

Abstract
Head and neck rhabdomyosarcoma occurs frequently in children and adolescents, and has been well studied in that population. In contrast, it is rare in adults and is not as well characterized clinically and pathologically. Seven cases of adult rhabdomyosarcoma occurring in head and neck were retrieved from the archives of Department of Pathology and Division of Oral Pathology at University of Washington. Radiologic findings and clinical history, as well as pathologic findings from hematoxylin and eosin slides and immunohistochemistry for myogenic markers were reviewed. A total of seven cases of rhabdomyosarcoma (two embryonal, three alveolar and two pleomorphic subtype) were reviewed. Patient ages ranged from 18 to 57 years (median 21 years). Classic and unique histologic features for each subtype, including post-treatment morphologic changes, were identified. Clinical follow-up information was available for 4 patients. 3 of 4 patients experienced recurrence, including two with distant metastasis. One patient died of disease progression 41 months after presentation. Head and neck rhabdomyosarcoma in adults can manifest both classic and unique histologic features for each subtype. In addition, recurrence and distant metastasis were observed, suggesting aggressive clinical behavior regardless of subtype.
Keywords: Rhabdomyosarcoma, Head and neck, Alveolar, Embryonal, Pleomorphic
Introduction
Rhabdomyosarcoma (RMS) is a soft tissue sarcoma pathologically characterized by abnormal myogenesis. It is the most common soft tissue sarcoma of children and adolescents, with an incidence of approximately 0.44/100,000 per year [1]. About 30% of pediatric RMS occurs in the head and neck [1]. In contrast, RMS is relatively rare in adults and occurs more frequently in the extremities and very infrequently in the head and neck region [2]. Due to the rarity of head and neck RMS in adults, most narratives of this disease have been published only as case reports [3–7].
Major histological subtypes of RMS include embryonal (ERMS), alveolar (ARMS), pleomorphic (PRMS), and spindle cell/sclerosing rhabdomyosarcoma. ERMS and ARMS are the major subtypes seen in the pediatric population. ERMS tends to occur in younger children, and carries a better prognosis. ARMS occurs more frequently in adolescents, and exhibits more aggressive biological behavior [8]. PRMS occurs in both children and adults, but the outcome is significantly worse in adults, with higher rates of recurrence and metastasis [9, 10]. Spindle cell RMS was traditionally included as a variant of ERMS but it is now provisionally listed as a separate spindle cell/sclerosing RMS subtype in the latest World Health Organization (WHO) classification (4th Edition, 2013). Recently, a rare epithelioid variant of RMS mimicking carcinoma or melanoma that frequently occurs in older patients has also been described [11]. Of the RMS subtypes, ARMS is the only one with known recurrent chromosomal translocations. In about 75% of ARMS cases, chromosomal translocation results in the fusion of two transcription factor-encoding genes: the PAX3 gene (or less frequently the PAX7 gene) and the FOXO1 gene [12, 13]. This fusion results in ARMS tumor cells expressing chimeric PAX3/7-FOXO1 protein. In contrast, ERMS frequently shows loss of heterozygosity at chromosome 11p15.5 [14], and recurrent mutations affecting the receptor tyrosine kinase/RAS/PIK3CA axis are present in >90% of cases [15]. Recurrent MYOD1(L122R) mutation has been identified in a subset of pediatric and adult spindle cell/sclerosing variant of RMS [16–19].
To provide a deeper insight into the clinical and pathologic features of RMS, this study presents seven cases of adult head and neck RMS. Each was treated at the University of Washington Medical Center between 2000 and 2015.
Materials and Methods
Cases were retrieved from the archives of the Department of Pathology and Department of Oral and Maxillofacial Surgery at University of Washington. H&E and immunohistochemistry slides were independently reviewed by 3 pathologists (EC, DO and RR). For immunohistochemical characterization, tissue sections (5 uM thickness) were de-paraffinized, and immunochemical staining was performed following standard antigen retrieval procedure. The following antibody dilutions were used: Desmin (1:800, Dako), MYOD1 (1:80, AbCam), Myogenin (1:25, Cell Marque).
