identical twins, as age increases [55]. In this study, we focus on human naive CD4+ T cells and identify age-associated DNA methylation changes and their relationship to cellular functions.
We observed that over half of the age-sensitive methylation sites identified by our study increased in methylation with age. This observation is consistent with other studies that report similar proportions of the age-associated CpG sites [13,14,42,43], although general tendency toward global age-associated hypomethylation has been noted [43,44]. These hypermethylated CpG sites were enriched in CpG islands and evolutionarily conserved regions of the human genome. CpG islands are CpG-rich homogeneous regions of the genome that are associated with transcriptional regulation [56]. Consistent with previous observations, our results support the notion that CpG sites hypermethylated with age are enriched in PRC2 signature, while hypomethylated CpGs are enriched in enhancers [17].
Consistent with the H3K27me3 enrichment, we identified an enrichment of binding sites of EZH2 and SUZ12, two members of PRC2 complex, as being enriched in CpG sites hypermethylated with age. Conversely, CpG sites hypomethylated with age were significantly depleted in EZH2 and SUZ12. Although it is tempting to speculate that the entire PRC2 complex preferentially binds at CpG sites hypermethylated with age, we observed enrichment of EED, another component of the PRC2 complex, in CpG sites both hypermethylated and hypomethylated with age. A prior study by our group suggests that EZH2 might play a role in epigenetic remodeling in naive CD4+ T cells that favors T cell activation, and might predispose to lupus flares [57]. Our observations warrant further studies of the role of PRC2 in autoimmunity, focusing on the complex interplay of the PRC2 components.
Despite this contrasting difference in EZH2 and SUZ12 binding sites enrichment, CpG sites hypermethylated or hypomethylated with age were similarly depleted in PHF8 and KDM5B transcription factor binding sites. Both PHF8 and KDM5B are members of the Jumonji family involved in histone demethylation and cell cycle progression [51,52]. This observation positions Jumonji family of proteins as ‘protected’ from age-associated methylation changes, suggesting their binding sites are preserved during aging. These results suggest that age-associated methylation differences affect distinct transcription factor binding site signatures, while methylation status of cell cycle and Jumonji family binding sites are largely unaffected during aging.
We localized the areas of methylation associated with aging to specific chromosomal bands. Chromosomal regions enriched with CpG sites of increasing DNA methylation levels with age included the top five most significant regions: 2q36.2, 2q24.1, 2q31.1, 6q16.3 and 3q24. These regions have been previously associated with aging or age-associated disorders like Alzheimer's disease and hearing loss in genome-wide association studies [58–61]. Chromosomal regions enriched with DNA methylation sites that decrease in methylation levels with age include: 6p21.32, 17q23.1, 3p21.2, 6p21.33 and 11q13.1. These regions have been associated with disease like amyotrophic lateral sclerosis, and immune-mediated diseases, such as multiple sclerosis and Crohn's disease [62–65]. These results suggest that CpG sites hypermethylated and hypomethylated with age are concentrated in distinct genomic locations, and might regulate functionally different genes by altering distinct epigenomic signatures.
To identify functional networks related to age-sensitive CpG sites, we first identified genes proximal to these sites. Notably, our results identified age-associated hypermethylation in the ELOVL2 gene, which encodes a protein involved in the synthesis of long polyunsaturated fatty acids, and previously reported to be strongly associated with age [14,18,42]. ELOVL2 methylation has been shown to be a biomarker for aging in whole blood by Garagnani et al. [66], particularly site cg16867657, which had the strongest association with age in our study (Pearson coefficient = 0.81). We observed a second CpG site associated with ELOVL2 (cg24724428) with a slightly lower, but significant association with age (Pearson coefficient = 0.72). A recent study of Elovl2 (-/-) mice has shown increased production of the inflammatory cytokines IFN-γ and IL-17, a sign of increased Th1 and Th17 activity, and a reduction in the number of Foxp3+ Treg cells compared with wild-type mice [67]. As hypermethylation is generally associated with transcriptional silencing, progressive age-dependent hypermethylation of ELOVL2 in naive CD4+ T cells might suggest a proinflammatory T-cell epigenotype with age. Other genes, such as APOE, FOXO3, NEFM, CCDC102B, MBP and CAPN2 have also been reported as age-associated in different cell- and tissue types (Supplementary Table 1) [14,18,42,68]. These observations suggest that some genes may show age-associated methylation changes across multiple tissues, including naive CD4+ T cells. The functional role of these methylation changes in T-cell maturation and activity and how it changes with age remains to be investigated.
