

Abstract
Background and Aims
Saturated free fatty acids (SFA) and their toxic metabolites contribute to hepatocyte lipotoxicity in nonalcoholic steatohepatitis (NASH). We previously reported that hepatocytes, under lipotoxic stress, express the potent macrophage chemotactic ligand C-X-C motif chemokine 10 (CXCL10) and release CXCL10-enriched extracellular vesicles by a mixed lineage kinase (MLK) 3-dependent mechanism. In the current study, we sought to examine the signaling pathway responsible for CXCL10 induction during hepatocyte lipotoxicity. Here, we demonstrate a role for signal transducer and activator of transcription (STAT) 1 in regulating CXCL10 expression.
Methods
Huh7 and HepG2 cells were treated with lysophosphatidylcholine (LPC), the toxic metabolite of the SFA palmitate.
Results
In LPC-treated hepatocytes, CXCL10 induction is mediated by a mitogen activated protein kinase (MAPK) signaling cascade consisting of a relay kinase module of MLK3, MKK3/6, and p38. P38 in turn induces STAT1 Ser727 phosphorylation and CXCL10 upregulation in hepatocytes, which is reduced by genetic or pharmacological inhibition of this MAPK signaling cascade. The binding and activity of STAT1 at the CXCL10 gene promoter were identified by chromatin immunoprecipitation and luciferase gene expression assays. Promoter activation was attenuated by MLK3/STAT1 inhibition or by deletion of the consensus STAT1 binding sites within the CXCL10 promoter.
Conclusion
In lipotoxic hepatocytes, MLK3 activates a MAPK signaling cascade, resulting in the activating phosphorylation of STAT1, and CXCL10 transcriptional upregulation. Hence, this kinase relay module and/or STAT1 inhibition may serve as a therapeutic target to reduce CXCL10 release, thereby attenuating NASH pathogenesis.
Keywords: MLK3, STAT1, CXCL10, p38 MAPK, lipotoxicity, NASH
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is currently recognized as the most common cause of liver disease worldwide [Younossi et al., 2016]. A subset of patients with NAFLD, have the more severe and inflammatory condition known as nonalcoholic steatohepatitis (NASH) which has the potential to progress to cirrhosis and end stage liver disease [Younossi et al., 2016]. Indeed, NASH-related cirrhosis is predicted to be the leading indication for liver transplantation by 2030 [Afzali et al., 2012]. Yet, the cellular and molecular mechanisms culminating in NASH pathogenesis remain poorly understood, and apart from lifestyle modification, no effective therapy has been validated and approved by regulatory agencies. Therefore, there is a critical need to identify specific therapeutic targets based on well-defined pathogenic signaling mechanisms.
Excess circulating saturated free fatty acids (SFA)s in NASH, contributes to hepatocyte injury, [Neuschwander-Tetri, 2010], and promotes hepatic inflammation characterized, in part, by macrophage hepatic recruitment and activation [Tosello-Trampont et al., 2012]. Palmitate is one of the more toxic SFAs. Excess palmitate delivered to hepatocytes in NASH is converted to lysophosphatidylcholine (LPC) which mimics all the toxicity of palmitate [Han et al., 2011; Kakisaka et al., 2012; Schattenberg and Lee, 2016]. Liver lipidomic analyses demonstrate that LPC content is increased in the liver specimens from NASH patients [Han et al., 2008]. Hence, we employed LPC in the current study as a proximal mediator of lipotoxic signaling cascades in hepatocytes.
Mixed lineage kinase 3 (MLK3) is the mitogen activated protein kinase kinase kinase (MAPKKK) which is activated by SFA, initiating stress signaling cascades [Jaeschke and Davis, 2007]. We have previously demonstrated that MLK3−/− mice are protected against steatohepatitis during an obesity-inducing diet [Ibrahim et al., 2014]. Moreover, we have reported that MLK3 mediates the release of C-X-C motif chemokine 10 (CXCL10)-enriched extracellular vesicles (EV)s from hepatocytes under lipotoxic conditions [Ibrahim et al., 2015]. CXCL10 is a potent chemotactic ligand that links hepatocyte lipotoxicity to macrophage-associated liver inflammation in NASH [Ibrahim et al., 2015; Tomita et al., 2016]. Furthermore, we and others have demonstrated increased CXCL10 hepatic expression and serum levels in patients with NASH; this increase correlates with disease severity [Ibrahim et al., 2015; Zhang et al., 2014]. However, the precise molecular mechanisms involved in CXCL10 induction under lipotoxic conditions in hepatocytes remain unclear.
Signal transducer and activator of transcription (STAT) 1 is known to mediate interferon gamma (IFN-γ) and lipopolysaccharide (LPS) induced chemokine expression in macrophages [Kopydlowski et al., 1999; Ohmori and Hamilton, 2001]. Furthermore, STAT1 was reported to bind to the CXCL10 promoter and enhance CXCL10 induction in islet cells in response to IFN-γ and interleukin (IL) 1 beta in a diabetes model, and in endothelial cells in response to LPS and IFN-γ in an atherosclerosis model [Burke et al., 2013; Chmielewski et al., 2014]. Current dogma suggests that STAT1 activation is mediated by the Janus kinase (JAK) family, which phosphorylates STAT1 at Tyr701, induces its dimerization, nuclear translocation and subsequent STAT1 Ser727 phosphorylation [Darnell et al., 1994]. STAT1 Ser727 phosphorylation regulates STAT1 transcriptional activity [Decker and Kovarik, 2000; Wen et al., 1995]. However, STAT1 Ser 727 phosphorylation without significant change in STAT1 Tyr701 or JAK1 phosphorylation is mediated in immune cells by p38 MAPK activation [Cuadrado and Nebreda, 2010; Kovarik et al., 1999; Kovarik et al., 1998]. Although the role of MLK3 in STAT1 activation in hepatocytes has not been explored, STAT1 appears to be a promising candidate regulator of CXCL10 induction during hepatocyte lipotoxic stress.
Here, we describe for the first time that lipotoxic stress induces the activation of a MAPK relay module in hepatocytes, resulting in STAT1 Ser727 phosphorylation and subsequent transcriptional upregulation of the potent macrophage chemotactic ligand CXCL10. These data have potential therapeutic implications for the treatment of NASH (e.g., STAT1 inhibitors).
MATERIALS AND METHODS
Materials
LPC (Sigma, MO, USA) was dissolved as previously described in detail [Kakisaka et al., 2012] and used at a concentration of 40 μM. Primary antisera employed for these studies include the following: Phospho-MLK3 (ab191530) and MLK3 (ab51068) from Abcam (UK); Phospho-MKK3/MKK6 (#12280), MKK3 (#8535), MKK6 (#9264), Phospho-p38 (#9211), p38 (#9212), Phospho-STAT1 (Ser) (#9177), Phospho-STAT1 (Tyr) (#7649), STAT1 (#9177), lamin B1 (#13435), α-tubulin (#3873) and Normal Rabbit IgG (#2729) from Cell Signaling Technology (MA, USA); CXCL10 (AF-466-NA) from R&D (MN, USA); actin (sc-1615) from Santa Cruz Biotechnology (TX, USA); glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (MAB 374) from Millipore (Germany). The MLK3 inhibitor URMC-099 (URMC) [Marker et al., 2013] was utilized for the in vitro experiments and kindly provided by Dr. Harris A. Gelbard (University of Rochester Medical Center, Rochester, NY). The STAT1 inhibitor fludarabine (flud) (S1491) [Sikorski et al., 2011] was obtained from Selleckchem (TX, USA). Phospho-p38 inhibitor SB203580 was obtained from Millipore (#559389 Germany) [Nash and Heuertz, 2005].