Immunohistochemical staining was scored using the 4-point system [20]:
Strong (3+): dark staining in >50% of cells.
Moderate (2+): focal darkly staining areas in <50% of cells.
Weak (1+): focal moderate staining in <50% of cells.
Negative (0): None of the above.
Results
Clinical Findings
Our adult patients with head and neck RMS included three females and four males ranging in age from 18 to 57 years (median 21 years). Three cases occurred in the maxillary sinus, two in the cheek, one in the alveolar ridge, and one in the palate. Bone destruction was observed radiologically on presentation in four cases (cases 1, 4, 6 and 7; see examples in Fig. 1a, b). Tumor sizes at the time of excision ranged from 3.4 to 6.2 cm. Four patients (cases 1, 3, 6 and 7) underwent adjuvant chemotherapy and radiation post-operatively. Post-operative follow-up information was available for 4 patients. ERMS in case 1 recurred 17 months after completion of surgery, radiation and chemotherapy with regional and distant metastasis to the lung. ARMS in case 3 recurred locally 11 months post-composite resection. The patient of PRMS in case 6 was disease free in a limited 3-month follow-up after surgery and adjuvant radiation and chemotherapy treatments. The patient with PRMS in case 7 died of progressive disease after several regimens of chemotherapy, 41 months after presentation. Clinical features are summarized in Table 1.
Fig. 1.
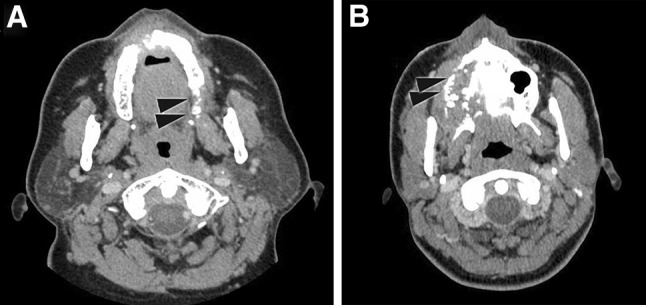
Radiologic features of RMS. CT scans of a ERMS (case 1); b ARMS (case 4), showing destructive invasion of bone by tumor cells as indicated by arrowheads
Table 1.
Clinical features
| Case # | RMS subtype | Age | Sex | Site | Size (cm) | Treatment | FU Duration (mo) | Recurrence | Metastasis | DOD |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Embryonal | 40 | F | Maxillary sinus | 3.4 | S, C, R | 17 | Y | Y | – |
| 2 | Embryonal | 18 | M | Cheek | – | S | – | – | – | – |
| 3 | Alveolar | 19 | F | Cheek | – | S, C, R | 11 | Y | – | – |
| 4 | Alveolar | 20 | F | Maxillary sinus | –– | S | – | – | – | – |
| 5 | Alveolar | 21 | M | Alveolar ridge | – | S, C | – | – | – | – |
| 6 | Pleomorphic | 55 | M | Maxillary sinus | 4.2 | S, C, R | 3 | N | N | – |
| 7 | Pleomorphic | 57 | M | Palate | 6.2 | S, C, R | 41 | Y | Y | Y |
Pathologic Findings
There were two cases of embryonal, three cases of alveolar and two cases of pleomorphic rhabdomyosarcoma with spindle cell features. Cases 1 and 2 showed characteristic features of ERMS including primitive mesenchymal cells recapitulating various stages of myogenesis with variable presence of rhabdomyoblasts (example in Fig. 2a). As cytodifferentiation progresses, the rhabdomyoblasts acquire increased amount of eosinophilic cytoplasm. Cytoplasmic striations and multinucleation can be seen occasionally in rhabdomyoblasts, but these features are not evident in the rhabdomyoblasts seen in the ERMS cases of this series. Cases 3, 4 and 5 showed conventional features of ARMS, characterized by fibrovascular septa separating primitive small round cells into discrete nests (example in Fig. 2b). Wreath-like multi-nucleated cells with rhabdomyoblastic differentiation, which may be seen in ARMS, are not present in these cases. Cases 6 and 7 showed features of PRMS characterized by large, atypical and occasionally multi-nucleated cells with prominent eosinophilic cytoplasm (example in Fig. 2c).
