

Abstract
Oncolytic viruses selectively lyse tumor cells, disrupt immunosuppression within the tumor and reactivate anti-tumor immunity, but they have yet to live up to their therapeutic potential. Immune checkpoint modulation has been efficacious in a variety of cancer with an immunogenic microenvironment, but is associated with toxicity due to nonspecific T-cell activation. Therefore, combining these two strategies would likely result in both effective and specific cancer therapy. To test the hypothesis, we first constructed oncolytic adenovirus Delta-24-RGDOX expressing the immune co-stimulator OX40 ligand (OX40L). Like its predecessor Delta-24-RGD, Delta-24-RGDOX induced immunogenic cell death and recruit lymphocytes to the tumor site. Compared to Delta-24-RGD, Delta-24-RGDOX exhibited superior tumor-specific activation of lymphocytes and proliferation of CD8+ T cells specific to tumor-associated antigens, resulting in cancer-specific immunity. Delta-24-RGDOX mediated more potent anti-glioma activity in immune-competent C57BL/6 but not immune-deficient athymic mice, leading to specific immune memory against the tumor. In order to further overcome the immune suppression mediated by programmed death-ligand 1 (PD-L1) expression on cancer cells accompanied with virotherapy, intratumoral injection of Delta-24-RGDOX and an anti-PD-L1 antibody showed synergistic inhibition of gliomas and significantly increased survival in mice. Our data demonstrate that combining an oncolytic virus with tumor-targeting immune checkpoint modulators elicits potent in situ autologous cancer vaccination, resulting in an efficacious, tumor-specific and long-lasting therapeutic effect.
Keywords: oncolytic adenovirus, Delta-24-RGD, Delta-24-RGDOX, OX40L, PD-L1
Introduction
Oncolytic viruses are a promising alternative therapy for refractory cancers, including glioblastoma whose prognosis, with a 5-year survival rate of 5%, has barely improved even after decades of preclinical and clinical research (1). Although virus-mediated oncolysis should, in theory, spread to all cancer cells within the tumor (2, 3), this has yet to play out in clinical trials. Thus far, clinical experience has shown that the antiviral immune response and limited antitumor immunity developed during virotherapy constrain the efficiency of the viruses when they are used alone (2).
The second-generation oncolytic adenovirus Delta-24-RGD, which targets aberrant RB pathway and αv integrins expression in cancer cells (4), caused potent oncolysis in human cancer xenografts in immunodeficient mice (4–7) and triggered anti-glioma immunity in a syngeneic immunocompetent mouse glioma model (8, 9). Importantly, the first phase I clinical trial of Delta-24-RGD in patients with recurrent glioblastoma revealed that an intratumoral viral injection solicited an inflammatory response and a durable complete response in about 12% of patients (10). We speculated that the suboptimal efficacy of Delta-24-RGD could be improved by developing strategies to enhance virus-mediated anti-tumor immunity since the paradigm in oncolytic virotherapy has shifted from oncolysis to immune-mediated eradication of the tumor (2, 11).
In the past two decades, immune checkpoint modulation has shown promise in treating a variety of malignancies (12). However, this type of therapy is less effective in cancers with an immunosuppressive microenvironment (13). Interestingly, during virotherapy, viral infection, replication, and subsequent tumor necrosis cooperate to disrupt the immunosuppressive tumor microenvironment, resulting in T cell reactivity against cancer neo-antigens (2, 14, 15). Thus, combining checkpoint blockade with oncolytic virotherapy is an attractive candidate anti-cancer strategy. Accordingly, pre-clinical and clinical studies are presently underway to investigate the potential of combining checkpoint blockade with oncolytic viral therapies (15, 16). A phase II, multi-center clinical trial for the combination of Delta-24-RGD and an antibody against programmed cell death 1 (PD-1), an immune checkpoint co-inhibitor, has just begun in patients with recurrent glioblastomas or gliosarcomas (ClinicalTrials.gov Identifier: NCT02798406).
We hypothesized that the efficacy of Delta-24-RGD would be improved by modifying it to express an immune co-stimulator, such as OX40 ligand (OX40L), to enhance the antigen-presenting function of the tumor cells and stimulate tumor-specific T cell activation. Unlike ligands CD80 and CD86 that bind both immune co-stimulator CD28 and co-inhibitor CTLA-4 (12), OX40L binds a unique co-stimulator OX40 on T cells (17), which makes it a better choice to arm the virus to enhance activation of T cells recognizing tumor antigens on tumor cells infected by the virus. Thus, due to cancer-selectivity of the virus, this strategy is more tumor-specific than immune checkpoint blockade. The latter can result in a diffuse T-cell repertoire expansion that reduces self-tolerance and damages healthy organs, causing immune-related adverse events (18). Moreover, the receptors of this type of co-stimulators are upregulated on many immune cells upon activation (19, 20) and their agonist antibodies have shown therapeutic benefit in both preclinical cancer models and cancer patients (20–23), especially when combined with strategies to shift the cytokine balance towards a Th1 milieu (20), which is a typical consequence of adenoviral therapy (8, 9). In addition, programmed death-ligand 1 (PD-L1), a ligand for PD-1, is commonly expressed on tumor cells (12), and what makes it even more relevant to oncolytic adenoviral treatment is that, as a negative feedback to inflammatory response, PD-L1 is upregulated by interferon γ (IFNγ) (24) which is induced by adenoviral infection (8, 9). Since blocking PD-L1 with its antibody mainly targets tumor cells and antigen-presenting cells, this strategy should have less toxicity than targeting T cells with CTLA-4 or PD-1 antibodies. We speculated that intratumoral injection of PD-L1 antibody, which confines its effect more localized within the tumor, in addition to oncolytic adenoviruses should block the inhibitory signal from the tumor cells and further potentiate the effect of the viruses.
Thus far, although oncolytic viruses have been combined with immune modulators to increase efficacy (2, 15, 16), the specificity and safety of this type of combination haven’t been well addressed. We hypothesized that we could improve them through taking advantage of the cancer-selectivity of oncolytic viruses and immune modulations mainly targeted to cancer cells. Therefore, in this study, we first constructed oncolytic adenovirus Delta-24-RGDOX whose genome includes a mouse OX40L expression cassette. The new virus efficiently expressed the ligand on the cell surface of a panel of human and mouse cancer cell lines without significant change in its replication capacity. Like Delta-24-RGD, Delta-24-RGDOX induced autophagy and immunogenic cell death that could trigger innate immune response. Compared to Delta-24-RGD, Delta-24-RGDOX was more efficient to increase both CD4+ and CD8+ T cells in the tumor site and induce lymphocyte activity against tumor cells in a mouse glioma model. Further studies revealed that Delta-24-RGDOX infection directly enhanced the ability of tumor cells to activate lymphocytes, including CD8+ T cells specific for TAAs, through expressing OX40L on the cell surface, resulting in tumor-specific immunity. Consequently, Delta-24-RGDOX was more potent to induce anti-glioma activity than Delta-24-RGD in immunocompetent but not in athymic mice, causing specific immune memory against the tumor which had been treated with the virus. Furthermore, we found IFNγ upregulated PD-L1 expression in a panel of human glioma stem cell lines and mouse glioma cell lines. Consistently, Delta-24-RGDOX infection also upregulated PD-L1 expression on the tumor cells from the mouse glioma model. Thus, we combined Delta-24-RGDOX with intratumoral injection of a PD-L1 antibody and found they synergized to reject gliomas. In summary, our findings demonstrate that oncolytic adenovirus combined with tumor-targeting immune modulations induces potent ongoing in situ cancer vaccination during therapy, resulting in efficacious, specific and long-lasting anti-cancer effect.
