

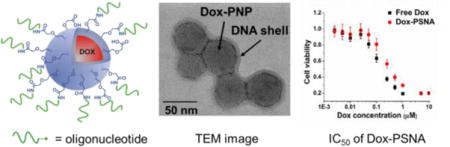
Abstract
Small-sized (~65 nm) doxorubicin (Dox)-loaded polymeric nanoparticles (PNPs) were modified with oligonucleotides to form colloidally stable Dox-loaded polymeric spherical nucleic acid (Dox-PSNA) nanostructures in biological media. The nucleic acid shell facilitates the cellular uptake of Dox-PSNA, resulting in in-vitro cytotoxicity against SKOV3 cancer cells.
Keywords: Drug-loaded nanoparticles, polymeric nanoparticles, spherical nucleic acids, doxorubicin, ring-opening metathesis polymerization
TOC figure
Polymeric nanoparticles (PNPs) prepared from amphiphilic ROMP block copolymers (ROMP = ring-opening metathesis polymerization) represent a versatile class of multifunctional nanoconstructs for biodetection,1,2 imaging,3 and drug delivery.4,5 In particular, the ease with which these nanoparticles can be surface-functionalized2 with a multitude of biologically active entities2,6 has shown promise in many drug delivery applications.5,7,8 Previously, we reported that ROMP-based doxorubicin-loaded PNPs can be modified with cationic surface moieties post-assembly to improve their stability in buffered media and in vitro cellular uptake.7 However, the large diameters of these particles (~100–200 nm) may limit their cellular uptake9 and tumor penetration when delivered in vivo.10,11 Thus, we set out to improve the bioavailability of the doxorubicin-loaded PNP platform by reducing nanoparticle size and covalently modifying the surface with negatively charged oligonucleotides, which have been shown to promote higher cellular uptake.12 Since sub-100 nm-sized nanoparticles have been shown to exhibit increased cellular uptake and better penetration into tumor tissue,13,14 these two modifications may allow for extended stability under physiologically relevant conditions and should enhance overall efficacy for in vivo applications.
The high-density surface modification of nanoparticles with oligonucleotides results in a three-dimensional architecture known as a spherical nucleic acid (SNA).12 The dense oligonucleotide shells in SNAs have been shown to stabilize a broad range of nanoparticle cores–from inorganic Au,15 Ag,16 and silica17 ones to organic structures such as ultra-small liposomes,18 proteins,19 and polymers20,21–under physiological conditions via electrostatic repulsion.18 The SNA architecture also promotes superior cellular internalization for the nanoparticle cores over PEG-functionalized analogs22 and linear nucleic acids,23 given its ability to bind to cellular membrane-bound scavenger receptors,23 which facilitate cellular internalization via endosomal pathways.12,24 As a result, SNA structures have been utilized in a wide variety of applications spanning biodetection,22 gene regulation25, drug delivery,26–28 and immunomodulation.12,29 By covalently modifying small (sub-100 nm) doxorubicin-loaded PNPs with oligonucleotides (Scheme 1), we show herein that the resulting doxorubicin-loaded polymeric SNA (Dox-PSNA) structures are stable under physiologically relevant conditions, are capable of active cellular internalization, and exhibit cytotoxicity with respect to SKOV-3 ovarian cancer cells. While the potency of these Dox-PSNA nanoparticles is comparable to that of the free drug, they should not be prone to the renal clearance issues that plague small molecules.30
Scheme 1.
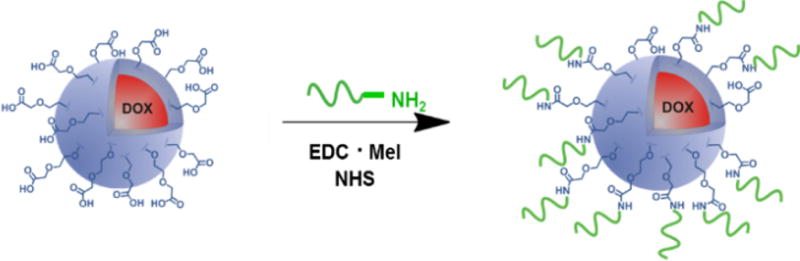
The covalent surface modification of carboxy-functionalized PNPs with amine-terminated oligonucleotides (green) via amide-coupling chemistry.
MATERIALS AND METHODS
Materials and Instrumentation
Please see SI, Section S1
Synthesis of block copolymer 215-b-115
In an inert-atmosphere glovebox, monomer 2 (25 mg, 45.2 μmol; see SI, Section S2 for the synthesis of monomer 2) was dissolved in an anhydrous mixture of CHCl3/MeOH (9:1 v/v, 1 mL) in a 20 mL scintillation vial equipped with a magnetic stir bar. A stock solution of Grubbs’ first-generation catalyst (5 mg) in CHCl3/MeOH (9:1 v/v, 5 mL) was prepared, a portion of which (2.48 mL, 3.01 μmol) was added to the vial containing the solution of monomer 2 under vigorous stirring. The resulting reaction mixture was stirred for 45 min at room temperature, at which time an aliquot (100 μL) was removed and quenched with excess ethyl vinyl ether. A portion of this quenched aliquot was evaporated to dryness, redissolved in CDCl3, and analyzed by 1H NMR spectroscopy, which indicated complete consumption of the monomer. The remaining portion was evaporated to dryness, dissolved in HPLC-grade THF, and subjected to GPC analysis (Mn = 9000 (theoretical Mn = 8000), PDI = 1.13).
