

Abstract
Agnoprotein is an important regulatory protein of the human polyoma JC virus (JCV) and plays critical roles during the viral replication cycle. It forms highly stable dimers and oligomers through its Leu/Ile/Phe-rich domain, which is important for the stability and function of the protein. We recently resolved the partial 3D structure of this protein by NMR using a synthetic peptide encompassing amino acids Thr17 to Gln52, where the Leu/Ile/Phe- rich domain was found to adopt a major alpha-helix conformation spanning amino acids 23 to 39. Here, we report the resolution of the 3D structure of full-length JCV agnoprotein by NMR, which not only confirmed the existence of the previously reported major α-helix domain at the same position but also revealed the presence of an additional minor α-helix region spanning amino acid residues Leu6 to Ala10. The remaining regions of the protein adopt an intrinsically unstructured conformation.
Keywords: Agnoprotein, NMR, dimer, oligomer, polyomavirus, JCV, SV40, BKV, Merkel cell, DNA replication, progressive multifocal leukoencephalopathy, alpha-helix, intrinsically unstructured
INTRODUCTION
JC virus (JCV) is a human polyomavirus and causes a fatal neurodegenerative disease of the human central nervous system (CNS), known as progressive multifocal leukoencephalopathy (PML), where the virus lytically and abortively infects the oligodendrocytes and astrocytes respectively. PML was once considered as a rare disease and usually develops in patients with underlying immunosuppressive conditions including Hodgkin’s lymphoma, lymphoproliferative diseases and AIDS [Berger and Concha, 1995]. During the era of AIDS epidemic, however, it became a commonly encountered brain disease in patients of a variety of different age groups with AIDS [Power et al., 2000]. Approximately, 5–7% of HIV patients develop PML [Berger and Houff, 2006]. PML has also recently been described in different groups of autoimmune disease patients, including, multiple sclerosis (MS) and Crohn’s disease [Chen et al., 2009; Kleinschmidt-DeMasters and Tyler, 2005; Langer-Gould et al., 2005; Van Assche et al., 2005], rheumatoid arthritis [Carson et al., 2009] and severe plaque psoriasis [Major, 2010], who were treated with immunomodulatory drugs that target specific cell surface receptors on either T or B cells to downregulate the harmful autoimmune effects of these cells on the target organs.
JCV encodes a limited number of regulatory proteins from its small DNA genome (approximately 5kb in size), one of which is a small and highly basic protein (71 amino acid long protein), named agnoprotein (Agno). This protein plays essential regulatory roles during the viral replication cycle, because, in the absence of its expression, JCV is unable to sustain its productive life cycle [Sariyer et al., 2011]. It is primarily localized to the cytoplasmic compartment of the infected cells with high concentrations accumulating in the perinuclear area. However, a small fraction of it also localizes to nucleus (15–20%) [Safak et al., 2002]. Agno was shown to be involved in JCV gene transcription and replication through interaction with JCV LT-Ag and the cellular transcription factor, YB-1 [Safak et al., 2001; Safak et al., 2002; Saribas et al., 2012]. In addition, we reported its phosphorylation by protein kinase C (PKC) at Ser7, Ser11 and Thr21 during the infection cycle [Sariyer et al., 2006]. These phosphorylation sites play critical roles in agnoprotein function. Similar findings have been also reported for BKV agnoprotein Ser11 residue [Johannessen et al., 2008], supporting the importance of phosphorylation in function of agnoprotein of polyomaviruses. We also reported the formation of highly stable and SDS-resistant dimeric/oligomeric (multimeric) complexes by agnoprotein [Saribas et al., 2011] where the central portion of the protein (Leu/Ile/Phe-rich region) plays an essential role in this process [Saribas et al., 2011]. Recent structural studies by NMR revealed that the Leu/Ile/Phe-rich region adopts an α-helix conformation and certain amino acids within this region are important for the stability and function of the protein [Saribas et al., 2013; Saribas et al., 2014]. Additionally, agnoprotein interacts with p53 [Darbinyan et al., 2002], FEZ1 [Suzuki et al., 2005] and adaptor protein complex 3 [Suzuki et al., 2013]. Gerits et al. also demonstrated the interaction of BKV agnoprotein with the cellular protein, PCNA [Gerits et al., 2015]. Recent studies by Suzuki et al. suggest that agnoprotein functions as a viroporin during the viral replication cycle [Suzuki et al., 2013; Suzuki et al., 2010].
Agnoprotein possesses a highly basic amino acid composition at its amino and carboxy-terminus, mostly dominated by Arg and Lys residues. The middle portion of the protein is, however, highly hydrophobic and is rich in Leu/Ile/Phe residues. Interestingly, two negatively charged residues Asp38 and Glu34 are also dispersed within this hydrophobic region. Agnoprotein Glu34 is highly conserved among the polyomaviruses but Asp38 is substituted for Glu38 and Gln38 in BKV and SV40 respectively. Another peculiarity within this region is that all three Phe residues of agnoprotein (Phe31, Phe35 and Phe39) localize to this hydrophobic pocket. Genetic and biochemical studies regarding Phe residues suggest that they not only positively influence viral DNA replication [Saribas et al., 2014; Saribas et al., 2012] but also play regulatory roles in the intracellular distribution of agnoprotein in infected cells [Saribas et al., 2012; Unterstab et al., 2010] mostly concentrating at the perinuclear area of the cytoplasm. Another unique residue of agnoprotein is the Cys40, which is juxtaposed to the Leu/Ile/Phe-rich domain and highly conserved among JCV, BKV and SV40.
Determining the three-dimensional (3D) structures of proteins is essential for studying their regulatory roles and developing therapeutic antiviral strategies. The 3D structure of full- length agnoprotein has yet to be resolved. However, we recently resolved the partial 3D structure of JCV agnoprotein by NMR using a partial synthetic peptide encompassing amino acid residues Thr17 to Gln52. Results revealed that the Leu/Ile/Phe-rich domain of the protein adopts an α-helix conformation spanning residues Lys23 to Phe39 but the remaining flanking amino and carboxy-terminal residues exhibits an intrinsically unstructured conformation [Coric et al., 2014]. Mutational analysis of the α-helix region revealed that it plays a critical role in the stability and function of the protein [Coric et al., 2014]. Furthermore, theoretical modeling studies showed that agnoprotein could form antiparallel dimers through its α-helix region which is consistent with our dimerization studies [Saribas et al., 2013; Saribas et al., 2011]
In this study, we have further extended our previously reported NMR studies and determined the complete 3D structure of the agnoprotein by NMR. These structural studies confirmed the location of the previously reported major α-helix region and also revealed the formation of an additional minor α-helix spanning amino acid residues Leu6 to Ala10. Interestingly, the 3D structure also revealed that the remaining regions of the protein adopt an intrinsically unstructured conformation.
MATERIALS AND METHODS
Hydrophobicity and secondary structure predictions for agnoprotein
The primary sequence of full-length agnoprotein peptide, Met1-Thr71, was analyzed to predict the secondary structure present in the peptide using two programs: University College London’s PSIPRED v3.3 (http://bioinf.cs.ucl.ac.uk/psipred/) [McGuffin et al., 2000] and the NPS@Web servers (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sspred.html) available from Pole Bioinformatique Lyonnais.
There are several ways of revealing the hydrophobic and hydrophilic features of a protein, one of which is the Kyte-Doolittle scale [Kyte and Doolittle, 1982]. This scale was used to predict the hydrophobicity of full-length agnoprotein and was implemented in protscale, on the Expasy server site (http://web.expasy.org/protscale/). The PSIPRED server (http://bioinf.cs.ucl.ac.uk/psipred/) [McGuffin et al., 2000] was used to analyze the primary sequence of the agnoprotein to predict the secondary structures present within the protein. PSIPRED is a simple and accurate secondary structure prediction method, incorporating two feed-forward neural networks which perform an analysis on output obtained from PSI- BLAST (Position Specific Iterated - BLAST).
Circular dichroism spectra for agnoprotein
Circular dichroism (CD) spectra were recorded at room temperature (20°C) in a 0.1 cm path length quartz cell on a Jobin-Yvon model C8 spectropolarimeter calibrated with d-camphor-10-sulfonate. The experiments were performed either in 100% H2O or in a H2O/TFE (2,2,2-trifluoroethanol) (90/10, 80/20, and 70/30, v/v) mixture at pH 3.0. The peptide concentration in each sample was kept at 50 μM to minimize the chance of aggregation by the full agnoprotein. Under this low concentration of the protein, it is not required to add NaCl or increase the temperature of the sample to improve the quality of the signal. Three averaged scans were collected in 0.5 nm interval steps using an integration time of 2 s and a light bandpass or slit width of 2 nm. Curves were smoothed from 200 to 260 nm using 25 data points. Respective intensities are expressed in mean residue molar ellipticity [θ], calculated from the equation [θ]=[θ]obs/(10.C.n.L), where [θ]obs is the observed ellipticity in millidegrees (mdeg). L is the optical path length in centimeters (0.1 cm), C is the final concentration of the peptide in molar and n represents the number of amino acid residues.
