

ABSTRACT
Reverse transcription–quantitative polymerase chain reaction (RT-qPCR) is one of the most sensitive, economical and widely used methods for evaluating gene expression. However, the utility of this method continues to be undermined by a number of challenges including normalization using appropriate reference genes. The need to develop tailored and effective strategies is further underscored by the burgeoning field of extracellular vesicle (EV) biology. EVs contain unique signatures of small RNAs including microRNAs (miRs). In this study we develop and validate a comprehensive strategy for identifying highly stable reference genes in a therapeutically relevant cell type, cardiosphere-derived cells. Data were analysed using the four major approaches for reference gene evaluation: NormFinder, GeNorm, BestKeeper and the Delta Ct method. The weighted geometric mean of all of these methods was obtained for the final ranking. Analysis of RNA sequencing identified miR-101-3p, miR-23a-3p and a previously identified EV reference gene, miR-26a-5p. Analysis of a chip-based method (NanoString) identified miR-23a, miR-217 and miR-379 as stable candidates. RT-qPCR validation revealed that the mean of miR-23a-3p, miR-101-3p and miR-26a-5p was the most stable normalization strategy. Here, we demonstrate that a comprehensive approach of a diverse data set of conditions using multiple algorithms reliably identifies stable reference genes which will increase the utility of gene expression evaluation of therapeutically relevant EVs.
KEYWORDS: Extracellular vesicles, microRNAs, cardiosphere-derived cells, CDCs, miRs, reference genes, qPCR, stem cells, RT-qPCR
Introduction
The assessment of gene expression using reverse transcription–quantitative polymerase chain reaction (RT-qPCR) is a central tool for evaluating gene expression and can inform mechanistic and therapeutic discovery. A fundamental prerequisite to the reliability of this method is the selection of appropriate endogenous reference genes; that is, genes that are constitutively active, abundant and stable across different conditions [1].
Extracellular vesicles (EVs) are a broad class of lipid bilayer particles that are secreted by nearly all prokaryotic and eukaryotic cells [2]. EVs represent versatile autocrine [3], paracrine [4] and endocrine [5] mediators of cell communication, and play central roles in physiology and disease [6]. Subpopulations of EVs, including exosomes and shedding vesicles (microvesicles, apoptotic bodies), have distinct origins, size distributions and molecular content, and play different physiological roles. EVs contain diverse signalling mediators including lipids, proteins and nucleic acids. Recent investigations suggest that the relative abundance and species of these macromolecules differ across the different classes of EVs. Furthermore, factors such as disease state and tissue type will affect the “signature” of secreted cargoes and therefore the physiological effect that they impart. These observations underscore the importance of identifying suitable reference genes to evaluate the changes in the EV portion of the secretome.
However, the identification of reference genes for EVs remains challenging, especially as the source of the EVs becomes more complex. For instance, identifying reference genes for multiple-tissue-derived EVs such as those isolated from bodily fluids (e.g. serum) is more challenging than identifying reference genes for EVs derived from single cell types. Thus, different groups have identified reference genes for their specific EV populations, including liver carcinoma [7], colorectal cancer [8] and cerebrospinal fluid [9]. In the majority of these studies, one reference gene is not sufficient but rather the mean of multiple genes is necessary. Furthermore, in the majority of these investigations, the primary candidates for consideration are microRNAs (miRs), evolutionarily conserved non-coding RNAs that regulate gene expression through specifically targeting nascent messenger RNA (mRNA) peptides [10] and mediating epigenetic modification [11]. The miRs are a promising class for reference gene investigation because of their extraordinary evolutionary conservation and fundamental involvement in homeostatic pathways such as metabolism.
The field of EV molecular biology is beginning to appreciate the need to identify reference genes for tissue- and disease-specific EVs; currently, most groups opt to use the U6 spliceosomal nuclear RNA (among others). Also highly conserved, this non-coding fragment of the ribonuclear protein of the same name mediates splicing of pre-mRNA [12]. However, recent investigation of serum RNA in healthy and diseased patients and animals revealed high variability [13,14]. This poses a challenge to both the basic and translational fields of EV biology. Using non-suitable reference genes further hampers our understanding of the role of the genetic constituents of EVs through aberrant expression profiles. Furthermore, the field of EV diagnostics relies on reference genes to identify diagnostic candidates and correlate trends. From the perspective of EV therapeutics, it is important to develop assays that vet manufacturing processes towards increased product purity and potency and, with the paucity of reliable in vitro assays, rely extensively on following the expression level of certain potency-linked EV-derived gene constituents.
Cardiosphere-derived cells (CDCs) are a population of adult cardiac tissue-derived cells that have been shown in multiple animal models and human trials to regenerate the myocardium after infarction [15]. Furthermore, the primary mechanism by which these cells impart their effect is indirect; that is, through the secretion of EVs which deliver signals, including miRs, to the injured microenvironment [16,17]. In the present study, we identify suitable reference genes in EVs isolated from CDCs (CDC-EVs) and compare them to conventional reference genes including U6, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The methods employed here include the four top methods for reference gene identification: NormFinder, GeNorm, BestKeeper and Delta Ct. NormFinder is a model-based approach that considers intragroup and intergroup variability when ranking stability [18]. GeNorm determines pairwise standard deviation values of all genes and eliminates the least stable genes until only two remain, which are considered the most stable [19]. BestKeeper generates an index of stability based on quantification cycle (Cq) values and amplification efficiencies followed by a pairwise correlation analysis to rank each of the candidates in the index [20]. The Delta Ct method assigns stability based on Cq standard deviation differences for each pairwise comparison [21].