Gene ontology and pathway analyses of genes near or containing age-sensitive CpG sites revealed several age-related functional groups. Hedgehog signaling pathway genes were enriched for CpG sites hypermethylated with age as well as signaling pathways for MAPK and the Wnt pathways. Gene ontologies for RNA polymerase II binding sites and system and structural development pathways were also enriched for in these sites. In contrast, CpG sites hypomethylated with age were enriched for pathways related to T-cell activity including T cell receptor, mTOR and chemokine signaling, as well as insulin signaling and apoptosis. Gene ontologies were enriched for cellular responses and immune regulation.
The age-associated methylation pattern we observed in naive CD4+ T cells corroborate in many ways findings observed in T cells isolated from lupus patients. Lupus is a prototype autoimmune disease characterized by T cell DNA methylation defect resulting in T-cell autoreactivity [24]. Indeed, lupus T cells are characterized by defective MAPK signaling [69], and our findings showed that MAPK signaling pathway appeared significantly hypermethylated with age and therefore likely silenced at the epigenetic level in aging naive CD4+ T cells. CD40LG and CD11a (encoded by ITGAL) which are regulated by DNA methylation and are hypomethylated and overexpressed in lupus CD4+ T cells and normal CD4+ T cells treated by DNA methylation inhibitors [70,71], were also hypomethylated with age in our study. Indeed, hypomethylation of ITGAL and CD11a overexpression has been previously demonstrated with aging in total T cells [26], consistent with our naive CD4+ T cell data. Lupus T cells are characterized by mTOR activation, which contributes to T cells autoreactivity and proinflammatory phenotype [72]. Similarly, mTOR signaling pathway appeared progressively poised for activation at the epigenetic level in naive CD4+ T cells from healthy individuals with increased age in our study. T cells in lupus patients and in aging are characterized by increased apoptosis, presumably providing the source of autoantigens for T-cell autoreactivity. We show that apoptosis-related pathways were hypomethylated in naive CD4+ T cells with age. Taken together, our data support that epigenetic dysregulation at the DNA methylation level in aging naive CD4+ T cells favor T-cell autoreactivity and increased apoptosis. These findings might contribute to increased risk of autoimmunity with age.
Previous enhancer-mapping studies have reported that active cis-regulatory modules in gene promoters are CpG-depleted [38], suggesting CpG sites negatively associated with age may be located in such enhancer-like cis-regulatory modules. These observations further strengthen our observation that CpG sites hypermethylated or hypomethylated with age may have different epigenomic signatures. Notably, CpG sites hypermethylated with age showed overall higher regulatory similarity despite the fact these sites were identified in different cell types (Figure 3), indicating similar location and regulatory impact of age-associated hypermethylated CpGs across cell types. These observations suggest age-associated hypermethylated regions may be conserved among cell and tissue types and are highly distinct from the cell type specific age-associated hypomethylated regions.
Conclusion
We demonstrate age-dependent DNA methylation changes in naive CD4+ T cells, and distinct genomic and regulatory enrichment patterns between loci hypermethylated and hypomethylated with age. Our findings suggest a progressive age-associated shift in T-cell epigenomes toward proinflammatory and T cell activating epigenotypes. These findings support a role for age-dependent DNA methylation changes in explaining increased autoimmunity with age. Future studies that examine longitudinal samples collected over time from the same individuals, and studies to examine other specific immune cell types will allow for a more comprehensive understanding of age-related autoimmune epigenotypes.