Agnoprotein synthesis and sample preparation for NMR
Purification of agnoprotein for NMR studies from expression systems was difficult, because NMR requires highly pure protein for analysis. Therefore, full-length agnoprotein was commercially synthesized by AnaSpect (Fremont, CA, USA) using standard Fmoc (fluoren-9-ylmethoxycarbonyl) chemistry on an automated solid-phase platform and purified by reverse-phase HPLC. The protein was analyzed by Mass Spectrometry (MS) and High Performance Liquid Chromatography (HPLC) and found to have purity greater than 98%. Agnoprotein showed a strong propensity to aggregate in pure water and thus different organic solvent conditions were used to determine the optimal conditions for the NMR study: 100% H2O; H2O/TFE, v/v, 70/30; CD3OH/CDCl3, v/v, 50/50) or in presence of 100 eq. dodecylphosphatidylcholine (DPC-d38) or 50 to 200 eq. Sodium Dodecyl Sulfate (SDS-d25) at pH 3.0. Under our experimental conditions, SDS or DPC did not prevent the aggregation of agnoprotein, which is supported by the previous findings by Saribas et al., [Saribas et al., 2011], where it was demonstrated that relatively high concentration of SDS does not prevent the dimer and oligomer formation by agnoprotein.
Analysis of agnoprotein structure by NMR
All NMR experiments were recorded on a sample at a concentration of 0.2 mM in 30% TFE, 200 mM NaCl, pH 3.0, at 293°K and 313°K, on an Avance III Bruker spectrometer operating at 600.13 MHz and equipped with a cryoprobe. Two-dimensional spectra were performed with 2048 real points in t2, a spectral width of 7002.8 Hz and 512 t1 increments. The transmitter frequency was set to the water signal. The solvent resonance was suppressed using a 3–9–19 pulse sequence [Piotto et al., 1992] with gradients during the relaxation delay of 1.6 s between free induction decays or by using excitation sculpting with gradients [Hwang and Shaka, 1995]. A mixing time of 250 ms has been used in the Nuclear Overhauser Effect Spectroscopy (NOESY) experiments [Jeener et al., 1979]. For all experiments, the temperature was controlled externally using a special temperature control system (BCU 05 Bruker). Agnoprotein includes 14 Arg/Lys and 7 Asp/Glu and at physiological pH, basic and acidic residues are positively and negatively charged, respectively. The intermolecular interactions through these charges could lead to additional aggregation of the protein and therefore we used 200 mM NaCl conditions. A pH of 3.0 was chosen, because at low pH, the protonation of the basic and acidic residues prevents intermolecular interactions. Moreover, the amide proton exchange rate is slower at low pH compared to the rate at a pH close to neutrality. In the case where agnoprotein has a tendency to aggregate, it allows to get a sharper signal and more intense peaks that facilitate the NMR spectra analysis.
Processing of the NMR data
All data were processed using Bruker Topspin 3.2 software. A π/6 phase-shifted sine bell window function was applied and data were zero filled once prior to Fourier transformation in both dimensions (t1 and t2). The final sizes of the frequency domains matrices were 2048 real points in both ω2 and ω1 dimensions. For all experiments, 1H frequency scale was directly referenced to water. The data were then analyzed with the CcpNmr Analysis program [Vranken et al., 2005].
Determining the 3D structure of the full-length agnoprotein
The calculations for the 3D structure determination of agnoprotein was performed iteratively with the software programs ARIA [Nilges, 1995] and CNS [Brunger et al., 1998]. ARIA used the initial data CcpNmr project, which contains a table of chemical shifts and a list of NOE peaks from NMR assigned spectra. A set of distance restraints obtained from NOE cross-signal volumes measured on a 250 ms mixing time NOESY at 313°K by integration of the peaks into distances by an R−6 dependency was used. The distances were calibrated using the distance between aromatic protons and no dihedral torsion angle restraints were included in the calculations. Eight iterations were performed by ARIA where the constraint violation threshold was defined at 0.2 Å and systematically violated constraints were corrected or eliminated. During the ninth iteration the structure was analyzed and refined by performing a molecular dynamics step in a water box. This process was repeated until the introduction of all the experimental data were consistent with the lowest energy structures obtained. The structures were evaluated with CING, an integrated residue-based structure validation program suite [Doreleijers et al., 2012] including PROCHECK [Laskowski et al., 1996]. Pymol [Delano, 2002] and UCSF Chimera [Pettersen et al., 2004] software programs were used to visualize the structures. Calculations were performed on a Dell Precision 690 workstation.
RESULTS
The full-length agnoprotein is predicted to contain two alpha-helices and intrinsically unstructured regions
Visual inspection of the agnoprotein primary sequences revealed that it is a highly basic protein. The basic residues are in general located at the amino and carboxy termini of the protein, mostly dominated by Arg and Lys residues. There is however a hydrophobic region in the middle of the protein, which is rich in Leu/Ile/Phe residues. Interestingly, two negatively charged residues Asp38 and Glu34 are also dispersed within this hydrophobic region.
We then analyzed the primary sequence of agnoprotein using the network protein sequence analysis server (http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sspred.html) [Combet et al., 2000] and the PSIPRED server (http://bioinf.cs.ucl.ac.uk/psipred/) [McGuffin et al., 2000], which allowed us to predict the presence of two α-helices in the full-length sequence of agnoprotein. One of these α-helices is located to far N-terminal region (Val2 to Ala10) of the protein and the other one is situated towards the middle portion of the protein spanning residues Gly20 to Cys40. The remaining portions of agnoprotein, spanning Ser11 to Ser19 and Thr41 to Thr71, are predicted to be intrinsically unstructured (Fig. 1).
Figure 1. Secondary structure prediction for full-length agnoprotein.
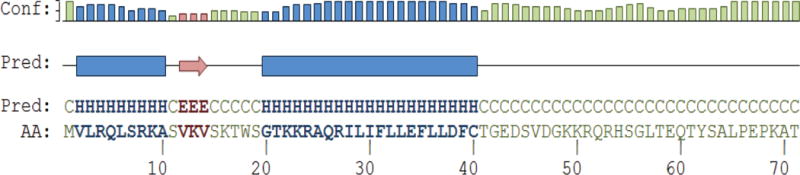
The full-length agnoprotein sequence was analyzed by the use of the PSIPRED [McGuffin et al., 2000] and the NPS@ [Combet et al., 2000] web server secondary structure predictions. These predictions suggest that agnoprotein has two α-helices (blue regions) encompassing residues Val2-Ala10 and Gly20-Cys40. There is also a short β-pleated region located at amino acids, Val12-Val14 and two regions, Ser15-Ser19 and Thr41-Thr71 that appears to be intrinsically unstructured. The confidence of the prediction is indicated by “Conf.”, the prediction of the secondary structure is designated by E = extended, C = Coil or H = Helix. “AA” indicates the amino acid sequence. Pred.: Prediction.
The hydrophilic and hydrophobic characteristics of agnoprotein
We are also interested in analyzing the potential hydrophobic and hydrophilic domains of agnoprotein utilizing a method established by Kyte and Doolittle [Snider et al., 2009] (http://web.expasy.org/protscale/) [Gasteiger et al., 2005] as shown in Fig. 1. This analysis allowed us to identify only one hydrophobic domain in agnoprotein, as previously predicted [Saribas et al., 2016], spanning residues Arg27 to Cys40 with a maximum peak at position Leu32 which encompasses approximately the second predicted α-helix (Ser19-Cys40). This region contains the Leu/Ile/Phe-rich domain of the protein, which is highly conserved in agnoprotein for JCV, SV40 and BKV and involved in formation of an amphipathic-like α- helix [Roy et al., 2010; Saribas et al., 2013; Saribas et al., 2011]. Data from the secondary structure predictions (Fig. 1) also correlate well with that of the hydrophobicity/hydrophilicity analysis of agnoprotein (Fig. 2) with respect to the position of a second predicted α-helix of the protein spanning residues Val2 to Ala10.
Figure 2. Hydrophobicity plot for the full-length agnoprotein.
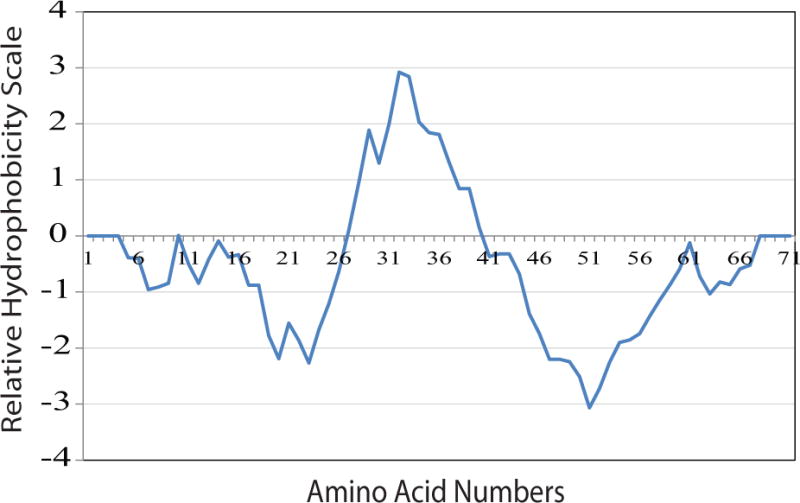
This plot was generated according the method established by Kyte and Doolittle, which was implemented in protscale, on the Expasy Server (http://web.expasy.org/protscale/). The curve displays the hydrophobic and hydrophilic regions of agnoprotein.