Methods
Human CDC culture
Atria and ventricular septa were obtained from healthy hearts of deceased tissue donors. Tissue was chopped, mixed in a 1:4 atrium to septum ratio, washed and seeded on CellBIND flasks (Corning, NY, USA). Explants were incubated at 37°C, 5% carbon dioxide (CO2), 5% oxygen (O2) in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 20% foetal bovine serum (FBS) for 2–3 weeks until outgrowth reached 80% confluence. Cells were then harvested using TrypLE Select (Thermo Fisher Scientific, Waltham, MA, USA), filtered through a 100 μm Steriflip unit (Millipore, Billerica, MA, USA) to remove explants, and resuspended in CryoStor CS10 (STEMCELL Technologies) before freezing in liquid nitrogen.When needed, a frozen vial was removed from the liquid nitrogen and seeded on Ultra-Low attachment flasks (Corning, NY, USA) to form cardiospheres.CDCs were formed by seeding cardiospheres on fibronectin-coated flasks and culturing at 37°C, 5% CO2, 5% O2 in IMDM supplemented with 10% FBS. Cells were conditioned at passage 5 or subjected to a second cardiosphere step and conditioned over two passages after reculturing on fibronectin-coated plates. Human heart biopsy specimens, from which CDCs were grown, were obtained under a protocol approved by the institutional review board for research on human subjects.
Dataset 1: sample preparation
This data set of 10 samples was prepared, comprising six unique human CDC-EVs. Each EV population was prepared from CDCs at passage 5 and conditioned for 5 days at 20% Ohuman CDC-EVs. Each EV population was prepared from CDCs at passage 5 and conditioned for 5 days at 20% O2. All samples were cultured at 37°C and 5% CO2 for growth and conditioning. An additional sample from donor 1 also had EVs prepared from the cardiospheres themselves. Two CDC donors also had additional samples that were conditioned for 15 days. Cells were cultured to confluence in a T175 flask precoated with 20 μg/ml of human fibronectin in IMDM basic medium (Gibco, Waltham, MA, USA) supplemented with 20% FBS (HyClone, Logan, UT, USA), 1% penicillin/streptomycin and 0.1 ml 2-mercaptoethanol.At confluence, flasks were rinsed three times with phosphate-buffered saline and 15 ml of IMDM (Gibco, Waltham, MA, USA). Five or 15 days after conditioning in serum-free medium (IMDM only), the medium was centrifuged for 15 min at 3000 × g to pellet dead cells and debris. Conditioned medium was then stored at −80°C for RNA isolation. Along with the CDC groups, one normal human dermal fibroblast-derived EV sample was also sequenced (as a therapeutically inert control) (Supplementary Table S1). All samples in this data set were isolated using the Norgen Urine Exosome Isolation Kit (Norgen Biotek Corp., Toronto, Canada). This is a column-based method that uses silica-based slurry to bind vesicles in the medium, followed by lysis and RNA extraction. This kit was selected for sequencing to ensure a pure and efficient retrieval of EV RNA. Previous studies have validated this non-target-specific bead-binding method against the ultracentrifugation method which remains the standard in EV isolation [22].
Exosome RNA sequencing
Sequencing was performed by the Cedars-Sinai Genomics Core (Los Angeles, CA, USA). Library construction was performed according to the manufacturer’s protocols using the Ion Total RNA-Seq Kit v2 (Life Technologies, Carlsbad, CA, USA). One microgram of total RNA was assessed for quality using the Agilent Bioanalyzer 2100, enriched with magnetic beads, fragmented, ligated with adapters and reverse transcribed to make complementary DNA (cDNA). The resulting cDNA was barcoded using the Ion Xpress™ RNA-Seq Barcode 1–16 Kit and then amplified. RNA-sequencing libraries were assessed for concentration (Qubit dsDNA HS Assay Kit; Invitrogen, Carlsbad, CA, USA) and size (DNA 1000 Kit; Agilent, Santa Clara, CA, USA).Samples were multiplexed and amplified (pooled libraries) on to proprietary sphere particles using the Ion PI Template OT2 200 Kit. Particles were then purified and prepared (Ion PI Sequencing 200 Kit) for sequencing on an Ion Proton sequencer. The raw sequencing signal (FastQ) was processed and quality control was conducted using FastQC [23]. The adaptor was trimmed (using Torrent Suite software) to obtain 5 million reads per sample.
Dataset 2: sample preparation
A data set of 19 samples was prepared, comprising seven unique donor CDC-EVs prepared under various conditions, including oxygen concentration at conditioning (2%, 5% and 20%) and days of conditioning (1 day, 5 days and 15 days) (Table5% and 20%) and days of conditioning (1 day, 5 days and 15 days) (Table S2).
NanoString
EVs were isolated from conditioned media using a 10 kDa Amicon regenerated cellulose filter (Millipore, Billerica, MA, USA). Total RNA was isolated from the filtrate using a miRNeasy Mini Kit (Qiagen, Hilden, Germany) as per the manufacturer’s instructions. RNA concentration was quantified (with the help of the Cedars-Sinai Genomics Core) using the AATI Fragment Analyzer (Advanced Analytical Technologies, Ankeny, IA, USA; supplementary Figure S1) or the Qubit MicroRNA assay (Thermo Fisher Scientific, Waltham, MA, USA).The NanoString (nCounter, Human v2 miRNA Assay; NanoString Technologies, Seattle, WA, USA) was used to analyze 15–150 ng of total RNA from each sample. The code set contained 800 probes for mature miR detection from humans based on miRBase version 17. Raw counts were obtained and used for analysis. All values that fell below the average negative control counts (background) were excluded from analysis.