Summary points.
The aim of this study was to characterize age-associated DNA methylation changes in human naive CD4+ T cells and their potential disease relevance.
Seventy four healthy female individuals (age 19–66 years) were recruited, and naive CD4+ T cells were collected from peripheral blood.
Genome-wide DNA methylation was assessed using the Infinium HumanMethylation450 BeadChip array (Illumina).
Age-related DNA methylation changes were defined using regression analysis and characterized for genomic and epigenomic enrichment patterns, and gene set functional enrichment.
We identified 11,431 age-associated CpG sites, 57% of which were hypermethylated with age in naive CD4+ T cells.
We observed distinct genomic locations and epigenomic enrichment patterns of CpG sites hypermethylated or hypomethylated with age.
CpG sites hypermethylated with age were enriched in EZH2 and SUZ12 binding sites, comprising the well-known polycomb repressive complex 2 signature.
CpG sites hypomethylated with age showed T cell specific enrichment in active enhancers marked by H3K27ac and H3K4me1 histone marks.
A total of 3022 genes associated with CpG sites hypomethylated with age were enriched in immune-related pathways, such as T-cell receptor signaling and the mammalian target of rapamycin pathway.
A total of 3183 genes associated with CpG sites hypermethylated with age were enriched in Hedgehog signaling, pathways in cancer, focal adhesion and Wnt signaling. The MAPK signaling pathway, which is defective in autoimmune diseases such as lupus, was also significantly hypermethylated with age.
Our findings extend the role of age-associated hypermethylation of polycomb repressive complex 2 binding signature to naive CD4+ T cells, and strengthen the notion that age-associated methylation changes may alter distinct regulatory mechanisms and signaling pathways that predispose to autoimmunity.
Supplementary Material
Footnotes
Financial & competing interests disclosure
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI097134 and award number U19AI110502. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The institutional review boards of the participating institutions approved this study and all participants provided written, informed consent prior to enrollment.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Pilling LC, Atkins JL, Bowman K, et al. Human longevity is influenced by many genetic variants: evidence from 75,000 UK Biobank participants. Aging (Albany NY) 2016 doi: 10.18632/aging.100930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brooks–Wilson AR. Genetics of healthy aging and longevity. Hum. Genet. 2013;132(12):1323–1338. doi: 10.1007/s00439-013-1342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christensen K, Johnson TE, Vaupel JW. The quest for genetic determinants of human longevity: challenges and insights. Nat. Rev. Genet. 2006;7(6):436–448. doi: 10.1038/nrg1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coolen MW, Statham AL, Qu W, et al. Impact of the genome on the epigenome is manifested in DNA methylation patterns of imprinted regions in monozygotic and dizygotic twins. PLoS ONE. 2011;6(10):e25590. doi: 10.1371/journal.pone.0025590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gertz J, Varley KE, Reddy TE, et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011;7(8):e1002228. doi: 10.1371/journal.pgen.1002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodriguez–Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011;17(3):330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 7.Johnson AA, Akman K, Calimport SR, Wuttke D, Stolzing A, De Magalhaes JP. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012;15(5):483–494. doi: 10.1089/rej.2012.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lopez–Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richardson BC. Role of DNA methylation in the regulation of cell function: autoimmunity, aging and cancer. J. Nutr. 2002;132(Suppl. 8):S2401–S2405. doi: 10.1093/jn/132.8.2401S. [DOI] [PubMed] [Google Scholar]
- 10.Teschendorff AE, Menon U, Gentry–Maharaj A, et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20(4):440–446. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hannum G, Guinney J, Zhao L, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 2013;49(2):359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• A landmark study identifying age-associated CpG sites and presenting a predictive model of age-associated methylation changes.