Analysis of full-length agnoprotein by Circular Dichroism (CD)
Circular Dichroism is an excellent tool for rapid evaluation of the secondary structure and folding properties of proteins. However, it does not give the residue-specific information that can be obtained by NMR or X-ray crystallography. CD is widely used to determine whether a protein is folded or if a mutation affects its conformation or stability. It can also be used to study protein-protein interactions. In this study, CD was primarily used to determine whether agnoprotein contains the secondary structures (α-helices) that were predicted by different programs.
Circular dichroism experiments performed on the agnoprotein showed that the protein adopts an alpha-helical structure in H2O, as well as in different organic solvent concentrations of TFE (H2O/TFE, v/v, 90/10, 80/20 and 70/30) (Fig. 3). As shown in Figure 3, the CD spectra of agnoprotein in H20 (red spectra), in various concentrations of H2O/TFE mixture [(90/10 (green), 80/20 (blue), and 70/30 (purple)] at pH 3.0 show the characteristic negative bands at 208 nm (parallel π → π* transitions) and at 222 nm, indicating the formation of an α- helix. The stability of the α-helix can be estimated by the depth of the minima at 208 nm and 222 nm. The part of the spectra containing the maximum at 190 nm is not completely represented because the signal was saturated. The two different solvents used demonstrate that the agnoprotein peptide adopts an α-helical structure in these two conditions. The helical content and the stability of the helix seem to slightly increase with the amount of TFE added.
Figure 3. Circular Dichroism (CD) spectra of the full-length agnoprotein.
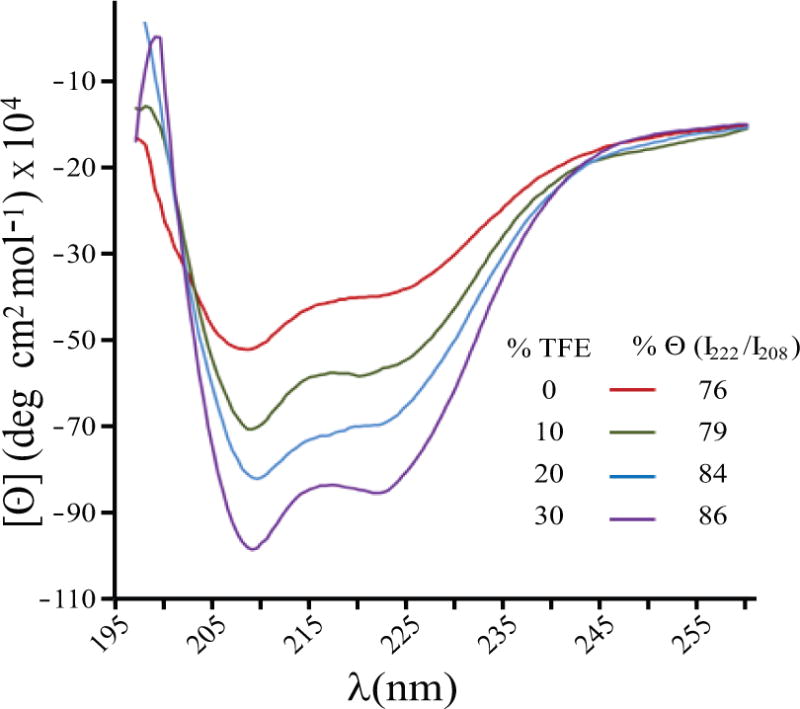
The spectra were recorded at room temperature (20°C) in a 0.1 cm path-length quartz cell on a Jobin-Yvon model C8 spectropolarimeter, calibrated with d-camphor-10-sulfonate. The experiments were performed at pH 3.0 in 100 % H20 (red spectra) as well as in various concentrations of the H2O/TFE mixture [H2O/TFE, (v/v), 90/10 (green), 80/20 (blue), and 70/30 (purple)]. The characteristic negative bands at 208 nm (parallel π → π* transitions) and at 222 nm indicate the formation of an α-helix stabilized by the TFE. The peptide concentration in each sample was 50 μM. The stability of the α-helix can be monitored by the value of the minima and maxima of Θ. The % (I222/I208) indicates the fraction of α-helical structure that slightly increases with the fraction of TFE used.
Optimization of the NMR spectroscopy conditions to determine agnoprotein structure
One-dimensional (1D) NMR experiments were recorded either in pure water or in presence of 30% TFE at two different temperatures, 293°K to 313°K (Fig. 4). Spectra in H2O showed the broaden peaks most likely due to the tendency of agnoprotein to oligomerize or to aggregate. Taking the same spectra recordings in an organic solvent (30% TFE) however, resulted in narrower linewidths and the spread of the resonances due to interaction with nearby nuclei, suggesting that the secondary structure of agnoprotein is stabilized. In parallel, an increase in temperature from 293°K to 313°K allowed even more reduction in the linewidths. In a previous NMR study performed on HIV-1 Vpr, an accessory protein of HIV-1, we used TFE and acetonitrile to solubilize the protein and to eliminate the protein oligomerization [Wecker et al., 2002]. Acetonitrile is less hydrophobic than TFE and allowed to maintain the tertiary structure of the protein due to hydrophobic interactions between its three helices [Bourbigot et al., 2005; Morellet et al., 2003; Wecker et al., 2002]. However, in this study, we only used 30% TFE to prevent aggregation of agnoprotein.
Figure 4. Comparison of the 1D1H NMR spectra performed on the full-length agnoprotein in various solvent conditions.
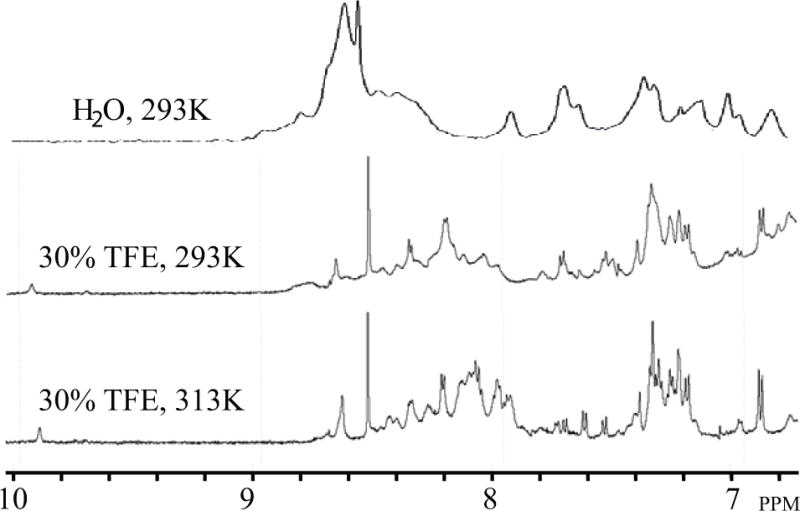
The spectra were acquired on an Avance Bruker spectrometer operating at 600.13 MHz, equipped with a cryoprobe. Various conditions were tested to prevent the aggregation and to increase the solubility of agnoprotein. The protein concentration was 0.2 mM in all experiments conducted. (Upper trace) The protein appeared to be aggregated with very broad peaks in pure water, at pH 3.0 and 293°K. (Middle trace) In presence of 30% (v/v) TFE and at pH 3.0 and 293°K, the protein started to solubilize with a large spreading of the chemical shifts but the linewidth remained relatively broad. (Lower trace) Under the same conditions as stated for “middle trace”, but at temperature of 313°K, we obtained spectra with a narrower linewidth.
Resolution of the full-length agnoprotein structure by NMR
Standard 2D NMR experiments were performed to analyze the structure of agnoprotein. Double-quantum-filtered correlated spectroscopy (DQF-COSY) [Marion and Wuthrich, 1983; Rance et al., 1983] and total correlated spectroscopy (TOCSY) [Griesinger et al., 1988] experiments were used to identify the spin systems while NOESY experiments [Jeener et al., 1979; Kumar et al., 1980] were used for sequential and long-range distance evaluation [Wuthrich, 1986]. The assignment step was very complicated due to overlaps occurring all over the spectra (Fig. 5). Characteristic connectivity dαN(i,i+3) and dαβ(i,i+3) and strong NOEs dNN(i,i+1) and dβN(i,i+1) have been identified indicating the presence of a short α-helix spanning residues Ser7-Ser11 and a longer α-helix spanning residues Lys22- Phe39 (Fig. 6). The regions, including Met1-Leu6, Val12-Thr21 and Thr41-Thr71 of the agnoprotein do not present any particular secondary structure element (Fig. 6). The full 1H resonance assignment was performed and is presented in Table 1.
Figure 5. Superimposition of the TOCSY and NOESY spectra of full-length agnoprotein.