Fig. 2.

Histologic features of head and neck RMS. a Rhabdomyoblasts (indicated by arrowheads) in ERMS (Case 2); b ARMS with alveolar arrangement of tumor cells (Case 4); c PRMS with pleomorphic cell morphology including multi-nucleated cells (Case 6); d ERMS (Case 1) with sub-epithelial condensation of tumor cells, resembling a cambium layer; e Nests and clusters of ERMS cells with abundant clear cytoplasm (Case 2); f Prominent cytodifferentiation of ARMS cells into myoblast-like cells post-radiation (3); g Nests of ARMs with prominent palisading of tumor cells in the periphery (Case 5); h PRMS with areas of spindle cell morphology (Case 7); i PRMS with pleomorphic tumor cells with prominent lymphocytic infiltrate (Case 6)
In addition to the conventional morphology as described for each RMS subtype, more unusual morphologic features and growth patterns were also present. While most of the ERMS in case 1 showed conventional features of ERMS, there were also focal areas of subepithelial condensation of tumor cells beneath the overlying squamous epithelium, reminiscent of the cambium layer seen in the botryoid variant of ERMS (Fig. 2d). The ERMS in case 2 showed an abundance of cells with clear cytoplasm, admixed with rhabdomyoblasts (Fig. 2e). ARMS in cases 3 and case 5 showed the presence of residual differentiated rhabdomyoblasts with eosinophilic cytoplasm, some binucleated, following adjuvant radiation (Fig. 2f). In addition to the characteristic nested arrangement of tumor cells (Fig. 2b), ARMS of case 4 also showed prominent palisading of tumor cells in the periphery of each tumor cell nest (Fig. 2g). In both cases 6 and 7 of PRMS, areas of spindle cell morphology with fascicular growth pattern were present in addition to the classic pleomorphic epithelioid morphology (Fig. 2h), suggesting that these two cases may represent a mixed subtype of pleomorphic and spindle cell RMS. Case 6 also demonstrates brisk lymphocytic infiltrate (Fig. 2i), in contrast to the minimal inflammatory infiltrate seen in other RMS described in this series.
Immunohistochemical Findings
By immunohistochemistry (IHC), all 7 cases demonstrated positive expression of myogenic markers, desmin, MyoD1 or Myogenin. In particular, the ARMS in cases 3–5 showed a diffuse positive staining (3+) of Myogenin or MyoD1, in contrast to the focal Myogenin staining (2+) seen in the cases of ERMS (cases 1 and 2) and PRMS (cases 6–7) (examples of Myogenin IHC in Fig. 3). This is in keeping with the previous findings that strong and diffuse expression of Myogenin is significantly associated with ARMS [21]. PRMS of case 7 with spindle cell areas showed negative Myogenin expression, strong expression of MyoD1 (3+), and focal expression of desmin (2+). The spindle cell/sclerosing variant of RMS has been described to show limited expression of Myogenin, strong MyoD1 expression and focal dot-like desmin expression [22]. The immunoprofile of case 7 further suggests that this tumor may represent a mixed subtype of pleomorphic and spindle cell RMS. Table 2 summarizes quantitative IHC scoring results.
Fig. 3.

Immunohistochemistry for Myogenin. a ERMS in case 1; b ARMS in case 4
Table 2.