RT-qPCR
Specificity and efficiency
For specificity, cDNA was prepared from CDC-EV RNA using the QuantiMir Kit (SBI) according to the manufacturer’s instructions. For qPCR, the sequence of miR-101-3p, miR-26a-5p and miR-23a-3p was used as forward primers, and QuantiTect SYBR Green (QiagenmiR-26a-5p and miR-23a-3p was used as forward primers, and QuantiTect SYBR Green (Qiagen, Hilden, Germany) was used as an intercalating DNA dye. Dissociation curves were prepared by increasing the temperature using the standard protocol on the QuantStudio 12K Flex system and measuring fluorescence. A single peak in the first derivative of the dissociation curve indicates specificity. The efficiency of the qPCR was assessed using a standard curve prepared from serial dilutions of CDC-EV RNA using TaqMan MicroRNA primers and kits (Thermo Fisher Scientific, Waltham, MA, USA) on a QuantStudio 6K system (Thermofisher Scientific, Waltham, MA, USA). Linear regression calculations were performed using GraphPad Prism 5. Efficiency was calculated from the slope of the standard curve using the equation: Efficiency = −1 + 10(−1/slope).
Total RNA was isolated from CDC-EVs using the Qiagen miRNeasy mini kit, following the manufacturer’s protocol. RNA concentration was quantified (with the help of the Cedars-Sinai Genomics Core) using the AATI Fragment Analyzer (Advanced Analytical Technologies, Ankeny, IA, USA) or the Qubit MicroRNA assay for the U6, miR-101-3p and miR-26a-5p samples or the miR-23a-3p samples, respectively. RT-qPCR was performed using TaqMan MicroRNA primers and kits (Thermo Fisher Scientific, Waltham, MA, USA), and the Applied Biosystems ViiA7 or QuantStudio 12K Flex real-time PCR systems in standard run mode for the U6, miR-101-3p and miR-26a-5p samples or the miR-23a-3p samples, respectively. The TaqMan MicroRNA assay kit contains a specific reverse transcription primer and a specific qPCR primer for each miRNA. Reverse transcription was performed using the TaqMan MicroRNA reverse transcription kit. Each reverse transcription reaction used 10 ng of input RNA, and the reaction was set up according to the manufacturer’s specifications. In brief, a master mix was prepared with 0.15 μl of 100 mM deoxynucleoside triphosphate (dNTP), 1 μl of MultiScribe reverse transcriptase (50 U/μl) (Thermo Fisher Scientific, Waltham, MA, USA), 1.5 μl of 10 × reverse transcription buffer, 0.19 μl of RNase inhibitor (20 U/μl) and 4.1 μl of nuclease-free water per reaction. Then, 7 μl of master mix was combined with 3 μl of 5 × reverse transcription TaqMan assay primer and 5 μl of RNA (10 ng total input). Thermal cycling conditions for reverse transcription were as follows: 16°C for 30 min, 42°C for 30 min, 85°C for 5 min and 4°C hold. Subsequent qPCR was performed using 1.33 μl of cDNA and the TaqMan Fast Advanced Master Mix according to the manufacturer’s specifications. In brief, each qPCR reaction consisted of 10 μl of 2 × TaqMan Fast Advanced Master Mix, 1 μl of 20 × qPCR TaqMan assay primer, 7.67 μl of nuclease-free water and 1.33 μl of cDNA. Thermal cycling for qPCR was performed in Fast mode as follows: 50°C for 2 min, 95°C for 20 s, and 40 cycles of 95°C for 1 s and 60°C for 20 s.Fast mode as follows: 50°C for 2 min, 95°C for 20 s, and 40 cycles of 95°C for 1 s and 60°C for 20 s.
1.
Primer sequences for miRs and U6.
| Gene | Sequence |
|---|---|
| hU6 | GUGCUCGCUUCGGCAGCACAUAUACUAAAAUUGGAACGAUACAGAGAAGAUUAGCAUGGCCCCUGCGCAAGGAUGACACGCAAAUUCGUGAAGCGUUCCAUAUUUU |
| hsa-miR-23a-3p | AUCACAUUGCCAGGGAUUUCC |
| hsa-miR-101-3p | UACAGUACUGUGAUAACUGAA |
| hsa-miR-26a-5p | UUCAAGUAAUCCAGGAUAGGCU |
Reference gene identification
To identify reference genes specific to CDC EVs, we evaluated two different data sets: next-generation sequencing data from six unique donor sources and miR expression data by the NanoString ncounter from seven unique donor sources. Candidates were identified using three algorithms for gene stability determination: NormFinder ncounter from seven unique donor sources. Candidates were identified using three algorithms for gene stability determination: NormFinder [18], GeNorm [19] and the comparative Delta Ct method [21]. The four algorithms used here were collectively available using RefFinder, a web-based tool developed for evaluating and screening reference genes from experimental data sets, then applying a weighted geometric mean to produce a consolidated list of the various ranking methods [24]. A link to the web-based software is provided at: http://fulxie.0fees.us/
Electron microscopy
Negative stain electron microscopy
Each sample was diluted 100 × using pure water. We used 2% uranyl acetate as the staining solution. Samples were mounted on a glow-discharge carbon-coated grid, using the PELCO easiGlow Glow Discharge Cleaning System (Ted Pella, Redding, CA, USA). The current used was 15 mA and grids were glow-discharged for 30 s. The glow-discharged grid was held using reverse-force anti-capillary forceps with the carbon-coated side facing upwards, then 2.5 μl of each sample was applied to the glow-discharge grid and incubated for 60 s. The sample was blotted off using filter paper and then the grid was briefly contacted twice with the stain, followed by additional blotting with filter paper. This was procedure was repeated a second time. The grid was then left to dry for 5 min.