Immediately after aliquot removal, a solution of monomer 1 (see SI, Section S1, 30.7 mg, 45.2 μmol) in CHCl3/MeOH (9:1 v/v, 1 mL) was added to the reaction vial and the resulting polymerization mixture was stirred for an additional 45 min before being terminated with the addition of ethyl vinyl ether (1 mL). The reaction mixture was added quickly into vigorously stirred cold (−10 °C) pentanes (200 mL), and the resulting precipitate was isolated via vacuum-filtration and washed thoroughly with fresh pentanes to afford the product copolymer quantitatively as a red solid. See the SI for the 1H NMR spectrum, Section S3). Due to the lack of complete polymerization of monomer 1 when being initiated as the first block,7,31 we did not attempt to synthesize the reverse block copolymer 115-b-215.
To maximize the number of chemical handles that can subsequently be used for surface functionalization of the PNPs, we employed a block copolymer with an equimolar ratio of monomers 2:1, while keeping the PNP diameter < 100 nm.
General procedure for the preparation of PNPs in the 60–75 nm size range
An aqueous suspension of the polymer nanoparticles was prepared by dialysis following a modification of the published procedure.7 An aliquot (2.5 mL) of a stock solution of block copolymer 215-b-115 (0.01 wt %) in DMSO was transferred to a 4 mL scintillation vial and stirred vigorously. Ultrapure deionized water was added to this stirring copolymer solution at a rate of 1 drop (50 μL, 1.75 wt %) every 1 s using a 20–100 μL micro-pipette until the mixture contained 18 wt % water. The resulting cloudy mixture was placed in a 3 mL dialysis cassette and dialyzed against ultrapure deionized water (500 mL), with the dialate changed every 2 h. Complete absence of DMSO in the dialysate after 48 h was verified by UV-vis spectroscopy as indicated by the disappearance of the UV cut-off for DMSO at 268 nm.
Characterization of PNPs derived from block copolymer 215-b-115
DLS analysis of the final PNP aqueous suspension derived from block copolymer 215-b-115 revealed relatively monodisperse PNPs with an average DH = 63 ± 7 nm and a PDI = 0.11 (Figure 1B). TEM analysis indicated a uniform size distribution for the PNPs in the solid-state, with an average diameter of ~65 nm (Figure 1C) that is consistent with the DLS data. The zeta potential of these PNPs was determined to be −6.83 ± 0.5 mV. The average concentration of PNP particles is ~ 1 × 1010 particles/mL, as determined by NTA. The doxorubicin loading for the PNPs is 64 wt %, as calculated based on the doxorubicin wt% in the 215-b-115 copolymer.
Figure 1.
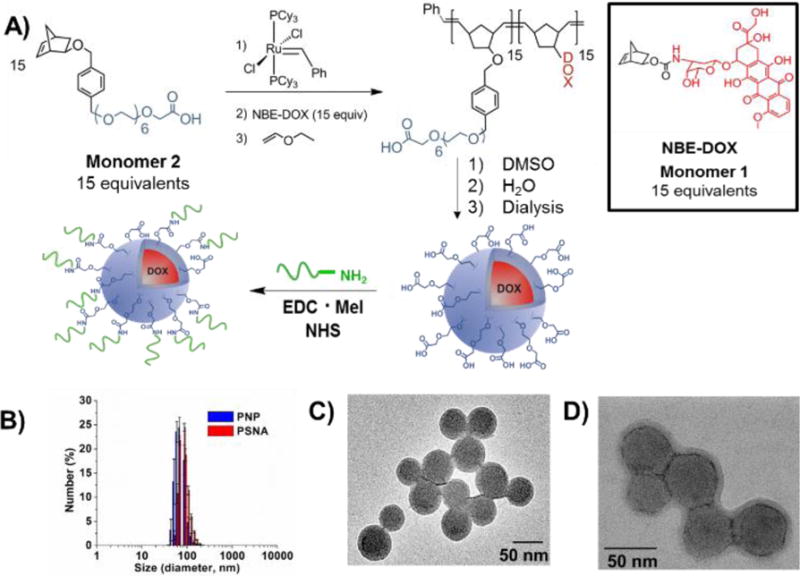
A) Synthesis of block copolymer 115-b-215 and subsequent formation of the corresponding PNPs. B) Dynamic light scattering (DLS) data for the PNPs before and after covalent DNA functionalization. C–D) TEM image of the PNPs before (C) and after (D) DNA functionalization.
DNA functionalization of doxorubicin-loaded PNPs and characterization data
In a 1.5 mL safe-lock Eppendorf tube, solutions of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC•HCl, final concentration in reaction solution = 5 μM) and N-hydroxysulfosuccinimide (sulfo-NHS, final concentration in reaction solution = 7.5 μM) in ultrapure deionized water were added to an aliquot (1 mL) of the carboxy-functionalized PNP suspension (1 × 1010 particles/mL, ~125,000 carboxylate groups) derived from the desired block copolymer. The reaction mixture was agitated (1000 rpm) on a multi-therm platform shaker (Benchmark Scientific, Inc., South Plainfield, NJ, USA) for 5 min at room temperature. A freshly prepared aliquot of 3′-amino-terminated oligonucleotides (100 μL of a 10 μM solution) was then added to the reaction mixture, and the resulting mixture was covered with aluminum foil and agitated on a multi-therm platform shaker for 4 h at room temperature.