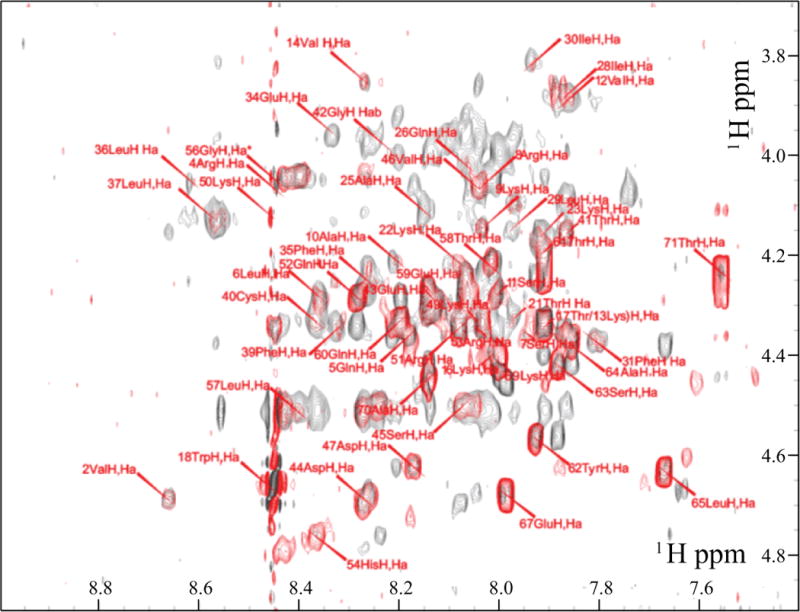
Only the region of the spectra showing the inter-residual correlations between alpha proton (i) and amide proton (i+1) is shown. The NOESY spectra (dark grey) was recorded with a mixing time of 250 ms and the TOCSY spectra (red) with a mixing time of 70 ms, in presence of 30% (v/v) TFE at pH 3.0 and a temperature of 313°K. The intra-residual correlations for each residue (i) are labeled in red.
Figure 6. Summary of the sequential and short-range NOE connectivities for the full-length agnoprotein.

(A) Amino acid sequence of agnoprotein. (B) Characteristic strong dNN(i,i+1), dαN(i,i+1) and medium range dαN(i,i+2; i,i+3; i,i+4), dαβ(i,i+3) signals identified by TOCSY and NOESY experiments. α, β and N are the alpha, beta and amide protons of each amino acid, respectively and dαβ, dNN and dβN are the distance between alpha and beta, between amide and amid, and between beta and amide protons of two distinct residues, respectively. The thickness of the lines is related to the estimated intensity of the NOE (strong, medium or weak). (C) The Chemical Shift Index (CSI) represented indicates the secondary structure formation according the Hα chemical shift. The chemical shift index (CSI) Δδ was used to display and identify the location of the secondary structures using 1Hα chemical shift data, which are shifted upfield in helices and downfield in beta strands relative to their random coil values. (D) Schematic organization the two alpha helices on agnoprotein deduced from the NOESY spectra information.
Table 1.
Proton chemical shifts of Agnoprotein in the presence of 30% TFE calibrated to H2O
| Position residue | Proton chemical shift(s) (ppm)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| NH | H-α | H-β | H-γ | H-δ | H-ε | H-ζ | H-η | |
| 1 Met | 4.15 | |||||||
| 2 Val | 8.66 | 4.69 | ||||||
| 4 Arg | 8.44 | 4.07 | 1.84,1.84 | 3.38, 3.38 | ||||
| 5 Gln | 8.18 | 4.36 | 2.03, 2.12 | 2.39, 2.39 | ||||
| 6 Leu | 8.36 | 4.28 | 1.63 | 0.90, 0.94 | ||||
| 7 Ser | 7.95 | 4.36 | 3.98, 3.98 | |||||
| 8 Arg | 8.05 | 4.06 | 1.99, 1.99 | 1.75, 1.75 | 3.22, 3.22 | |||
| 9 Lys | 8.03 | 4.14 | 1.94, 1.94 | 1.50, 1.50 | 1.63, 1.63 | 3.01, 3.01 | ||
| 10 Ala | 8.21 | 4.21 | 1.50 | |||||
| 11 Ser | 8.01 | 4.28 | 4.00, 4.07 | |||||
| 12 Val | 7.86 | 3.88 | 2.23 | 0.99, 1.09 | ||||
| 13 lys | 7.91 | 4.33 | 1.67, 1.74 | 1.50, 1.50 | 3.03, 3.03 | |||
| 14 Val | 8.27 | 3.86 | 2.14 | 0.91, 1.05 | ||||
| 15 Ser | 8.04 | 4.34 | 3.92, 3.92 | |||||
| 16 Lys | 8.02 | 4.40 | ||||||
| 17 Thr | 7.91 | 4.35 | 4.21 | 1.24 | ||||
| 18 Trp | 8.45 | 4.67 | 3.42, 3.42 | 7.27 | 7.64 | 7.13, 7.47 | 7.21 | |
| 19 Ser | 8.27 | 4.22 | 3.86, 3.86 | |||||
| 20 Gly | 8.10 | 3.96, 3.96 | ||||||
| 21 Thr | 7.97 | 4.32 | 4.10 | 1.30 | ||||
| 22 Lys | 8.09 | 4.21 | 1.91, 1.91 | 1.44, 1.44 | ||||
| 23 Lys | 7.92 | 4.16 | 1.82, 1.82 | 1.49, 1.49 | 1.73, 1.73 | |||
| 24 Arg | 7.88 | 4.09 | 1.70, 1.73 | 1.64, 1.64 | 3.23, 3.23 | 7.23 | ||
| 25 Ala | 8.15 | 4.11 | 1.51 | |||||
| 26 Gln | 8.06 | 4.03 | 2.18, 2.26 | 2.38, 2.54 | 6.62, 6.91 | |||
| 27 Arg | 7.74 | 4.07 | 1.83, 2.02 | 1.74, 1.83 | 3.23, 3.23 | 7.28 | ||
| 28 Ile | 7.86 | 3.88 | 2.03 | 0.99, 1.38, 1.51, 1.76 | 0.87 | |||
| 29 Leu | 7.98 | 4.15 | 1.64, 1.64 | 0.87, 0.94 | ||||
| 30 Ile | 7.93 | 3.81 | 1.96 | 0.94, 1.30, 1.76 | 0.87 | |||
| 31 Phe | 7.81 | 4.37 | 3.29, 3.40 | |||||
| 32 Leu | 8.57 | 4.11 | 1.74, 1.74 | 1.66 | 0.91, 0.94 | |||
| 33 Leu | 8.57 | 4.13 | 1.92, 1.92 | 1.64 | 0.88, 0.93 | |||
| 34 Glu | 8.33 | 3.96 | 2.08, 2.25 | 2.40, 2.65 | ||||
| 35 Phe | 8.27 | 4.25 | 2.98, 3.24 | 7.17 | ||||
| 36 Leu | 8.62 | 4.06 | 1.64, 1.98 | 1.64 | 0.95 | |||
| 37 Leu | 8.57 | 4.13 | 1.87, 1.87 | 1.53 | 0.88, 0.94 | |||
| 38 Asp | 8.15 | 4.47 | 2.67, 2.88 | |||||
| 39 Phe | 8.33 | 4.34 | 2.89, 3.12 | 7.20 | ||||
| 40 Cys | 8.37 | 4.34 | 2.98, 3.08 | |||||
| 41 Thr | 7.87 | 4.15 | 4.36 | 1.29 | ||||
| 42 Gly | 8.22 | 3.92, 4.00 | ||||||
| 43 Glu | 8.14 | 4.28 | 2.08, 2.14 | 2.36, 2.36 | ||||
| 44 Asp | 8.27 | 4.69 | 2.76, 2.80 | |||||
| 45 Ser | 8.08 | 4.51 | ||||||
| 46 Val | 8.04 | 4.06 | 2.18 | 1.00, 1.00 | ||||
| 47 Asp | 8.17 | 4.62 | 2.78, 2.78 | |||||
| 48 Gly | 8.21 | 3.93, 4.00 | ||||||
| 49 Lys | 8.05 | 4.33 | 1.73, 1.73 | 1.46, 1.54 | 1.65, 1.65 | 3.01, 3.01 | ||
| 50 Lys | 8.46 | 4.12 | 1.91, 2.03 | 1.52, 1.75 | 1.84, 1.84 | 3.15, 3.15 | ||
| 51 Arg | 8.08 | 4.35 | 1.90, 1.90 | 1.72, 1.81 | 3.24, 3.24 | 7.23 | ||
| 52 Gln | 8.28 | 4.29 | 2.08, 2.15 | 2.41, 2.41 | 6.70, 7.39 | |||
| 53 Arg | 8.04 | 4.35 | 1.90, 1.90 | 1.73, 1.80 | 3.25, 3.25 | |||
| 54 His | 8.37 | 4.76 | 3.24, 3.37 | 7.33 | 8.56 | |||
| 55 Ser | 8.24 | 4.14 | 3.85, 3.85 | |||||
| 56 Gly | 8.45 | 4.05, 4.05 | ||||||
| 57 Leu | 8.40 | 4.51 | 1.66, 1.66 | 0.90 | ||||
| 58 Thr | 8.02 | 4.22 | 4.35 | 1.23 | ||||
| 59 Glu | 8.14 | 4.27 | 2.17, 2.17 | 2.44, 2.44 | ||||
| 60 Gln | 8.20 | 4.33 | 2.06, 2.14 | 6.63, 7.35 | ||||
| 61 Thr | 7.91 | 4.19 | 4.25 | 1.17 | ||||
| 62 Tyr | 7.93 | 4.56 | 3.01, 3.12 | 7.13 | 6.83 | |||
| 63 Ser | 7.88 | 4.42 | 3.86, 3.90 | |||||
| 64 Ala | 7.86 | 4.38 | 1.43 | |||||
| 65 Leu | 7.67 | 4.63 | 1.59, 1.60 | 1.74 | 0.94, 0.94 | |||
| 66 Pro | 4.45 | 2.23, 2.23 | 1.94, 2.04 | 3.61, 3.82 | ||||
| 67 Glu | 7.99 | 4.68 | 1.94, 2.10 | 2.43, 2.43 | ||||
| 68 Pro | 4.44 | 2.28, 2.28 | 2.00, 2.08 | 3.71, 3.78 | ||||
| 69 Lys | 8.00 | 4.41 | 1.74, 1.91 | 1.49, 1.49 | 3.04, 3.04 | |||
| 70 Ala | 8.14 | 4.45 | 1.45 | |||||
| 71 Thr | 7.56 | 4.23 | 4.30 | 1.20 | ||||
Structure calculation of the monomeric form of agnoprotein
A total of 572 interproton distance restraints were identified on the 250-ms-mixing- time NOESY spectra recorded at pH 3.0 and 313°K in the presence of 30% (v/v) TFE and were used for the structure calculation of agnoprotein using the visualization program CcpNmr analysis [Vranken et al., 2005] and the software ARIA to assign NOE constraints and manage the resulting ambiguities. The Ambiguous Distance Restraint method was introduced by Nilges [Nilges, 1995] and allows the use of ambiguous distance constraints iteratively in molecular modeling. Among these interproton distance restraints, 387 were intraresidual, 150 were sequential and 35 were medium range. The unstructured regions are located at the N- and C-terminus of the protein and a well-defined α-helix spans residues, Arg24 to Phe39. The structures were calculated by restrained simulated annealing and energy minimization and selected according to their lowest total energy and number of NOE restraint violations. Among an ensemble of 200 generated structures, twelve structures were selected for structural analysis (Table 2) and used to generate the average structure. The 12 best structures of the protein were superimposed on the backbone atoms of the domain Arg24- Phe39 of the average structure (Fig. 7).
