

Abstract
Herein, conditions are provided for the formation and use of the oxazolone enolate for the nucleophilic substitution of highly fluorinated (hetero) arenes, which after unmasking yield highly fluorinated non-natural amino acids and derivatives. In addition, the properties and chemical behavior of this new class of amino acids are explored. The utility is demonstrated in the one pot synthesis of medicinally relevant 2-aminohydantoins.
Multifluorinated arenes and heteroarenes represent an important motif, owing to the ability of fluorine to substantially alter the performance of molecules in various applications (Scheme 1A) . However, in sharp contrast to their importance, there is a relative dearth of methods that rapidly yield multifluorinated arenes, and thus, there is a need for methods to access multifluorinated (hetero) arenes.
Scheme 1.

Multifluorinated Arenes, Defluorination as a Strategy, and Arylation of Oxazolones
Selective installation of fluorine is possible, but often is lengthy and requires a unique strategy for every fluorination pattern on every arene (Scheme 1B, right) ,1 making it less than ideal for discovery chemistry, which demands the ability to rapidly synthesize analogs. Alternatively, C F functionalization of highly fluorinated or perfluorinated (hetero) arenes is an attractive approach, since perfluorinated arenes are relatively inexpensive and already possess the difficult to install fluorines in the desired locations (Scheme 1B, left) . The potential advantage of the C F functionalization approach has been recognized by others, and inroads are being made.2 Taken in conjunction with recently developed photocatalytic C–F functionalization,3 considerable access to these important motifs can be realized.
Perfluoroarenes are particularly well suited for nucleophilic aromatic substitution, since the difficult step in such a reaction is typically nucleophilic attack on the carbon by the incoming nucleophile. The small size and the electronegative nature of the fluoride facilitates the addition step, and additional ring fluorination (except para-fluorination) further accelerates the addition through inductive effects.1 However, despite the feasibility and the potential for enhancing the pool of fluorinated aryl building blocks, many of these potential reactions remain undeveloped.
Amino acids and derivatives are indispensable building blocks within mainstream organic chemistry. The addition of an oxazolone enolate to an electrophile is a common strategy to elaborate the alpha amino acid motif.4 α-Arylation of the oxazolone enolate has been accomplished via Ag-mediated arylation with diaryliodonium salts (Scheme 1C, eqn 1) ,4d Pd-catalyzed cross-coupling of aryl-bromides (eqn 2) ,5 and pyridine addition via nucleophilic addition to N-tosylated pyridines.4a However, these methods do little to access amino acids that possess highly fluorinated arenes, because the requisite fluorinated starting materials are not available. Additionally, while these methods proved useful for accessing tertiary carbons, secondary carbons were never obtained. We posited that the oxazolone enolate might undergo uncatalyzed substitution of the perfluoroarenes, via nucleophilic aromatic substitution (SNAr) , and because of the anticipated diminishment of nucleophilicity of the product, would cleanly provide the product without over arylation. Importantly, if successful, this would provide rapid and facile access to non-natural multifluoroarylated amino acids.
The oxazolone is a versatile nucleophile which can conceivably undergo addition at C2, C4, and even the C5-oxygen bond.6 The only example of SNAr-type reactions with oxazolones is the addition to 2-nitroarenes, which undergoes subsequent cyclization with the nitro functional group to give indazole products (eqn 3) .7 Though the reaction leads to a different product, it suggests that oxazolone enolate might be a competent nucleophile in the addition to perfluoroarenes.