Primary cells, murine NASH model, cell lines and stable clones
Primary mouse hepatocytes (PMH) s were isolated as previously described by us [Hirsova et al., 2013]. Briefly, PMHs were isolated from chow–fed adult mice by collagenase perfusion and purified by Percoll (Sigma, St. Louis, MO) gradient centrifugation. In related study, PMH were isolated from mice fed a NASH-inducing diet for 20 weeks, as previously described in details [Charlton et al., 2011a; Krishnan et al., 2017]. Isolated hepatocytes were plated onto collagen-coated dishes. The human hepatocellular carcinoma Huh7 cells and HepG2 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Gibco, CA, USA), and primocin (100 μg/ml) (InvivoGen, CA, USA). To generate clones stably expressing shRNA against STAT1, HEK293T cells were transfected with STAT1 shRNA lentivirus plasmid using Lipofectamin 2000 (Invitrogen, CA, USA) in OptiMEM I (GibcoInvitrogen, Carlsbad, CA). Virus was harvested 48 h after transfection and passed through at 0.45μM pore cellulose acetate filter (Millipore). Huh7 cells were transduced with STAT1-targeting lentivirus in the presence of 8μg/ml Polybrene. Successfully transfected cells were selected under 2μg/ml puromycin (Invitrogen, CA, USA) selection medium. Expression of STAT1 in the clones was assessed by Immunoblot analysis.
Immunoblot analysis
Whole cell lysates (WCL) were obtained using lysis buffer (50 mM Tris-HCl, pH 7.4; 1% Nonidet P-40; 0.25% sodium deoxycholate; 150 mM NaCl; 1mM EDTA with protease inhibitors) followed by centrifugation at 15,000 g for 15 minutes at 4°C. Protein concentrations of the lysates were measured by the Bradford assay method (Sigma, MO, USA). Equal amounts of protein were loaded onto SDS-PAGE gel, transferred to nitrocellulose membrane (Bio-Rad, CA, USA) or Immobilion-FL PVDF membrane (EMD Millipore) and incubated overnight with the primary antibody of interest. The horseradish peroxidase-conjugated secondary antibody (1:3000) (Santa Cruz Biotechnologies, TX, USA) or the IRDye 680RD and 800CW secondary antibodies (1:2000) (LI-COR, NE, USA) were used. Proteins of interest were detected using enhanced chemiluminescence reagent (Amersham, IL, USA) and Kodak X-OMAT film or with an infrared imaging system (LI-COR, NE, USA). Actin, GAPDH, α-tubulin and lamin B1 protein levels were used as loading controls.
RNA interference
A specific RNA sequence complementary to the target message was used to silence human MLK3, p38, MKK3 and MKK6 expression. Validated small interfering RNAs (siRNA), targeting MLK3, p38 and MKK6 were purchased from OriGene Technologies (MD, USA); siRNAs targeting MKK3 was purchased from Cell Signaling Technology (MA, USA). As a control, cells were transfected with a scrambled RNA duplex from OriGene Technologies (MD. USA). Briefly, cells grown in 6-cm dishes were transiently transfected with siRNA using Lipofectamin RNAiMAX (Invitrogen, CA, USA). Target protein expression was assessed by immunoblot analysis 48 hours post transfection with the siRNA.
Immunocytochemistry
Cells were seeded on Chamber SlideTM (Thermo Fisher Scientific Inc.) at 50% confluence and fixed with 3% paraformaldehyde following LPC treatment. After permeabilization using 100% methanol, the slides were blocked using blocking buffer (5% bovine serum albumin, 0.1% glycine in PBS) for 1 hour at room temperature, then incubated with primary antibody overnight at 4°C. Antibodies were diluted in PBS containing 5% bovine serum albumin. Primary antibodies and their dilutions were as follows: anti-Phospho-Ser727 STAT1 antibody (1:200); anti-Phospho-Tyr701 STAT1 antibody (1:200). After washing, slides were incubated with corresponding secondary antibodies in the dark for 1 h at room temperature. Cells were mounted using Prolong Gold Antifade reagent with DAPI (Life technology, CA, USA) to visualize the nuclei. The slides were examined by fluorescent confocal microscopy equipped with an ultraviolet laser (LSM 780; Zeiss, Jena, Germany).
Chromatin immunoprecipitation (ChIP) assays
To examine potential STAT1-CXCL10 promoter interactions we initially performed in silico search for putative STAT1 binding sites within the promoter region of human CXCL10 using the MatInspector software (Genomatix, Munich, Germany). We employed an anti-STAT1 antibody (#9172), a normal rabbit IgG (#2729) (Cell Signaling Technology MA, USA) as a control and a commercially available chromatin immmunoprecipitation (ChIP) assay kit ab500 (Abcam, UK). The ChIP assay was performed according to the manufacturer’s instructions. Briefly, Huh7 cells under the different treatment conditions of interest were cross-linked with 1% formaldehyde. Cells were lysed using the supplied buffer (Abcam, UK). Cell lysates were subsequently sonicated, resulting in DNA fragments of 200–1000 bp. Protein-bound, immunoprecipitated DNA was reverse cross-linked and then purified using supplied buffer. DNA extracts were amplified by PCR using HotStarTaq (QIAGEN, Germany) for 35 cycles. The following primers 5- GTTAGAATGGATTGCAACCTTTG -3 (forward) and 5- CTCTGCTGTAGGCTCAGAATA -3 (reverse) were employed to amplify the STAT1 predicted binding sites.
Luciferase assays
CXCL10 promoter activity was examined by employing a LightSwitch Luciferase Assay System that consists of a transfection-ready luciferase reporter CXCL10 promoter construct (S711565, active motif). The construct contains the three putative STAT1 binding sites. The predicted STAT1 binding sites in the human CXCL10 gene promoter region corresponding to the sequence from −907 to +27 (relative to the transcriptional start site) were cloned into the LightSwitch_ Prom construct, purchased from SwitchGear Genomics (SwitchGear Genomics, CA, USA). Mutations in the three putative STAT1 binding sites [proximal STAT1 binding site (BS1), intermediate STAT1 binding site (BS2) and distal STAT1 binding site (BS3)] were generated using standard site-directed mutagenesis deletion techniques (New England BioLabs). Italic, uppercase letters in the following sequences denote deletion sites: for STAT1 BS1, 5′-cacgttTTCTgagacattc-3′, for STAT1 BS2, 5′-gactTCCCcaggaacagcc-3′ and for STAT1 BS3, 5′-tggaaagtGAAAcctaattcactat-3′. Huh7 cells were transiently transfected with each CXCL10 promoter construct using FuGENE HD Transfection Reagent (Promega, WI, USA) as per the manufacturer’s instructions. Forty eight hours post-transfection, and under the desired treatment conditions, luciferase activities generated by binding of STAT1 to the luciferase reporter CXCL10 promoter construct in transfected Huh7 cells were measured by a luminometer after adding LightSwitch Assay Reagents (SwitchGear Genomics, CA, USA) according to the manufacturer’s instructions. Data were expressed as fold increase in stimulated cells over control. The results were averaged from triplicate wells per each condition.