Immunohistochemical study of myogenic markers, MyoD, Myogenin and Desmin
| Case # | MyoD | Myogenin | Desmin |
|---|---|---|---|
| 1 | 3+ | 2+ | 3+ |
| 2 | – | 3+ | 3+ |
| 3 | 3+ | – | 3+ |
| 4 | – | 3+ | 3+ |
| 5 | – | 3+ | 3+ |
| 6 | – | 2+ | 3+ |
| 7 | 3+ | 0 | 2+ |
IHC was scored as the following categories: Strong (3+), dark staining in >50% of cells; moderate (2+), focal darkly staining areas in <50% of cells; weak (1+), focal moderate staining in <50% of cells; negative (0), none of the above. Staining not performed is indicated as “–”
Discussion
This study described clinical and pathologic characteristics of seven cases of adult head and neck RMS, including alveolar, embryonal and pleomorphic subtypes with spindle cell features. In children, RMS occurring in the head and neck tends to have favorable outcome except for tumors arising in parameningeal sites such as the nasal cavity, paranasal sinuses, infratemporal fossa and the mastoid. Poorer outcomes for tumors in these sites are due in part to difficulty in achieving complete resection from these areas. In contrast, the cases of head and neck RMS in adults in our study manifested more aggressive biological behavior compared to pathologically similar RMS in pediatric patients. Of our 4 currently described adult cases with available post-operative clinical follow-up, case 1 (ERMS) in the maxillary sinus, case 3 (ARMS) in the cheek and case 7 (PRMS) in the palate showed local recurrence within 3 years. The case 1 ERMS and case 7 PRMS patients also experienced distant metastases, and one patient with PRMS (case 7) died of progressive disease within 4 years from the time of diagnosis.
All adult head and neck RMS cases described in this series showed conventional histologic features for each subtype. Some of the cases demonstrated more unusual morphologic features. In particular, ERMS cells in case 2 showed foci of tumor cells with abundant clear cytoplasm admixed with typical rhabdomyoblasts, a morphologic feature not previously described in ERMS. In cases 3 and case 5 of ARMS, we noted maturation of myogenic tumor cells post adjuvant chemotherapy and radiation. Cytodifferentiation of RMS cells post-treatment has been previously described in a subset of cases, but more frequently in the ERMS subtype and appears to correlate with decreased proliferative activity of tumor cells [23]. Finally, the two PRMS cases (case 6 and case 7) demonstrate focal areas of spindle cells admixed with tumor cells showing the more characteristic pleomorphic morphology. The findings raise the possibility that these cases may represent a mixed subtype of pleomorphic and spindle cell RMS. In all, the additional morphologic features seen in adult head and neck RMS likely reflect tumor heterogeneity in cell differentiation and pathogenesis distinct from their pediatric counterpart.
Other malignant tumors with rhabdomyosarcomatous differentiation need to be considered on the differential diagnosis. For example, malignant peripheral nerve sheath tumor (MPNST) with heterologous rhabdomyosarcomatous differentiation (“malignant Triton tumor”) should be considered, particularly in patients with history of type 1 neurofibromatosis (NF1) and in cases where there is gross or radiologic finding showing a close association of the tumor with a peripheral nerve. Histologically, the association with a peripheral nerve or a benign nerve sheath tumor, e.g. neurofibroma, may be seen in a subset of MPNSTs. Immunohistochemistry for S100 and SOX10 can sometimes be helpful diagnostically. However, the staining pattern for these markers is focal and positive expression is seen in approximately 50–60% of MPNST [24, 25].
Sarcomatoid carcinoma of cutaneous, mucosal or salivary gland origin needs to be considered on the differential diagnosis when dealing with a limited tissue biopsy and a mucosal biopsy from an older patient, particularly in the setting of prior carcinoma or radiation. Thorough sampling of the resection specimen to cover the surface component and heterogeneous areas of the tumor is crucial. Immunohistochemistry for multiple cytokeratins and p40/p63 can be utilized to further support the diagnosis of sarcomatoid carcinoma.
RMS, in particular the ERMS and ARMS subtypes, can take on the appearance of small, primitive and round cells, therefore differential diagnosis in this context needs to include other small round cell tumors. Examples are Ewing’s sarcoma, small cell melanoma, and lymphoblastic lymphoma. Immunohistochemistry for markers such as CD99 (diffuse membranous pattern in Ewing’s sarcoma [26]), S100, Melan A and HMB-45 (melanoma) and TdT, CD43 and CD79a (lymphoblastic lymphoma [27, 28]) will be helpful in the diagnostic work-up. Ancillary tests such as Fluorescent In Situ Hybridization (FISH) or Reverse Transcription Polymerase Chain Reaction (RT-PCR) assays to show the presence of EWS gene rearrangement in Ewing’s sarcoma and flow cytometry assay to show clonal lymphoid population will also be useful.
