

Abstract
Using amber suppression in coordination with a mutant pyrrolysyl-tRNA synthetase-tRNAPyl pair, azidonorleucine is genetically encoded in E. coli. Its genetic incorporation followed by traceless Staudinger ligation with a phosphinothioester allows convenient synthesis of a protein with a site-specifically installed lysine acylation. By simply changing the phosphinothioester identity, any lysine acylation type could be introduced. Using this approach, we demonstrated that both lysine acetylation and lysine succinylation can be installed selectively in ubiquitin and synthesized histone H3 with succinylation at its K4 position (H3K4su). Using an H3K4su-H4 tetramer as a substrate, we further confirmed that Sirt5 is an active histone desuccinylase. Lysine succinylation is a recently identified novel posttranslational modification. The reported technique makes it possible to explicate regulatory functions of this modification in proteins.
Keywords: lysine succinylation, lysine malonylation, lysine glutarylation, azidonorleucine, amber suppression
TOC image
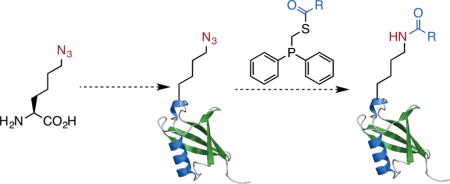
An azide containing amino acid, azidonorlucine is genetically encoded. Its incorporation followed by traceless Staudinger ligation potentiates the synthesis of proteins with a myriad of site-specific lysine acylations.
As the only amino acid with a side chain amine, lysine undergoes a myriad of posttranslational acylations (Figure 1A).[1,2] The most studied lysine acylation is acetylation. It was initially discovered in histones and occurs widespread in transcription factors and cytosolic proteins.[3] Two other well-known lysine acylations are biotinylation and lipoylation. Both modifications anchor a catalytic cofactor to enzymes.[4] With the advent of sophisticated proteomic techniques, there has been an increased diversity of lysine acylations revealed in histones and non-histone proteins. Three of these novel acylations, malonylation, succinylation, and glutarylation, reverse the charge state of the lysine side chain and potentiates a unique way of controlling protein functions.[2] So far, more than ten lysine acylation types have been discovered. However, functions of novel acylations are largely unexplored. One impediment is the difficulty of obtaining proteins with site-specific lysine acylations. The enzymatic synthesis of proteins with novel lysine acylations is not applicable since enzymes responsible for these modifications are elusive.[5] Alternatively, native chemical ligation/expressed protein ligation (NCL/EPL), a cysteine-based alkylation approach, and a recently developed dehydroalanine-based coupling approach may be applied for the synthesis of proteins with lysine acylations.[6] However, ECL/EPL requires a cysteine for ligation and the structures generated by the later two methods are slightly different from the natural modifications. Here we report the synthesis of proteins with genuine lysine acylations by coupling the amber suppression based mutagenesis approach[7] with traceless Staudinger ligation.
Figure 1. A proposed versatile approach to access proteins with lysine acylations.
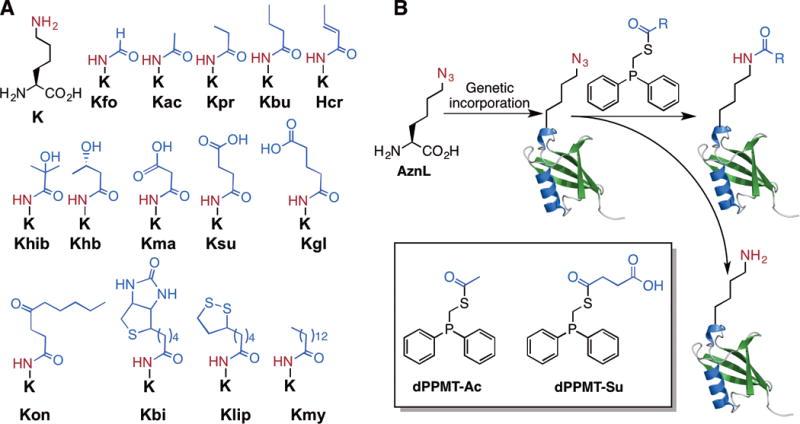
(A) Posttranslationally acylated lysines and their abbreviations. Acylations with small proteins such as ubiquitin and ubiquitin like proteins are not shown. (B) The genetic incorporation of AznL followed by traceless Staudinger ligation with a phosphinothioester to install a site-specific lysine acylation in a protein. The reducing nature of the phosphinothioester leads to the partial conversion of AznL to lysine as a side product. Shown in the inlet are two initially designed phosphinothioesters.