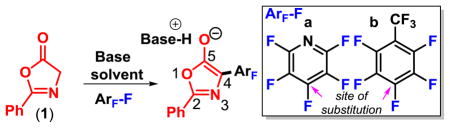
We started our investigation using 2-phenyloxazol-5(4H) -one (1, Table 1) derived from glycine with pentafluoropyridine and diisopropylamine (DIPEA) in acetonitrile which were optimal conditions in the analogous addition of Meldrum’s acid enolate.8 Initially, we ran the reaction at 45 °C (entry 1) but found the reaction gave full conversion at room temperature, giving complete conversion to the C4-arylation product within just 20 min (entry 2) . Owing to the relatively fast addition, we were able to lower the amount of the arene to 1.025 equiv (entry 3) without any detectable diminishment of conversion. Previously, we have observed DIPEA can undergo a slow N substitution/dealkylation sequence when subjected to highly electrophilic perfluoroarenes at elevated temperatures.3d Consequently, by keeping the amount of excess perfluoroarene low (0.025 equiv) and reactions at room temperature, the amine substituted product was not observed. A solvent screen (entries 2–7) revealed polar aprotic solvents work best, while ethereal, hydrocarbon, and halogenated solvents gave little to no conversion (entries 5–7) . To facilitate reaction scale up, we simultaneously reduced the arene loading and concentrated the reaction to 1 M (entry 8) which smoothly proceeded to full conversion. Next, we evaluated a less activated arene, octafluorotoluene (b) , instead of pentafluoropyridine (entry 9) , but unfortunately, it gave very low conversion under these conditions.
Table 1.
Optimization of Reaction Conditions
| |||||
|---|---|---|---|---|---|
| entry | conditions | Arene (equiv) | Amine (equiv) | T (°C) | % conv |
| 1. | 0.3 M ACN | a (2) | DIPEA (3) | 45 | 100 |
| 2. | 0.3 M ACN | a (2) | DIPEA (3) | 22 | 100 |
| 3. | 0.3 M DMF | a (2) | DIPEA (3) | 22 | 100 |
| 4. | 0.3 M DMSO | a (2) | DIPEA (3) | 22 | 100 |
| 5. | 0.3 M THF | a (2) | DIPEA (3) | 22 | 24 |
| 6. | 0.3 M Tol | a (2) | DIPEA (3) | 22 | <5 |
| 7. | 0.3 M DCM | a (2) | DIPEA (3) | 22 | 25 |
| 8. | 1 M ACN | a (1.025) | DIPEA (3) | 22 | 100 |
| 9. | 1 M ACN | b (2) | DIPEA (3) | 22 | <5 |
| 10. | 1 M ACN | b (2) | TMG (2.4) | −20 | 100 |
| 11. | 1 M ACN | b (1.025) | TMG (2.05) | −20 | 100 |
| 12. | 1 M ACN | a (1.025) | TMG (2.05) | −20 | 100 |
Conversion determined by 19F NMR after 30 min.
However, upon switching to stronger base, tetramethyl guanidine (TMG-H+, pKa(MeCN) = 23.3 compared to ~18.8 for DIPEA-H+) .9 We observed complete conversion, even at −20 °C (entry 10) . Lower temperatures were necessary to avoid TMG addition to the perfluoroarenes (not shown) .
Thus, with the use of tetramethylguanidine (TMG) at −20 °C, we were able achieve full conversion of 1, using just 1.025 equivalents of octafluorotoluene (b, entry 11) . When we applied these reaction conditions to pentafluoropyridine (a, entry 12) we observed quantitative conversion to the product by 19F NMR, suggesting that the TMG conditions would have a broader scope.
With the optimal conditions in hand (entry 12) , we were prepared to explore the scope. Typically, oxazolones are not easily isolated due to their predisposition for ring opening, and instead are usually derivatized immediately to a more stable molecule.4b This is easily accomplished under acidic conditions, since the oxazolone behaves as an activated ester. We explored the ring opening of the oxazolone with alcohols to form perfluoroaryl N-benzoyl protected esters (Table 2) . We found protonation of the oxazolone under anhydrous conditions was the key to high yield and that this was best accomplished using catalytic amounts trifluoroacetic acid in dry alcohol.