Table 2.
Structural statistics for the structure of agnoprotein
| Parametersa | Values | |
|---|---|---|
| Restraints for calculation | 572 | |
| unambiguous | 468 | |
| ambiguous | 104 | |
| Total NOE restraints | 605 (33 merged) | |
| Intraresidue | 387 | |
| Sequential (|i−j|=1)a | 150 | |
| Medium range (|i−j| < 4) | 35 | |
| RMSD Structure statistics | ||
| Bonds(Å) | 2.85×10−3 – 3.09×10−3 | |
| Bond angles (°) | 0.44 – 0.49 | |
| Improper torsions (°) | 1.04 – 1.09 | |
| NOE restraints (Å) | 2.36 × 10−2 – 2.45 × 10−2 | |
| Final energies (kcal/mol) | ||
| Total | −1 931 to −1 770 | |
| Bonds | 9.55 – 11.20 | |
| Angles | 61.78 – 77.37 | |
| Improper angles | 23.56 – 26.32 | |
| dihedrals | 332.61 – 327.2 | |
| Van der Waals | −128.29 to −114 | |
| elec | −2245.75 to −2115.27 | |
| NOE | 15.9 – 17.15 | |
| Ramachandran plot | ||
| Residues in most favored regions (%) | 61.8 | |
| Residues in additional allowed regions (%) | 33.5 | |
| Residues in generously allowed regions (%) | 1.5 | |
| Residues disallowed regions (%) | 3.3 | |
| Atomic r.m.s.d. (Å) on backbone atoms | (I28-F39) | (R24-F39) |
| Pairwise | 0.605 ± 0.180 | 1.48 ± 0.63 |
| To mean structure | 0.590 ± 0.100 | 1.25 ± 0.44 |
j represents an amino acid different from amino acid i. Residues represented by i and j may be consecutive in the sequence or separated by 1, 2, or 3 residues.
Figure 7. NMR structure of the full-length agnoprotein.
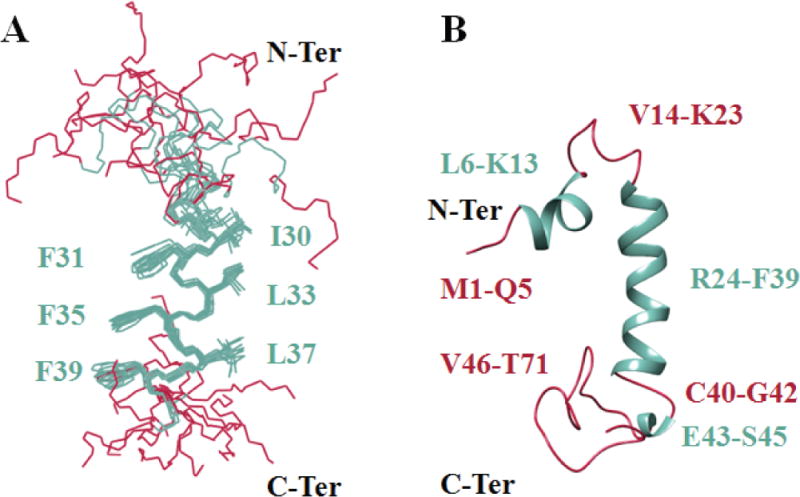
Structure was determined in presence of 30% (v/v) TFE at pH 3.0 and 313°K, using CNS [Brunger et al., 1998] and ARIA [Nilges, 1995] programs. Alpha helical regions and intrinsically unstructured regions are colored in green and red respectively. (A) Superimposition of the 12 energetically most favorable structures of the protein on the backbone atoms of domain Lys24-Phe39 of the average structure. The backbone bonds were drawn with lines. The well-defined sidechains (Phe31, Phe35, Phe39, Ile30, Leu33 and Leu37) were displayed in the stick representation. (B) Average structure of agnoprotein contains two α-helical structures Leu6- Lsy13 and Arg24-Phe39 (colored green) and two principal unstructured regions spanning residues Val14-Lys23 and Cys40-Thr71 (colored red). In some structures, a short α-helix region, Glu43-Ser45, is also formed.
The structure is composed of one short (Leu6-Lys13) and one relatively long (Arg24-Phe39) α-helix, represented in green and surrounded by unstructured regions (Met1-Leu6), (Val12-Thr21) and (Thr41-Thr71) in its N- and C-terminal regions, (Fig. 7, colored in red). The root mean square deviation (RMSD) with respect to the mean coordinates was evaluated to 0.59 ± 0.10 Å and 1.25 ± 0.44 Å while the pairwise RMSD was 0.61 ± 0.18 Å and 1.48 ± 0.63 Å calculated on the backbone atoms for all non-hydrogen atoms of the helix (Ile28- Phe39) and (Arg24-Phe39) respectively (Table 2).
The Leu/Ile/Phe-rich region shows hydrophobic properties with the structural characteristics of a partial amphipathic helix (Fig. 8). This region shows two faces of opposite polarity, one side of the helix is rather narrow and covers one-quarter (¼) of the surface of the helix, which is composed of the hydrophilic residues Lys23, Gln26, Arg27, Glu34 and Asp38, providing this face a hydrophilic property. The remaining face is much wider, covers three-quarters (¾) of the α-helical surface and is constituted of hydrophobic residues, including Ala25, Ile28, Leu29, Ile30, Phe31, Leu32, Leu33, Phe35, Leu36, Leu37 and Phe39 (Fig. 8). The structure of agnoprotein contains three unstructured regions, spanning residues Met1-Leu6, Val12-Thr21 and Thr41-Thr71. Of those, the Thr41-Thr71 region was observed to assume a short α-helix in some selected NMR structures, which is not found systematically in all structures (Fig. 8). The hydrophobicity and partial amphipathicity of this helical domain could explain the propensity of agnoprotein to dimerize/oligomerize [Coric et al., 2014; Saribas et al., 2011; Saribas et al., 2016].
Figure 8. The average structure of agnoprotein in a hydrophobic surface representation.
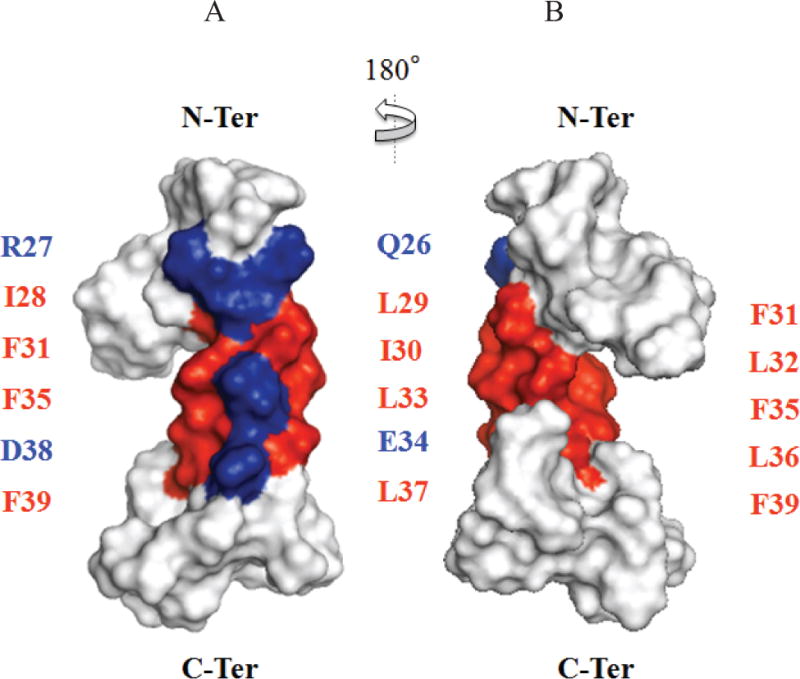
Residues of the main alpha helix region were colored according to their hydrophobicity following the Eisenberg hydrophobicity scale (red for positive values corresponding to hydrophobic residues and blue for negative values corresponding to hydrophilic residues). The partial amphipathic helix region composed of the Leu/Ile/Phe-rich residues exhibits an opposite polarity and shows two faces: A narrow hydrophilic face which is composed of hydrophilic residues (Gln26, Arg27, Glu34 and Asp38) (A) and a wider hydrophobic face which is constituted with hydrophobic residues (Ile28, Leu29, Ile30, Phe31, Leu32, Leu33, Phe35, Leu36, Leu37, Phe39) (B).