Following the pioneer work by Chin et al., several groups have successfully engineered the native pyrrolysine incorporation system for the genetic incorporation of acyl-lysines into proteins, including Kac, Kpr, Kbu, Kcr, and Khib.[8] Although convenient, the genetic acyl-lysine incorporation approach requires that a pyrrolysyl-tRNA synthetase (PylRS) mutant recognizing a particular acyl-lysine but not any native amino acid must be identified. The process of identifying a mutant PylRS is tedious and doesn’t always work. So far, our efforts to search PylRS mutants for Kma, Ksu, and Kgl have ended with no success. Given large sizes of Kon, Kbi, Klip, and Kmy, their direct incorporation at amber codon is undoubtedly difficult if not impossible. Inspired by the traceless Staudinger ligation reaction developed by Bertozzi, Raines, and coworkers for the conversion of an azide to an acyl amide,[9, 10] we perceived that the genetic incorporation of azidonorleucine (AznL, Figure 1B) followed by a reaction with a phosphinothioester will effectively formulate a “one-size-fits-all” approach for the installation of a large variety of lysine acylations in proteins by simply changing the acyl group in the phosphinothioester. For example, using two phosphinothioesters dPPMT-Ac and dPPMT-Su will in theory allow the synthesis of proteins with site-specific acetylation and succinylation. Although the intrinsic reductive nature of the phosphinothioester[10, 11] will inevitably convert part of AznL to lysine, this side reaction leads to the formation of unacylated proteins that could be resolved from acylated ones using lysine acylation-specific antibodies for immunoaffinity separation. AznL was previously installed at methionine positions of proteins using the residue-replacement method.[12] Although advantageous in analyzing nascent proteomes in cells, its global substitution of methionines makes the approach undesirable for the purpose of synthesizing proteins with selective lysine acylations. For this reason, we chose the amber suppression-based mutagenesis approach for coding AznL. The synthesis of AznL followed procedures in SI Schemes 1&2. A bulk amount around 5 g was easily made. To identify AznL-specific PylRS mutants, a PylRS gene library with randomization at five active site residues, Y306, L309, C348, Y384, and W411 was constructed. This library was subjected to a widely adopted double-sieve selection,[13] yielding a highly efficient AznL-specific mutant Y306L/C348I/Y384F that is coined as AznLRS. For optimal expression, the AznLRS gene was codon-optimized and then, in coordination with tRNAPyl, used in the E. coli BL21 cell to drive the expression of full-length superfolder green fluorescent protein (sfGFP) where an amber codon was introduced at the D134 coding position of its gene. When cells were grown in the LB medium supplemented with 5 mM AznL, full-length sfGFP with an expression level of 20 mg/L was achieved, which was markedly contrasted with non-detectable full-length sfGFP expression in the absence of AznL (Figure 2A). The electrospray ionization mass spectrometry (ESI-MS) analysis of the expressed protein sfGFP-D134AznL displayed one major peak at 27,866 Da and one minor peak at 27,735 Da, which agree well with theoretic molecular weights of full-length sfGFP with AnzL installed at its 134 position (27,866 Da) and its N-terminal methionine cleavage product (27,835 Da) (Figure 2B). The existence of the azide functionality in sfGFP-D134AznL was further confirmed by its selective labeling with an alkyne-fluorescein dye (SI Figure 1). These combined results validated the specificity of AznLRS toward AznL and the selective incorporation of AznL at amber codon.
Figure 2. The genetic incorporation of AznL into a model protein sfGFP.