Single-particle cryo-electron microscopy
CDC-EV samples were prepared for cryo-electron microscopy as previously described [25] and imaged using a T12 Quick CryoEM and CryoET (FEI) at the UCLA Electron Imaging Center for NanoMachines (EICN) facility.
Western blot
Western blot was used to detect the presence of specific EV proteins in CDC-EV preparations. In brief, CDC-EV preparations were lysed using radioimmunoprecipitation assay (RIPA) buffer at a 1:1 ratio of EV sample to RIPA buffer. Total protein was quantified using the DC protein assay (Bio-Rad, Hercules, CA, USA) to normalize the amount of protein loaded into each well. Samples were separated using gel electrophoresis with Bolt 4–12% gradient Bis-Tris Plus Gels (Thermofisher Scientific, Waltham, MA, USA) under non-reducing conditions. Separated proteins were transferred to a nitrocellulose membrane using the iBlot Western system (Thermofisher Scientific, Waltham, MA, USA). Membranes were then stained with SYPRO Ruby blot stain (Thermofisher Scientific, Waltham, MA, USA) to ensure uniform transfer of protein from the gel to the membrane in all lanes. Finally, membranes were destained using the iBind blocking buffer and probed for various proteins using the iBind system. All secondary antibodies were horseradish peroxidase conjugated and the signal was generated using the West Pico substrate. Images were acquired using the Universal Hood III with the Chemi high-sensitivity setting (Bio-Rad, Hercules, CA, USA).
Acetylcholinesterase assay
Acetylcholinesterase (AChE) activity was measured using a fluorescence-based FluoroCet assay kit (SBI, Palo Alto, CA, USA) according to the manufacturer’s instructions. Fluorescence values were quantitated using an MDC SpectraMax Gemini EM Microplate Fluorescence reader (Molecular Devices, Sunnyvale, CA, USA). Samples were measured in triplicate and all fluorescence values were background subtracted (Plasma-Lyte vehicle; Shire, Los Angeles, CA, USA) and graphed as AChE activity per microgram of EV input. An input amount of 40 pg purified AChE from the kit was used as a positive control for the assay (data not shown). The protein quantification was performed using the Quick Start Bradford Protein Assay (Bio-Rad, Hercules, CA, USA).
Nanoparticle tracking analysis
The number and size of EV particles were evaluated using nanoparticle tracking analysis on a NanoSight NS300 instrument (Malvern Instruments, Malvern, UK) as per the manufacturer’s instructions. The following parameters were used for acquisition: camera level = 15, detection level = 4, number of videos taken = 5 and video length = 60 s.
Results
EVs were isolated from CDCs under serum-free conditions and concentrated using a centrifugation-based ultrafiltration system. Isolated particles showed typical size distribution, as measured by diffraction light scattering using nanoparticle tracking analysis of three different CDC donor sources (Figure 1(a)). EV output was also similar between different donors. This was further confirmed by negative stain and cryo-electron microscopy showing the size and morphology of vesicles described previously in the literature (Figure 1(b)) [26]. The presence of EV proteins and the absence of cellular cytoplasmic proteins were confirmed using CD81 and the endoplasmic reticulum marker calnexin, respectively. Periostin is a protein enriched in EVs and shown to bind integrins (Figure 1(c)). Finally, an AChE assay was performed to demonstrate functionality and confirm enzymic activity (Figure 1(d)).
Figure 1.
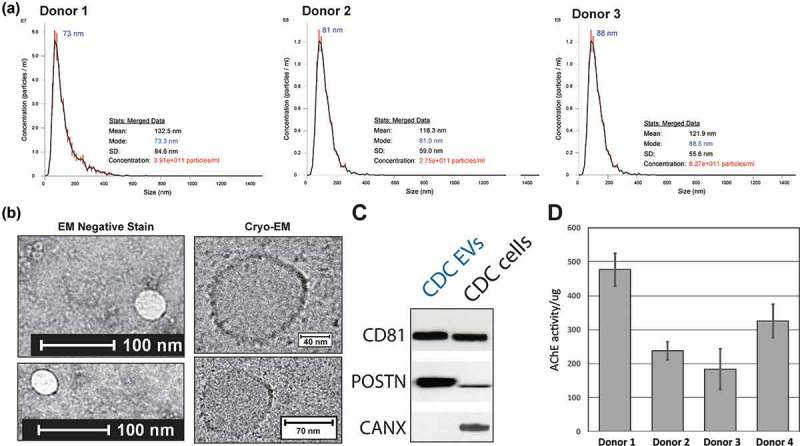
Characterization of cardiosphere-derived cell (CDC) extracellular vesicles (EVs). (a) Nanotracking analysis of EVs derived from three donor sources. (b) Electron microscopy (EM) and cryo-EM of CDC-EVs (negative-stain images in left-hand panels). (c) Western blot showing the presence of conserved tetraspanin CD81 and Periostin, and the absence of the endoplasmic reticulum marker Calnexin. (d) Bioactivity of CDC EVs as shown by acetylcholinesterase (AChE) activity. All fluorescence values were background-subtracted (Plasma-Lyte vehicle) and graphed as AChE activity per microgram of EV input. An input amount of 40 pg purified AChE from the kit was used as a positive control for the assay.