Materials and Methods
Cell lines and culture conditions
Human glioblastoma-astrocytoma U-87 MG (2005–2010) and lung carcinoma A549 cells (2005–2010, ATCC), mouse glioma GL261 cells (NCI-Frederick Cancer Research Tumor Repository, 2011), GL261-5 cells (an isolated GL261 cell clone that resulted in a longer life span of the mice than did the parental GL261 cells when implanted intracranially); GL261- enhanced green fluorescent protein (EGFP) cells (a kind gift from Dr. Kaminska, Nencki Institute of Experimental Biology, Warsaw, Poland, 2011), and GL261-OVA cells (8) were cultured in Dulbecco’s modified Eagle’s medium-nutrient mixture F12 (DMEM/F12) supplemented with 10% fetal bovine serum (HyClone Laboratories, Inc.), 100 μg/ml penicillin, and 100 μg/ml streptomycin, except in the GL261-OVA culture, to which 1 μg/ml puromycin (Life Technologies) was also added as described (8). Mouse melanoma cell line B16-F10 (ATCC, 2012) was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum and antibiotics. Human embryonic kidney 293 (Qbiogene, Inc., 1990s), mouse glioma CT-2A (generously donated by Dr. Thomas Seyfried, Boston College, Boston, MA, 2016) and mouse lung carcinoma CMT64 (Culture Collections, Public Health England, UK, 2014) cells were maintained in DMEM supplemented with 10% fetal bovine serum and antibiotics. Mouse primary astrocytes (AllCells, LLC, 2015) were grown in AGM Astrocyte Growth Medium (Lonza). Human glioblastoma stem cell lines (GSCs) had been established from acute cell dissociation of human glioblastoma surgical specimens (2005–2015). The study was approved by the Institutional Review Board at The University of Texas MD Anderson Cancer Center and in accordance with Belmont Report. Written informed consent was required for every patient. The GSCs were maintained in DMEM/F12 medium supplemented with B27 (Invitrogen), epidermal growth factor, and basis fibroblast growth factor (20 ng/mL each, Sigma-Aldrich) according to the procedures described elsewhere (6). All cells were kept at 37°C in a humidified atmosphere containing 5% CO2. All GSC lines were verified through short-tandem repeat (STR) fingerprinting (in 2012). Experiments were carried out within 6 months after the cell lines were obtained from a cell bank (B16-F10 and CMT64) or after the verification (GSCs). U-87 MG cells were reauthenticated with STR in 2016. GL261 cells were re-verified through karyotyping in 2016. All cell lines were tested as mycoplasma-free.
Mice
C57BL/6 and athymic mice were provided by the MD Anderson Cancer Center Mouse Resource Facility. OT-I mice (C57BL/6-Tg[TcraTcrb]1100Mjb/J) were purchased from The Jackson Laboratory.
Animal studies
For tumor implantation, GL261 cells and its derivatives (5 × 104 cells/mouse) cells were grafted into the caudate nucleus of the 7 to 10-week old mice using a guide-screw system as previously described (5). The mice with implanted tumors were randomly assigned to experimental groups. Then the viruses (5 × 107 plaque-forming units (PFU)/mouse), the OX40 agonist antibody OX86 (25 μg/mouse; provided by the Monoclonal Antibody Core Facility at MD Anderson Cancer Center), the anti-mouse PD-L1 antibody and/or rat IgG (25 μg/mouse; Bio X Cell) were injected intratumorally. For rechallenging the surviving mice, GL261-5 (5 × 104 cells/mouse) or B16-F10 (1× 103 cells/mouse) cells were implanted in the same hemisphere previously implanted with the cured tumor or in the contralateral hemisphere of the mouse brain. All animal studies (except one survival study in athymic mice) were conducted in C57BL/6 mice. All experimental procedures involving the use of mice were done in accordance with protocols approved by the Animal Care and Use Committee of MD Anderson Cancer Center and followed National Institutes of Health and United States Department of Agriculture guidelines.
Flow cytometry analysis
To monitor the disruption of the cell membrane (cell death) induced by the viruses, cells (2–5 × 105) were stained with 8 μM ethidium homodimer 1 (Sigma-Aldrich) in PBS solution for 15 minutes at room temperature. To analyze cell surface protein expression, cells (2–5 × 105) were first incubated in 100 μl primary antibody solution diluted in PBS plus 3% bovine serum albumin and 1 mM ethylenediaminetetraacetic acid. After incubation at 4°C in the dark for 30 minutes, the cells were washed once with 1 ml cold PBS. If a secondary antibody was to be applied, the incubation procedure was repeated with the secondary antibody. After being washed once with PBS, the cells were finally resuspended in 0.5 ml PBS. The stained cells were then analyzed using flow cytometry. The antibodies used in the studies were as follows: anti-mouse CD252 (OX40L) adenomatous polyposis coli (APC, 17-5905-80), anti-mouse CD45 APC-eFluor 780 (47-0451-82), anti-mouse CD3 fluorescein isothiocyanate (FITC, 11-0031-81), Anti-Mouse CD4 eFluor® 450 (48-0041-80), anti-mouse CD8a PerCP-Cyanine 5.5 (45-0081-82), anti-human PD-L1 APC (17-5983-41), and anti-mouse CD8a APC (17-0081-81), Anti-Mouse CD279 (PD-1) PE-Cyanine7 (25-9985-80), Anti-Mouse CD152 (CTLA-4) APC (17-1522-80) were obtained from eBioscience; Goat Anti-Rat IgG -FITC (ab6115) was from abcam; and anti-mouse CD252 (OX40L) purified (108802) and anti-mouse PD-L1 APC (124311) were obtained from BioLegend.
ATP and HMGB1 release analyses
The medium was collected from the cell cultures. The amount of ATP in the medium was determined with an ENLITEN ATP Assay System (Promega). The HMGB1 in the medium was quantitated with an HMGB1 enzyme-linked immunosorbent assay (ELISA) kit (IBL International).
Preparation of splenocytes and CD8+ T lymphocytes
Mouse spleens were collected, placed in a 100-μm cell strainer set in petri dishes with RPMI 1640 medium, and then smashed through the cell strainer into the dish. The mixture in the dish was gently pipetted up and down and brought up to 5 ml/spleen. The cells were pelleted by centrifugation at 350 g for 7 minutes at room temperature and then resuspended in Red Blood Cell Lysing Buffer Hybri-Max (Sigma-Aldrich) to lyse the red blood cells, according to the manufacturer’s instructions. Finally, CD8+ T cells were enriched with a Mouse CD8a+ T Cell Isolation Kit (Miltenyi Biotec, Inc.).
Preparation of brain-infiltrating lymphocytes (BILs)
Brain-infiltrating leukocytes (from a group of 5 to 9 mouse brain hemispheres) were separated from myelin debris using Percoll (GE Healthcare Bio-Sciences) and gradient centrifuged as described previously (25). BILs were then enriched using a gradient centrifuge with Lympholyte-M Cell Separation Media (Cedarlane) as instructed by the manufacturer.
Preparation of bone marrow-derived mouse dendritic cells (mDCs)
mDCs were isolated from mouse bone marrow of femurs and tibias as described (26). After 7 days in culture, mDCs were collected and 1 μg/ml Lipopolysaccharides (Sigma-Aldrich) was added for the final 18h of culture to induce maturation. The matured mDCs were primed with 10 μg/ml OVA (257-264) peptide for one hour at 37 °C.
Stimulation of immune cells
To prepare the target cells, GL261 or GL261-OVA cells were infected with virus at 100 PFU per cell. Four hours later, 100 units/ml of mouse IFNγ (Prospec Protein Specialists) was added to the culture. Forty-eight hours after viral infection, the cells were fixed with 1% paraformaldehyde. To activate immune cells, pre-fixed target cells (2 × 104/well) were incubated with splenocytes (5 × 105/well) or brain-infiltrating lymphocytes (BILs) (5 × 104/well). To measure the OVA-specific T-cell reaction, CD8+ T-cells (3 × 105/well) were stimulated with pre-fixed mouse dendritic cells (1 × 105/well) primed with the OVA 257-264 peptide (InvivoGen). Forty hours after the co-culture in a round-bottom 96-well plate, the concentration of IFNγ in the supernatant was assessed with an ELISA (Mouse IFNγ DuoSet, R&D Systems).
In vitro lymphocyte cell proliferation
OVA-specific CD8+ T-cells were isolated from the spleens of OT-I mice (C57BL/6-Tg[TcraTcrb]1100Mjb/J, The Jackson Laboratory) and labeled with 5 μM carboxyfluorescein succinimidyl ester (CFSE, Life Technologies) for 5 minutes (27). The labeled T-cells (1 × 105/well) were stimulated with target cells (5 × 104/well) in a round-bottom 96-well plate. Four days later, the cells were stained with anti-mouse CD8a allophycocyanin (APC) and analyzed with flow cytometry for green fluorescence (CFSE amount) in CD8+ cells. Proliferating cells were defined as those exhibiting lower CFSE amount than un-stimulated cells.
Statistics
In quantitative studies of cultured cells, each group consisted of triplicate samples. Each study was repeated at least once. The differences between groups were evaluated using a 2-tailed Student’s t-test. The animal survival curves were plotted according to the Kaplan–Meier method. Survival rates in the different treatment groups were compared using the log-rank test. The synergistic interactions of the agents in survival studies were analyzed based on the normal survival model using the function survreg in R, and a residual plot was used to examine the parametric assumption of the model (28). P values < .05 were considered significant.
Results
Oncolytic adenovirus Delta-24-RGDOX expresses OX40L and preserves replication capacity
We first generated Delta-24-RGDOX, a replication-competent adenovirus that included an expression cassette for mouse OX40L (mOX40L) on Delta-24-RGD backbone (5, 7) (Figure 1A). The new virus efficiently expressed mOX40L on the cell membranes of living cultured mouse and human cancer cells (P < 0.0001, Figure 1B and Supplementary Figure S1). In in vivo settings, we analyzed mOX40L expression in cells from gliomas arising from intracranial injection of GL261 cells stably expressing enhanced green fluorescent protein (GL261-EGFP, Figure 1C), which revealed that approximately 10% of the tumor cells were mOX40L+ 3 days after Delta-24-RGDOX intratumoral injection (P = 0.02, Figure 1C). The modification in the viral genome did not significantly change its replication capability in human U-87 MG glioma cells (P = 0.05) and mouse GL261 glioma cells (P = 0.44) (Figure 1D).