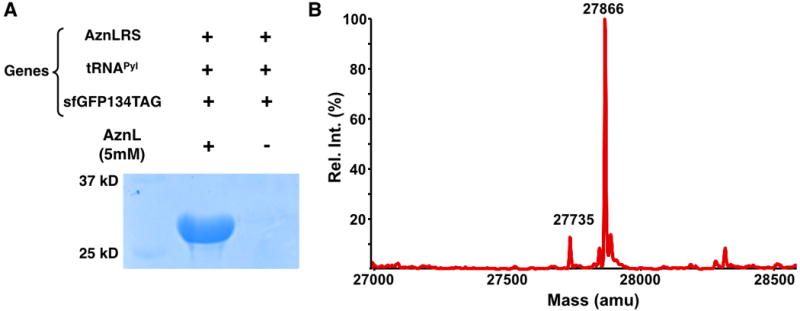
(A) The site-specific incorporation of AznL into sfGFP at its D134 position to produce sfGFP-D134AznL. Cells were transformed with two plasmids coding genes for AznLRS, tRNAPyl, and sfGFP with an amber mutation at its D134 position and grown in the LB medium with or without 5 mM AnzL. (B) The deconvoluted ESI-MS spectrum of purified sfGFP-D134AznL.
After demonstrating the selective incorporation of AznL, we went further to prove its application in the synthesis of proteins with site-specific acylations. Due to our ready access to a MALDI-TOF-MS instrument that is not optimal for analyzing large proteins such as sfGFP, we switched to work with ubiquitin. A ubiquitin variant Ub-K48AznL with a C-terminal 6×His tag and AznL incorporated at the original K48 position was produced similarly as sfGFP-D134AznL. Expression levels around 10 mg/L were routinely obtained (SI Figure 2). Ub-K48AznL was then reacted with excessive dMMPT-Ac. DMMPT-Ac and later used dMMPT-Su were synthesized according to procedures in SI Schemes 3&4. Due to the slow kinetics of traceless Staudinger ligation (a second-order rate constant around 0.001 M−1s−1),[14] 5 mM dMMPT-Ac and 37 °C at pH 6.0 were chosen as reaction conditions. Products from different reaction times were analyzed by SDS-PAGE and then probed by a pan anti-Kac antibody in the Western blot analysis. Ub-K48ac that was produced using a previously identified AcKRS-tRNAPyl pair was used as a positive control. The Western blot analysis clearly showed improved acetylation when the incubation time increased (Figure 3A). The reaction is deemed close to completion at 48 h since the acetylation level at this reaction time was not significantly higher than at 36 h. In contrast to the intense acetylation detected in reaction products of Ub-K48AznL, a similar reaction with wild type Ub did not yield a detectable acetylation level. This result indicates a much slower intermolecular S-to-N acyl transfer between dMMPT-Ac and Ub than traceless Staudinger ligation, assuring the selectivity of using traceless Staudinger ligation to convert AznL in a protein specifically to Kac. The conversion of Ub-K48AznL to its corresponding acetylation product Ub-K48ac was further confirmed with the MALDI-TOF-MS analysis of the 48 h reaction product of Ub-K48AznL. The spectrum displayed two major peaks at 9,386 and 9,428 Da, representing correspondingly the Staudinger reduction product (a wild type 6×His-tagged Ub) and the traceless Staudinger ligation product (Ub-K48ac), whose theoretical molecular weights are 9,387 and 9,429 Da, respectively (Figure 3B). In the final products, Ub-K48ac is slightly more than wild type Ub. In order to confirm that the acetylation is at K48, the 48 h reaction product was trypsinized and analyzed by the tandem mass spectrometry (MS/MS) analysis. The K48ac-containing fragment was clearly observed. Its further fragmentation clearly indicated the presence of acetylation at K48 (Figure 3C).
Figure 3. The synthesis of Ub-K48ac.