RNA sequencing data (data set 1) and NanoString data (data set 2) were assessed using NormFinder, GeNorm, BestKeeper and the Delta Ct method. Commonly identified miRs were then chosen to undergo validation by RT-qPCR (Figure 2). Analysis of the sequencing data set revealed differences in RNA class distribution among the analysed donors. Each CDC-EV sample was derived from cells from a unique donor, which may contribute to the different distributions of RNA (Figures S1.1 and 1.2, and Table S1). Indeed, we have shown previously that CDCs vary in a number of characteristics (e.g. CD90 expression), which could affect their therapeutic efficacy in vivo [27]. Reference gene analyses revealed similar results among the NormFinder, GeNorm and Delta Ct methods (Figure 3(a–c)). All three methods identified miR-101-3p, miR-23a-3p, miR-23b-3p, miR-107a, miR-376a-3p and miR-376c-3p as being among the most stable genes. BestKeeper yielded significantly different results, with miR-101-3p and miR-376c-3p being the only species in common with the other three methods (Figure 3(d)). The geometric mean of all four methods was used as the final list of candidates from this data set (Figure 3(e)). All candidates showed greater stability than the current standard reference gene, U6. In addition, miR-26a-5p was identified as one of the most stable genes. Previous studies have identified miR-26a-5p and miR-23a/b-3p in searching for reference genes for serum EVs [28] as well as other tissue [29–31]. Abundance values for the top candidate genes were then obtained, as low abundance is likely to yield a high Cq value and inconsistent results, or fall below the level of detection (Figure 3(f)). The gene miR-23a/b showed the highest abundance of all miR candidates identified.
Figure 2.
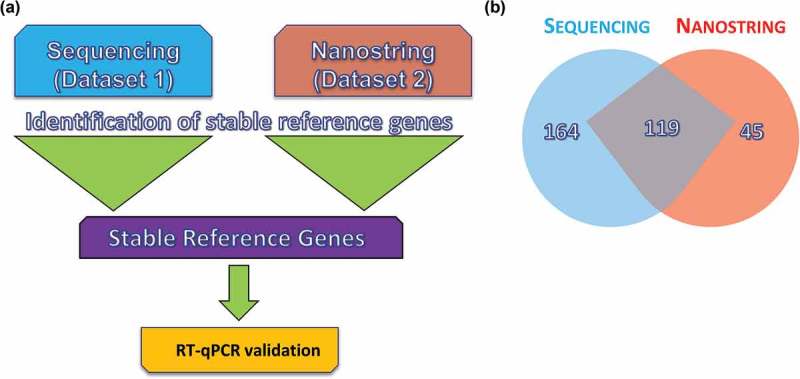
Workflow for the reference gene identification method. Reference genes were identified using small RNA sequencing and a chip-based method. Each data set was unique and included different donors and diverse conditions. Identification of reference genes from each data set was conducted in parallel using the four major algorithms for reference gene identification (NormFinder, GeNorm, BestKeeper and Delta Ct). (a) Common microRNAs (miRs) were selected from each set for further validation using reverse transcription–quantitative polymerase chain reaction (RT-qPCR) in a third unique sample set. (b) Venn diagram showing miRs identified by sequencing compared to those identified by NanoString. Data are representative of the two donors in common between data sets 1 and 2.
Figure 3.
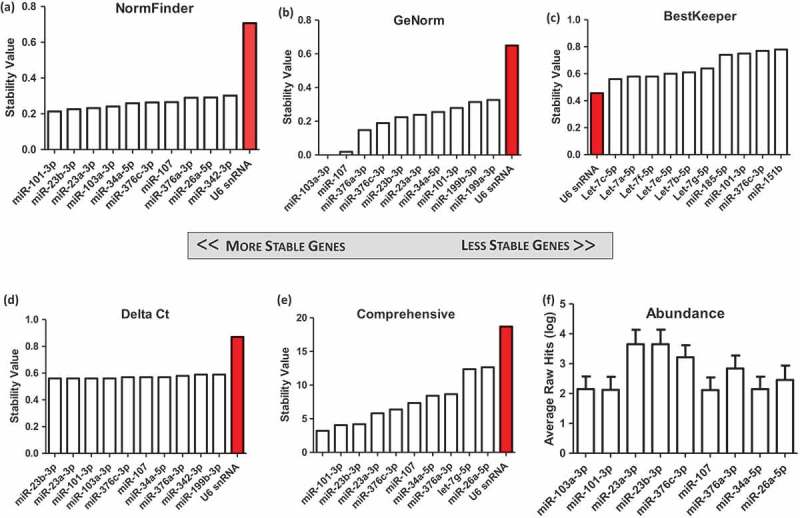
Identification of candidate genes from next-generation small RNA sequencing of nine samples, from six unique cardiosphere-derived cell (CDC) donors and a fibroblast line. All CDC extracellular vesicle (EV) samples were harvested from cells at passage 5, conditioned for 5 days in serum-free media in 20% O2. Two donors also had samples isolated from cells conditioned for 15 days. One donor also had exosomes isolated from cardiospheres conditioned for 5 days. Data were analysed using (a) NormFinder, (b) GeNorm, (c) BestKeeper, and (d) Delta Ct. (e) The weighted geometric mean of each of these samples was taken to provide a consolidated list of the most stable genes. (f) The average of non-normalized hits for each gene was obtained as a measure of abundance.