To increase the density of oligonucleotide packing on the PNP surface, EDC and NHS solutions (to 5 μM and 7.5 μM final concentrations, respectively, assuming that all the initially added reagents have been consumed/decomposed) were added to the PNP solution, and the resulting mixture was subjected to a salt-aging process.32 Aliquots of NaCl (8 μL of a 5 M solution to attain a final [NaCl] = 50 mM) were added to the reaction mixture every 30 min, and the Eppendorf tube was sonicated for 15 s (Branson 2510 ultrasonic cleaner, Branson Ultrasonics, Danbury, CT, USA) after each salt addition. Upon reaching a total NaCl concentration of 300 mM, the reaction mixture was agitated on a multi-therm platform shaker for an additional 12 h at room temperature. The resulting PSNAs were then purified using size-exclusion chromatography (Sepharose CL-4B, Sigma Aldrich). The average concentration of PSNA particles was ~ 109/mL, as determined by NTA.
Characterization of oligonucleotides-conjugated doxorubicin-loaded PNPs
DLS analysis of the final DNA-functionalized PNP aqueous suspension derived from block copolymer 315-b-515 revealed relatively monodisperse PNPs with an average DH = 88 ± 3 nm and a PDI = 0.13. The zeta potential of the oligonucleotide-modified PNPs was determined to be −31.2 ± 3.2, indicating that negatively charged oligonucleotides are conjugated to the PNP surface. For TEM analysis, the PNPs were pre-stained with uranyl acetate solution (10 μL, 2% w/v).
Quantification of oligonucleotides strands on the surface of PSNAs
To assess the degree of oligonucleotide surface modification, an aliquot (20 μL) of the purified Cy5-oligonucleotide-modified PNPs was added to DMSO solution and placed on a multi-therm shaker for 2 h at 37 °C to dissolve the PSNAs. An aliquot (50 μL) of the resulting solution was further mixed with DMSO (final volume = 200 μL), and its fluorescence at the emission wavelength of Cy5 (λex = 649 nm, λem = 670 nm) was recorded using a Synergy HT multi-mode microplate reader (BioTek, Winooski, VT, USA). This fluorescence was compared against a calibration curve constructed from absorbance values for solutions of the Cy5-labeled oligonucleotides in DMSO (concentrations = 0, 0.1, 0.25, 0.5,1, 2.5, 5 μM). From this analysis and the number of PSNA nanoparticles in solution (obtained from NTA), each cholesterol-containing PSNA particle was estimated to have ~1900 oligonucleotides strands and each doxorubicin-loaded PSNA particle was estimated to have ~1200 oligonucleotides strands. The doxorubicin loading is 63.8 wt %, as calculated based on the doxubicin wt % in the 215-b-115 copolymer and the total weight of the DNA, assuming an average density of 1 for the whole construct.
Stability study of PNPs and PSNAs
Two separate aliquots of the doxorubicin-containing PNPs and PSNAs (500 μL) were transferred to a 1.5 mL safe-lock Eppendorf tube and centrifuged for 30 min at 10K rpm to a solid pellet. The supernatant was removed and the PNPs were re-suspended in PBS (500 μL, 10 mM, pH 7.4, 150 mM [NaCl]). As expected, characterization of the PNP suspension in PBS resulted in loss of the original well-defined morphology of the parent PNPs as observed by TEM and DLS. The size of PSNAs was analysed periodically over a two month period using DLS. We note in passing that while single-stranded DNA has been adsorbed to the surface of polymer nanoparticles through non-covalent modification,33,34 such constructs have not been tested for stability in cell culture media.
Melt Analysis
Melt analysis experiments were carried out using a Cary 5000 UV-vis spectrometer equipped with a programmable heating stage (ThermoFisher Scientific, Carlsbad, CA, USA). Doxorubicin-containing PSNAs functionalized with Melt A strands were prepared as described above. 13 nm Au-core SNAs functionalized with the complementary Melt B strands (Table S1) were prepared following literature protocols.32 The aggregates were formed by combining these two materials in a 1:1 ratio (total DNA concentration = 1.5 μM, total volume = 1 mL). The aggregates were then subjected to a temperature ramp from 20 to 65 °C (rate of 0.25 °C/min) while the absorbance for the aggregates was continuously monitored at 260 nm.
Cell culture experiments
SKOV-3 cells were purchased from American Tissue Culture Collection (ATCC, Manassas, VA, USA) and grown in McCoy’s 5A medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 IU/mL), and streptomycin (50 μg/mL) and maintained at 37 °C with 5% CO2 as per ATCC instructions. For cellular studies, the cells were plated at 60% confluency 24 h prior to the treatment.
Confocal microscopy of PSNAs
In a typical experiment, SKOV-3 cells were plated on a 35 mm FluoroDish (World Precision Instruments, Saratosa, FL, USA) and incubated for 24 h before being transfected with the appropriate oligonucleotide formulation (final DNA concentration = 0.1 μM). The treated cells were incubated for 24 h before being washed three times with 1× phosphate-buffered saline (PBS, Invitrogen) and imaged under a Zeiss LSM 510 inverted laser-scanning confocal microscope using Hoechst 33342 as a nuclear stain. The fluorescence excitation for Cy5 was set as 630 nm and emission 650–710 nm. For Z-stack images, the images were collected in different planes with a depth of 0.3 μM.