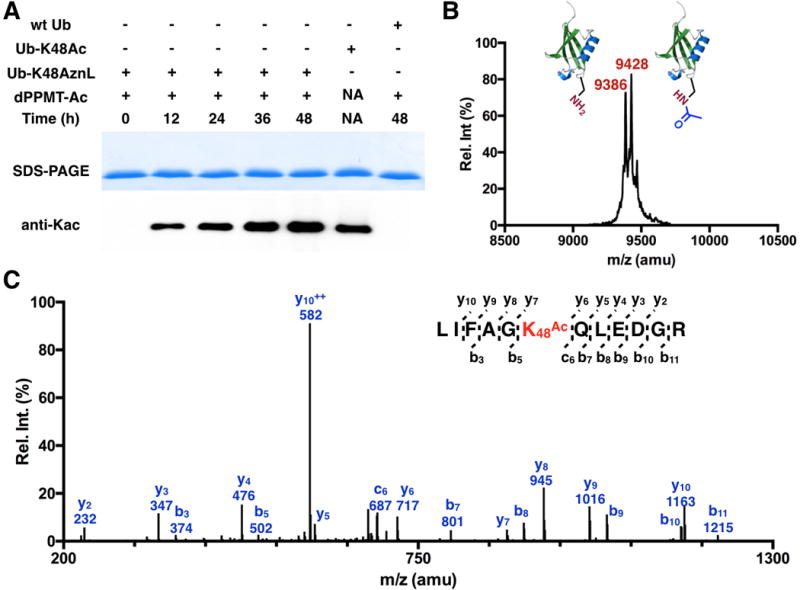
(A) Using dPPMT-Ac to convert Ub-K48AznL to Ub-K48ac. Reaction conditions: 20 μM Ub-K48AznL samples were incubated with 5 mM dPPMT-Ac in a PBS buffered H2O/DMSO (1:1) solution (pH 6.0) at 37 °C for various hours. After reactions, the samples were analyzed by SDS-PAGE and probed by a pan-anti-Kac antibody in Western blot. A control reaction with wild type (wt) Ub was carried out and analyzed similarly. Ub-K48ac was provided as a postive control. (B) The MALDI-TOF-MS spectrum of the reaction product of Ub-K48AznL after its 48 h incubation with dMMPT-Ac. (C) The tandem MS analysis of the trypsinized Kac-containing fragment of the 48 h reaction product of Ub-K48AznL.
Encouraged by the acetylation results, we proceeded to test the application of dPPMT-Su for the conversion of Ub-K48AznL to Ub-K48su that has succinylation at the K48 position. Reactions were set up almost identically as for the synthesis of Ub-K48ac. Products at different reaction times were analyzed by SDS-PAGE and probed by a pan anti-Ksu antibody. Although the formation of succinylation on Ub-K48AznL was clearly detected, the control reaction with wild type Ub also exhibited a high level of lysine succinylation (Figure 4A), indicating non-selective succinylation of seven Ub lysines by dPPMT-Su. Since dPPMT-Su has a carboxylate that potentially triggers an intramolecular S-to-O acyl transfer reaction with the thioester to form more reactive succinic anhydride for succinylating Ub lysines, this result was not a total surprise (Figure 4B). Indeed, we observed that dPPMT-Su in an oil form was completely eliminated to succinic anhydride after preserving at −20°C for several weeks. To avoid the formation of succinic anhydride, we conceived dPPMT-NB-Su (Figure 4C). In dPPMT-NB-Su, a photocleavable nitrobenzyl group[15] shields the carboxylate from the intramolecular S-to-O acyl transfer reaction and also allows its easy recovery via UV photolysis.
Figure 4. The nonselect ive succinylat ion by dPPMT -Su.
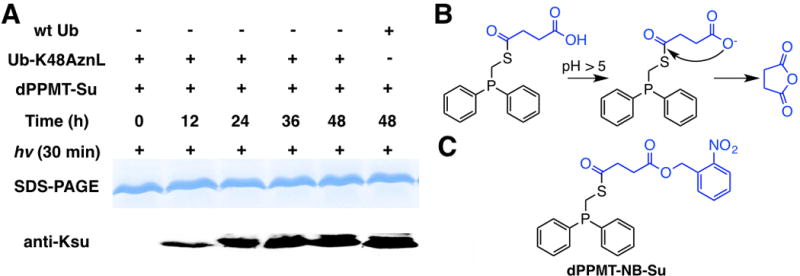
(A) Ub-K48AznL reactions with to dPPMT-Su. Reactions conditions were similar to the synthesis of Ub-K48ac. (B) An intramolecular S-to-O acyl tranfer reaction of dMMPT-Su. (C) The structure of dPPMT-NB-Su.