Figure 1.
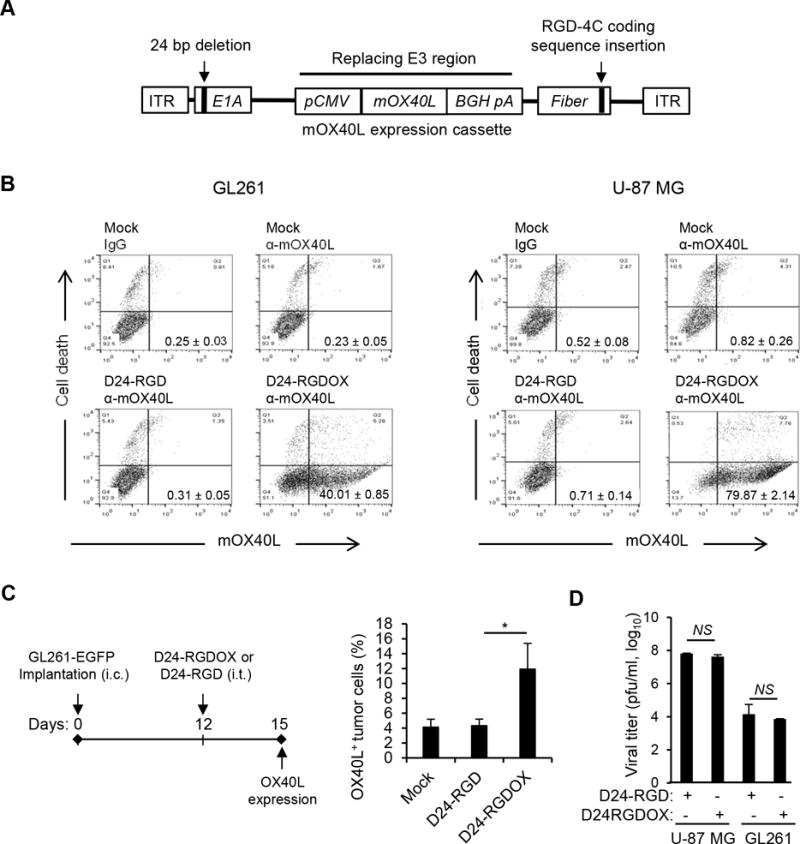
Construction and characterization of Delta-24-RGDOX. A, Schematic representation of the Delta-24-RGDOX genome, including a 24-base pair deletion in the E1A gene that encodes an RB-binding region and an insertion in the fiber gene that encodes an integrin-binding motif (RGD-4C) in the HI loop of the protein. The mouse OX40L (mOX40L) expression cassette, including the cytomegalovirus (CMV) promoter (pCMV), mOX40L cDNA, bovine growth hormone poly-adenylation signal (BGH pA), replaces the E3 region of the human Adenovirus 5 genome. ITR: inverted terminal repeat. B, Expression of mOXO40L by Delta-24-RGDOX in mouse GL261 and human U-87 MG glioma cells. Cells were infected with Delta-24-RGD or Delta-24-RGDOX at 100 (GL261) or 10 (U-87 MG) PFU/cell. After 48 hours, the cells were harvested, and mOX40L expression and cell death (cells with broken membrane stained with ethidium homodimer-1) were analyzed with flow cytometry. Representative dot plots for each analysis are shown. The numbers at the lower right corners indicate the percentages of live cells expressing mOX40L on their cell membrane. C, (left panel) A cartoon depiction of the treatment scheme. i.c.: intracranial; i.t.: intratumoral. (right panel). Expression of mOX40L on tumor cells from virus-treated tumors. The hemispheres with tumors from treated mice (3 mice per group) were harvested, and the cells were dissociated and stained with anti-mOX40L-APC. The stained cells were analyzed using flow cytometry. Tumor cells were gated for EGFP+. The results from two independent experiments are shown. D, Replication of Delta-24-RGDOX or Delta24-RGD in U-87 MG and GL261 cells. Values represent the mean ± standard deviation (n = 3). * P = 0.02; NS: not significant (P ≥ 0.05), 2-tailed Student’s t test. Mock: PBS; D24-RGD: Delta-24-RGD; D24-RGDOX: Delta-24-RGDOX.
Delta-24-RGDOX induces autophagy and immunogenic cell death
Adenoviruses potently induce autophagic cell death (29). Accordingly, we found this capability in Delta-24-RGDOX, which induced autophagy and cell lysis more robustly than Delta-24-RGD, as shown by the increased LCII/I ratio (Figure 2A) and rupture in cell membrane (Figure 2B). It was reported that this type of cell death attracted immune cells via the extracellular release of damage- (or danger-)associated molecular pattern (DAMP) molecules, such as adenosine triphosphate (ATP) and the high-mobility group protein B1 (HMGB1) (30, 31). Thus, we found that both Delta-24-RGD and Delta-24-RGDOX induced the release of ATP and HMGB1 from infected cells (P < 0.0001, Figure 2C and D) and that Delta-24-RGDOX mediated HMGB1 release more efficiently than Delta-24-RGD (P = 0.001, Figure 2D), most likely because of its enhanced ability to induce autophagy and lysis in infected cells (P < 0.0002, Figure 2A and B).
Figure 2.
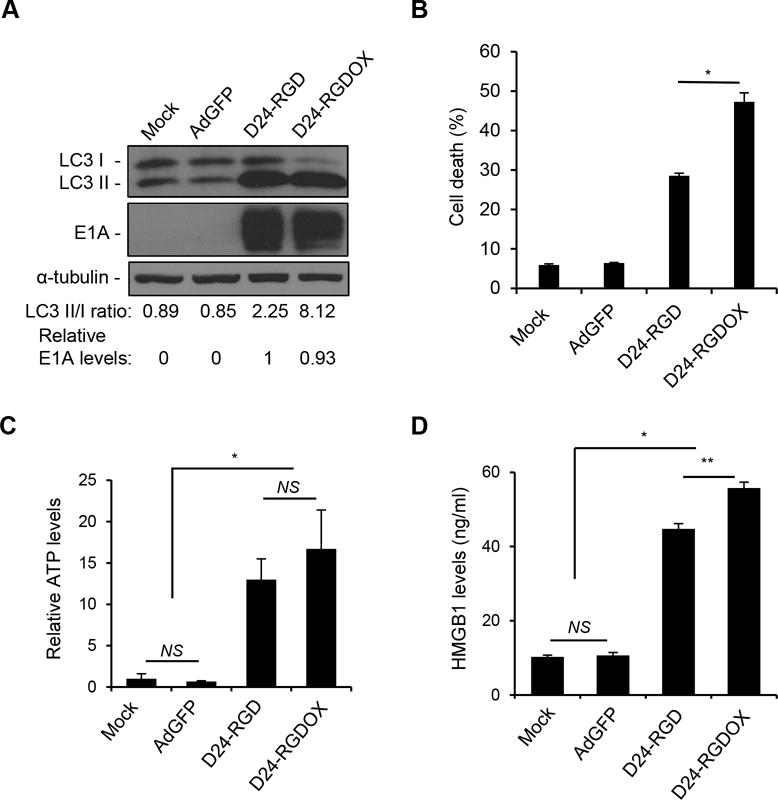
Immunogenic cell death induced by Delta-24-RGDOX. A, GL261 cells were infected with indicated viruses at 100 PFU/cell. 72 hours later, the cell lysates were analyzed with immunoblotting for the cytosolic form of microtubule-associated protein 1A/1B-light chain 3 (LC3 I), or its phosphatidylethanolamine conjugate (LC3 II). The LC3 II/I ratio is used to monitor autophagy. The E1A levels were used as an indicator of the relative viral dose and normalized to the value in the D24-RGD group, which was set to 1. α-tubulin levels are shown as a protein loading control. AdGFP was used as a replication-deficient viral vector control. B, GL261 cells were infected with the indicated viruses at 100 PFU/cell. Cells were harvested after 72 hours and cell lysis (cell death) was monitored with ethidium homodimer-1 staining and analyzed with flow cytometry. C and D, To assess immunogenic cell death induced by the viruses, GL261 cells were infected with the indicated viruses at 100 PFU/cell. After 72 hours, the culture medium was collected and assayed for the amount of ATP (C) or HMGB1 (D). The relative ATP levels (C, 1 = average amount of ATP in mock-treated cells) and HMGB1 concentrations are shown (D). Values represent the mean ± standard deviation (n = 3). NS: not significant (P ≥ 0.05); * P < 0.0002, ** P = 0.001, 2-tailed Student’s t test. Mock: PBS; D24-RGD: Delta-24-RGD; D24-RGDOX: Delta-24-RGDOX.