Quantitative real-time polymerase chain reaction
Total RNA was isolated with the RNeasy Mini Kit (Qiagen, CA, USA) and was reverse transcribed with Moloney murine leukemia virus reverse transcriptase and oligo-dT random primers (both from Invitrogen, CA, USA). Quantification of gene expression was performed by real-time polymerase chain reaction (PCR) using SYBR green fluorescence on a Light Cycler 480 instrument (Roche Applied, IN, USA). Target gene expression was calculated using the ΔΔCt method and expression was normalized to 18s rRNA. The following human primers were used; CXCL10, forward (5′-3′) GTGGCATTCAAGGAGTACCTC, reverse (5′-3′) GCCTTCGATTCTGGATTCAG, 18S, forward (5′-3′) CGCTTCCTTACCTGGTTGAT, reverse (5′-3′) GAGCGACCAAAGGAACCATA.
Statistical analysis
Data are expressed as the means ± SEM and represent at least three independent experiments. Differences between two groups were compared using the two-tailed Student t-test. Differences between multiple groups were compared using one-way analysis of variance followed by Bonferroni’s multiple comparison test. *, **, indicate statistical significance with P < 0.05 and P < 0.01, respectively. All analyses were performed using GraphPad Prism 6 software (CA, USA).
RESULTS
MLK3 mediates lipotoxic stress-induced STAT1 phosphorylation via a MAPK signaling cascade
We employed immunoblot analysis to examine the kinase signaling pathways involved in MLK3-mediated STAT1 activating phosphorylation in Huh7 cells treated with LPC. LPC activates a signaling cascade that induces the phosphorylation of MLK3, MKK3/6, p38 and STAT1 (Fig. 1A, B). Interestingly, LPC induced increased phosphorylation of STAT1 at Ser727 in a time dependent manner (Fig. 2B), whereas increased STAT1 phosphorylation at Tyr701 was not observed; also we did not observe an increase in activating JAK1 phosphorylation which mediates Tyr701 phosphorylation of STAT1 (Fig. 2B). To confirm that these in vitro findings correlate with the in vivo observation, we assessed STAT1 Ser 727 phosphorylation and CXCL10 protein levels in PMH isolated from mice-fed a NASH-inducing diet [Charlton et al., 2011b]. We also observed an increase in STAT1 Ser727 phosphorylation and CXCL10 upregulation in the PMH derived from the FFC-fed mice when compared to chow-fed mice (Fig. 1C). These observations suggest that lipotoxic stimulus induces CXCL10 upregulation via a MAPK signaling cascade, resulting in increased STAT1 Ser727 phosphorylation (Fig. 1A). We next interrogated this pathway employing loss of function paradigms by both genetic and pharmacological inhibition. We first confirmed that phosphorylation of MKK3/6, the downstream MAPKK of MLK3 [Han et al., 1996], is attenuated by the MLK3 inhibitor URMC-099 (Fig. 1D). This observation was further confirmed by employing siRNA targeting MLK3, demonstrating that siRNA targeting MLK3 resulted in reduced LPC-induced MKK3/6 phosphorylation (Fig. 1E). Likewise, siRNA targeting both MKK3 and MKK6 resulted in reduced LPC-induced p38 phosphorylation (Fig. 1F). Taken together, these findings define a MAPK relay module triggered by lipotoxic stress in hepatocytes.
Figure 1. MLK3 mediates lipotoxic stress-induced STAT1 phosphorylation via a MAPK signaling cascade in hepatocytes.
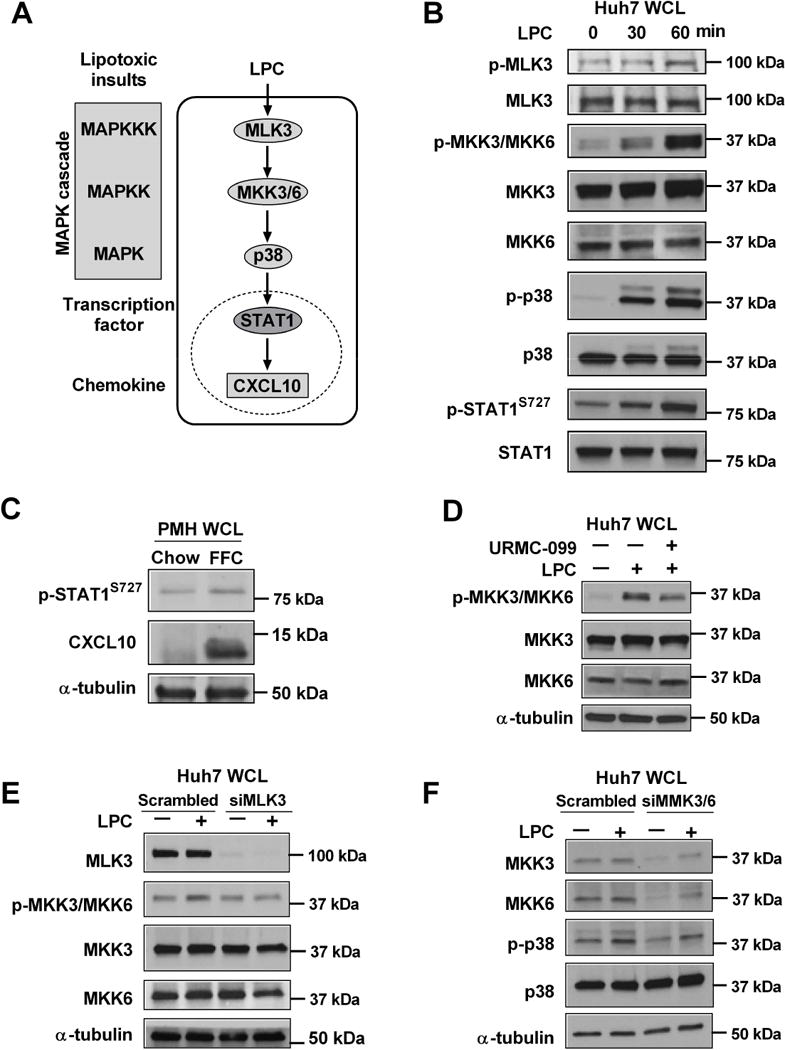
(A) Schematic representation of CXCL10 induction under lipotoxic conditions. MLK3, a mitogen activated protein kinase kinase kinase (MAPKKK), mediates CXCL10 induction by triggering a MAPK signaling pathway consisting of the mitogen activated protein kinase kinase (MAPKK) MKK3/6, and the mitogen activated protein kinase (MAPK) p38, which in turn induces STAT1 phosphorylation and transcriptional activation with subsequent CXCL10 induction. Immunoblot was used to assess: (B) Phosphorylated MLK3, MKK3/MKK6, p38, and STAT1 Ser727 and their respective total protein levels in whole cell lysates (WCL) from Huh7 cells treated with 40 μM LPC; (C) Phospho-STAT1 Ser727 and CXCL10 protein levels in primary mouse hepatocytes (PMH) isolated from high fat, fructose and cholesterol (FFC) or chow fed mice; (D) Phospho-MKK3/MKK6, total MKK3/MKK6 and α-tubulin levels on whole cell lysates from Huh7 cells treated with either vehicle, or 40 μM LPC for 1hr with and without 1 μM URMC-099 (URMC); (E) Phospho MKK3/6, total MLK3, MKK3, MKK6 and α-tubulin on whole cell lysates from Huh7 cells transfected with siMLK3 or scrambled siRNA and treated with vehicle or 40 μM LPC for 1 hr.; (F) Phospho-p38, total p38, total MKK3, MKK6 and α-tubulin in whole cell lysates from Huh7 cells transfected with siMKK3/6 or scrambled siRNA and treated with vehicle or 40 μM LPC for 1 hr.