The scope of the one pot perfluoroarylation and esterification of the oxazolone was explored (Scheme 2) . Generally, the reaction scope was very good and worked well with many of the commercially available perfluoroarenes. No undesired reactivity was observed with esters (2a) or ketones (2b) . Less activated octafluorotoluene, octafluoronapthalene, and de-cafluorobiphenyl all reacted to give the expected product in excellent yields (2c, 2f, and 2g) . Both benzoxazoles (2h) and benzonitriles (2i) underwent arylation and esterification in good yield. Comparison of pyridine products 2d and 2e revealed the preference for substitution of the 4-fluoro over that of 3-chloro leaving group. This is consistent with a SNAr reaction, and is expected, as fluorine is the preferred nucleofuge and attack of the 4-position maximizes delocalization of the charge in the Meisenheimer complex.
Scheme 2.

Arylation and Esterification of Oxazolone
However, substitution of 4-chlorotetrafluoropyridine (see ESI) leads primarily to substitution of the chloride, rather than substituting at the 2-fluoro position, to give 2d as the major product. This suggests, in this case, that the regioselectivity is primarily dictated by the electronics of the arene rather than the nucleofuge.
Next, we explored the ring opening of oxazolones with water which would provide direct access to non-natural N-benzoyl protected amino acids in a rapid and facile manner (Scheme 3) . We were pleased to see that the perfluoroarylated oxazolone intermediates could be efficiently hydrolyzed (3a–3e) . However, since facilitating access to more elaborate fluorinated building blocks is our primary objective, we also developed chromatography-free conditions that were amenable to scaling, and allowed us to synthesize the amino acids derivatives on a gram scale with minimal effort. After initial removal of MeCN, the crude acids were extracted with CHCl3, washed with acidic brine, and stripped of solvent in vacuo. Next, the acid was washed with hexanes and trace dichloromethane to selectively extract the impurities and yield pure acids. This process was applied successfully on a gram scale for several substrates (3a–3c) , suggesting the method may be useful for making more sizeable quantities of the N-benzoyl protected acids.
Scheme 3.

Multifluoroaryl Amino Acids
Next, we probed the deprotection of the N-benzoyl groups in hopes of accessing the free amino acids. Unfortunately, in the case of pyridine derived 3a, standard deprotection conditions of refluxing in concentrated HCl led to the deprotected and decarboxylated ammonium salts. The amino acid derivatives apparently underwent a thermal decarboxylation, however it was not clear whether debenzoylation occurred prior to or following decarboxylation. Fortunately, after lowering the reaction temperature to 60 °C, we found that the amino acids underwent smooth debenzoylation, but did not undergo the decarboxylation. Thus, by subjecting acids (3a–c, Scheme 4) to aqueous HCl carefully heated to 60 °C, we acquired the unprotected amino acids in quantitative yields. NMR doping experiments were performed, in which the isolated hydrochloride salts of amino acids (4a–c) were added after prolonged heating in HCl (see ESI) , and confirmed that debenzoylation occurs prior to decarboxylation.
Scheme 4.

Debenzoylation of Amino Acids and Schiff Base Mediated Decarboxylation
Alternatively, upon exposure to acetone the amino acid salts (4a–c) undergo rapid and quantitative decarboxylative protonation. Presumably, this takes place through a transiently formed Schiff base, which facilitates the decarboxylation and then undergoes hydrolysis to form (5a–c) .12 Given the high yielding and facile nature of each step, the addition of oxazolone to perfluoroarenes is an attractive strategy to access highly fluorinated benzylic amine derivatives such as 5a–c.
Having generated a strategy to access a novel class of fluorinated amino acids and our own experience with the unexpected decarboxylation of the amino acid derivatives, we thought it appropriate to explore the thermal stability of several derivatives. Thus, we performed thermal gravimetric analysis on several of the products we had formed (Scheme 5) . As expected the benzoyl protected esters (2d) were thermally stable and gradually sublimed. However, the benzoyl protected acids 3a–c all underwent decarboxylation at 108 °C, 132 °C, and 159 °C, respectively. The differences in decarboxylation temperature seem to reflect the stability of a benzylic anion, however, it may also correlate to basicity of the amide motif. Comparison of 3b and 4b shows that the HCl salts actually undergo a more facile thermal decarboxylation than the N-benzoylated substrate, and is consistent with our previous findings in Scheme 4. However, 4c failed to undergo decarboxylation and instead gradually sublimed. It may be possible that the 4c is sufficiently acidic that it instead sublimed through a small equilibrium of neutral (or potentially zwitterionic) amino acid.