Delta-24-RGDOX increases lymphocyte infiltration at the tumor site and enhances anti-glioma immunity through OX40L expression
During viral therapy, the DAMPs induced by intratumoral viral injections attract immune cells to the tumor site and elicit an innate immune response that results in the development of adaptive anti-tumor immunity (30). To test this, we used a syngeneic GL261-C57BL/6 immunocompetent glioma model with tumor-infiltrating OX40+ T cells (21). We treated mice with 3 intratumoral viral injections to partially compensate for deficient viral replication in GL261 cells (Figure 3A) (8). In mice injected with either Delta-24-RGD or Delta-24-RGDOX, more T lymphocytes (CD45+/CD3+), T helper cells (CD45+/CD3+/CD4+), and cytotoxic T cells (CD45+/CD3+/CD8+) were present at the tumor site than in mice injected with phosphate-buffered saline (PBS) (P < 0.05). Moreover, significantly more of these cells were present in Delta-24-RGDOX- than in Delta-24-RGD-injected mice (P < 0.001, Figure 3B). Next, we examined the anti-tumor activity of the immune cells through assessing the interferon-γ (IFNγ) secretion by these cells when they were stimulated with tumor cells. Thus, the brain-infiltrating lymphocytes (BILs) from the hemispheres with Delta-24-RGDOX-injected tumor showed significantly higher activity against the tumor cells with or without viral infection than the BILs from the Delta-24-RGD- or PBS-treated groups (P ≤ 0.0001, Figure 3C left panel), indicating that Delta-24-RGDOX mediated a stronger antitumor immune response at the tumor site than did Delta-24-RGD. The same effect was observed in splenocytes from the treatment groups (P < 0.05, Figure 3C right panel) although the increment of the activation induced by Delta-24-RGDOX is not as great as in BILs. Consistent with the co-stimulating activity of OX40L, Delta-24-RGDOX-infected tumor cells triggered higher activation of the BILs than Delta-24-RGD-infected cells (P < 0.001, Figure 3C left panel), and this effect was blocked by the presence of the anti-OX40L antibody (P = 0.004, Figure 3D).
Figure 3.
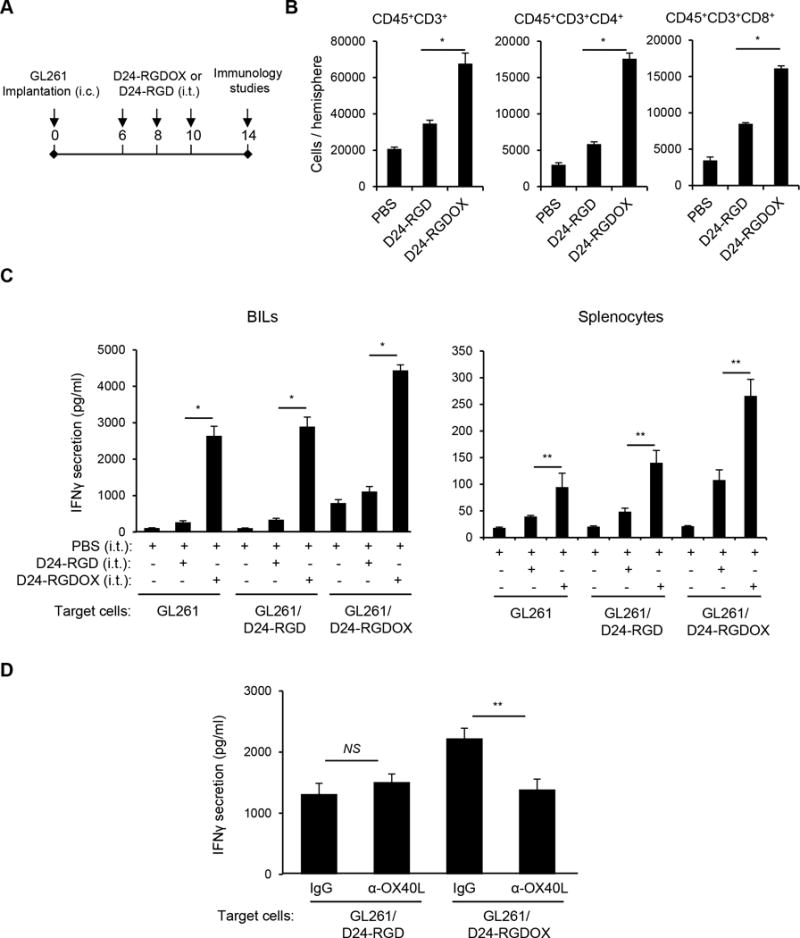
Anti-glioma immunity mediated by Delta-24-RGDOX. A, A cartoon depiction of the treatment scheme. i.c.: intracranial; i.t.: intratumoral. B, Brain-infiltrating leukocytes (BILs) from brain hemispheres with tumors of glioma-bearing mice treated with PBS or the indicated viruses (6 mice per group) were isolated. The BILs were examined using flow cytometry for the indicated cell surface markers to assess numbers of tumor-infiltrating lymphoctyes (CD45+CD3+), helper T lymphocytes (CD45+CD3+CD4+) and cytotoxic T lymphocytes (CD45+CD3+CD8+) at the tumor site. C, BILs (left panel) or splenocytes (right panel) taken from the three groups of mice described in above (5 mice per group), were stimulated with pre-fixed GL261 cells that were uninfected, or had been infected with the indicated virus. Forty hours later, the concentration of IFNγ in the supernatant was assessed with ELISA. D, Inhibition of Delta-24-RGDOX-mediated activation of BILs by an anti-mOX40L antibody. BILs from hemispheres (taken from 9 mice) with Delta-24-RGDOX-infected tumors were isolated and stimulated with pre-fixed GL261 cells that had been infected with Delta-24-RGD or Delta-24-RGDOX in the presence of control immunoglobulin G (IgG) or an anti-mOX40L antibody (4 μg/ml). The concentration of IFNγ in the supernatant was assessed with ELISA. Values represent the mean ± standard deviation (n = 3). NS: not significant (P ≥ 0.05); * P < 0.0001, ** P < 0.05, 2-tailed Student’s t test. D24-RGD: Delta-24-RGD; D24-RGDOX: Delta-24-RGDOX.
Delta-24-RGDOX stimulates a tumor- specific immune response
To further demonstrate the capability of Delta-24-RGDOX to stimulate immunity against tumor-associated antigens (TAAs), we used ovalbumin (OVA) protein as a model antigen (32). Using CFSE staining to track T cell proliferation, we found that GL261-OVA cells (8) infected with Delta-24-RGDOX induced proliferation of OVA-specific CD8+ T-cells more robustly than GL261-OVA cells infected with Delta-24-RGD (P = 0.0002, Figure 4A). Accordingly, CD8+ T-cells isolated from mice harboring GL261-OVA gliomas that had been treated with Delta-24-RGDOX displayed significantly higher activity against mouse dendritic cells primed with an OVA (257–264) peptide (32) than cells from mice treated with Delta-24-RGD (P = 0.001, Figure 4B left panel). This virus-elicited immunity against OVA correlated with the tumor cell-stimulated activation of splenocytes from virus-treated glioma-bearing mice, which was not observed when the splenocytes were co-cultured with primary mouse astrocytes (P = 0.0002, Figure 4B right panel), indicating that Delta-24-RGDOX-elicited immunity is tumor specific.
Figure 4.