Cellular uptake experiments
The cellular uptake of the PSNAs was compared to that of the free drug in SKOV3 cells. In preparation for this analysis, the cells were seeded at 60% confluency in a 96 well plate 24 h before treatment with PSNAs and free doxorubicin. The treatments were carried out at 0.1, 0.5, 1, and 2.5 μM of doxorubicin. The cells were then incubated with either free drug or the PSNAs for 24 h before washing three times with 1× PBS. The cells were then detached from the plate surface by exposing to trypsin (20 μL) for 5 mins and fixed using a 4 % para-formaldehyde solution (in 1× PBS). The intracellular delivery of doxorubicin was quantified via flow cytometry on a Guava easyCyte 8HT instrument (Millipore, Billerica, MA, USA) using the doxorubicin channel and normalized based on the intensity of the untreated cells.
Cell viability studies for doxorubicin and Dox-PSNAs
SKOV-3 (ovarian cancer) cells were seeded in 96 well plates at a density of ~5000 cells per well 24 h before the experiment. Cells were incubated with a fixed amount (100 μL) of solution containing either doxorubicin or Dox-PSNAs that were prepared at varying concentrations in OptiMEM (Invitrogen) at 37 °C. After overnight incubation, the media was replaced with regular growth medium (McCoy’s 5A supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 IU/mL), and streptomycin (50 μg/mL)), and the cells were incubated at 37 °C for an additional 48 h. Cell viability was assessed using the alamarBlue® cell viability assay (ThermoFisher Scientific, Carlsbad, CA, USA) according to the manufacturer’s recommended protocol. Briefly, 10% alamarBlue® reagent in regular growth medium was added to cells (100 μL per well), and the treated cell plate was incubated at 37 °C for 4 h before being analyzed on a Synergy H4 Multimode Microplate Reader. Fluorescence data at 570 nm were collected and normalized to that for an untreated control. Reported values represent the mean ± standard deviation of 5 replicates.
RESULTS AND DISCUSSION
As a general strategy,35 ROMP-based PNPs were assembled from short amphiphilic block copolymers consisting of one hydrophobic doxorubicin-conjugated block and a hydrophilic carboxy-terminated, poly(ethylene oxide) (PEO)-loaded block (Figure 1). With this design, the doxorubicin-loaded block constitutes the core of the PNPs, and the PEO-loaded segment forms a shell with terminal carboxy groups that can be used for further surface modification. The doxorubicin payload was linked to the hydrophobic segment through an acid-cleavable linkage,4 which can be released in the low-pH environment of the late endosome36 or in the tumor tissue.37
The amphiphilic block copolymers needed for PNP preparation were synthesized by block-copolymerizing the carboxy-terminated, PEO-conjugated monomer 2 and the doxorubicin-conjugated monomer 1 (Figure 1A). A 15:15 molar ratio for monomers 2:1 was chosen to limit the molecular weight of the polymer < 45 kDa, thus facilitating renal clearance of the polymers post drug release.38 Monomer 2 was successfully homo-polymerized using Grubbs’ first-generation olefin metathesis ((PCy3)2Cl2Ru=CHPh) catalyst (3) as confirmed by 1H NMR spectroscopy and gel-permeation chromatography (GPC) analyses (Mn = 9000 (theoretical Mn = 8000), polydispersity index (PDI) = 1.17; Supporting Information (SI), Section S2). Sequential addition of monomer 1 to the homo-polymerized block 215, followed by quenching with ethyl vinyl ether resulted in the formation of our desired monodisperse amphiphilic block copolymer 215-b-115 (final polymer Mn = 15000 (theoretical Mn = 14000), PDI = 1.12; see Materials and Methods section and SI, Section S3).
Following our previously established strategy,7 dropwise addition of water to a DMSO solution of copolymer 215-b-115, followed by exhaustive dialysis against ultrapure deionized water, afforded an aqueous suspension of carboxy-functionalized, doxorubicin-loaded PNPs (Figure 1A). These nanoparticles have small hydrodynamic diameters (DH = 63 ± 7 nm, Figure 1B) and exhibit a narrow size distribution (PDI = 0.11 ± 0.02), as measured by dynamic light scattering (DLS). Transmission electron microscopy (TEM) also confirmed their homogeneous spherical morphology (Figure 1C) with an average DH of 65 ± 5 nm. Key to the realization of these small nanoparticle sizes was a fast rate of water addition over a shorter time period than previously reported7 (see Materials and Methods section).