dMMPT-NB-Su was synthesized according to procedures in SI Scheme 5. The setup of using dMMPT-NB-Su to trigger the conversion of Ub-K48AznL to Ub-K48su was similar to the synthesis of Ub-K48ac except that there was an additional step of 365 nm UV treatment for 30 min before reaction samples were analyzed by SDS-PAGE and probed by the pan anti-Ksu antibody. As shown in Figure 5A, the anti-Ksu probed succinylation formation on Ub-K48AznL was time dependent and appeared reaching completion at 48 h. Although the control reaction with wild type Ub did show detectable lysine succinylation formation, its level is minimal. Since we did not observe a similar side reaction with dMMPT-Ac, it is possible that the electron withdrawing inductive effect of the ester in dMMPT-NB-Su activates its thioester for more favorable S-to-N acyl transfer with Ub lysines than dMMPT-Ac. To alternatively confirm the formation of Ub-K48su, the 48 h incubation and UV treated product of Ub-K48AznL was analyzed by MALDI-TOF-MS. The spectrum exhibited two major peaks, one at 9,388 Da and the other at 9488 Da, representing correspondingly wild type Ub and Ub-K48su, whose theoretic molecular weights are 9,387 and 9,487 Da, respectively (Figure 5B). Wild type Ub is a little more than Ub-K48su. To confirm the installation of succinylation at the K48 position, the final product was further trypsinized and analyzed by tandem MS analysis. The K48su-containing fragment was detected. Its fragmentation showed evidently the existence of succinylation at K48 (Figure 5C).
Figure 5. The synthesis of Ub-K48su.
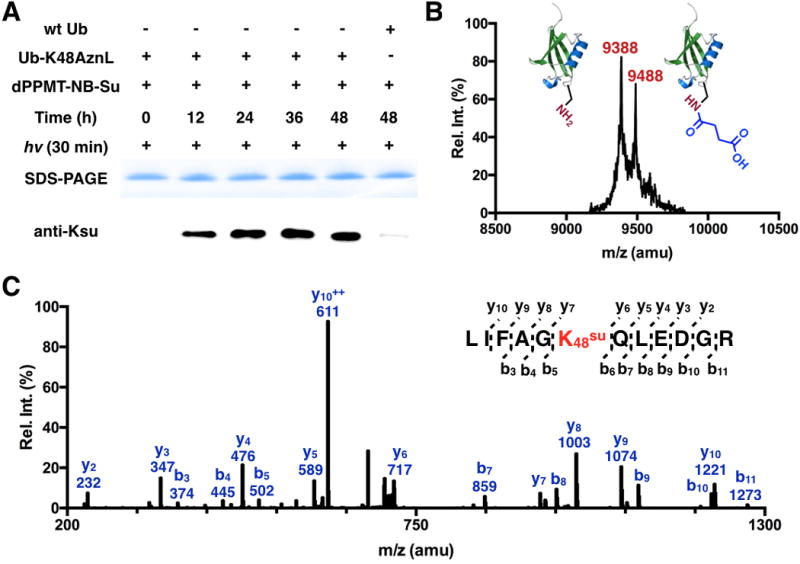
(A) Using dPPMT-NB-Su to convert Ub-K48AznL to Ub-K48su. Reaction conditions were similar to the synthesis of Ub-K48ac except that an additional step of 365 nm UV treatment. (B) The MALDI-TOF-MS of the 48 h reaction and UV treated product of Ub-K48AznL. (C) The tandem MS spectrum of the trypsinized Ksu-containing fragment of the 48 h reaction and UV treated product of Ub-K48AznL.
Similarly as lysine acetylation, lysine succinylation has been discovered in histones.[16] Since lysine succinylation averts the charge state of lysine, its occurrence in histones is expected to substantially impact interactions between the histone octamer and its associated DNA. Sirt5 is a NAD-dependent protein deacylase that has been suspected to remove succinylation from histones and regulate chromatin functions.[17] To demonstrate Sirt5 is an active histone desuccinylase, we decided to use the above-demonstrated approach to synthesize histone H3 with succinylation at K4 (H3K4su) and its tetramer with H4 as a substrate of Sirt5 for its activity test. H3 with AznL incorporated at K4 (H3K4AznL) was synthesized similarly as Ub-K48AznL. Its reaction with dMMPT-NB-Su was carried out similarly as for the synthesis of Ub-K48su. As shown in Figure 6A, H3K4AznL was successfully converted to H3K4su that was intensely detected by the pan anti-Ksu antibody. The control reaction with wild type H3 led to a very low level of lysine succinylation. To confirm K4 succinylation, the 48 h incubation and UV treated product of H3K4AznL was trypsinized and analyzed by the MS/MS analysis. The tandem MS spectrum of the Ksu-containing fragment clearly showed succinylation at K4 (Figure 6B). After confirming the succinylation site, we directly used this 48 h incubation and UV treated product of H3K4AznL to assemble an H3K4su-H4 tetramer that was subsequently used as a substrate to test Sirt5 activity. As shown in Figure 6C, when Sirt5 and its cofactor NAD+ were provided to the H3K4su-H4 solution, the succinylation was actively removed from H3. On the contrary, a control reaction with Sirt1 showed no succinylation removal from H3. These combined results unequivocally approve that Sirt5 but not Sirt1 is a histone desuccinylase.