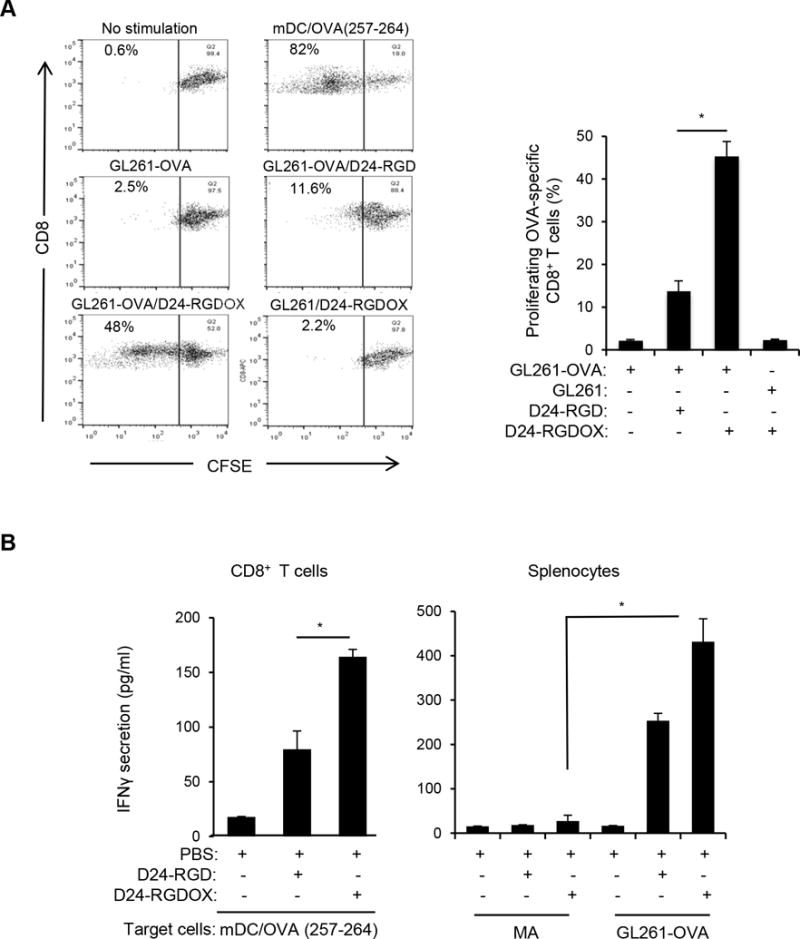
Tumor-specific immunity mediated by Delta-24-RGDOX. A, In vitro proliferation of T-cells recognizing tumor-associated antigen induced by Delta-24-RGDOX. OVA-specific CD8+ T-cells (from the spleens of OT-I mice) pre-stained with CFSE were incubated with the indicated pre-fixed target cells. After 4 days, the cells were analyzed using flow cytometry for CFSE amount to measure cell proliferation. (left panel), Cells were gated for CD8+, and representative dot plots are shown. The numbers at the upper left corners indicate the percentage of proliferating T-cells. Unstimulated T-cells (no stimulation) were used as a negative control, and T cells stimulated with pre-fixed mouse dendritic cells (mDCs) primed with OVA (257-264) peptide (mDC/OVA(257-264)) were used as a positive control. (right panel), Quantification of the proliferating T-cells. Shown are the percentages of the proliferating CD8+ cells after stimulation with the indicated pre-fixed target cells: GL261-OVA cells with or without infection of Delta-24-RGD or Delta-24-RGDOX, GL261 cells infected with Delta-24-RGDOX. B, Tumor-specific immunity induced by Delta-24-RGDOX. (left panel), CD8+ T-cells from the spleens of GL261-OVA glioma-bearing mice treated intratumorally with PBS, Delta-24-RGD or Delta-24-RGDOX (5 mice per group) as in Figure 3A were isolated and stimulated with pre-fixed mDCs primed with OVA (257-264) peptide for 40 hours. (right panel), splenocytes from the above treatment groups were stimulated with pre-fixed mouse primary astrocytes (MAs) or GL261-OVA cells for 40 hours. The concentration of IFNγ in the supernatant was assessed with ELISA. Values represent the mean ± standard deviation (n = 3). * P ≤ 0.001, 2-tailed Student’s t test. D24-RGD: Delta-24-RGD; D24-RGDOX: Delta-24-RGDOX.
Delta-24-RGDOX effectively induces anti-glioma activity in syngeneic immunocompetent mouse glioma models
Next, we performed survival studies using the GL261-C57BL/6 mouse glioma model to evaluate the anti-glioma activity of Delta-24-RGDOX (Figure 5A). The results revealed that GL261 tumors treated with three doses of Delta-24-RGD alone did not affect survival compared to PBS (median survivals: 17 vs 16 days, P = 0.08, Figure 5B left panel). However, the addition of the OX40 agonist antibody OX86 significantly prolonged survival (median survivals: 24 vs 17 days, P = 0.0002, Figure 5B left panel). The treatment of tumors with Delta-24-RGDOX further extended the median survival (median survival: 28.5 vs 17 days, P < 0.0001), producing a 20% long-term survival rate (Figure 5B left panel).
Figure 5.
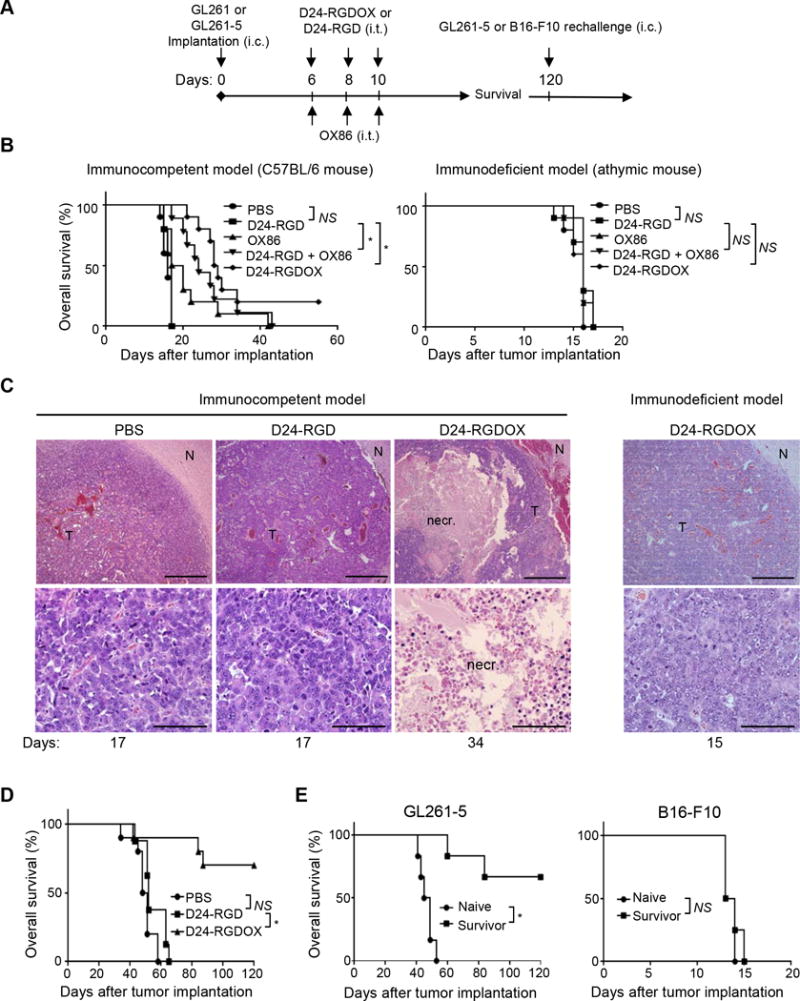
Anti-glioma activity of Delta-24-RGDOX. A, A cartoon depiction of the treatment scheme. i.c.: intracranial; i.t.: intratumoral. B, Survival plots of the different treatment groups in C57BL/6 (immunocompetent, left) or athymic (immunodeficient, right) mice (n = 10 per group, except n = 9 per group for OX86 + Delta-24-RGD in left panel). C, Delta-24-RGDOX-induced necrosis (necr.) in gliomas taken from C57BL/6 mice. Upper panel: representative hematoxylin and eosin-stained sections of the brains from treatment groups showing tumor (T) and normal brain (N) tissue. Lower panel: enlarged images of areas within the tumor. Representative results from at least 6 mice from each group in B are shown. The numbers at the bottom indicate the number of days between tumor implantation and the sacrificing of the mice. Scale: upper panel, 200 μm; lower panel, 50 μm. D, Survival plots of mice in the treatment groups bearing slow-growing GL261-5 gliomas. n = 10, except for Delta-24-RGD, where n = 8. E, Survival plots for mice treated with Delta-24-RGDOX after being re-challenged with GL261-5 (left panel, n = 6) or B16-F10 (right panel, n = 4) cells. NS: not significant (P ≥ 0.05); * P < 0.001, log-rank test. D24-RGD: Delta-24-RGD; D24-RGDOX: Delta-24-RGDOX.
To determine the effect of anticancer immunity on survival rates, we repeated the treatments in immunodeficient athymic mice. Neither Delta-24-RGDOX nor the combination of Delta-24-RGD with OX86 showed a therapeutic benefit when compared to PBS (median survival: 16 vs 16 days, P > 0.05, Figure 5B right panel). The dramatic difference in the therapeutic effect of Delta-24-RGDOX between immunocompetent and immunodeficient mice underscores the essential role played by virotherapy-induced immunity.
Consistent with these results, histopathologic studies of the mouse brains revealed that Delta-24-RGDOX induced tumor necrosis in C57BL/6 mice, which was not observed in either C57BL/6 mice treated with Delta-24-RGD or athymic mice treated with Delta-24-RGDOX (Figure 5C), indicating the necrosis was induced by anti-tumor immunity but not by oncolysis. Moreover, the morphology and histology of the brains of the Delta-24-RGDOX-treated mice showed no signs of acute or chronic inflammation in the normal brain tissue (Figure 5C and Supplementary Figure S2). These data are consistent with our observations of the tumor-specific immunity induced by Delta-24-RGDOX (Figure 4B right panel).