It is well known that many intrinsically disordered proteins (IDPs) initiate various interactions with their biological partners and become active by a binding- induced folding mechanism. This mechanism may induce a folded conformation from an initial random state or by selecting a pre-formed state already present in the protein. This could be the case for agnoprotein which has a very well-defined major alpha-helix spanning residues Lys23-Phe39 and two other potential minor alpha-helical domains identified by several secondary structure prediction programs, spanning residues Leu3- Arg8 and Lys50-His54. In addition, there are unstructured regions on the protein spanning Lys9-Thr22 and Cys40-Thr71. We believe that the two potential minor helical domains could constitute a pre-formed state leading to a starting point for the progressive transition from unstructured to the ordered domains during the interaction of agnoprotein with its partners.
Comparison of the experimental and theoretical structures of agnoprotein
Iterative Threading Assembly Refinement (I-TASSER) is a tool for the prediction of protein 3D structure and function [Roy et al., 2010]. For predictions, first, templates were identified from the protein data bank (PDB) by multiple threading approaches and full-length agnoprotein models were built by simulations of assembly of template fragments. We used the I-TASSER software to predict the 3D structure of JCV agnoprotein. Our model predicts that agnoprotein contains three α-helices located at position Leu3-Arg8, Lys22-Gly42 and Lys50-His54 and two unstructured regions Lys9-Thr21 and Ser55-Thr71 (Fig. 9A). These results are consistent with our experimentally obtained structures, where we obtained two short [(Leu6-Lys13) and (Glu43-Ser45)] and one relatively long (Arg24-Phe39) α-helices (Fig. 9B).
Figure 9. Comparison of a predicted (A) and the average NMR (B) structures of the full-length agnoprotein.
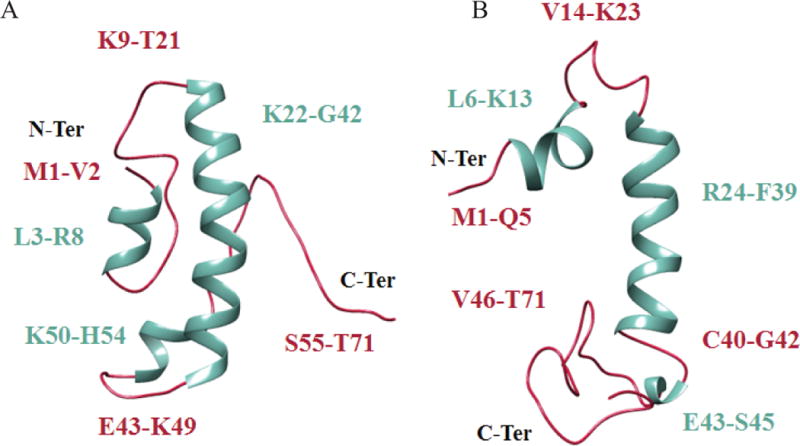
The secondary structures are colored in green while the intrinsically unstructured regions are colored in red. The predicted model has been obtained with I- TASSER, a tool for protein structure and function prediction [Roy et al., 2010; Yang et al., 2015]. This model predicts that agnoprotein contains three α-helices located at position Leu3- Arg8, Lys22-Gly42 and Lys50-His54 and two intrinsically unstructured regions Lys9-Thr21 and Ser55-Thr71. These predictions are consistent with our NMR structure findings, in which two alpha helices, one short (Leu6-Lys13) and a relatively long one (Arg24-Phe39) are found almost identical regions on agnoprotein. In some NMR structures, another short α-helix also was found at position, Glu43-Ser45.
The previously determined structure of the dimerization domain of agnoprotein, which was deposited to the Protein Data Bank (PDB) under identification code 2MJ2 was used as a template by I-TASSER program to predict the secondary structures and to propose different theoretical 3D models for the full-length agnoprotein, but the I-TASSER program had to search the full PDB for other templates to predict the rest of the protein as well.
During the use of I-TASSER, there was a possibility of excluding some of templates from I-TASSER template library and the 3D structure prediction of the full-length agnoprotein was performed excluding the structure 2MJ2. Obviously, the knowledge of the 3D structure of the dimerization domain of the agnoprotein helped us to better predict the 3D structure of the full-length agnoprotein. When we compared the confidence score for estimating the quality of predicted models by I-TASSER (C- score), the TM-score and RMSD to evaluate the structural similarity between the structures, we showed that the predicted results were better when the 2MJ2 is included in the I-TASSER library (Table 3). We have recently deposited the full structure of agnoprotein to the PDB under identification code 5NHQ.
Table 3.
Agnoprotein structure and function prediction results using I-TASSER template library
| Model | C-score | Exp.TM- Score | Exp.RMSD | No.of decoys | Cluster density |
|---|---|---|---|---|---|
| Including PDB:2MJ2
| |||||
| 1 | 1.25 | 0.56 ± 0.15 | 5.8 ± 3.6 | 4959 | 0.1444 |
| 2 | 2.25 | 1756 | 0.0532 | ||
| 3 | 4.43 | 262 | 0.0060 | ||
| 4 | 5.00 | 118 | 0.0029 | ||
| 5 | 5.00 | 108 | 0.0029 | ||
| Excluding PDB: 2MJ2
| |||||
| 1 | −2.75 | 0.40 ± 0.13 | 9.1 ± 4.6 | 4801 | 0.1125 |
| 2 | −3.05 | 2856 | 0.0835 | ||
| 3 | −2.99 | 3050 | 0.0888 | ||
| 4 | −5.00 | 401 | 0.0106 | ||
| 5 | −5.00 | 52 | 0.0027 | ||
C-score: A confidence score for estimating the quality of predicted models by I-TASSER. TM-score and RMSD: Both are known standards for measuring structural similarity between two structures which are usually used to measure the accuracy of structure modeling when the native structure is known. For each target, I-TASSER simulations generate tens of thousands conformations called decoys. To select the final models, I-TASSER uses SPICKER program to cluster all the decoys based on the pair-wise structure similarity, and report up to five models which corresponds to the five largest structure clusters (http://zhanglab.ccmb.med.umich.edu/I-TASSER/FAQ.html).
DISCUSSION
The NMR data show that the 3D structure of JCV agnoprotein contains two α-helices at positions, Leu6-Lys13 (short, minor) and Arg24-Phe39 (long, major) (Fig. 7) and the positions of each α- helix is consistent with the predicted secondary structure for agnoprotein (Fig. 1). The flanking sequences of main and minor α-helices exhibit intrinsically disordered conformation. Our current NMR data also confirm the position of our previously reported α-helix by NMR [Coric et al., 2014]. In contrast to the amphipathic characteristics of the long α-helix, the short α-helix spanning amino acids Leu6 to Lys13 does not exhibit any such property and is relatively dynamic since we do not find it systematically in all calculated structures (Fig. 7).
NMR studies also revealed that agnoprotein has a strong propensity to aggregate in pure water (Fig. 3) and therefore the use of an organic solvent such as 30% TFE (v/v) was necessary to solubilize and prevent its aggregation in order to make NMR studies possible. Additional aggregation of the protein may take place through intermolecular interactions caused by charged residues due to the presence of 14 Arg/Lys and 7 Asp/Glu. Therefore, a relatively high concentration of salt (200 mM NaCl) was used to prevent these interactions. At low pH, the protonation of the basic and acidic residues helps to prevent intermolecular interactions and the amide proton exchange rate is slower than that observed at neutral pH. Therefore, more intense peaks were obtained to facilitate the NMR spectra analysis. Thus, these were the reasons why a low pH value (pH 3.0) was chosen to perform the NMR experiments. Finally, a sharper signal was obtained when the temperature was increased to 313K. It was also our experience that the concentration of agnoprotein (200 μM) used in NMR experiments has to be optimized to the certain levels to get a good signal to noise ratio.
In addition, we also previously reported that the major α-helix region plays an essential role both in formation of highly stable dimers/oligomers and in the stability of the protein [Coric et al., 2014; Saribas et al., 2013; Saribas et al., 2011]. A complete deletion of the region resulted in a rapid degradation of the protein and therefore no dimers/oligomer was formed [Saribas et al., 2013; Saribas et al., 2011]. Moreover, amino acid substitution studies within the same region demonstrated that certain amino acids, including Leu29 and Leu36 play critical regulatory roles in the stability and function of the protein [Coric et al., 2014].