Figure 6. The synthesis of H3K4su and its application to test the desuccinylation activity of Sirt5.
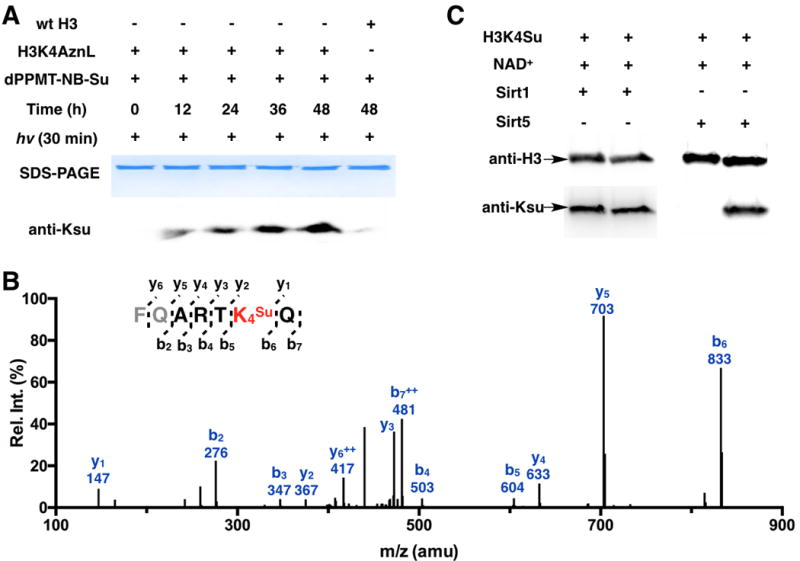
(A) Using dPPMT-NB-Su to convert H3K4AznL to H3K4su. Reaction conditions and analysis were as same as for the synthesis of Ub-K48su. (B) The tandem MS analysis of the trypsinized Ksu-containing fragment of the 48 h reaction and UV treated product of H3K4AznL. (C) Desuccinylation of the H3K4su-H4 tetramer by Sirt1 and Sirt5. 180 μM H3K4su-H3 tetramer was incubated with or without 0.5 μM Sirt1/Sirt5 in the presence of 10 mM NAD+ for 4 h. After reactions, proteins were analyzed in the Western blot analysis using anti-H3 and pan anti-Ksu antibodies.
In summary, a versatile approach has been developed for the site-specific installation of lysine acylations in proteins. We demonstrated that this approach can be readily applied for the synthesis of proteins with site-selective lysine acetylation and succinylation. Succinylation is a recently discovered novel lysine acylation. Its proteomes have been profiled in E. coli, Saccharomyces cerevisiae, Toxoplasma gondii, Vibrio parahemolyticus, Mycobacterium tuberculosis, human cells, and mouse liver tissues.[18] Many proteins in these organisms that undergo lysine succinylation are metabolic enzymes. Their catalytic alteration by lysine succinylation is anticipated. Xie et al. recently reported lysine succinylation on histone proteins.[16] One key feature of lysine succinylation of histones is that it mainly takes place on lysines in the nucleosome core region. The majority of these lysines involve direct charge-charge interactions with DNA. Their succinylation not just removes these charge-charge interactions but also creates repulsion with DNA for loosing the nucleosome structure. Whether or not cells use this strategy to unwrap DNA and Sirt5 for its reversal is an interesting aspect to explore. With our current method available, many questions related to lysine succinylation can be addressed Since the method potentiates the synthesis of proteins with any lysine acylation types, it provides a myriad of opportunities to understand functional roles of other novel posttranslational lysine acylations such as malonylation, glutarylation, myristoylation, etc. Although not explored in the current study, the method reported here could be further improved using water-soluble phosphinothioesters at high concentrations for accelerating the traceless Staudinger ligation reaction.[11] One may also consider to add substituents to phenyl groups of phosphinothioesters to retard the hydrolysis of the P-N ylide to reduce the side Staudinger reduction product.[19]
Supplementary Material
Acknowledgments
Support of this work was provided from National Institute of Health (grants R01CA161158, R01GM098596, and R01GM121584) and Welch Foundation (grant A-1715).