NanoString miR counts from 19 samples comprising seven unique CDC donor sources (Table S3) revealed a notably different set of candidate genes (Figure 4(a–c)). GeNorm and BestKeeper identified miR-149-5p, miR-1972-5p, miR-127-5p and let-7c-5p as the most stable genes (Figure 4(b,c)), while NormFinder and Delta Ct identified miR-23a, miR-382, let-7c, miR-873 and miR-1972 as the top five most stable genes. The geometric mean of the four methods identified miR-23a-3p as the strongest candidate reference gene (Figure 4(d)) and, in this data set, this was also the most abundant of all candidate genes (Figure 4(f)).
Figure 4.
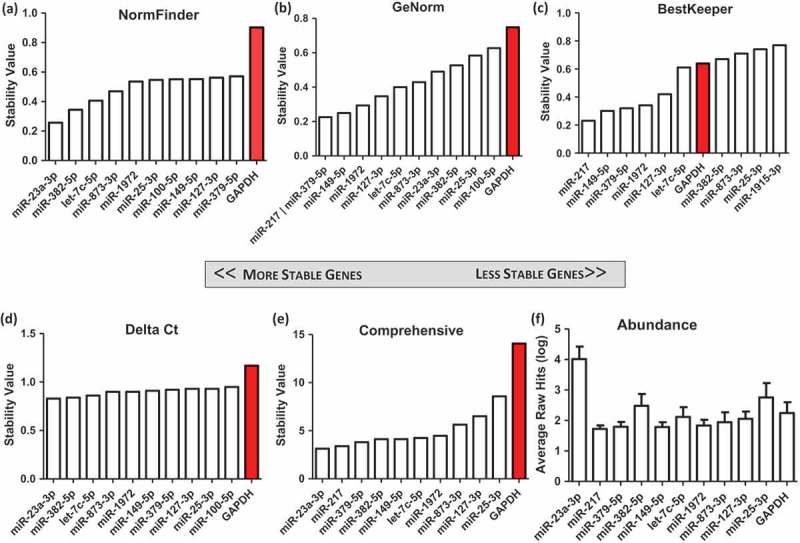
Identification of candidate genes from NanoString absolute microRNA (miR) quantification data of 19 samples, from seven unique cardiosphere-derived cell (CDC) donors and a fibroblast control. All CDC extracellular vesicle (EV) samples were harvested from cells at passage 5, conditioned for 15 days in serum-free media in 5% O2. One donor also had two extra samples of EVs from cells conditioned for 24 h in 2% and 5% O2. Data were analysed using (a) NormFinder, (b) GeNorm, (c) BestKeeper, and (d) Delta Ct. (e) The weighted geometric mean of each of these samples was taken to provide a consolidated list of the most stable genes. (f) The average of non-normalized hits for each gene was obtained as a measure of abundance.
Comparisons were drawn between the results of both analyses to establish common genes. In comparing the two data sets, miR-23a-3p was consistently identified as a strong reference candidate. Between the two approaches, sequencing is the more sensitive approach for gene identification, and because of the variable RNA input into the NanoString system, the abundance of some miRs may not be detected by this probe-based approach. Therefore, along with validating miR-23a-3p by RT-qPCR, we also included miR-26a-5p, as it was identified by sequencing data and previously identified in the literature. In addition, miR-101-3p was included as it was identified as being more stable than miR-23a-3p in the sequencing data set. Central to choosing multiple reference genes is avoiding the possibility of co-regulation. To minimize this possibility, we chose genes that belong to different miR families and had no genomic clustering (defined by miRBase as 10 kb or less). For this reason, miR-23b was excluded from further analysis as it belongs to the miR-23 family (Table S4).
To assess the quality of miR-23a-3p, miR-26a-5p and miR-101-3p qPCR assays, efficiency and specificity were first analysed. Efficiencies were 94.3% (miR-23a-3p), 98.4% (miR-26a-5p) and 115.6% (miR-101-3p) (Figure S2.1). The SYBR Green dissociation curve shows one peak for each miR, indicating the specificity of the primers (Figure S2.2). The three candidates were investigated in a third group of samples to assay gene stability. This sample set included EVs prepared under diverse conditions including oxygen concentration at the time of conditioning, donor source, days of conditioning and cell passage at time of conditioning (Table S3). Figure 5(a) shows the stability of Cq values across the nine samples. To evaluate the suitability of multiple reference miRs compared to single miRs, we included permutations of the arithmetic means of miR-23a-3p, miR-101-3p and miR-26a-5p. U6 showed the greatest fluctuation, while the combination of miR-23a-3p, miR-26a-5p and miR-101-3p showed the greatest stability. Analysis by all four methods confirmed the greater stability of these three reference genes compared to each of them individually (Figure 5(b–f)).
Figure 5.