The covalent surface modification of the carboxy-functionalized PNPs with 3′-amine-terminated DNA was achieved using EDC/NHS coupling (Figure 1A). A mixture of the two components was stirred together with the EDC and NHS reagents for 4 h before being subjected to a salt-aging process to further increase the DNA density on the PNP surface32 (see Materials and Methods section). After the total salt concentration reached 0.3 M, this mixture was allowed to react further overnight before being purified by size-exclusion chromatography. A TEM image (Figure 1D) of the isolated PSNA clearly shows the retention of the discrete and spherical morphology of the parent PNP (diameter ~ 87 nm). Their DLS-derived size distribution plot (Figure 1B) shows an increase in the hydrodynamic diameter (DH = 88 ± 3 nm and PDI = 0.13 ± 0.03) that can be attributed to the addition of a dense DNA shell on the PNP surfaces. The shift in zeta potential (−5.1 ± 1.5 mV for the parent PNPs) to a higher negative value (−31.2 ± 3.2 mV for the PSNAs) also corroborates the successful conjugation of DNA to the PNP surface. Notably, the presence of the DNA shell on the surface of the PNP can be clearly observed upon staining of the PSNAs with uranyl acetate (z-contrast and transmission modes; Figure 1D). While not a primary focus of the current work, the thickness of the DNA shell can be changed by altering the length and composition of the oligonucleotide strands.39 On average, ~1200 DNA strands/PNP were conjugated to the surface of each PNP as determined by fluorescence spectroscopy and nanoparticle tracking analysis (NTA) to quantify the number of PSNAs in a given sample via light scattering and Brownian motion. This DNA surface density (~7 pmol/cm2, see Materials and Methods section for calculation) is comparable to but slightly higher than the loadings obtained for our previously reported liposomal SNA constructs.40
The doxorubicin-loaded PSNAs show excellent stability in comparison to the parent PNPs, a property that enables their use in cellular systems. For example, the PSNAs remain colloidally stable after 2 months of incubation in phosphate-buffered saline (1× PBS) at room temperature (Figure 2A). In contrast, subjecting the unmodified PNPs to the same treatment for only a week resulted in formation of polydisperse aggregates irreversibly agglomerated, presumably through interparticle fusion. Most relevant for physiological applications, no change was observed in the DLS distribution plot for the PSNAs after 3 days of incubation at 37 °C in 1× PBS (Figures 2C and 2D). We attribute this enhanced stability to the electrostatic repulsion between the negatively charged oligonucleotides that comprise the shell of the PSNA, which inhibits particle-particle fusion under physiologically relevant conditions.18 Notably, Dox-PSNAs remain stable in the presence of human serum while still maintaining their acid-triggered drug-release properties.31 The doxorubicin-based fluorescence profiles for Dox-PSNAs after being incubated at 37 °C in PBS and human serum solution (10% v/v) are very similar as evidenced by minimal leakage of the drug (SI, Figure S8). In contrast, exposing Dox-PSNAs to 0.1 M HCl (aq) leads to a six-fold increase in drug release after one hour (SI, Figure S8).
Figure 2.
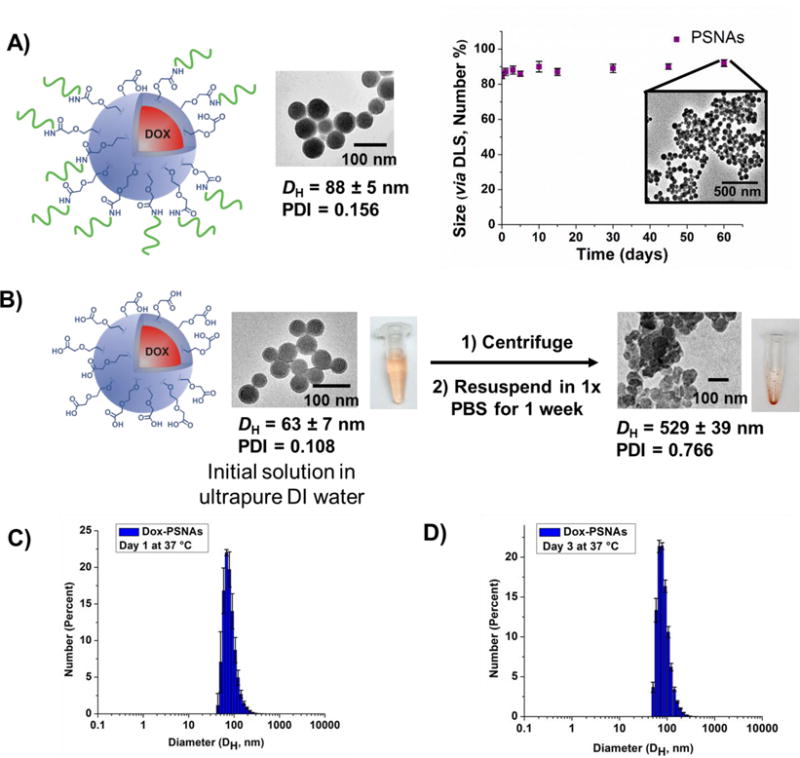
A) Doxorubicin-loaded PSNAs remain stable for over two months in 1× PBS as analysed by TEM and DLS. B) Doxorubicin-loaded PNPs aggregate when incubated in 1× PBS for a week. C and D) DLS profiles for Dox-PSNAs confirming their stablity in 1× PBS at 37 °C for 3 days.
Consistent with the assigned SNA structure, PSNAs cooperatively hybridize with complementary Au core SNAs. For example, when PSNAs were combined with complementary 13 nm Au-core SNAs (Figure 3A), visible aggregates can be observed and isolated. TEM analysis (Figures 3B and 3C) of these hybridized materials clearly shows a complete coverage of the larger PSNAs by the smaller Au-core SNAs. The increase in the melting temperature (Tm of the PNP-AuNP hybrid = 54.7 °C vs Tm = 52 °C (as calculated at 150 mM NaCl and 1.5 uM oligonucleotides using IDT OligoAnalyzer41) for the free DNA duplex analog) of these aggregates and their narrow thermal denaturation profile (full width at half-maximum (fwhm) of the first derivative is ∼2.3 °C) confirms cooperative binding behavior that is characteristic of SNAs15 (Figure 3D).