Figure 2. LPC-induced MAPK activation mediates p38 activation and subsequent STAT1 Ser727 phosphorylation.
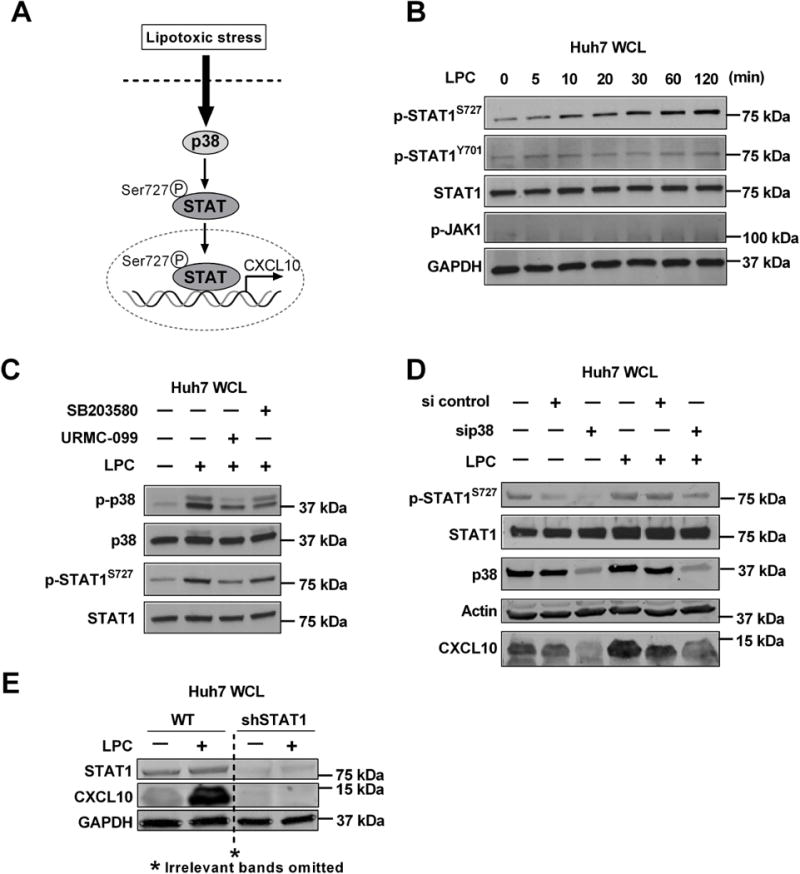
(A) Schematic representation of LPC-induced stress response resulting in p38 MAPK activation and STAT1 Ser727 phosphorylation, nuclear localization and transcriptional activation with subsequent CXCL10 induction. Immunoblot was used to assess (B) Phospho-STAT1 Ser727 and Tyr701, total STAT1, Phospho-JAK1 and GAPDH levels in whole cell lysates from Huh7 treated with 40 μM LPC at the time points indicated; (C) Phospho-p38 and Phospho-STAT1 Ser727and their respective total protein levels in whole cell lysates from Huh7 cells treated with either vehicle, or 40 μM LPC for 1 hr. with and without 1 μM URMC-099 or 10 μM SB203580; (D) Phospho-STAT1 Ser727, total STAT1, p38, actin and CXCL10 protein levels in whole cell lysates from Huh7 cells transfected with sip38 or scrambled siRNA and treated with vehicle or 40 μM LPC for 1 hr.; (E) STAT1, GAPDH, and CXCL10 protein levels in whole cell lysates from WT and shSTAT1 Huh7 cells treated with vehicle or 40 μM LPC for 1 hr.
LPC-induced MAPK activation mediates p38 activation and subsequent STAT1 Ser727 phosphorylation
We employed immunoblot to examine LPC-induced p38 activation and subsequent STAT1 Ser 727 phosphorylation (Fig 2A). To assess STAT1 activating phosphorylation at Tyr701 versus Ser727 under lipotoxic conditions in hepatocytes, we examined the level of both Ser727 phospho-STAT1 and Tyr701 phospho-STAT1in whole cell lysates derived from LPC-treated Huh7 over time (Fig. 2B). Ser727 STAT1 phosphorylation was detected within 5 minutes of treatment and reached a maximum level at 2 hours, whereas no significant change in the level of phospho-Tyr701 or phospho-JAK1 (Fig. 2B) was observed. These results indicate that LPC induces STAT1 Ser727 phosphorylation (Fig. 2B). We next examined the role of p38 in STAT1 Ser727 phosphorylation. Only two out of the four p38 isoforms, p38α and p38β, are expressed in hepatocytes and STAT1 is a known substrate of these two isoforms [Cuenda and Rousseau, 2007]. The pharmacological p38 inhibitor SB203580 inhibits both isoforms and genetic knockdown of p38α or p38β is compatible with cell viability [de la Cruz-Morcillo et al., 2012]. In our model, both the p38 inhibitor SB203580 and the MLK3 inhibitor URMC-099 reduced LPC-induced p38 activation, and STAT1 Ser727 phosphorylation (Fig. 2C). We next confirmed the above findings obtained by employing siRNA targeted knockdown of p38α and p38β. Likewise, p38α and p38β siRNA reduced LPC-induced STAT1 Ser727 phosphorylation and CXCL10 protein upregulation (Fig. 2D). In addition, STAT1 genetic knockdown by shRNA technology reduced LPC-induced CXCL10 protein upregulation (Fig. 2E). Taken together, these data suggest that LPC induces p38 activation and subsequent STAT1 Ser727 phosphorylation resulting in upregulation of CXCL10 in hepatocytes under lipotoxic stress.
LPC-induced nuclear localization of Phospho-STAT1 (Ser727) is MLK3/p38-dependent
We employed immunocytochemistry for phospho-STAT1 to assess its nuclear localization in LPC-treated Huh7 cells. Indeed, LPC significantly promoted the nuclear localization of phospho-STAT1 Ser727, without significant influence on phospho-STAT1 Tyr701 nuclear localization (Fig. 3A). Phospho-STAT1 Ser727 nuclear localization was significantly reduced in the presence of either the MLK3 inhibitor URMC-099 or the p38 inhibitor SB203580 (Fig. 3A). These observations were further confirmed by cell fractionation and western blot analysis, demonstrating an increase in Ser727 phosphorylation in the nuclear fraction of LPC-treated Huh7 cells, which was again attenuated by MLK3 or p38 inhibition (Fig. 3B). Taken together, these data suggest that LPC-induced STAT1 phosphorylation at Ser727 and its nuclear localization are MLK3/p38-dependent.
Figure 3. LPC-induced nuclear localization of phospho-STAT1 (Ser727) is MLK3/p38-dependent.