Footnotes
Supporting information (synthesis of AznL, dPPMT-Ac, dPPMT-Su, and dPPMT-NB-Su, the identification of AznLRS, expression of sfGFP-D134AznL, Ub-K48AznL, and H3K4AznL, reactions of Ub-K48AznL with dPPMT-Ac, dPPMT-Su, and dPPMT-NB-Su, the synthesis of H3K4su, MS analysis, and desuccinylation of H3K4su by Sirt1 and Sirt5) and the ORCID identification number(s) for the author(s) of this article can be found under http://.
Contributor Information
Zhipeng A. Wang, Department of Chemistry, Texas A&M University, College Station, TX 77843, USA.
Yadagiri Kurra, Department of Chemistry, Texas A&M University, College Station, TX 77843, USA.
Xin Wang, Department of Plant Pathology and Microbiology, Institute for Plant Genomics, Office of the Texas State Chemist, Department of Veterinary Pathobiology, College Station, TX 77843, USA.
Yu Zeng, Department of Chemistry, Texas A&M University, College Station, TX 77843, USA.
Yan-Jiun Lee, Department of Chemistry, Texas A&M University, College Station, TX 77843, USA.
Vangmayee Sharma, Department of Chemistry, Texas A&M University, College Station, TX 77843, USA.
Hening Lin, Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853, USA.
Susie Y. Dai, Department of Plant Pathology and Microbiology, Institute for Plant Genomics, Office of the Texas State Chemist, Department of Veterinary Pathobiology, College Station, TX 77843, USA
Wenshe R. Liu, Department of Chemistry, Texas A&M University, College Station, TX 77843, USA
References
- 1.Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. Nat Rev Mol Cell Bio. 2014;15:536–550. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]; Wisniewski JR, Zougman A, Mann M. Nucleic Acids Res. 2008;36:570–577. doi: 10.1093/nar/gkm1057. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dai L, Peng C, Montellier E, Lu Z, Chen Y, Ishii H, Debernardi A, Buchou T, Rousseaux S, Jin F, Sabari BR, Deng Z, Allis CD, Ren B, Khochbin S, Zhao Y. Nat Chem Biol. 2014;10:365–370. doi: 10.1038/nchembio.1497. [DOI] [PubMed] [Google Scholar]; Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, Falck JR, Peng J, Gu W, Zhao Y. Mol Cell Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, Zhao Y. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; Xie Z, Zhang D, Chung D, Tang Z, Huang H, Dai L, Qi S, Li J, Colak G, Chen Y, Xia C, Peng C, Ruan H, Kirkey M, Wang D, Jensen LM, Kwon OK, Lee S, Pletcher SD, Tan M, Lombard DB, White KP, Zhao H, Li J, Roeder RG, Yang X, Zhao Y. Mol Cell. 2016;62:194–206. doi: 10.1016/j.molcel.2016.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, Hang HC, Hao Q, Lin H. Nature. 2013;496:110–113. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jin J, He B, Zhang XY, Lin HN, Wang Y. J Am Chem Soc. 2016;138:12304–12307. doi: 10.1021/jacs.6b04977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirschey MD, Zhao Y. Mol Cell Proteomics. 2015;14:2308–2315. doi: 10.1074/mcp.R114.046664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phillips DM. Biochem J. 1961;80:40. doi: 10.1042/bj0800189. [DOI] [PMC free article] [PubMed] [Google Scholar]; Spange S, Wagner T, Heinzel T, Kramer OH. Int J Biochem Cell Biol. 2009;41:185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]; Manohar M, Mooney AM, North JA, Nakkula RJ, Picking JW, Edon A, Fishel R, Poirier MG, Ottesen JJ. J Biol Chem. 2009;284:23312–23321. doi: 10.1074/jbc.M109.003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kothapalli N, Camporeale G, Kueh A, Chew YC, Oommen AM, Griffin JB, Zempleni J. J Nutr Biochem. 2005;16:446–448. doi: 10.1016/j.jnutbio.2005.