Figure 3.
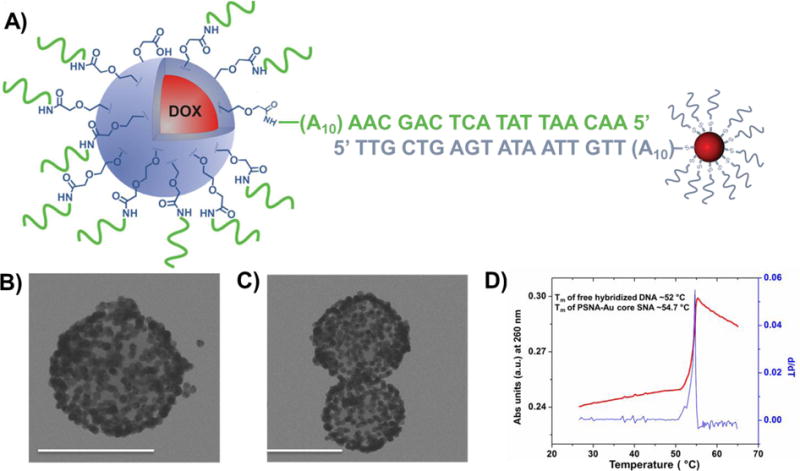
A) A schematic drawing of the Dox-PSNAs hybridized to complementary 13 nm Au-core SNAs. B–C) TEM images of the Dox-PSNAs surrounded by complementary Au-core SNAs conjugates. Scale bar is 100 nm. D) The thermal denaturation profile (red) of the [Dox-PSNAs + Au-core SNAs] hybridized aggregates monitored as a change in extinction at 260 nm. The overlaid 1st derivative (blue) of this profile shows the sharp melting transition. See SI, section S10 for the control experiment.
As expected, the dense oligonucleotide shell of the Dox-PSNAs greatly facilitates their entrance into cells. As shown by confocal microscopy, SKOV-3 cells (ovarian cancer ascites) readily uptake Dox-PSNAs that were surface-modified with 5′-Cy5-labelled oligonucleotides (Figure 4A). This is in stark contrast to the non-functionalized PNP analog, which settles out of cell culture media due to aggregation and shows low and non-uniform uptake.31 Such enhanced cellular internalization of SNA structures has been attributed by Mirkin and coworkers to the interaction of the oligonucleotide shell with Class A scavenger receptors (SR-A) present on the cell surface, followed by caveolin-mediated endocytosis.23 Because our goal was to enhance the delivery of the therapeutically active payload of Dox-PSNAs to diseased cells, a generic T20 DNA sequence was sufficient for this purpose. We note in passing that although a detailed mechanistic study comprising competitive and endocytosis inhibition assays can be carried out to ascertain the exact uptake mechanism, such a study is beyond the scope of the present work.
Figure 4.
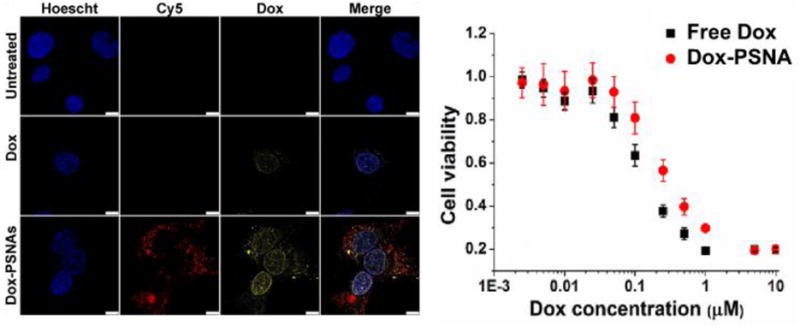
A) Confocal laser-scanning microscopy images of SKOV-3 cells that have been incubated for 24 h with free doxorubicin (Dox) and doxorubicin-loaded PSNAs (Dox-PSNAs) at 2.5 μM Dox concentration. Given the qualitative nature of the confocal image (e.g., change in focal point between sample wells), the various signal intensities should not be compared across rows. In addition, each panel is limited to a few cells and do not represent the entire population. A statistically significant flow cytometry study (SI, Figure S10) does show higher doxorubicin fluorescence signal for the cells that have been incubated with free doxorubicin. B) Cytotoxicity profiles for SKOV-3 cells that have been incubated for 48 h with dox and Dox-PSNA formulations at 5 nM-10 μM [ Dox]. The similarity in toxicity between the doxorubicin-loaded NPs and free doxorubicin is not surprising given previous studies from our group,31,42 and the fact that IC50 measurements are highly dependent on many variables (treatment time, incubation time post-treatment, release of payload from the delivery vehicle, cell type and confluency) and can vary by several fold, depending on conditions.
The cellular internalization properties of the Dox-loaded PSNAs (Dox-PSNAs) lead to in-vitro cytotoxicity against SKOV3 cancer cells that is comparable to that of free doxorubicin (Figure 4B). Such a study could not have been properly carried out with the parent PNPs due to their aggregation behavior in biological media. Indeed, PNPs have been observed to settle out of cell culture media over time, leading to low and non-uniform uptake in SKNSH wild-type cells.31 The observed cytotoxicity of the Dox-PSNAs can be attributed to the acid-induced cleavage of the carbamate linkage between the block copolymer backbone and the doxorubicin, allowing for the release of the latter into the cytoplasm.6,31 Supporting this conclusion is the observation that the analogous PSNAs with cholesterol-loaded cores does not show any cytotoxicity against SKOV3 cells (SI, Section S5).