Because Delta-24-RGDOX only induced 20% long-term survival in the GL261-C57BL/6 model, we speculated that its therapeutic efficacy had been compromised by the rapid growth of the tumor. Accordingly, Delta-24-RGDOX demonstrated much greater therapeutic efficacy in the slow-growing GL261-5 glioma model than Delta-24-RGD (median survival: undefined vs 50–52 days, P ≤ 0.0003), resulting in a 70% long-term survival rate (Figure 5D).
Re-challenging the survivors of Delta-24-RGDOX-treated mice with GL261-5 cells failed to produce gliomas in 4 of 6 animals, whereas all naive mice showed signs of intracranial disease and died of gliomas (median survivals: undefined vs 47 days, P = 0.0006, Figure 5E left panel). However, re-challenging mice with B16-F10 melanoma cells caused brain tumors in both the survivor and naive mice (median survivals: 13.5 vs 13.5 days, P = 0.6, Figure 5E right panel). These results suggest that Delta-24-RGDOX effectively induces specific immune memory against the same type of tumor that has been treated with the virus, which is potentiated by the virus-mediated OX40L expression (33).
Delta-24-RGDOX and anti-PD-L1 antibody synergize to inhibit gliomas
The immune checkpoint inhibitor PD-1 ligand PD-L1 is commonly overexpressed in many different tumor types, including gliomas, where it inhibits tumor-directed T-cell activity (12). Moreover, adenovirus-induced IFNγ may further upregulate the expression of PD-L1 during virotherapy (8, 12). To explore this, we examined 8 human glioma stem cell (GSC) lines and found that in all cases, relatively low levels of PD-L1 were significantly up-regulated by IFNγ (Figure 6A, P < 0.002). Similarly, GL261-5 cells expressed a low level of PD-L1 (median fluorescence intensity (MFI) = 37.4), which was slightly enhanced by infection with Delta-24-RGDOX (MFI = 59.7, Figure 6B). However, IFNγ dramatically increased PD-L1 expression in GL261-5 cells both with (MFI = 529) and without (MFI = 661) Delta-24-RGDOX infection (Figure 6B). Basal PD-L1 expression levels were slightly high in GL261-EGFP cells and also increased in response to IFNγ treatment (Supplementary Figure S3). Moreover, Delta-24-RGDOX injection in the gliomas derived from GL261-EGFP cells further upregulated PD-L1 levels in the tumor cells that was already higher than in the cultured cells (MFI increased from 750 to 1176, Figure 6C). Furthermore, after Delta-24-RGDOX treatment, the expression of PD-1 on tumor-infiltrating CD8+ T cells increased by 58% (P = 0.0007), whereas the expression of another immune checkpoint inhibitor, CTLA-4, remained unchanged (P = 0.4) (Figure 6D). These results suggest that the virotherapy results in a feedback activation of PD-L1/PD-1 pathway to compromise the anti-tumor immunity induced by the virus.
Figure 6.
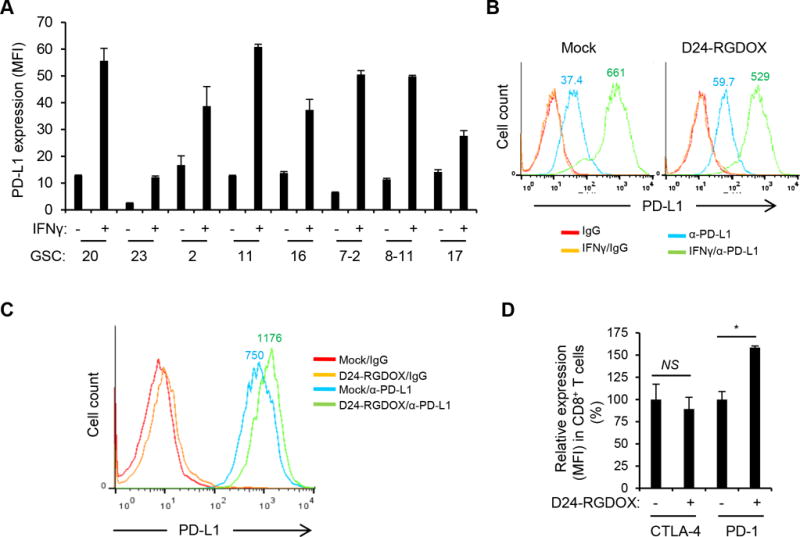
PD-L1/PD-1 expression in glioma or T cells. A, PD-L1 expression in human glioblastoma stem cells (GSCs with serial numbers). Cells were cultured with or without human IFNγ (200 U/ml) for 48 hours and then analyzed with flow cytometry for PD-L1 expression measured by median fluorescence intensity (MFI). Values represent the mean ± standard deviation (n = 3). B, PD-L1 expression in mouse glioma GL261-5 cells. Cells were mock-infected or infected with Delta-24-RGDOX (D24-RGDOX, 100 PFU/cell) in the presence or absence of mouse IFNγ (100 U/ml) for 48 hours and then analyzed with flow cytometry for PD-L1 expression. C, PD-L1 expression in glioma cells from implanted tumors. Fourteen days after the implantation of GL261 cells expressing enhanced green-fluorescent protein (EGFP), Delta-24-RGDOX (D24-RGDOX) was injected intratumorally. After 24 hours, the tumors (taken from 3 mice/group) were harvested, dissociated, and analyzed with flow cytometry for PD-L1 expression. Tumor cells were gated for EGFP+. IgG staining was used as a negative control. The colored numbers indicate the MFI for the curve of the same color in B and C. D, Effect of Delta-24-RGDOX on CTLA-4 and PD-1 expression in CD8+ T cells. Expression of CTLA-4 or PD-1 on the T cells from BILs in glioma-bearing mice treated with PBS or Delta-24-RGDOX as shown in Figure 3A was assessed with flow cytometry. The relative expression levels are shown. The values from the mock-treated (PBS) group were set to 100%. Values represent the mean ± standard deviation (n =3). NS: not significant (P ≥ 0.05); * P = 0.0007, 2-tailed Student’s t test.
To further potentiate efficacy, we combined Delta-24-RGDOX with an anti-PD-L1 antibody to treat the gliomas derived from GL261-5 cells in C57BL/6 mice. We intratumorally injected the antibody to confine its effect mainly in the tumor, 2 days after the first viral dose and 3 days after the second to diminish the potential adverse effects of the antibody on the virus (Figure 7A). The combination resulted in a long-term survival rate of 85%, whereas 2 injections of the virus alone extended the median survival time by 19 days, which corresponded to a long-term survival rate of only 28% (median survival: undefined vs 57 days, P = 0.0001); the antibody alone extended the median survival time by 11 days, which corresponded to a long-term survival rate of only 15% (median survival: undefined vs 49 days, P < 0.0001). The results demonstrated that these two agents synergized to reject the tumor (Figure 7B, P = 0.036).
Figure 7.
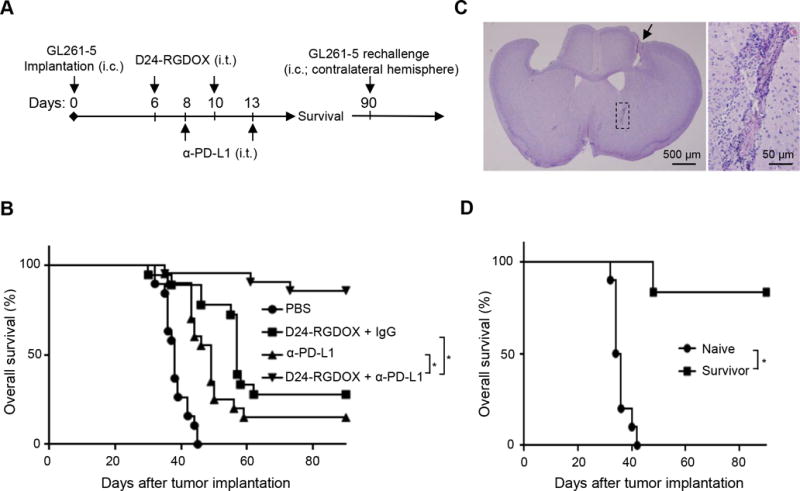
Therapeutic effect of combining Delta-24-RGDOX and anti-PD-L1 antibody. A, A cartoon depiction of the treatment scheme. i.c.: intracranial; i.t.: intratumoral. B, Survival plots of the treatment groups (n = 20, except n = 19 for PBS, n = 18 for D24-RGDOX + IgG). C, Complete tumor regression induced by the combination of Delta-24-RGDOX and anti-PD-L1 antibody (α-PD-L1) in long-term surviving mice. A representative hematoxylin and eosin-stained, whole-mount coronal mouse brain section (left, sacrificed on day 104 after tumor implantation) from the long-term surviving mice treated with the combination is shown. The arrow marks a residue dent left by the screw for guiding the tumor implantation and viral injections. Tumor sequel (marked with dashed lines in the left panel, also enlarged image on the right) is present at the tumor implantation site. D, Survival plots of the group treated with Delta-24-RGDOX together with α-PD-L1 in B when re-challenged with GL261-5 in the contralateral hemisphere rather than the hemisphere with the originally treated tumor. Naive: n = 10; Survivor: n = 6. * P ≤ 0.0001, log-rank test. D24-RGDOX: Delta-24-RGDOX.