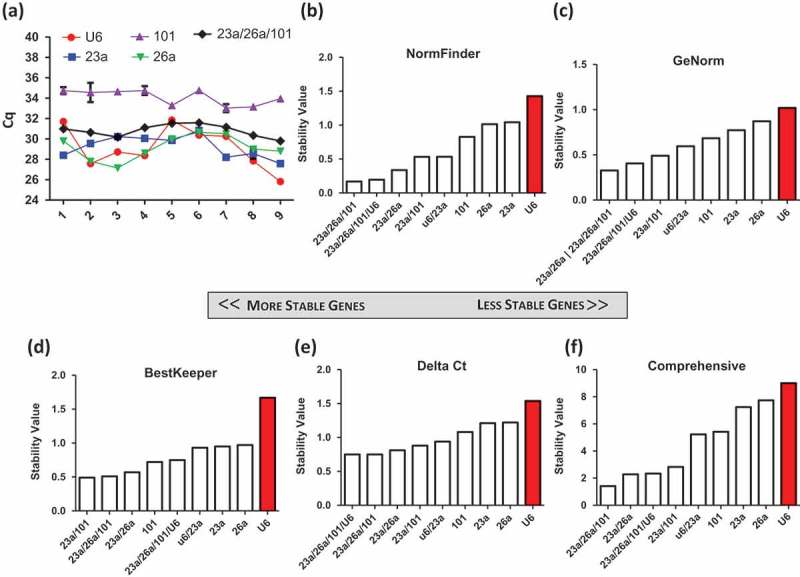
Validation of candidate genes from small RNA sequencing of nine samples, from five unique cardiosphere-derived cell (CDC) donors and a fibroblast line. Four conditions were variable across each sample, including oxygen concentration (2%, 5% and 20%), days of conditioning (5 and 15 days), three different passages (3–5) and a fibroblast control. (a) The Cq values for each sample were plotted to show the fluctuation of expression across donors and conditions.Data were analysed using (b) NormFinder, (c) GeNorm, (d) BestKeeper, and (e) Delta Ct. (f) The weighted geometric mean of each of these samples was taken to provide a consolidated list of the most stable genes.
Sequencing data availability
Sequencing data have been submitted to the NCBI Bioproject Database (submission ID SUB2675413, BioProject ID PRJNA386719, http://www.ncbi.nlm.nih.gov/bioproject/386719).
Discussion
In this article, we describe a novel and rigorous method of identifying and validating reference genes in CDC-EVs. CDC-EVs are a therapeutically relevant source and were used as a model for this method. There is a paucity of reference gene investigations in the literature. Rarer still are studies of reference genes from EVs, which have garnered much attention over the past decade for their translational potential in the field of diagnostics and, more recently, therapeutic applications. Indeed, the explosion of basic and translational research on EVs underscores the importance of identifying suitable reference genes for elucidating the signalling mechanisms of these vesicles, alterations in payload based on different conditions of EV preparation, and the differences in content among various EV populations.
The most prevalent reference gene for small RNAs is the U6 small nuclear RNA (snRNA). Being a nuclear transcript involved in the spliceosome complex of mRNA transcription, it is likely that it has no post-transcriptional signalling functions. Previous work by Villarroya-Beltri et al. [32] and Gu et al. [33] identified key sorting proteins of miRs (hnRPA2B1) and proteins (VPS33b), respectively. Furthermore, the previously mentioned hnRNPa2b1 is also a nuclear transcript and is present in low amounts in EVs.
In this study, we integrated sequencing- and chip-based methods to identify suitable reference candidates. Validation strategies were guided by the properties of a suitable reference gene. miR-23a-3p is present in all CDC-EV samples analysed, which reflects its active packaging into EVs irrespective of conditioning period, oxygen concentration during the conditioning period or the donor source of the CDCs. It is stable across all the aforementioned conditions, as shown by sequencing, NanoString and RT-qPCR validation. Finally, it is present with enough abundance to be assayed reliably. Because of the variety of conditions that alter the content of EVs, we investigated whether an arithmetic mean of other miRs would perform better than miR-23a-3p on its own. We therefore selected miR-101-3p, which was identified as being more stable than miR-23a-3p (but in lower abundance), and miR-26a-5p, which was also identified by our analysis and by other groups as a stable reference gene in EVs.
Using the arithmetic mean of these three genes yielded a stronger reference for normalization than any one species on its own. The efficiency of the qPCR for miR-101-3p was over 110%. Despite this, the combination of miR-101-3p, miR-23a-3p and miR-26a-5p was more stable than that of miR-23a-3p and miR-26a-5p alone. One of the challenges to the applicability of the NanoString study was the low input of RNA in some of the samples, which may have disqualified some of the less abundant miR candidates from analysis. Perhaps expectedly, U6 did not contribute to further stability as a reference gene. This may be due to the fact that U6 snRNA biogenesis is mechanistically separated from miR biogenesis, which is processed not by a spliceosome (as in pre-mRNA processing) [34] but by the Drosha complex (and later by Dicer) [35]. This underscores the dependence of the selection of the reference RNA on the class of RNAs being investigated. Evaluations of mRNA would be likely to support the use of U6 or ribosomal RNA, whereas using reference miR genes would be indicated for miRs and other small RNAs. More studies are needed to investigate the utility of miR reference genes with the growing list of other RNA classes found in EVs, most of which remain to be fully described. For translational development, the selection of appropriate reference genes will further strengthen gene expression studies for productive development, scale-up and quality control. From the basic science and discovery perspective, understanding EV signalling will further the translational potential of EVs.
Supplementary Material
Acknowledgements
We thank the Cedars-Sinai Genomics Core, specifically Jie Tang, for expert help with RNA sequencing and data analysis.
Responsible Editor Kenneth W. Witwer, Johns Hopkins University, USA
Funding Statement
Work in the Marban lab was supported by NIH R01124074; Work at Capricor Therapeutics was supported by DOD PRMRP PR150618.
Disclosure statement
A.I. is an employee of Capricor Therapeutics. E.M. is founder of, unpaid advisor to, and owns equity in Capricor Therapeutics. No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed here.