CONCLUSIONS
In summary, we have demonstrated that the successful modification of doxorubicin-loaded PNPs with a dense oligonucleotide shell greatly increases their colloidal stability in biological media under physiological conditions. The dense shell of oligonucleotides allows the resulting Dox-PSNAs to have enhanced cellular uptake, manifesting in cytotoxicity that is comparable to the free drug. Importantly, this SNA-based strategy should be generalizable to other therapeutic-loaded nanoparticles for in vivo applications in the treatment of cancer and other diseases.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the Center for Cancer Nanotechnology Excellence initiative of the National Cancer Institute of the National Institutes of Health under Award Number U54CA151880 and U54CA199091. This material is also based upon work supported by the AFOSR under Award No. FA9550-12-1-0280. CAM additionally acknowledges support from the NTU-NU Institute for NanoMedicine located at the International Institute for Nanotechnology, Northwestern University, USA and the Nanyang Technological University, Singapore. Instruments in the Northwestern University IMSERC, Keck Biophysics, and EPIC/NUANCE facilities were purchased with grants from NSF-NSEC (NSF EEC-0647560), NIH-CCNE, NSF-MRSEC (NSF DMR-1121262), the Keck Foundation, the State of Illinois, and Northwestern University. We additionally thank the reviewers of the initial version of this manuscript for comments that help improving its quality. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the AFOSR.
ABBREVIATIONS
- Dox
doxorubicin
- DH
hydrodynamic diameters
- DLS
dynamic light scattering
- NTA
nanoparticle tracking analysis
- PDI
polydispersity index
- PEG
poly(ethylene glycol)
- PNP
polymeric nanoparticles
- PSNA
polymeric spherical nucleic acids
- ROMP
ring-opening metathesis polymerization
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.
General experimental and instrumentation details; procedures for the syntheses of monomers, homopolymers, block copolymers, and cholesterol-loaded PNPs; characterization data for monomers, homopolymers, block copolymers, and cholesterol-loaded PSNAs; stability data for Dox-PSNAs in serum and cell culture media; pH-dependent drug release profiles for Dox-PSNAs; quantification of cellular uptake of Dox-PSNA and free doxorubicin (PDF).
Notes: The authors declare no competing financial interest.
References
- 1.Rush AM, Nelles DA, Blum AP, Barnhill SA, Tatro ET, Yeo GW, Gianneschi NC. J Am Chem Soc. 2014;136:7615–7618. doi: 10.1021/ja503598z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertin PA, Gibbs JM, Shen CK, Thaxton CS, Russin WA, Mirkin CA, Nguyen ST. J Am Chem Soc. 2006;128:4168–4169. doi: 10.1021/ja056378k. [DOI] [PubMed] [Google Scholar]
- 3.Allen MJ, Raines RT, Kiessling LL. J Am Chem Soc. 2006;128:6534–6535. doi: 10.1021/ja061383p. [DOI] [PubMed] [Google Scholar]
- 4.Bertin PA, Smith D, Nguyen ST. Chem Commun. 2005;41:3793–3795. doi: 10.1039/b504643b. [DOI] [PubMed] [Google Scholar]
- 5.Callmann CE, Barback CV, Thompson MP, Hall DJ, Mattrey RF, Gianneschi NC. Adv Mater. 2015;27:4611–4615. doi: 10.1002/adma.201501803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krovi SA, Smith D, Nguyen ST. Chem Commun. 2010;46:5277–5279. doi: 10.1039/c0cc00232a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krovi SA, Swindell EP, O’Halloran TV, Nguyen ST. J Mater Chem. 2012;22:25463–25470. doi: 10.1039/C2JM35420A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukherjee S, Patra D, Dinda H, Chalkraborty I, Shashank L, Bhattacharyya R, Das Sarma J, Shunmugam R. Macromolecules. 2016;49:2411–2418. [Google Scholar]
- 9.Huang K, Ma H, Liu J, Huo S, Kumar A, Wei T, Zhang X, Jin S, Gan Y, Wang PC, He S, Zhang X, Liang XJ. ACS Nano. 2012;6:4483–4493. doi: 10.1021/nn301282m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi JS, Cao J, Naeem M, Noh J, Hasan N, Choi HK, Yoo JW. Colloids Surf, B. 2014;122:545–551. doi: 10.1016/j.colsurfb.2014.07.030. [DOI] [PubMed] [Google Scholar]
- 11.Tang L, Gabrielson NP, Uckun FM, Fan TM, Cheng J. Mol Pharmaceutics. 2013;10:883–892. doi: 10.1021/mp300684a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cutler JI, Auyeung E, Mirkin CA. J Am Chem Soc. 2012;134:1376–1391. doi: 10.1021/ja209351u. [DOI] [PubMed] [Google Scholar]