In the long-term surviving mice treated with the combination, tumor remnant was found in the brains at the tumor implantation site (Figure 7C), suggesting that the treatment induced complete regression. Moreover, five of the six surviving mice in the combination treatment group also survived a re-challenge with the same tumor cells in the contralateral hemisphere, whereas all naive mice died of gliomas (median survival: undefined vs 35 days, P = 0.0001, Figure 7D). These findings suggest that the combination treatment induced the development of an immune memory that prevented tumor growth at a distant site.
Discussion
In this work, we have developed an effective cancer-targeting immunotherapeutic strategy through constructing an oncolytic adenovirus to express immune co-stimulator OX40L and combining it with an intratumoral injection of an anti-PD-L1 antibody. For the first time, we demonstrate that this strategy is both efficacious and specific for cancer therapy.
Oncolytic viruses have emerged as promising immunotherapeutics for cancer treatment (2). Viral infection of the tumor cells within the tumor mass not only disrupts the immunosuppression to cause local inflammation and activate the innate response but also lead to adaptive anti-tumor immunity (8, 15). To improve the relative dismal efficacy of oncolytic viruses as a single agent in cancer patients, the viruses have been modified to express cytokines or combined with immune checkpoint blockade to upregulate the activity of immune cells (15, 34–37). To this end, in 2015, Amgen’s T-Vec (talimogene laherparepvec), a modified herpes simplex virus type 1 with cancer-selective replication and GM-CSF expression, has shown efficacy in melanoma patients and became the first oncolytic virus to gain approval by US FDA to treat surgically unresectable skin and lymph node lesions in patients with advanced melanoma (38, 39). However, since the cytokines expressed by the viruses can be released to the vicinity of the cells and transported to the whole body through blood and lymphatic circulation, the effect is not cancer cell-specific and poses the risk to globally activate the immune cells, resulting in toxicity. On the contrary, Delta-24-RGDOX expresses an immune co-stimulator on cancer cells, which enhances the capability of the cells to activate T cells recognizing TAAs presented on the cell surface. Therefore, the effect induced by Delta-24-RGDOX is more localized, resulting in specific immunity to cancer cells.
To pursue cancer-specific therapy, efforts have been made to develop therapeutic cancer vaccines. So far, the therapeutic vaccination can only be clinically successful as monotherapy in premalignant or minimal residual disease but not in established cancers (40). Although vaccine strategies have been successful in increasing the frequency and activity of tumor-specific T cells, they have failed to ensure that these T cells could home to tumors and/or exert their function within the tumor because of the immunosuppressive environment within the tumor (40). Unlike just presenting antigens through professional antigen-presenting cells in cancer vaccine therapy, the effect of Delta-24-RGDOX is multiplex. The infection of the cancer cells by the virus releases PAMPs and DAMPs to induce innate immune response within the tumor, changing the tumor microenvironment from immunosuppressive to immune active (8, 9, 41). Before the TAAs from the debris of the cancer cells lysed by the virus are presented through professional antigen-presenting cells, the OX40L expression and IFNγ-mediated expression of MHCs on the tumor cells induced by the virus enhance the role of cancer cells as ad hoc antigen-presenting cells (8). Moreover, since the vaccine strategy only covers a part of the cancer antigen repertoire, after immune editing during the therapy, cancer cells with different antigens can escape and give rise to new tumor cell populations that are resistant to the vaccine therapy (40, 42). On the other hand, Delat-24-RGDOX is designed to infect the whole cancer cell population and can mediate the presentation of the entire cancer antigens to the immune system during the therapy (5–7). This makes the virus promising to overcome the resistance of cancers due to their heterogeneity and therapy-induced evolution of the tumor cells, which are the main challenges in developing targeted cancer therapies.
It has been reported that replication-deficient adenoviruses expressing immunostimulator and cytotoxic genes showed efficacy in a syngeneic rat multifocal glioma model (43–45), suggesting an involvement of anti-tumor immunity induced by the viruses. Furthermore, replication-competent adenoviruses can induce autophagy and cell lysis, which result in immunogenic cell death (29), triggering an innate immune response within the tumor that leads to adaptive anticancer immunity (30). Additionally, unlike a replication-deficient adenoviral vector expressing OX40L (26), Delta-24-RGDOX can preferentially replicate its viral genome in tumor cells (5, 7), suggesting that OX40L expression is more specific to cancer cells. We have demonstrated that Delta-24-RGDOX-infected tumor cells was more efficient than Delta-24-RGD-infected cells to stimulate the proliferation of CD8+ T cells recognizing TAA, suggesting the virus is more potent to enhance in situ expansion of cancer-specific T-cell populations within a tumor (46). Hence, within the tumor microenvironment, Delta-24-RGDOX should be more efficient to enlarge the pool of tumor-specific T cells from the naive repertoire and reactivate existing tumor-specific T cells that may be in a dormant or anergic state. This may be partially responsible for the more remarkable increase of the antitumor activity in BILs than in the splenocytes induced by Delta-24-RGDOX treatment.
PD-L1 expression in glioma cells or tumor-associated macrophages mediates immunosuppression within gliomas (47, 48). To increase the in situ activation of tumor-specific T cells, instead of delivering it systemically in our experimental mouse models, we intratumorally injected the anti-PD-L1 antibody to block this immune checkpoint. The anti-PD-L1 antibody within the tumor should further increase the antigen-presenting function of the tumor cells, and enhance the presentation of TAAs by dendritic cells and macrophages after they engulf and process the debris of cancer cells lysed by the virus. Moreover, viral infection increases the number of nature killer cells at the tumor site (8, 49), which bind the Fc region of anti-PD-L1 antibody and can kill PD-L1-expressing tumor cells via antibody-dependent cellular cytotoxicity (50). Together, this combination of Delta-24-RGDOX with anti-PD-L1 antibody synergistically increases antitumor efficacy and promotes the development of a systemic immune memory that can attack cancer cells in a distant location. This is particularly important in glioblastoma where post-surgical resection and temozolomide therapy, recurrence at distant sites is common, if not inevitable (1).
Because human adenoviruses replicate less efficiently in mouse cells, the ability of our study to recapitulate the actual effect of an oncolytic adenoviral therapy in human patients is limited. To partially compensate for this, we intratumorally injected the virus multiple times over two- or three-day intervals to mimic the viral infection-cell lysis-re-infection cycle. However, the amount of virions does not exponentially escalate as in the human host at the early stage of the viral infection. Thus, the efficacy of the oncolytic adenoviruses may be compromised in the mouse model because fewer virions are available for subsequent re-infection and immunity against viral antigens presented on tumor cells is weaker. Therefore, we expect Delta-24-RGDOX to be even more potent in human patients than in the mouse models.
In summary, the oncolytic adenoviruses expressing immune co-stimulator ligands demonstrate higher anticancer efficacy than their predecessor Delta-24-RGD. Intratumoral injection of Delta-24-RGDOX and an anti-PD-L1 antibody induced a synergistic therapeutic effect in a mouse glioma model. The effect is more targeted and specific to the tumor. This new tumor-targeting combination strategy has exhibited exceptional anti-tumor efficacy and immune memory, and may be translated to other solid tumors or metastatic tumors to offer safer and effective alternative therapies for patients with refractory cancers.
Supplementary Material
Acknowledgments
We thank Xuejun Fan, Verlene K. Henry, Joy Gumin, Kathryn E. Ruisaard and Andrew Dong for technical help and Amy L. Ninetto and Donald R. Norwood for editing the manuscript.
Grant Support: NIH/NCI (P50CA127001 to J. Fueyo and H. Jiang, R01NS069964 to C. Gomez-Manzano, Cancer Center Support Grant P30CA016672), the University Cancer Foundation via the Institutional Research Grant program at The University of Texas MD Anderson Cancer Center (H. Jiang), the Marnie Rose Foundation (J. Fueyo), the Will Power Foundation (J. Fueyo), the Broach Foundation (J. Fueyo), J.P. Harris Brain Tumor Research Fund (J. Fueyo), and DNAtrix Therapeutics (J. Fueyo).
Footnotes
Disclosure of Potential Conflict of Interest: H. Jiang, J. Fueyo and C. Gomez-Manzano are inventors of intellectual property “Adenovirus Expressing Immune Cell Stimulatory Receptor Agonist(s)” licensed by The University of Texas MD Anderson Cancer Center to DNAtrix Therapeutics. J. Fueyo and C. Gomez-Manzano are advisers and shareholders of DNAtrix Therapeutics. The authors have no additional financial interests.