As noted, our previous NMR work on an agnoprotein peptide encompassing residues Thr17 to Gln52 provided a higher definition of the structure with better convergence. The current NMR work, on the other hand, when compared to the previous one is fairly disordered and the major α-helix located at position, Arg24-Phe39, appears to be very dynamic. The pairwise RMSD varies from 0.61 ± 0.18 Å to 1.48 ± 0.63 Å when calculated on domains of Ile28-Phe39 and Arg24-Phe39 respectively. Perhaps, this is due to a phenomenon of exchange occurring between the monomeric, dimeric and oligomeric forms of the protein, despite the use of 30% TFE to facilitate the protein solubility. The dynamic nature of this α- helix (Arg24-Phe39) and the presence of the unstructured domains of the protein suggest that those regions allow agnoprotein to interact with multiple targets in a given time, and therefore amplifies its biological activities during the viral infection cycle.
As mentioned above, the NMR structure of agnoprotein further revealed that the flanking sequences of short and long α-helices adopt intrinsically disordered conformations. In other words, except those two helices, both N-terminal and C-terminal regions of agnoprotein shown in figure 7B do not display a stable tertiary structure. This type of disordered conformation results from a combination of the low hydrophobicity and high net charge in those regions providing the protein considerable flexibility to interact with various targets. Through this mechanism, agnoprotein is thought to diversify and amplify its functions during the viral replication cycle [Dyson and Wright, 2005; Fink, 2005]. There are several small viral proteins that also possess such features. For example, E4-ORF3 (adenovirus small protein) has been shown to form multivalent functional matrices in infected cells by exploiting its intrinsically disordered structure [Ou et al., 2012]. This interaction then results in the inactivation of various tumor suppressor proteins including, TIM24, MRE11/Rad50/NBS1 (MRN), p53 and promyelocytic leukemia factor. Similarly, intrinsically disordered regions of agnoprotein may play important regulatory roles in agnoprotein function in interactions with its binding-partners in infected cells and thereby contribute to the overall viral replication cycle.
Our group has extensive experience to work with the proteins that have solubility issues under physiological conditions to determine their 3D structure by NMR. To obtain the structural data for such difficult proteins, the normal physiological conditions are not sufficient. In order to solubilize agnoprotein, for example, we adopted the conditions that are not compatible with normal physiological conditions. Even under harsh conditions, the NMR structure that we obtained for agnoprotein is in good agreement with the theoretical model of agnoprotein established by bioinformatics predictions. Moreover, it was interesting to observe that the agnoprotein has much in common with the HIV-1 protein Vpr with respect to forming dimers and oligomers and aggregation properties. Our group has extensively studied the structural properties of Vpr by NMR and determined its structure using nonphysiological conditions, including 30% TFE or acetonitrile, low pH (pH 3.0) and relatively high temperature (50°C) [Morellet et al., 2003]. Recently, the structure of Vpr was determined again by co-crystallization studies [Wu et al., 2016]. This new crystallographic structure of Vpr exactly matches with the one obtained by our group by NMR under nonphysiological conditions. Thus, we believe that the NMR structure that we obtained for JCV agnoprotein under harsh conditions most likely resembles the Vpr case as mentioned above.
As mentioned above, the importance of the main α-helix region of agnoprotein has been extensively studied with respect to its contribution to dimer/oligomer formation. The significance of the minor α-helix region and the some parts of the intrinsically unstructured regions were also investigated in part by mutagenesis analysis. The minor α-helix region of agnoprotein appears to make two full-turns and contains two phosphorylation sites (Ser7 and Ser11) for the Ser- and Thr-specific protein kinase C [Sariyer et al., 2006]. We have previously characterized the significance of these phosphorylation sites [Sariyer et al., 2006] and found that when Ser7 and Ser11 were mutated to Ala either singly or combinatorically (Ser7Ala or Ser7Ala plus Ser11Ala), JCV was unable to sustain its productive life cycle. In other words, these mutants were able partially replicate during the first round of replication cycle but unable to continue their replication thereafter [Sariyer et al., 2006]. Similar studies have been also carried out by Johannessen et al. on the BKV agnoprotein Ser11 residue, which is a highly conserved between JCV virus, BKV and SV40. Mutation of Ser11 to either Ala or Asp significantly altered the ability of BKV propagation in Vero cells [Johannessen et al., 2008] similar to our findings from JCV agnoprotein phosphorylation mutants [Sariyer et al., 2006]. In addition, an interesting study has been reported by Suzuki et al. concerning this region, where JCV agnoprotein was suggested to function as a viroporin and the basic residues (Arg8 and Lys9) within the minor α-helix region plays a critical role in this process [Suzuki et al., 2010]. Moreover, agnoprotein was reported to interact with a cellular factor, heterochromatin protein 1 (HPα) through its N-terminus residues (aa 1–18), which is partially overlapping with those that are found in the minor α-helix region. Agnoprotein was suggested to disrupt the interaction between HPα and a nuclear receptor called lamin B (LBR) and thereby potentiates the egress of virions from the nucleus of infected cells [Okada et al., 2005]. Together these findings underscore the significance of the minor α-helix region of agnoprotein for its function and for viral propagation.
Regarding the intrinsically unstructured regions of agnoprotein, a previous study by Akan et al. demonstrated that a deletion mutant [Agno Δ(52–68)], which eliminates a considerable length of the C-terminus unstructured region of agnoprotein, significantly but negatively affected the viral gene expression and replication, suggesting that this unstructured region may play a critical role in the function of agnoprotein [Akan et al., 2006].
Three-dimensional structures of proteins offer opportunities to study their regulatory and structural roles. In this work, we resolved the 3D structure of the full-length JCV agnoprotein by NMR. We and others have previously reported that this protein plays critical regulatory roles during the viral replication cycle [Sariyer et al., 2011]. In addition, our recent detailed analysis of the dimerization domain of agnoprotein (the main α-helix region) demonstrated that if this domain is deleted or altered by mutagenesis, then the stability of the protein is severely compromised [Saribas et al., 2013] suggesting that this region can be targeted by small molecules to disrupt the functions of agnoprotein so that infection by JCV in PML patients can be controlled.
Acknowledgments
This work was supported by the grants awarded to M. Safak by National Institute of Health (Grant #: RO1NS090949), USA and by the Temple University Drug Discovery Initiative (Grant #: 161398), Philadelphia, USA. In addition, this work was also made possible by support money awarded to S. Bouaziz by the Centre National de la Recherche Scientifique, and by the University Paris Descartes, Paris, France.
Footnotes
CONFLICT OF INTEREST: Authors declare no conflict of interest.
References
- Akan I, Sariyer IK, Biffi R, Palermo V, Woolridge S, White MK, Amini S, Khalili K, Safak M. Human polyomavirus JCV late leader peptide region contains important regulatory elements. Virology. 2006;349:66–78. doi: 10.1016/j.virol.2006.01.025. [DOI] [PubMed] [Google Scholar]
- Berger JR, Concha M. Progressive multifocal leukoencephalopathy: the evolution of a disease once considered rare. J Neurovirol. 1995;1:5–18. doi: 10.3109/13550289509111006. [DOI] [PubMed] [Google Scholar]
- Berger JR, Houff S. Progressive multifocal leukoencephalopathy: lessons from AIDS and natalizumab. Neurol Res. 2006;28:299–305. doi: 10.1179/016164106X98198. [DOI] [PubMed] [Google Scholar]
- Bourbigot S, Beltz H, Denis J, Morellet N, Roques BP, Mely Y, Bouaziz S. The C- terminal domain of the HIV-1 regulatory protein Vpr adopts an antiparallel dimeric structure in solution via its leucine-zipper-like domain. Biochem J. 2005;387:333–41. doi: 10.1042/BJ20041759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, Laubach J, Bawn SD, Gordon LI, Winter JN, Furman RR, Vose JM, Zelenetz AD, Mamtani R, Raisch DW, Dorshimer GW, Rosen ST, Muro K, Gottardi-Littell NR, Talley RL, Sartor O, Green D, Major EO, Bennett CL. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood. 2009;113:4834–40. doi: 10.1182/blood-2008-10-186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bord E, Tompkins T, Miller J, Tan CS, Kinkel RP, Stein MC, Viscidi RP, Ngo LH, Koralnik IJ. Asymptomatic reactivation of JC virus in patients treated with natalizumab. N Engl J Med. 2009;361:1067–74. doi: 10.1056/NEJMoa0904267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combet C, Blanchet C, Geourjon C, Deleage G. NPS@: network protein sequence analysis. Trends Biochem Sci. 2000;25:147–50. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- Coric P, Saribas AS, Abou-Gharbia M, Childers W, White MK, Bouaziz S, Safak M. Nuclear magnetic resonance structure revealed that the human polyomavirus JC virus agnoprotein contains an alpha-helix encompassing the Leu/Ile/Phe-rich domain. Journal of virology. 2014;88:6556–75. doi: 10.1128/JVI.00146-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbinyan A, Darbinian N, Safak M, Radhakrishnan S, Giordano A, Khalili K. Evidence for dysregulation of cell cycle by human polyomavirus, JCV, late auxiliary protein. Oncogene. 2002;21:5574–81. doi: 10.1038/sj.onc.1205744. [DOI] [PubMed] [Google Scholar]