References
- [1].Bustin SA, Benes V, Garson JA, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–11. [DOI] [PubMed] [Google Scholar]
- [2].Deatherage BL, Cookson BT.. Membrane vesicle release in bacteria, eukaryotes, and archaea: a conserved yet underappreciated aspect of microbial life. Infect Immun. 2012;80:1948–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Luga V, Zhang L, Viloria-Petit AM, et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. 2012;151:1542–1556. [DOI] [PubMed] [Google Scholar]
- [4].Gangoda L, Boukouris S, Liem M, et al. Extracellular vesicles including exosomes are mediators of signal transduction: are they protective or pathogenic? Proteomics. 2015;15:260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lawson C, Vicencio JM, Yellon DM, et al. Microvesicles and exosomes: new players in metabolic and cardiovascular disease. J Endocrinol. 2016;228:R57–R71. [DOI] [PubMed] [Google Scholar]
- [6].Iraci N, Leonardi T, Gessler F, et al. Focus on extracellular vesicles: physiological role and signalling properties of extracellular membrane vesicles. Int J Mol Sci. 2016;17:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Li N, Li Q. Identification and characterization of endogenous viral elements for the three key schistosomes of humans. Pak J Pharm Sci. 2015;28:375–382. [PubMed] [Google Scholar]
- [8].Chiba M, Kimura M, Asari S. Exosomes secreted from human colorectal cancer cell lines contain mRNAs, microRNAs and natural antisense RNAs, that can transfer into the human hepatoma hepg2 and lung cancer a549 cell lines. Oncol Rep. 2012;28:1551–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zanello BC SB, Skog J. Brain gene expression signatures from cerebrospinal fluid exosome RNA profiling. NASA Human Research Program Investigators’ Workshop; 2014 Feb 12–13; Galveston, TX, USA; 2014. [Google Scholar]
- [10].Kim VN, Nam J-W. Genomics of microRNA. Trends Genet TIG. 2006;22:165–173. [DOI] [PubMed] [Google Scholar]
- [11].Chuang JC, Jones PA. Epigenetics and micrornas. Pediatr Res. 2007;61:24R–29R. [DOI] [PubMed] [Google Scholar]
- [12].Brow DA, Guthrie C. Spliceosomal RNA U6 is remarkably conserved from yeast to mammals. Nature. 1988;334:213–218. [DOI] [PubMed] [Google Scholar]
- [13].Benz F, Roderburg C, Vargas Cardenas D, et al. U6 is unsuitable for normalization of serum miRNA levels in patients with sepsis or liver fibrosis. Exp Mol Med. 2013;45:e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lamba V, Ghodke-Puranik Y, Guan W, et al. Identification of suitable reference genes for hepatic microRNA quantitation. BMC Res Notes. 2014;7:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Marban E. Breakthroughs in cell therapy for heart disease: focus on cardiosphere-derived cells. Mayo Clinic Proc. 2014;89:850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].de Couto G, Liu W, Tseliou E, et al. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Invest. 2015;125:3147–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ibrahim AG, Cheng K, Marban E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep. 2014;2:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64:5245–5250. [DOI] [PubMed] [Google Scholar]
- [19].Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pfaffl MW, Tichopad A, Prgomet C, et al. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26:509–515. [DOI] [PubMed] [Google Scholar]
- [21].Silver N, Best S, Jiang J, et al. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol Biol. 2006;7:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cheng L, Sharples RA, Scicluna BJ, et al. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J Extracell Vesicles. 2014;3:237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Simon A. FastQC: a quality control tool for high throughput sequence data. Babraham, UK: Babraham Bioinformatics; 2010. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc [Google Scholar]
- [24].Xie F, Xiao P, Chen D, et al. Mirdeepfinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol Biol. 2012;80:75–84. [DOI] [PubMed] [Google Scholar]
- [25].Yu X, Qiao M, Atanasov I, et al. Cryo-electron microscopy and three-dimensional reconstructions of hepatitis C virus particles. Virology. 2007;367:126–134. [DOI] [PubMed] [Google Scholar]
- [26].Yuana Y, Koning RI, Kuil ME, et al. Cryo-electron microscopy of extracellular vesicles in fresh plasma. J Extracell VesiclesExtracell Vesicles. 2013;2:21494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cheng K, Ibrahim A, Hensley MT, et al. Relative roles of CD90 and c-kit to the regenerative efficacy of cardiosphere-derived cells in humans and in a mouse model of myocardial infarction. J Am Heart Assoc. 2014;3:e001260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Li Y, Zhang L, Liu F, et al. Identification of endogenous controls for analyzing serum exosomal miRNA in patients with hepatitis B or hepatocellular carcinoma. Dis Markers. 2015;2015:893594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Timoneda O, Balcells I, Cordoba S, et al. Determination of reference microRNAs for relative quantification in porcine tissues. PLoS One. 2012;7:e44413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zheng G, Wang H, Zhang X, et al. Identification and validation of reference genes for qPCR detection of serum microRNAs in colorectal adenocarcinoma patients. PLoS One. 2013;8:e83025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mahdipour M, Van Tol HTA, Stout TAE, et al. Validating reference microRNAs for normalizing Qrt-Pcr data in bovine oocytes and preimplantation embryos. BMC Dev Biol. 2015;15:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, et al. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun. 2013;4:2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gu H, Chen C, Hao X, et al. Sorting protein vps33b regulates exosomal autocrine signaling to mediate hematopoiesis and leukemogenesis. J Clin Invest. 2016;126:4537–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Will CL, Luhrmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol. 2011;3:a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee Y, Ahn C, Han J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.