References
- 1.Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, Langer CE, et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol. 2014;16:896–913. doi: 10.1093/neuonc/nou087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14:559–67. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 3.Bambury RM, Morris PG. The search for novel therapeutic strategies in the treatment of recurrent glioblastoma multiforme. Expert Rev Anticancer Ther. 2014;14:955–64. doi: 10.1586/14737140.2014.916214. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki K, Fueyo J, Krasnykh V, Reynolds PN, Curiel DT, Alemany R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin Cancer Res. 2001;7:120–6. [PubMed] [Google Scholar]
- 5.Fueyo J, Alemany R, Gomez-Manzano C, Fuller GN, Khan A, Conrad CA, et al. Preclinical characterization of the antiglioma activity of a tropism-enhanced adenovirus targeted to the retinoblastoma pathway. J Natl Cancer Inst. 2003;95:652–60. doi: 10.1093/jnci/95.9.652. [DOI] [PubMed] [Google Scholar]
- 6.Jiang H, Gomez-Manzano C, Aoki H, Alonso MM, Kondo S, McCormick F, et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: role of autophagic cell death. J Natl Cancer Inst. 2007;99:1410–4. doi: 10.1093/jnci/djm102. [DOI] [PubMed] [Google Scholar]
- 7.Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 8.Jiang H, Clise-Dwyer K, Ruisaard KE, Fan X, Tian W, Gumin J, et al. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS One. 2014;9:e97407. doi: 10.1371/journal.pone.0097407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kleijn A, Kloezeman J, Treffers-Westerlaken E, Fulci G, Leenstra S, Dirven C, et al. The in vivo therapeutic efficacy of the oncolytic adenovirus Delta24-RGD is mediated by tumor-specific immunity. PLoS One. 2014;9:e97495. doi: 10.1371/journal.pone.0097495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lang FF, Conrad CA, Gomez-Manzano C, Tufaro F, Yung WKA, Sawaya R, et al. First-in-human Phase I clinical trial of oncolytic Delta-24-RGD (DNX-2401) with biological endpoints: implications for viro-immunotherapy. Neuro Oncol. 2014;16:iii39. [Google Scholar]
- 11.Jiang H, Gomez-Manzano C, Rivera-Molina Y, Lang FF, Conrad CA, Fueyo J. Oncolytic adenovirus research evolution: from cell-cycle checkpoints to immune checkpoints. Curr Opin Virol. 2015;13:33–39. doi: 10.1016/j.coviro.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 14.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 15.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6:226ra32. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dias JD, Hemminki O, Diaconu I, Hirvinen M, Bonetti A, Guse K, et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012;19:988–98. doi: 10.1038/gt.2011.176. [DOI] [PubMed] [Google Scholar]
- 17.Yao S, Zhu Y, Chen L. Advances in targeting cell surface signalling molecules for immune modulation. Nat Rev Drug Discov. 2013;12:130–46. doi: 10.1038/nrd3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. 2015;33:1974–1982. doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134) Annu Rev Immunol. 2010;28:57–78. doi: 10.1146/annurev-immunol-030409-101243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaer DA, Murphy JT, Wolchok JD. Modulation of GITR for cancer immunotherapy. Curr Opin Immunol. 2012;24:217–24. doi: 10.1016/j.coi.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kjaergaard J, Tanaka J, Kim JA, Rothchild K, Weinberg A, Shu S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res. 2000;60:5514–21. [PubMed] [Google Scholar]
- 22.Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013;73:7189–98. doi: 10.1158/0008-5472.CAN-12-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moran AE, Kovacsovics-Bankowski M, Weinberg AD. The TNFRs OX40, 4-1BB, and CD40 as targets for cancer immunotherapy. Curr Opin Immunol. 2013;25:230–7. doi: 10.1016/j.coi.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen LHX. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–91. doi: 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LaFrance-Corey RG, Howe CL. Isolation of brain-infiltrating leukocytes. J Vis Exp. 2011:e2747. doi: 10.3791/2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inaba K, Swiggard WJ, Steinman RM, Romani N, Schuler G. Current Protocols in Immunology. John Wiley & Sons, Inc.; 1998. Isolation of Dendritic Cells; pp. 3.7.1–3.7.15. [DOI] [PubMed] [Google Scholar]
- 27.Quah BJ, Parish CR. New and improved methods for measuring lymphocyte proliferation in vitro and in vivo using CFSE-like fluorescent dyes. J Immunol Methods. 2012;379:1–14. doi: 10.1016/j.jim.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 28.Team RC. R: A language and environment for statistical computing. 2013 Available from: http://www.R-project.org/
- 29.Jiang H, White EJ, Rios-Vicil CI, Xu J, Gomez-Manzano C, Fueyo J. Human adenovirus type 5 induces cell lysis through autophagy and autophagy-triggered caspase activity. J Virol. 2011;86:4720–29. doi: 10.1128/JVI.02032-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–75. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 31.Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6:e10. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipford GB, Hoffman M, Wagner H, Heeg K. Primary in vivo responses to ovalbumin. Probing the predictive value of the Kb binding motif. J Immunol. 1993;150:1212–22. [PubMed] [Google Scholar]
- 33.Mousavi SF, Soroosh P, Takahashi T, Yoshikai Y, Shen H, Lefrancois L, et al. OX40 costimulatory signals potentiate the memory commitment of effector CD8+ T cells. J Immunol. 2008;181:5990–6001. doi: 10.4049/jimmunol.181.9.5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freytag SO, Barton KN, Zhang Y. Efficacy of oncolytic adenovirus expressing suicide genes and interleukin-12 in preclinical model of prostate cancer. Gene Therapy. 2013;20:1131–9. doi: 10.1038/gt.2013.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du T, Shi G, Li YM, Zhang JF, Tian HW, Wei YQ, et al. Tumor-specific oncolytic adenoviruses expressing granulocyte macrophage colonystimulating factor or anti-CTLA4 antibody for the treatment of cancers. Cancer Gene Therapy. 2014;21:340–8. doi: 10.1038/cgt.2014.34. [DOI] [PubMed] [Google Scholar]
- 36.Grossardt C, Engeland CE, Bossow S, Halama N, Zaoui K, Leber MF, et al. Granulocyte-macrophage colony-stimulating factor-armed oncolytic measles virus is an effective therapeutic cancer vaccine. Human Gene Therapy. 2013;24:644–54. doi: 10.1089/hum.2012.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JH, Roh MS, Lee YK, Kim MK, Han JY, Park BH, et al. Oncolytic and immunostimulatory efficacy of a targeted oncolytic poxvirus expressing human GM-CSF following intravenous administration in a rabbit tumor model. Cancer Gene Therapy. 2010;17:73–9. doi: 10.1038/cgt.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol. 2015;33:2780–8. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 39.Sheridan C. First oncolytic virus edges towards approval in surprise vote. Nat Biotechnol. 2015;33:569–70. doi: 10.1038/nbt0615-569. [DOI] [PubMed] [Google Scholar]
- 40.van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer. 2016;16:219–33. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- 41.Jiang H, Fueyo J. Healing after death: antitumor immunity induced by oncolytic adenoviral therapy. Oncoimmunology. 2014;3:e947872. doi: 10.4161/21624011.2014.947872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 43.Ali S, King GD, Curtin JF, Candolfi M, Xiong W, Liu C, et al. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer Res. 2005;65:7194–204. doi: 10.1158/0008-5472.CAN-04-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.King GD, Muhammad AK, Curtin JF, Barcia C, Puntel M, Liu C, et al. Flt3L and TK gene therapy eradicate multifocal glioma in a syngeneic glioblastoma model. Neuro Oncol. 2008;10:19–31. doi: 10.1215/15228517-2007-045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.King GD, Kroeger KM, Bresee CJ, Candolfi M, Liu C, Manalo CM, et al. Flt3L in Combination With HSV1-TK-mediated Gene Therapy Reverses Brain Tumor-induced Behavioral Deficits. Mol Ther. 2008;16:682–690. doi: 10.1038/mt.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thompson ED, Enriquez HL, Fu YX, Engelhard VH. Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J Exp Med. 2010;207:1791–804. doi: 10.1084/jem.20092454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–8. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 48.Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res. 2013;19:3165–75. doi: 10.1158/1078-0432.CCR-12-3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brandstadter JD, Yang Y. Natural killer cell responses to viral infection. J Innate Immun. 2011;3:274–9. doi: 10.1159/000324176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Furness AJ, Vargas FA, Peggs KS, Quezada SA. Impact of tumour microenvironment and Fc receptors on the activity of immunomodulatory antibodies. Trends Immunol. 2014;35:290–8. doi: 10.1016/j.it.2014.05.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.