Scheme 5.

Thermal Gravimetric Analysis of α-Multifluoroaryl Amino Acid Derivatives.
Next, we evaluated the perfluoroarylations of substituted oxazolones, which yield fully substituted, non-natural amino acid derivatives (Scheme 6) . Upon additional substitution of the oxazolone, the carbonyl is permanently formed, which is prone to opening by nucleophiles, including TMG. We were pleased to see that even the substituted oxazolone enolate underwent selective C4-addition to give the desired amino acids, rather than C2-addition which has been observed for substituted oxazolones.6, 13 However, in order to be able to derivatize in the second step, we found it necessary to utilize DIPEA as the base. Perfluoroarylated amino acid esters were derived from alanine, valine, methionine, and phenylalanine in good yield (6a–d) .
Scheme 6.

Fully Substituted Amino Acid Esters
When we utilized TMG as the base, we found that after arylation the TMG would undergo nucleophilic ring opening to form N-acylated guanidines, which upon heating in HCl underwent debenzoylation and cyclization with extrusion of dimethyl amine. This sequence yields 2-aminohydantoins which are currently being studied14 as possible inhibitors of BACE1, an enzyme believed to play a central role in the formation β-amyloid formation associated with Alzheimer’s disease. Traditionally, 2-aminohydantoins have been synthesized by adding an activated carbon to the N-terminus which then serves to bridge the N- and C-termini of the amino acid. In contrast, the guanidine serves in place of the activated carbon and is attached to the C-terminus. To our knowledge, this cyclization approach is novel and demonstrates the potential for a one-pot, three component coupling that could be used to rapidly explore the chemical space surrounding this important motif.
In summary, we have shown that the oxazolone enolate is capable of nucleophilic aromatic substitution and can be utilized to rapidly form non-natural fluorinated amino acid derivatives. Importantly, the reaction is highly selective both in terms of C F and oxazolone regioselectivity. Furthermore, we have provided conditions that allow transformation of the product to valuable building blocks, namely, N-Bz esters, N-Bz acids, acid-HCl salts, and by way of Schiff-base decarboxylation, the benzylic amines. Furthermore, we have reported the behavior of these compounds towards thermal decarboxylation, which indicate that the compounds should be stable at room temperature. Finally, we demonstrate the utility of the reaction and its product by demonstrating its use in a 3-component reaction, which allows rapid access 2-aminohydantoins, a biologically relevant motif.
Supplementary Material
Scheme 7.

One Pot Multicomponent Synthesis of 2-Amino Hydantoins
Footnotes
Electronic Supplementary Information (ESI) available: See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Fluorinated heterocyclic compounds: synthesis, chemistry, and applications. Hoboken, N.J: Wiley, Hoboken, N.J; 2009. For recent reviews see Pattison G, Sandford G, Wilson I, Yufit D, Howard JAK, Christopher JA, Miller DD, editors. Tetrahedron. 2017;73:437–454.