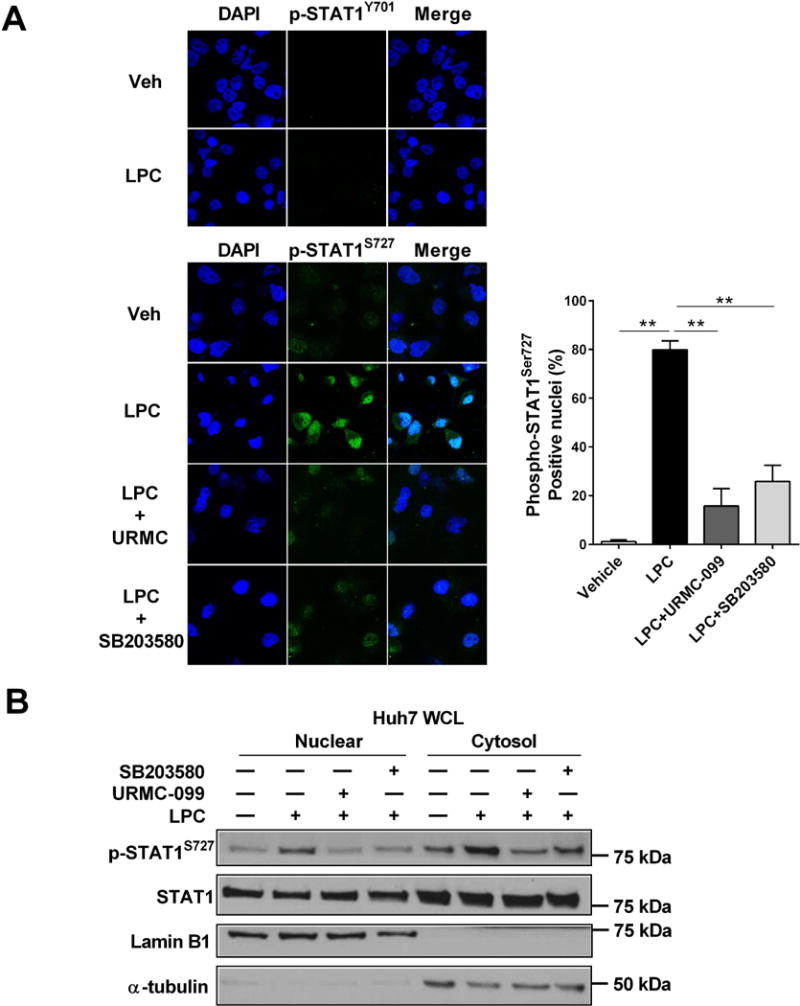
(A) Huh7 cells were treated with either vehicle or 40 μM LPC with or without 1 μM URMC-099 or 10 μM SB203580, a pharmacological p38 inhibitor, for 30 minutes. Nuclear localization of Phospho-Tyr707 and Phospho-Ser727 STAT1 were examined by immunocytochemistry and confocal microscopy. The Phospho-Ser727 STAT1 positive nuclei cells were quantified in five random 20 × microscopic fields. (B) Immunoblot analysis was used to assess Phospho-Ser727 STA1, STAT1, and GAPDH protein levels in the cytosolic fraction and Phospho-Ser727 STAT1, STAT1, and lamin B1 protein levels in the nuclear fraction from Huh7 cells treated with vehicle or LPC for 2 hours with or without 1 μM URMC-099 or 10 μM SB203580, a pharmacological p38 inhibitor. Bar columns represent mean ± standard error of the mean. ** p < 0.01
LPC induces STAT1 binding to CXCL10 promoter and its expression
We identified a significant increase in CXCL10 messenger RNA (mRNA) levels in HepG2 cells treated with LPC in a time dependent manner, which was significantly reduced in the presence of either the MLK3 inhibitor URMC-099 or p38 inhibitor SB203580 (Fig. 4A). These data suggest that CXCL10 is regulated at the transcriptional level by an MLK3/p38 kinase cascade. As this kinase cascade results in an activating phosphorylation of STAT1. We examined the CXCL10 promoter for STAT1 consensus binding sites (BS), and identified three such sequences (Fig. 4B). Therefore, we first ascertained if STAT1 directly binds to the CXCL10 gene promoter under lipotoxic conditions by employing a ChIP assay. We employed an anti-STAT1 antibody to immunoprecipitate the CXCL10 promoter. We amplified the area that encompasses two predicted STAT1 binding sites (BS2 and BS3) at the human CXCL10 gene promoter region using PCR and demonstrated that LPC significantly increased STAT1 binding to the CXCL10 promoter (Fig. 4B). Next, we employed a luciferase assay to determine whether STAT1 directly regulates CXCL10 gene promoter activation and transcription under lipotoxic conditions. We used the luciferase reporter CXCL10 promoter construct which contains the three putative STAT1 binding sites (Fig. 5A). LPC treatment significantly increased CXCL10 promoter activity, which was attenuated in the presence of either the MLK3 inhibitor URMC-099 or the STAT1 inhibitor fludarabine (Fig. 5B). To further investigate the specificity of STAT1-induced CXCL10 gene promoter activation, the three STAT1 binding sites in the luciferase reporter CXCL10 promoter construct were mutated using standard site-directed mutagenesis deletions, generating a triple mutant construct (ΔBS1-3). A significant reduction in LPC-induced luciferase activity was observed when Huh7 cells were transfected with the triple mutant construct, compared to the full construct (Fig. 5B). To further investigate the individual role of the three STAT1 binding sites in the CXCL10 promoter activity, we designed three luciferase reporter CXCL10 promoter mutant constructs, each with a deletion involving one of the three STAT1 binding sites (ΔBS1, ΔBS2 or ΔBS3) (Fig. 5A). Interestingly, while ΔBS2 or ΔBS3 construct demonstrated a significant attenuation of LPC-induced luciferase activity, ΔBS1 construct induced a significant increase in luciferase activity (Fig. 5B). We speculate that the BS1 is a repressor binding site that is inactivated with the deletion, resulting in increased CXCL10 promoter activity. Taken together, these data suggest STAT1 directly regulates CXCL10 promoter activity, and STAT1 binding site 3 (BS3) appears to be the prominent STAT1 binding site responsible for CXCL10 induction in our model of hepatocyte lipotoxicity.
Figure 4. LPC-induces STAT1 binding to the CXCL10 gene promoter.
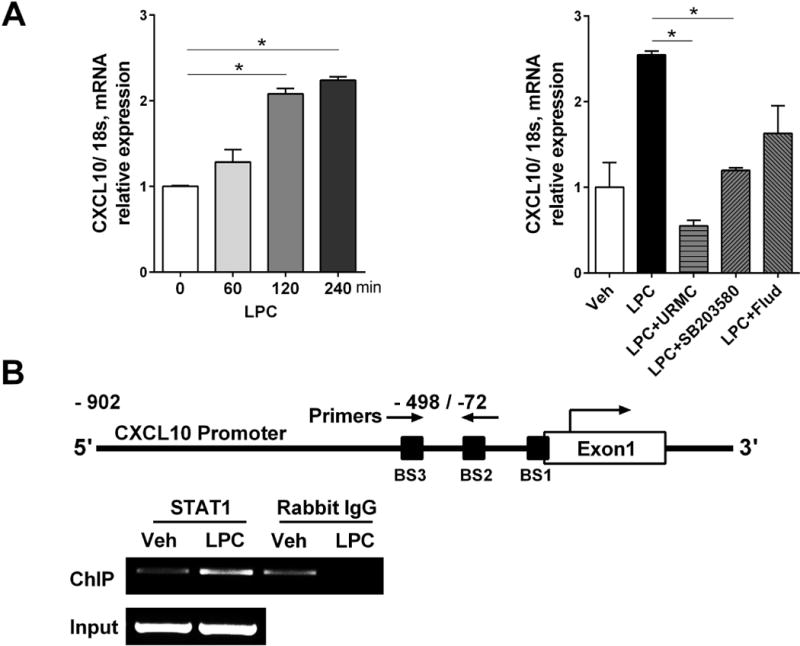
(A) Total RNA was extracted from HepG2 cells treated with either vehicle or 40 μM LPC at the time points indicated, cells were also treated with 40 μM LPC with or without 1 μM MLK3 inhibitor URMC-099 (URMC), 10 μM SB203580 and 100 μM STAT1 inhibitor fludarabine (Flud) for 4 hours. The mRNA expression of CXCL10 was evaluated by real-time qPCR. Fold change was determined after normalization to 18s mRNA expression, and expressed as fold change to that observed in vehicle-treated hepatocytes. (B) Schema of the human CXCL10 promoter, showing the STAT1 binding sites that were amplified by PCR. ChIP assay was performed on Huh7 cells treated with vehicle or LPC for 1 hr. Immunoprecipitated DNA was amplified by PCR demonstrating binding of STAT1 to the CXCL10 promoter with LPC treatment. Bar columns represent mean ± standard error of the mean. * p < 0.05
Figure 5. LPC-induced activation of the CXCL10 gene promoter requires STAT1 binding.