- 13.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K. Nat Nanotechnol. 2011;6:815–823. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 14.Biondi M, Guarnieri D, Yu H, Belli V, Netti PA. Nanotechnology. 2013;24:045101. doi: 10.1088/0957-4484/24/4/045101. [DOI] [PubMed] [Google Scholar]
- 15.Mirkin CA, Letsinger RL, Mucic RC, Storhoff JJ. Nature. 1996;382:607–609. doi: 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- 16.Lee JS, Lytton-Jean AK, Hurst SJ, Mirkin CA. Nano Lett. 2007;7:2112–2115. doi: 10.1021/nl071108g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young KL, Scott AW, Hao L, Mirkin SE, Liu G, Mirkin CA. Nano Lett. 2012;12:3867–3871. doi: 10.1021/nl3020846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banga RJ, Chernyak N, Narayan SP, Nguyen ST, Mirkin CA. J Am Chem Soc. 2014;136:9866–9869. doi: 10.1021/ja504845f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brodin JD, Auyeung E, Mirkin CA. Proc Natl Acad Sci U S A. 2015;112:4564–4569. doi: 10.1073/pnas.1503533112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peterson AM, Heemstra JM. Wiley Interdiscip Rev: Nanomed Nanobiotechnol. 2015;7:282–297. doi: 10.1002/wnan.1309. [DOI] [PubMed] [Google Scholar]
- 21.Jia F, Lu X, Tan X, Zhang K. Chem Commun. 2015;51:7843–7846. doi: 10.1039/c5cc01934f. [DOI] [PubMed] [Google Scholar]
- 22.Giljohann DA, Seferos DS, Patel PC, Millstone JE, Rosi NL, Mirkin CA. Nano Lett. 2007;7:3818–3821. doi: 10.1021/nl072471q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi CH, Hao L, Narayan SP, Auyeung E, Mirkin CA. Proc Natl Acad Sci U S A. 2013;110:7625–7630. doi: 10.1073/pnas.1305804110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu XA, Choi CH, Zhang C, Hao L, Mirkin CA. J Am Chem Soc. 2014;136:7726–7733. doi: 10.1021/ja503010a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giljohann DA, Seferos DS, Prigodich AE, Patel PC, Mirkin CA. J Am Chem Soc. 2009;131:2072–2073. doi: 10.1021/ja808719p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dhar S, Daniel WL, Giljohann DA, Mirkin CA, Lippard SJ. J Am Chem Soc. 2009;131:14652–+. doi: 10.1021/ja9071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan X, Li BB, Lu X, Jia F, Santori C, Menon P, Li H, Zhang B, Zhao JJ, Zhang K. J Am Chem Soc. 2015;137:6112–6115. doi: 10.1021/jacs.5b00795. [DOI] [PubMed] [Google Scholar]
- 28.Tan XY, Lu XG, Jia F, Liu XF, Sun YH, Logan JK, Zhang K. J Am Chem Soc. 2016;138:10834–10837. doi: 10.1021/jacs.6b07554. [DOI] [PubMed] [Google Scholar]
- 29.Radovic-Moreno AF, Chernyak N, Mader CC, Nallagatla S, Kang RS, Hao L, Walker DA, Halo TL, Merkel TJ, Rische CH, Anantatmula S, Burkhart M, Mirkin CA, Gryaznov SM. Proc Natl Acad Sci U S A. 2015;112:3892–3897. doi: 10.1073/pnas.1502850112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kadam RS, Bourne DWA, Kompella UB. Drug Metab Dispos. 2012;40:1380–1388. doi: 10.1124/dmd.112.044925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith D, Clark SH, Bertin PA, Mirkin BL, Nguyen ST. J Mater Chem. 2009;19:2159–2165. doi: 10.1039/B817511J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurst SJ, Lytton-Jean AK, Mirkin CA. Anal Chem. 2006;78:8313–8318. doi: 10.1021/ac0613582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li HL, Sun XP. Chem Commun. 2011;47:2625–2627. doi: 10.1039/c0cc05394e. [DOI] [PubMed] [Google Scholar]
- 34.Wang L, Zhang Y, Tian J, Li H, Sun X. Nucleic Acids Res. 2011;39:e37. doi: 10.1093/nar/gkq1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bertin PA, Watson KJ, Nguyen ST. Macromolecules. 2004;37:8364–8372. [Google Scholar]
- 36.Deng X, Wang Y, Zhang F, Yin Z, Hu Q, Xiao X, Zhou Z, Wu Y, Sheng W, Zeng Y. Chem Commun. 2016;52:3243–3246. doi: 10.1039/c5cc10396g. [DOI] [PubMed] [Google Scholar]
- 37.Poon Z, Chang D, Zhao X, Hammond PT. ACS Nano. 2011;5:4284–4292. doi: 10.1021/nn200876f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nori A, Kopeček J. Adv Drug Delivery Rev. 2005;57:609–636. doi: 10.1016/j.addr.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 39.Narayan SP, Choi CH, Hao L, Calabrese CM, Auyeung E, Zhang C, Goor OJ, Mirkin CA. Small. 2015;11:4173–4182. doi: 10.1002/smll.201500027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill HD, Millstone JE, Banholzer MJ, Mirkin CA. ACS Nano. 2009;3:418–424. doi: 10.1021/nn800726e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.https://www.idtdna.com/calc/analyzer; accessed on April 2, 2016.
- 42.Lee S-M, Chen H, O’Halloran TV, Nguyen ST. J Am Chem Soc. 2009;131:9311–9320. doi: 10.1021/ja9017336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.