- 13.Alisch RS, Barwick BG, Chopra P, et al. Age-associated DNA methylation in pediatric populations. Genome Res. 2012;22(4):623–632. doi: 10.1101/gr.125187.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marttila S, Kananen L, Hayrynen S, et al. Ageing-associated changes in the human DNA methylome: genomic locations and effects on gene expression. BMC Genomics. 2015;16:179. doi: 10.1186/s12864-015-1381-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mcclay JL, Aberg KA, Clark SL, et al. A methylome-wide study of aging using massively parallel sequencing of the methyl-CpG-enriched genomic fraction from blood in over 700 subjects. Hum. Mol. Genet. 2014;23(5):1175–1185. doi: 10.1093/hmg/ddt511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rakyan VK, Down TA, Maslau S, et al. Human aging–associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20(4):434–439. doi: 10.1101/gr.103101.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dozmorov MG. Polycomb repressive complex 2 epigenomic signature defines age-associated hypermethylation and gene expression changes. Epigenetics. 2015;10(6):484–495. doi: 10.1080/15592294.2015.1040619. [DOI] [PMC free article] [PubMed] [Google Scholar]; • A meta-analysis study identifying common age-associated CpG sites from nine independent studies, and their common epigenomic context.
- 18.Steegenga WT, Boekschoten MV, Lute C, et al. Genome-wide age-related changes in DNA methylation and gene expression in human PBMCs. Age (Dordr.) 2014;36(3):9648. doi: 10.1007/s11357-014-9648-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weidner CI, Lin Q, Koch CM, et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014;15(2):R24. doi: 10.1186/gb-2014-15-2-r24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teschendorff AE, Marabita F, Lechner M, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013;29(2):189–196. doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koch CM, Wagner W. Epigenetic-aging-signature to determine age in different tissues. Aging (Albany NY) 2011;3(10):1018–1027. doi: 10.18632/aging.100395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15(2):R31. doi: 10.1186/gb-2014-15-2-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moro-García MA, Alonso-Arias R, López-Larrea C. When aging reaches CD4+ T-cells: phenotypic and functional changes. Front. Immunol. 2013;4:107. doi: 10.3389/fimmu.2013.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeffries MA, Sawalha AH. Autoimmune disease in the epigenetic era: how has epigenetics changed our understanding of disease and how can we expect the field to evolve? Expert Rev. Clin. Immunol. 2015;11(1):45–58. doi: 10.1586/1744666X.2015.994507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sawalha AH, Harley JB. Antinuclear autoantibodies in systemic lupus erythematosus. Curr. Opin. Rheumatol. 2004;16(5):534–540. doi: 10.1097/01.bor.0000135452.62800.8f. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Z, Deng C, Lu Q, Richardson B. Age-dependent DNA methylation changes in the ITGAL (CD11a) promoter. Mech. Ageing Dev. 2002;123(9):1257–1268. doi: 10.1016/s0047-6374(02)00014-3. [DOI] [PubMed] [Google Scholar]
- 27.Coit P, Jeffries M, Altorok N, et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J. Autoimmun. 2013;43:78–84. doi: 10.1016/j.jaut.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coit P, Ognenovski M, Gensterblum E, Maksimowicz–Mckinnon K, Wren JD, Sawalha AH. Ethnicity-specific epigenetic variation in naive CD4+ T cells and the susceptibility to autoimmunity. Epigenetics Chromatin. 2015;8:49. doi: 10.1186/s13072-015-0037-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen YA, Lemire M, Choufani S, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrari S, Cribari-Neto F. Beta regression for modelling rates and proportions. J. Appl. Stat. 2004;31(7):799–815. [Google Scholar]
- 31.Cribari-Neto F, Zeileis A. Beta regression in R. J. Stat. Softw. 2010;34(2):1–24. [Google Scholar]
- 32.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24(13):1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 33.Dozmorov MG, Cara LR, Giles CB, Wren JD. GenomeRunner web server: regulatory similarity and differences define the functional impact of SNP sets. Bioinformatics. 2016;32(15):2256–2263. doi: 10.1093/bioinformatics/btw169. [DOI] [PMC free article] [PubMed] [Google Scholar]; • A tool introducing, among others, epigenomic enrichment analysis method, used for in silico validation of age-associated CpG sites.