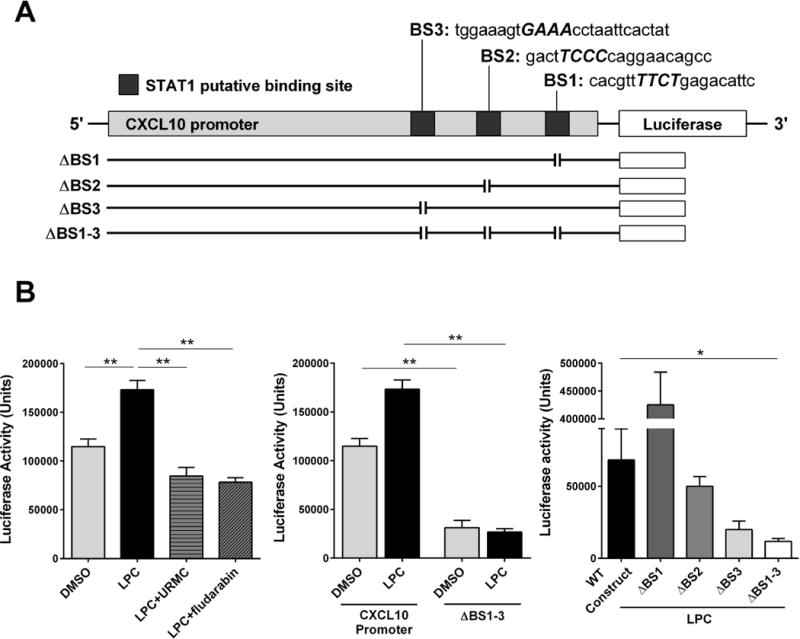
(A) Schematic diagram of CXCL10 promoter constructs, including the three putative STAT1 binding sites. Mutations in the three putative STAT1 promoter binding sites sequences were generated using standard site-directed mutagenesis deletion techniques. Italic, uppercase letters in the sequences denote deletion sites. (B) Luciferase activity was determined using the LightSwitch Luciferase Assay System. After transfection with the respective luciferase constructs, Huh7 cells were treated with either vehicle or 40 μM LPC with or without 1 μM URMC-099 (URMC) or 100 μM STAT1 inhibitor fludarabine. Bar columns represent mean ± standard error of the mean. ** p < 0.01; * p < 0.05
DISCUSSION
The present study provides mechanistic insights regarding the signaling pathway responsible for CXCL10 induction in LPC-treated hepatocytes (Fig. 6). The principal findings of the current study indicate that during hepatocyte lipotoxic stress in vitro: i) MLK3 activation triggers a MAPK signaling cascade resulting in STAT1 Ser727 phosphorylation; ii) STAT1 Ser727 phosphorylation, increases its nuclear localization, and transcriptional activity; and iii) STAT1 binds to and activates the CXCL10 gene promoter resulting in CXCL10 transcriptional upregulation. To our knowledge, our observations are the first to describe the molecular regulation of the CXCL10 gene in response to lipotoxic stress in hepatocytes. These observations are more thoroughly discussed below.
Figure 6. Schematic representation of CXCL10 induction under lipotoxic conditions.
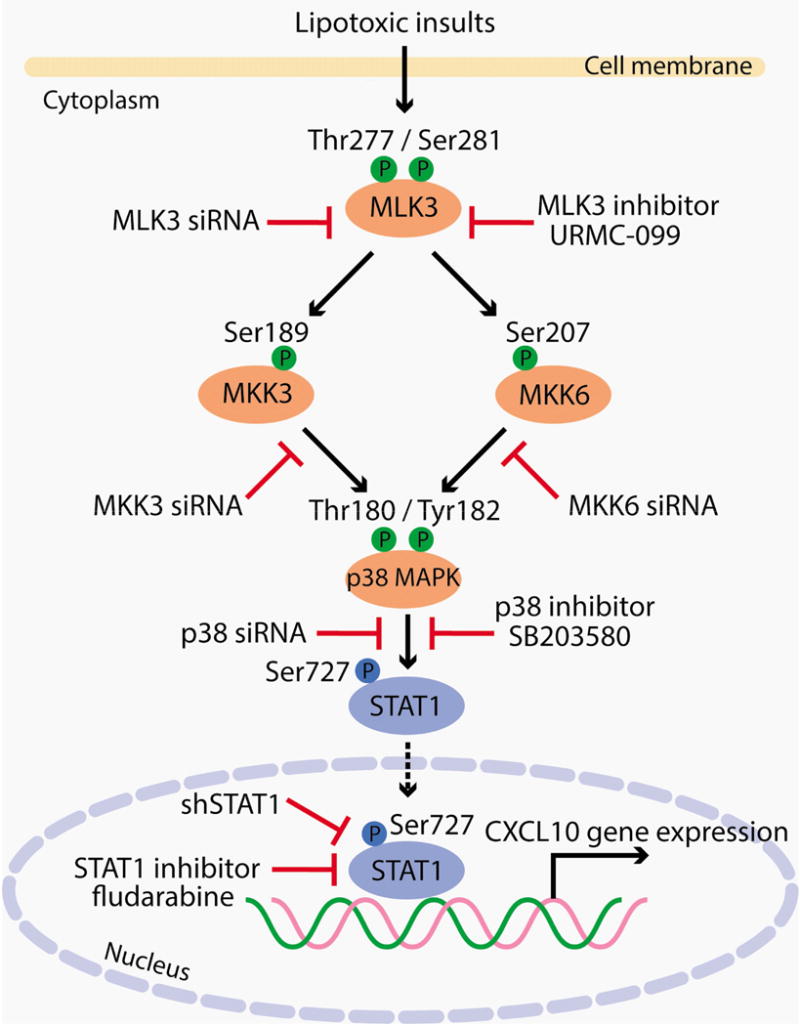
MLK3 mediates CXCL10 induction during hepatocyte lipotoxicity by triggering a MAPK signaling pathway that induces the activating phosphorylation of MKK3/6, p38 resulting in STAT1 phosphorylation at Ser727, and its nuclear localization. Phospho-Ser727 STAT1 binds to and activates the CXCL10 gene promoter. These events lead to transcriptional upregulation of CXCL10. We employed both genetic and pharmacological inhibition as indicated in the figure to demonstrate the sequence of events in the signaling cascade, and to test our hypothesis in a loss of function paradigm.
We previously reported that MLK3 mediates LPC-induced hepatocyte release of CXCL10-enriched extracellular vesicles [Ibrahim et al., 2015]. In the current study, we advance this observation by exploring the signaling pathway mediating MLK3-induced CXCL10 upregulation in hepatocytes under lipotoxic condition. We identified MMK3/6 as the MAPKK activated downstream of MLK3 in response to LPC treatment in hepatocytes, and p38 as the MAPK activated downstream of MKK3/6. In accordance with our observation, activation of this MAPK signaling cascade is described in other models of cell injury [Dong et al., 2016; van der Houven van Oordt et al., 2000; Zhou et al., 2014].