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Proc Natl Acad Sci U S A. 2004;101:2259–2264. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muir TW, Sondhi D, Cole PA. Proc Natl Acad Sci U S A. 1998;95:6705–6710. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dawson PE, Muir TW, Clark-Lewis I, Kent SB. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]; Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yang A, Ha S, Ahn J, Kim R, Kim S, Lee Y, Kim J, Soll D, Lee HY, Park HS. Science. 2016;354:623–626. doi: 10.1126/science.aah4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, Brock A, Herberich B, Schultz PG. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 8.Neumann H, Peak-Chew SY, Chin JW. Nat Chem Biol. 2008;4:232–234. doi: 10.1038/nchembio.73. [DOI] [PubMed] [Google Scholar]; Lee YJ, Wu B, Raymond JE, Zeng Y, Fang X, Wooley KL, Liu WR. ACS Chem Biol. 2013;8:1664–1670. doi: 10.1021/cb400267m. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gattner MJ, Vrabel M, Carell T. Chem Commun (Camb) 2013;49:379–381. doi: 10.1039/c2cc37836a. [DOI] [PubMed] [Google Scholar]; Xiao H, Xuan W, Shao S, Liu T, Schultz PG. ACS Chem Biol. 2015;10:1599–1603. doi: 10.1021/cb501055h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saxon E, Armstrong JI, Bertozzi CR. Org Lett. 2000;2:2141–2143. doi: 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]; Nilsson BL, Kiessling LL, Raines RT. Org Lett. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]
- 10.Weisbrod SH, Marx A. Synlett. 2010:787–789. [Google Scholar]; Sowa S, Muhlberg M, Pietrusiewicz KM, Hackenberger CP. Bioorg Med Chem. 2013;21:3465–3472. doi: 10.1016/j.bmc.2013.02.045. [DOI] [PubMed] [Google Scholar]
- 11.Tam A, Soellner MB, Raines RT. J Am Chem Soc. 2007;129:11421–11430. doi: 10.1021/ja073204p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanrikulu IC, Schmitt E, Mechulam Y, Goddard WA, 3rd, Tirrell DA. Proc Natl Acad Sci U S A. 2009;106:15285–15290. doi: 10.1073/pnas.0905735106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, Schultz PG. Chem Biol. 2001;8:883–890. doi: 10.1016/s1074-5521(01)00063-1. [DOI] [PubMed] [Google Scholar]; Wang YS, Wu B, Wang Z, Huang Y, Wan W, Russell WK, Pai PJ, Moe YN, Russell DH, Liu WR. Mol Biosyst. 2010;6:1557–1560. doi: 10.1039/c002155e. [DOI] [PubMed] [Google Scholar]
- 14.Soellner MB, Nilsson BL, Raines RT. J Am Chem Soc. 2006;128:8820–8828. doi: 10.1021/ja060484k. [DOI] [PubMed] [Google Scholar]
- 15.Kim MS, Diamond SL. Bioorg Med Chem Lett. 2006;16:4007–4010. doi: 10.1016/j.bmcl.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 16.Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, Boeke JD, Zhao Y. Mol Cell Proteomics. 2012;11:100–107. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, Choudhary C. Cell Rep. 2013;4:842–851. doi: 10.1016/j.celrep.2013.07.024. [DOI] [PubMed] [Google Scholar]; Yang M, Wang Y, Chen Y, Cheng Z, Gu J, Deng J, Bi L, Chen C, Mo R, Wang X, Ge F. Mol Cell Proteomics. 2015;14:796–811. doi: 10.1074/mcp.M114.045922. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pan J, Chen R, Li C, Li W, Ye Z. J Proteome Res. 2015;14:4309–4318. doi: 10.1021/acs.jproteome.5b00485. [DOI] [PubMed] [Google Scholar]; Colak G, Xie Z, Zhu AY, Dai L, Lu Z, Zhang Y, Wan X, Chen Y, Cha YH, Lin H, Zhao Y, Tan M. Mol Cell Proteomics. 2013;12:3509–3520. doi: 10.1074/mcp.M113.031567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin FL, Hoyt HM, van Halbeek H, Bergman RG, Bertozzi CR. J Am Chem Soc. 2005;127:2686–2695. doi: 10.1021/ja044461m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.