- 2.(a) Lentz D, Braun T, Kuehnel MF. Angew Chem Int Ed Engl. 2013;52:3328–3348. doi: 10.1002/anie.201205260. [DOI] [PubMed] [Google Scholar]; (b) Ahrens T, Kohlmann J, Ahrens M, Braun T. Chem Rev. 2015;115:931–972. doi: 10.1021/cr500257c. [DOI] [PubMed] [Google Scholar]; (c) Weaver J, Senaweera S. Tetrahedron. 2014;70:7413–7428. [Google Scholar]; (d) Kiplinger JL, Richmond TG, Osterberg CE. Chem Rev. 1994;94:373–431. [Google Scholar]; (e) Amii H, Uneyama K. Chem Rev. 2009;109:2119–2183. doi: 10.1021/cr800388c. [DOI] [PubMed] [Google Scholar]
- 3.(a) Singh A, Fennell CJ, Weaver JD. Chem Sci. 2016;7:6796–6802. doi: 10.1039/c6sc02422j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Senaweera SM, Weaver JD. J Am Chem Soc. 2016;138:2520–2523. doi: 10.1021/jacs.5b13450. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Singh A, Kubik JJ, Weaver JD. Chem Sci. 2015;6:7206–7212. doi: 10.1039/c5sc03013g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Senaweera SM, Singh A, Weaver JD. J Am Chem Soc. 2014;136:3002–3005. doi: 10.1021/ja500031m. [DOI] [PubMed] [Google Scholar]; (e) Lu J, Khetrapal NS, Johnson JA, Zeng XC, Zhang J. J Am Chem Soc. 2016;138:15805–15808. doi: 10.1021/jacs.6b08620. [DOI] [PubMed] [Google Scholar]; (f) Xie J, Yu J, Rudolph M, Rominger F, Hashmi ASK. Angew Chem Int Ed Engl. 2016;55:9416–9421. doi: 10.1002/anie.201602347. [DOI] [PubMed] [Google Scholar]
- 4.(a) Johnson TC, Marsden SP. Org Lett. 2016;18:5364–5367. doi: 10.1021/acs.orglett.6b02731. [DOI] [PubMed] [Google Scholar]; (b) Fisk JS, Mosey RA, Tepe JJ. Chem Soc Rev. 2007;36:1432–1440. doi: 10.1039/b511113g. [DOI] [PubMed] [Google Scholar]; (c) Hewlett NM, Hupp CD, Tepe JJ. Synthesis. 2009:2825–2839. [Google Scholar]; (d) Chai Z, Wang B, Chen J-N, Yang G. Adv Synth Catal. 2014;356:2714–2718. [Google Scholar]
- 5.Liu X, Hartwig JF. Org Lett. 2003;5:1915–1918. doi: 10.1021/ol034570q. [DOI] [PubMed] [Google Scholar]
- 6.Wang T, Yu Z, Hoon DL, Phee CY, Lan Y, Lu Y. J Am Chem Soc. 2016;138:265–271. doi: 10.1021/jacs.5b10524. [DOI] [PubMed] [Google Scholar]
- 7.D’Anello M, Erba E, Gelmi ML, Pocar D. Ber. 1988;121:67–73. [Google Scholar]
- 8.Senaweera S, Weaver JD. J Org Chem. 2014;79:10466–10476. doi: 10.1021/jo502075p. [DOI] [PubMed] [Google Scholar]
- 9.Margetic D. Superbases for Organic Synthesis. John Wiley & Sons, Ltd; 2009. pp. 9–48. [DOI] [Google Scholar]
- 10.Lv H, Zhan J-H, Cai Y-B, Yu Y, Wang B, Zhang J-L. J Am Chem Soc. 2012;134:16216–16227. doi: 10.1021/ja305204y. [DOI] [PubMed] [Google Scholar]
- 11.Gung BW, Amicangelo JC. J Org Chem. 2006;71:9261–9270. doi: 10.1021/jo061235h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Al-Sayyab AF, Lawson A. J Chem Soc C. 1968:406–410. doi: 10.1039/J39680000406. [DOI] [PubMed] [Google Scholar]
- 13.Alemán J, Milelli A, Cabrera S, Reyes E, Jørgensen KA. Chem Eur J. 2008;14:10958–10966. doi: 10.1002/chem.200802030. [DOI] [PubMed] [Google Scholar]
- 14.(a) Cruz DS, Castilho MS. Med Chem. 2014;10:162–173. doi: 10.2174/15734064113099990002. [DOI] [PubMed] [Google Scholar]; (b) Malamas MS, Erdei J, Gunawan I, Barnes K, Hui Y, Johnson M, Robichaud A, Zhou P, Yan Y, Solvibile W, Turner J, Fan KY, Chopra R, Bard J, Pangalos MN. Bioorg Med Chem Lett. 2011;21:5164–5170. doi: 10.1016/j.bmcl.2011.07.057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.