We further identified that STAT1 activating phosphorylation at Ser727 is p38-dependant during lipotoxic stress. Various ligands, including cytokines and growth factors, bind to cell surface receptors and activate JAK1 resulting in STAT1 phosphorylation at Tyr701 residue with subsequent dimerization, nuclear translocation and induction of transcription of target genes [Levy and Darnell, 2002]. Additionally, within the conventional JAK-STAT pathway, STAT1 Ser727 phosphorylation occurs in the nucleus after STAT1 Tyr701 activating phosphorylation to increase STAT1 transcriptional efficacy [Darnell et al., 1994]. However, in our model, STAT1 Ser727 phosphorylation was augmented under LPC treatment in a time dependent manner, without significant augmentation in STAT1 Tyr701, or JAK1 phosphorylation. Presumably, the level of constitutive STAT1 Tyr701 phosphorylation is sufficient to permit STAT1 Ser 727 phosphorylation. Consistent with our observation in hepatocyte lipotoxicity, direct STAT1 Ser727 phosphorylation is recognized in different models of cell injury in response to UV light, TNF-alpha and LPS, and described to be mediated by the MAPK p38 [Cuadrado and Nebreda, 2010; Kovarik et al., 1999]. LPC treatment in hepatocytes, significantly promoted the nuclear localization of phospho-STAT1 Ser727, without appreciated influence on phospho-STAT1 Tyr701 nuclear localization. Furthermore, increased STAT1 Ser727 phosphorylation in response to LPC treatment was also observed in the cytosolic cellular fraction. This observation is thought to be secondary to the nucleocytoplasmic shuttling of STAT1 [Marg et al., 2004], which is apparently dependent in part on STAT1 Ser727 phosphorylation in hepatocytes.
Consistent with the upregulation of CXCL10 protein level in response to LPC treatment in hepatocytes, CXCL10 transcription and mRNA levels were increased in hepatocytes under lipotoxic stress by an MLK3/p38/STAT1-dependant mechanism. We further demonstrate that lipotoxic treatment induces STAT1 binding to the CXCL10 gene promoter, and that STAT1 binding directly and specifically activates the CXCL10 gene promoter, as reported by others in islet cells in a diabetes model [Burke et al., 2013].
In summary, we demonstrate that in hepatocytes under lipotoxic stress, MLK3 triggers a MAPK signaling cascade resulting in p38 MAPK activation, STAT1 Ser727 phosphorylation, and transcriptional activation with subsequent induction of the potent chemotactic ligand CXCL10 (Fig. 6). Furthermore, we demonstrate that lipotoxic stress activates p38 MAPK/STAT1 Ser727 without significant engagement of the JAK-STAT signaling pathway. These findings provide mechanistic insights with potential therapeutic ramifications for a common, yet untreatable, hepatic disorder, and could be beneficial for developing strategies to diminish inflammation during hepatic lipotoxicity. For instance, blocking CXCL10 induction by employing a STAT1 inhibitor or a MAPK inhibitor may serve as a promising therapeutic strategy, to attenuate the sterile inflammatory component of NASH.
Acknowledgments
We thank Courtney N. Hoover for providing excellent secretarial support and Dr. Gregory J. Gores for his thorough review of the manuscript. The small molecule MLK3 inhibitor, URMC-099, used in this study is the proprietary asset of the University of Rochester Medical Center. (US Patents: 8,846,909, 8,877,772, and 9,181,247, and international patents/applications; References: PMCID: PMC3682381; PMC4032177)
Grant information: This work was supported by the Mayo Clinic Center for Clinical and Translational Science (CCaTS), KL2 program KL2TR000136-09 (to SHI) and pediatric small grant program (to SHI), a Pilot and Feasibility Award and the Optical Microscopy Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567) (to SHI), North American Society of Pediatric Gastroenterology Hepatology and Nutrition Young Investigator Award/Nestle Nutrition Award (to SHI), American Liver Foundation (to PH), and the Mayo Clinic, Rochester, MN. Research reported in this publication was supported by the National Institute of Diabetes And Digestive And Kidney Diseases of the National Institutes of Health under Award Number K08DK111397 (to SHI). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Non-standard abbreviations
- CXCL10
(C-X-C motif) ligand 10
- STAT
signal transducer and activator of transcription
- SFA
saturated free fatty acid
- NASH
nonalcoholic steatohepatitis
- MLK3
mixed lineage kinase 3
- LPC
lysophosphatidylcholine
- MAPK
mitogen activated protein kinase
- MAPKKK
mitogen activated protein kinase kinase kinase
- MKK
dual specificity mitogen-activated protein kinase kinase
- MAPKK
MAPK kinases
- IFN
interferon
- LPS
lipopolysaccharide
- TNF
tumor necrosis factor
- ChIP
chromatin immunoprecipitation
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- flud
fludarabine
- IP
immunoprecipitation
- JAK
Janus kinase
- PCR
polymerase chain reaction
- siRNA
small interfering RNA
- UV
ultraviolet
- PMH
primary mouse hepatocyte
Footnotes
Author contributions statement
K.T., B. L. F, S.F. B, P.H., S.H.I. conducted the experiment(s); KT and SHI designed the experiments, analyzed the data, and wrote the manuscript. A.K. and P.H. designed the experiment(s), and edited the manuscript. All authors reviewed the manuscript.