In conclusion, we have described a series of 7 adult head and neck RMS. The major subtypes share morphologic features with their pediatric counterpart, but also show more unusual features. The biological behavior appears to be aggressive regardless of histologic subtype.
Compliance with Ethical Standards
Conflict of interest
No conflict of interest to disclose.
Contributor Information
Eleanor Chen, Phone: (206) 616-9118, Email: eleanor2@uw.edu.
Dolphine Oda, Phone: (206) 616-4748, Email: doda@uw.edu.
References
- 1.Perez EA, Kassira N, Cheung MC, Koniaris LG, Neville HL, Sola JE. Rhabdomyosarcoma in children: a SEER population based study. J Surg Res. 2011;170(2):e243–e51. doi: 10.1016/j.jss.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. 2009;27(20):3391–3397. doi: 10.1200/JCO.2008.19.7483. [DOI] [PubMed] [Google Scholar]
- 3.Arul AS, Verma S, Arul AS, Verma R. Oral rhabdomyosarcoma-embryonal subtype in an adult: a rarity. J Nat Sci Biol Med. 2014;5(1):222–225. doi: 10.4103/0976-9668.127347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arya K, Vij H, Vij R, Rao NN. Rhabdomyosarcoma of mandible: a diagnostic predicament. J Oral Maxillofac Pathol. 2011;15(3):320–325. doi: 10.4103/0973-029X.86707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Astekar M, Metgud R, Sharma P, Ramesh G. Oral alveolar rhabdomyosarcoma: a case report with immunohistochemical analysis. Clin Pract. 2012;2(1):e17. doi: 10.4081/cp.2012.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin XY, Wang Y, Yu JH, Liu Y, Wang L, Li QC, et al. Sclerosing rhabdomyosarcoma presenting in the masseter muscle: a case report. Diagn Pathol. 2013;8:18. doi: 10.1186/1746-1596-8-S1-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou DN, Yang QQ, Li ZL, Pan ZY, Deng YF. Head and neck rhabdomyosarcoma: follow-up results of four cases and review of the literature. Int J Clin Exp Pathol. 2015;8(5):4277–4283. [PMC free article] [PubMed] [Google Scholar]
- 8.Malempati S, Hawkins DS. Rhabdomyosarcoma: review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr Blood Cancer. 2012;59(1):5–10. doi: 10.1002/pbc.24118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furlong MA, Fanburg-Smith JC. Pleomorphic rhabdomyosarcoma in children: four cases in the pediatric age group. Ann Diagn Pathol. 2001;5(4):199–206. doi: 10.1053/adpa.2001.26970. [DOI] [PubMed] [Google Scholar]
- 10.Furlong MA, Mentzel T, Fanburg-Smith JC. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod Pathol. 2001;14(6):595–603. doi: 10.1038/modpathol.3880357. [DOI] [PubMed] [Google Scholar]
- 11.Jo VY, Marino-Enriquez A, Fletcher CD. Epithelioid rhabdomyosarcoma: clinicopathologic analysis of 16 cases of a morphologically distinct variant of rhabdomyosarcoma. Am J Surg Pathol. 2011;35(10):1523–1530. doi: 10.1097/PAS.0b013e31822e0907. [DOI] [PubMed] [Google Scholar]
- 12.Barr FG, Galili N, Holick J, Biegel JA, Rovera G, Emanuel BS. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;3(2):113–117. doi: 10.1038/ng0293-113. [DOI] [PubMed] [Google Scholar]
- 13.Davis RJ, D’Cruz CM, Lovell MA, Biegel JA, Barr FG. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994;54(11):2869–2872. [PubMed] [Google Scholar]
- 14.Scrable HJ, Witte DP, Lampkin BC, Cavenee WK. Chromosomal localization of the human rhabdomyosarcoma locus by mitotic recombination mapping. Nature. 1987;329(6140):645–647. doi: 10.1038/329645a0. [DOI] [PubMed] [Google Scholar]