- 34.Rosenbloom KR, Sloan CA, Malladi VS, et al. ENCODE data in the UCSC genome browser: year 5 update. Nucleic Acids Res. 2013;41:D56–D63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harrow JL, Steward CA, Frankish A, et al. The vertebrate genome annotation browser 10 years on. Nucleic Acids Res. 2014;42:D771–D779. doi: 10.1093/nar/gkt1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooper GM, Stone EA, Asimenos G, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15(7):901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu H, Caffo B, Jaffee HA, Irizarry RA, Feinberg AP. Redefining CpG islands using hidden Markov models. Biostatistics. 2010;11(3):499–514. doi: 10.1093/biostatistics/kxq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andersson R, Gebhard C, Miguel–Escalada I, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507(7493):455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roadmap Epigenomics C, Kundaje A, Meuleman W, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slieker RC, van Iterson M, Luijk R, et al. Age-related accrual of methylomic variability is linked to fundamental ageing mechanisms. Genome Biol. 2016;17(1):191. doi: 10.1186/s13059-016-1053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reynolds LM, Taylor JR, Ding J, et al. Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat. Commun. 2014;5:5366. doi: 10.1038/ncomms6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heyn H, Li N, Ferreira HJ, et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl Acad. Sci. USA. 2012;109(26):10522–10527. doi: 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson VL, Jones PA. DNA methylation decreases in aging but not in immortal cells. Science. 1983;220(4601):1055–1057. doi: 10.1126/science.6844925. [DOI] [PubMed] [Google Scholar]
- 45.Florath I, Butterbach K, Muller H, Bewerunge-Hudler M, Brenner H. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum. Mol. Genet. 2014;23(5):1186–1201. doi: 10.1093/hmg/ddt531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 47.Schraml BU, Hildner K, Ise W, et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460(7253):405–409. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dovat S. Ikaros: the enhancer makes the difference. Blood. 2013;122(18):3091–3092. doi: 10.1182/blood-2013-09-526343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshida T, Landhuis E, Dose M, et al. Transcriptional regulation of the Ikzf1 locus. Blood. 2013;122(18):3149–3159. doi: 10.1182/blood-2013-01-474916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu Y, Wang J, Khaled W, et al. Bcl11a is essential for lymphoid development and negatively regulates p53. J. Exp. Med. 2012;209(13):2467–2483. doi: 10.1084/jem.20121846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu W, Tanasa B, Tyurina OV, et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466(7305):508–512. doi: 10.1038/nature09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X, Liu L, Yang S, et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc. Natl Acad. Sci. USA. 2014;111(19):7096–7101. doi: 10.1073/pnas.1324036111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Creyghton MP, Cheng AW, Welstead GG, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl Acad. Sci. USA. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zampieri M, Ciccarone F, Calabrese R, Franceschi C, Bürkle A, Caiafa P. Reconfiguration of DNA methylation in aging. Mech. Ageing Dev. 2015;151:60–70. doi: 10.1016/j.mad.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 56.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25(10):1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coit P, Dozmorov MG, Merrill JT, et al. Epigenetic reprogramming in naive CD4+ T cells favoring T cell activation and non-Th1 effector T cell immune response as an early event in lupus flares. Arthritis Rheumatol. 2016;68(9):2200–2209. doi: 10.1002/art.39720. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Revealed a proinflammatory epigenetic reprogramming in naive CD4+ T cells upon lupus flares, prior to changes in gene expression. Evidence was presented to implicate increased EZH2 as a mediator of this epigenetic shift. The current work shows that EZH2 binding sites are enriched in CpG sites hypermethylated with age, similar to what was observed in lupus flares.