Disclosure: The authors have no potential conflict of interest in regards to this manuscript
References
- Afzali A, Berry K, Ioannou GN. Excellent posttransplant survival for patients with nonalcoholic steatohepatitis in the United States. Liver Transpl. 2012;18:29–37. doi: 10.1002/lt.22435. [DOI] [PubMed] [Google Scholar]
- Burke SJ, Goff MR, Lu D, Proud D, Karlstad MD, Collier JJ. Synergistic expression of the CXCL10 gene in response to IL-1beta and IFN-gamma involves NF-kappaB, phosphorylation of STAT1 at Tyr701, and acetylation of histones H3 and H4. Journal of immunology. 2013;191:323–36. doi: 10.4049/jimmunol.1300344. [DOI] [PubMed] [Google Scholar]
- Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, Masuoko H, Gores G. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. American journal of physiology Gastrointestinal and liver physiology. 2011a;301:G825–34. doi: 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, Masuoko H, Gores G. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol. 2011b;301:G825–34. doi: 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielewski S, Olejnik A, Sikorski K, Pelisek J, Blaszczyk K, Aoqui C, Nowicka H, Zernecke A, Heemann U, Wesoly J, Baumann M, Bluyssen HA. STAT1-dependent signal integration between IFNgamma and TLR4 in vascular cells reflect pro-atherogenic responses in human atherosclerosis. Plos One. 2014;9:e113318. doi: 10.1371/journal.pone.0113318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. The Biochemical journal. 2010;429:403–17. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- Cuenda A, Rousseau S. P38 MAP-Kinases pathway regulation, function and role in human diseases. Biochimica Et Biophysica Acta-Molecular Cell Research. 2007;1773:1358–1375. doi: 10.1016/j.bbamcr.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–21. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- de la Cruz-Morcillo MA, Valero MLL, Callejas-Valera JL, Arias-Gonzalez L, Melgar-Rojas P, Galan-Moya EM, Garcia-Gil E, Garcia-Cano J, Sanchez-Prieto R. P38MAPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: implication in resistance. Oncogene. 2012;31:1073–1085. doi: 10.1038/onc.2011.321. [DOI] [PubMed] [Google Scholar]
- Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–37. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- Dong W, Embury CM, Lu Y, Whitmire SM, Dyavarshetty B, Gelbard HA, Gendelman HE, Kiyota T. The mixed-lineage kinase 3 inhibitor URMC-099 facilitates microglial amyloid-beta degradation. J Neuroinflammation. 2016;13:184. doi: 10.1186/s12974-016-0646-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee JD, Jiang Y, Li Z, Feng L, Ulevitch RJ. Characterization of the structure and function of a novel MAP kinase kinase (MKK6) The Journal of biological chemistry. 1996;271:2886–91. doi: 10.1074/jbc.271.6.2886. [DOI] [PubMed] [Google Scholar]
- Han MS, Lim YM, Quan W, Kim JR, Chung KW, Kang M, Kim S, Park SY, Han JS, Cheon HG, Dal Rhee S, Park TS, Lee MS. Lysophosphatidylcholine as an effector of fatty acid-induced insulin resistance. Journal of lipid research. 2011;52:1234–46. doi: 10.1194/jlr.M014787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MS, Park SY, Shinzawa K, Kim S, Chung KW, Lee JH, Kwon CH, Lee KW, Lee JH, Park CK, Chung WJ, Hwang JS, Yan JJ, Song DK, Tsujimoto Y, Lee MS. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J Lipid Res. 2008;49:84–97. doi: 10.1194/jlr.M700184-JLR200. [DOI] [PubMed] [Google Scholar]
- Hirsova P, Ibrahim SH, Bronk SF, Yagita H, Gores GJ. Vismodegib suppresses TRAIL-mediated liver injury in a mouse model of nonalcoholic steatohepatitis. Plos One. 2013;8:e70599. doi: 10.1371/journal.pone.0070599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim SH, Gores GJ, Hirsova P, Kirby M, Miles L, Jaeschke A, Kohli R. Mixed lineage kinase 3 deficient mice are protected against the high fat high carbohydrate diet-induced steatohepatitis. Liver international: official journal of the International Association for the Study of the Liver. 2014;34:427–37. doi: 10.1111/liv.12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, Goodfellow VS, Malhi H, Gores GJ. Mixed Lineage Kinase 3 Mediates Release of C-X-C Motif Ligand 10-Bearing Chemotactic Extracellular Vesicles from Lipotoxic Hepatocytes. Hepatology. 2015 doi: 10.1002/hep.28252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke A, Davis RJ. Metabolic stress signaling mediated by mixed-lineage kinases. Molecular cell. 2007;27:498–508. doi: 10.1016/j.molcel.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW, Mott JL, Gores GJ. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. American journal of physiology Gastrointestinal and liver physiology. 2012;302:G77–84. doi: 10.1152/ajpgi.00301.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopydlowski KM, Salkowski CA, Cody MJ, van Rooijen N, Major J, Hamilton TA, Vogel SN. Regulation of macrophage chemokine expression by lipopolysaccharide in vitro and in vivo. Journal of immunology. 1999;163:1537–44. [PubMed] [Google Scholar]
- Kovarik P, Stoiber D, Eyers PA, Menghini R, Neininger A, Gaestel M, Cohen P, Decker T. Stress-induced phosphorylation of STAT1 at Ser727 requires p38 mitogen-activated protein kinase whereas IFN-gamma uses a different signaling pathway. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:13956–61. doi: 10.1073/pnas.96.24.13956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovarik P, Stoiber D, Novy M, Decker T. Stat1 combines signals derived from IFN-gamma and LPS receptors during macrophage activation. The EMBO journal. 1998;17:3660–8. doi: 10.1093/emboj/17.13.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A, Abdullah TS, Mounajjed T, Hartono SP, McConico A, White TA, LeBrasseur NK, Lanza IR, Nair S, Gores GJ, Charlton M. A Longitudinal Study of Whole Body, Tissue and Cellular Physiology in a Mouse Model of Fibrosing NASH with High Fidelity to the Human Condition. Am J Physiol Gastrointest Liver Physiol. 2017 doi: 10.1152/ajpgi.00213.2016. ajpgi 00213 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nature reviews Molecular cell biology. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Marg A, Shan Y, Meyer T, Meissner T, Brandenburg M, Vinkemeier U. Nucleocytoplasmic shuttling by nucleoporins Nup153 and Nup214 and CRM1-dependent nuclear export control the subcellular distribution of latent Stat1. The Journal of cell biology. 2004;165:823–33. doi: 10.1083/jcb.200403057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marker DF, Tremblay ME, Puccini JM, Barbieri J, Marker MAG, Loweth CJ, Muly EC, Lu SM, Goodfellow VS, Dewhurst S, Gelbard HA. The New Small-Molecule Mixed-Lineage Kinase 3 Inhibitor URMC-099 Is Neuroprotective and Anti-Inflammatory in Models of Human Immunodeficiency Virus-Associated Neurocognitive Disorders. Journal of Neuroscience. 2013;33:9998–10010. doi: 10.1523/JNEUROSCI.0598-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash SP, Heuertz RM. Blockade of p38 map kinase inhibits complement-induced acute lung injury in a murine model. International immunopharmacology. 2005;5:1870–80. doi: 10.1016/j.intimp.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology. 2010;52:774–88. doi: 10.1002/hep.23719. [DOI] [PubMed] [Google Scholar]
- Ohmori Y, Hamilton TA. Requirement for STAT1 in LPS-induced gene expression in macrophages. Journal of Leukocyte Biology. 2001;69:598–604. [PubMed] [Google Scholar]
- Schattenberg JM, Lee MS. Extracellular Vesicles as Messengers Between Hepatocytes and Macrophages in Nonalcoholic Steatohepatitis. Gastroenterology. 2016;150:815–8. doi: 10.1053/j.gastro.2016.02.064. [DOI] [PubMed] [Google Scholar]
- Sikorski K, Chmielewski S, Przybyl L, Heemann U, Wesoly J, Baumann M, Bluyssen HAR. STAT1-mediated signal integration between IFN gamma and LPS leads to increased EC and SMC activation and monocyte adhesion. American Journal of Physiology-Cell Physiology. 2011;300:C1337–C1344. doi: 10.1152/ajpcell.00276.2010. [DOI] [PubMed] [Google Scholar]
- Tomita K, Freeman BL, Bronk SF, LeBrasseur NK, White TA, Hirsova P, Ibrahim SH. CXCL10-Mediates Macrophage, but not Other Innate Immune Cells-Associated Inflammation in Murine Nonalcoholic Steatohepatitis. Sci Rep. 2016;6:28786. doi: 10.1038/srep28786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. The Journal of biological chemistry. 2012;287:40161–72. doi: 10.1074/jbc.M112.417014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Houven van Oordt W, Diaz-Meco MT, Lozano J, Krainer AR, Moscat J, Caceres JF. The MKK(3/6)-p38-signaling cascade alters the subcellular distribution of hnRNP A1 and modulates alternative splicing regulation. The Journal of cell biology. 2000;149:307–16. doi: 10.1083/jcb.149.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–50. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- Zhou F, Xu Y, Hou XY. MLK3-MKK3/6-P38MAPK cascades following N-methyl-D-aspartate receptor activation contributes to amyloid-beta peptide-induced apoptosis in SH-SY5Y cells. J Neurosci Res. 2014;92:808–17. doi: 10.1002/jnr.23354. [DOI] [PubMed] [Google Scholar]