- 15.Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4(2):216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agaram NP, Chen CL, Zhang L, LaQuaglia MP, Wexler L, Antonescu CR. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes Chromosomes Cancer. 2014;53(9):779–787. doi: 10.1002/gcc.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kohsaka S, Shukla N, Ameur N, Ito T, Ng CK, Wang L, et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nat Genet. 2014;46(6):595–600. doi: 10.1038/ng.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rekhi B, Upadhyay P, Ramteke MP, Dutt A. MYOD1 (L122R) mutations are associated with spindle cell and sclerosing rhabdomyosarcomas with aggressive clinical outcomes. Mod Pathol. 2016 doi: 10.1038/modpathol.2016.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szuhai K, de Jong D, Leung WY, Fletcher CD, Hogendoorn PC. Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. J Pathol. 2014;232(3):300–307. doi: 10.1002/path.4307. [DOI] [PubMed] [Google Scholar]
- 20.Adams EJ, Green JA, Clark AH, Youngson JH. Comparison of different scoring systems for immunohistochemical staining. J Clin Pathol. 1999;52(1):75–77. doi: 10.1136/jcp.52.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dias P, Chen B, Dilday B, Palmer H, Hosoi H, Singh S, et al. Strong immunostaining for myogenin in rhabdomyosarcoma is significantly associated with tumors of the alveolar subclass. Am J Pathol. 2000;156(2):399–408. doi: 10.1016/S0002-9440(10)64743-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Folpe AL, McKenney JK, Bridge JA, Weiss SW. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. Am J Surg Pathol. 2002;26(9):1175–1183. doi: 10.1097/00000478-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Coffin CM, Rulon J, Smith L, Bruggers C, White FV. Pathologic features of rhabdomyosarcoma before and after treatment: a clinicopathologic and immunohistochemical analysis. Mod Pathol. 1997;10(12):1175–1187. [PubMed] [Google Scholar]
- 24.Kang Y, Pekmezci M, Folpe AL, Ersen A, Horvai AE. Diagnostic utility of SOX10 to distinguish malignant peripheral nerve sheath tumor from synovial sarcoma, including intraneural synovial sarcoma. Mod Pathol. 2014;27(1):55–61. doi: 10.1038/modpathol.2013.115. [DOI] [PubMed] [Google Scholar]
- 25.Wick MR, Swanson PE, Scheithauer BW, Manivel JC. Malignant peripheral nerve sheath tumor. An immunohistochemical study of 62 cases. Am J Clin Pathol. 1987;87(4):425–433. doi: 10.1093/ajcp/87.4.425. [DOI] [PubMed] [Google Scholar]
- 26.Scotlandi K, Serra M, Manara MC, Benini S, Sarti M, Maurici D, et al. Immunostaining of the p30/32MIC2 antigen and molecular detection of EWS rearrangements for the diagnosis of Ewing’s sarcoma and peripheral neuroectodermal tumor. Hum Pathol. 1996;27(4):408–416. doi: 10.1016/S0046-8177(96)90115-X. [DOI] [PubMed] [Google Scholar]
- 27.Lucas DR, Bentley G, Dan ME, Tabaczka P, Poulik JM, Mott MP. Ewing sarcoma vs lymphoblastic lymphoma. A comparative immunohistochemical study. Am J Clin Pathol. 2001;115(1):11–17. doi: 10.1309/K1XJ-6CXR-BQQU-V255. [DOI] [PubMed] [Google Scholar]
- 28.Ozdemirli M, Fanburg-Smith JC, Hartmann DP, Azumi N, Miettinen M. Differentiating lymphoblastic lymphoma and Ewing’s sarcoma: lymphocyte markers and gene rearrangement. Mod Pathol. 2001;14(11):1175–1182. doi: 10.1038/modpathol.3880455. [DOI] [PubMed] [Google Scholar]