- 58.Fransen E, Bonneux S, Corneveaux JJ, et al. Genome-wide association analysis demonstrates the highly polygenic character of age-related hearing impairment. Eur. J. Hum. Genet. 2015;23(1):110–115. doi: 10.1038/ejhg.2014.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lunetta KL, D'agostino RB, Karasik D, et al. Genetic correlates of longevity and selected age-related phenotypes: a genome-wide association study in the Framingham Study. BMC Med. Genet. 2007;8(Suppl. 1):S13. doi: 10.1186/1471-2350-8-S1-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walter S, Atzmon G, Demerath EW, et al. A genome-wide association study of aging. Neurobiol. Aging. 2011;32(11):2109.e2115–2109.e2128. doi: 10.1016/j.neurobiolaging.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kamboh MI, Barmada MM, Demirci FY, et al. Genome-wide association analysis of age-at-onset in Alzheimer's disease. Mol. Psychiatry. 2012;17(12):1340–1346. doi: 10.1038/mp.2011.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xie T, Deng L, Mei P, et al. Genome-wide association study combining pathway analysis for typical sporadic amyotrophic lateral sclerosis in Chinese Han populations. Neurobiol. Aging. 2014;35(7):1778.e1779–1778.e1723. doi: 10.1016/j.neurobiolaging.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 63.Perez-Andreu V, Roberts KG, Xu H, et al. A genome-wide association study of susceptibility to acute lymphoblastic leukemia in adolescents and young adults. Blood. 2015;125(4):680–686. doi: 10.1182/blood-2014-09-595744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baranzini SE, Wang J, Gibson RA, et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum. Mol. Genet. 2009;18(4):767–778. doi: 10.1093/hmg/ddn388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Festen EaM, Goyette P, Green T, et al. A meta-analysis of genome-wide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for Crohn's disease and celiac disease. PLoS Genet. 2011;7(1):e1001283. doi: 10.1371/journal.pgen.1001283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garagnani P, Bacalini MG, Pirazzini C, et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell. 2012;11(6):1132–1134. doi: 10.1111/acel.12005. [DOI] [PubMed] [Google Scholar]
- 67.Chiurchiù V, Leuti A, Dalli J, et al. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci. Transl. Med. 2016;8(353):353ra111–353ra111. doi: 10.1126/scitranslmed.aaf7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dashti M, Peppelenbosch MP, Rezaee F. Hedgehog signalling as an antagonist of ageing and its associated diseases. Bioessays. 2012;34(10):849–856. doi: 10.1002/bies.201200049. [DOI] [PubMed] [Google Scholar]
- 69.Gorelik G, Fang JY, Wu A, Sawalha AH, Richardson B. Impaired T cell protein kinase C delta activation decreases ERK pathway signaling in idiopathic and hydralazine-induced lupus. J. Immunol. 2007;179(8):5553–5563. doi: 10.4049/jimmunol.179.8.5553. [DOI] [PubMed] [Google Scholar]
- 70.Lu Q, Kaplan M, Ray D, et al. Demethylation of ITGAL (CD11a) regulatory sequences in systemic lupus erythematosus. Arthritis Rheum. 2002;46(5):1282–1291. doi: 10.1002/art.10234. [DOI] [PubMed] [Google Scholar]
- 71.Lu Q, Wu A, Tesmer L, Ray D, Yousif N, Richardson B. Demethylation of CD40LG on the inactive X in T cells from women with lupus. J. Immunol. 2007;179(9):6352–6358. doi: 10.4049/jimmunol.179.9.6352. [DOI] [PubMed] [Google Scholar]
- 72.Perl A. mTOR activation is a biomarker and a central pathway to autoimmune disorders, cancer, obesity, and aging. Ann. NY. Acad. Sci. 2015;1346(1):33–44. doi: 10.1111/nyas.12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.