- Delano W. THe PyMOL Molecular Graphics System. DeLano Scientific 2002 [Google Scholar]
- Doreleijers JF, Sousa da Silva AW, Krieger E, Nabuurs SB, Spronk CA, Stevens TJ, Vranken WF, Vriend G, Vuister GW. CING: an integrated residue-based structure validation program suite. J Biomol NMR. 2012;54:267–83. doi: 10.1007/s10858-012-9669-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud Se Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. Proteomics protocols handbook. Tottowa, New Jersey: Humana Press; 2005. pp. 571–607. [Google Scholar]
- Gerits N, Johannessen M, Tummler C, Walquist M, Kostenko S, Snapkov I, van Loon B, Ferrari E, Hubscher U, Moens U. Agnoprotein of polyomavirus BK interacts with proliferating cell nuclear antigen and inhibits DNA replication. Virol J. 2015;12:7. doi: 10.1186/s12985-014-0220-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesinger C, Otting G, Wuthrich K, Ernst RR. Clear TOCSY for 1H spin system identification in macromolecules. J Am Chem Soc. 1988;110:7870–7872. [Google Scholar]
- Hwang TL, Shaka AJ. Water suppression that works. Excitation sculpting using arbitrary wawe-forms and pulsed-field gradients. Journal of Magnetic Resonance, Series A. 1995;112:275–9. [Google Scholar]
- Jeener J, Meier BH, Bachmann P, Ernst RR. Investigation of exchange processes by two-dimensional NMR spectroscopy. Journal of Chemical Physics. 1979;71:5246–53. [Google Scholar]
- Johannessen M, Myhre MR, Dragset M, Tummler C, Moens U. Phosphorylation of human polyomavirus BK agnoprotein at Ser-11 is mediated by PKC and has an important regulative function. Virology. 2008;379:97–109. doi: 10.1016/j.virol.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt-DeMasters BK, Tyler KL. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353:369–74. doi: 10.1056/NEJMoa051782. [DOI] [PubMed] [Google Scholar]
- Kumar A, Ernst RR, Wuthrich K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem Biophys Res Commun. 1980;95:1–6. doi: 10.1016/0006-291x(80)90695-6. [DOI] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. Journal of molecular biology. 1982;157:105–32. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353:375–81. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–86. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu Rev Med. 2010;61:35–47. doi: 10.1146/annurev.med.080708.082655. [DOI] [PubMed] [Google Scholar]
- Marion D, Wuthrich K. Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem Biophys Res Commun. 1983;113:967–74. doi: 10.1016/0006-291x(83)91093-8. [DOI] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000;16:404–5. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- Morellet N, Bouaziz S, Petitjean P, Roques BP. NMR structure of the HIV-1 regulatory protein VPR. J Mol Biol. 2003;327:215–27. doi: 10.1016/s0022-2836(03)00060-3. [DOI] [PubMed] [Google Scholar]
- Nilges M. Calculation of protein structures with ambiguous distance restraints. Automated assignment of ambiguous NOE crosspeaks and disulphide connectivities. J Mol Biol. 1995;245:645–60. doi: 10.1006/jmbi.1994.0053. [DOI] [PubMed] [Google Scholar]
- Okada Y, Suzuki T, Sunden Y, Orba Y, Kose S, Imamoto N, Takahashi H, Tanaka S, Hall WW, Nagashima K, Sawa H. Dissociation of heterochromatin protein 1 from lamin B receptor induced by human polyomavirus agnoprotein: role in nuclear egress of viral particles. EMBO Rep. 2005;6:452–7. doi: 10.1038/sj.embor.7400406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou HD, Kwiatkowski W, Deerinck TJ, Noske A, Blain KY, Land HS, Soria C, Powers CJ, May AP, Shu X, Tsien RY, Fitzpatrick JA, Long JA, Ellisman MH, Choe S, O’Shea CC. A Structural Basis for the Assembly and Functions of a Viral Polymer that Inactivates Multiple Tumor Suppressors. Cell. 2012;151:304–19. doi: 10.1016/j.cell.2012.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Piotto M, Saudek V, Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J Biomol NMR. 1992;2:661–5. doi: 10.1007/BF02192855. [DOI] [PubMed] [Google Scholar]
- Power C, Gladden JG, Halliday W, Del Bigio MR, Nath A, Ni W, Major EO, Blanchard J, Mowat M. AIDS- and non-AIDS-related PML association with distinct p53 polymorphism. Neurology. 2000;54:743–6. doi: 10.1212/wnl.54.3.743. [DOI] [PubMed] [Google Scholar]
- Rance M, Sorensen OW, Bodenhausen G, Wagner G, Ernst RR, Wuthrich K. Improved spectral resolution in cosy 1H NMR spectra of proteins via double quantum filtering. Biochem Biophys Res Commun. 1983;117:479–85. doi: 10.1016/0006-291x(83)91225-1. [DOI] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–38. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Barrucco R, Darbinyan A, Okada Y, Nagashima K, Khalili K. Interaction of JC virus agno protein with T antigen modulates transcription and replication of the viral genome in glial cells. J Virol. 2001;75:1476–86. doi: 10.1128/JVI.75.3.1476-1486.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safak M, Sadowska B, Barrucco R, Khalili K. Functional interaction between JC virus late regulatory agnoprotein and cellular Y-box binding transcription factor, YB-1. J Virol. 2002;76:3828–38. doi: 10.1128/JVI.76.8.3828-3838.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Abou-Gharbia M, Childers W, Sariyer IK, White MK, Safak M. Essential roles of Leu/Ile/Phe-rich domain of JC virus agnoprotein in dimer/oligomer formation, protein stability and splicing of viral transcripts. Virology. 2013;443:161–176. doi: 10.1016/j.virol.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Arachea BT, White MK, Viola RE, Safak M. Human polyomavirus JC small regulatory agnoprotein forms highly stable dimers and oligomers: implications for their roles in agnoprotein function. Virology. 2011;420:51–65. doi: 10.1016/j.virol.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Coric P, Hamazaspyan A, Davis W, Axman R, White MK, Abou-Gharbia M, Childers W, Condra JH, Bouaziz S, Safak M. Emerging From the Unknown: Structural and Functional Features of Agnoprotein of Polyomaviruses. Journal of cellular physiology. 2016;231:2115–27. doi: 10.1002/jcp.25329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, Mun S, Johnson J, El-Hajmoussa M, White MK, Safak M. Human polyoma JC virus minor capsid proteins, VP2 and VP3, enhance large T antigen binding to the origin of viral DNA replication: evidence for their involvement in regulation of the viral DNA replication. Virology. 2014;449:1–16. doi: 10.1016/j.virol.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saribas AS, White MK, Safak M. JC virus agnoprotein enhances large T antigen binding to the origin of viral DNA replication: Evidence for its involvement in viral DNA replication. Virology. 2012;433:12–26. doi: 10.1016/j.virol.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Akan I, Palermo V, Gordon J, Khalili K, Safak M. Phosphorylation mutants of JC virus agnoprotein are unable to sustain the viral infection cycle. J Virol. 2006;80:3893–903. doi: 10.1128/JVI.80.8.3893-3903.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariyer IK, Saribas AS, White MK, Safak M. Infection by agnoprotein-negative mutants of polyomavirus JC and SV40 results in the release of virions that are mostly deficient in DNA content. Virol J. 2011;8:255. doi: 10.1186/1743-422X-8-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider C, Jayasinghe S, Hristova K, White SH. MPEx: a tool for exploring membrane proteins. Protein Sci. 2009;18:2624–8. doi: 10.1002/pro.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Okada Y, Semba S, Orba Y, Yamanouchi S, Endo S, Tanaka S, Fujita T, Kuroda S, Nagashima K, Sawa H. Identification of FEZ1 as a protein that interacts with JC virus agnoprotein and microtubules: role of agnoprotein-induced dissociation of FEZ1 from microtubules in viral propagation. J Biol Chem. 2005;280:24948–56. doi: 10.1074/jbc.M411499200. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Orba Y, Makino Y, Okada Y, Sunden Y, Hasegawa H, Hall WW, Sawa H. Viroporin activity of the JC polyomavirus is regulated by interactions with the adaptor protein complex 3. Proc Natl Acad Sci U S A. 2013;110:18668–73. doi: 10.1073/pnas.1311457110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Orba Y, Okada Y, Sunden Y, Kimura T, Tanaka S, Nagashima K, Hall WW, Sawa H. The human polyoma JC virus agnoprotein acts as a viroporin. PLoS Pathog. 2010;6:e1000801. doi: 10.1371/journal.ppat.1000801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterstab G, Gosert R, Leuenberger D, Lorentz P, Rinaldo CH, Hirsch HH. The polyomavirus BK agnoprotein co-localizes with lipid droplets. Virology. 2010;399:322–31. doi: 10.1016/j.virol.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med. 2005;353:362–8. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–96. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Wecker K, Morellet N, Bouaziz S, Roques BP. NMR structure of the HIV-1 regulatory protein Vpr in H2O/trifluoroethanol. Comparison with the Vpr N-terminal (1–51) and C-terminal (52–96) domains. Eur J Biochem. 2002;269:3779–88. doi: 10.1046/j.1432-1033.2002.03067.x. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhou X, Barnes CO, DeLucia M, Cohen AE, Gronenborn AM, Ahn J, Calero G. The DDB1-DCAF1-Vpr-UNG2 crystal structure reveals how HIV-1 Vpr steers human UNG2 toward destruction. Nature structural & molecular biology. 2016;23:933–940. doi: 10.1038/nsmb.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuthrich K. NMR of Proteins and Nucleic Acids. Wiley Interscience; Hoboken, NJ: 1986. [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nature methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]