

Abstract
Three new guaiazulene alkaloids muriceidines A–C (1–3) and one new bis-sesquiterpene muriceidone A (4), were isolated from the South China Sea gorgonian Muriceides collaris. Muriceidines are the first examples structurally architected by guaiazulene coupling with an inner-salt Δ 1-pipecolic acid via a unique sp2 methine-bridged linkage, and the bis-sesquiterpene was comprised by a guaiazulene and a indene units linked through a unprecedented carbon-carbon σ-bond between the high steric bridgehead carbon C-10 of guaiazulene moiety and C-2′ of indene moiety. The chiral compounds 2–4 were obtained initially as racemates and further separated by chiral HPLC methods. The inner-salt structures of 1–3 and absolute configurations of 2–4 were fully elucidated by calculated 13C NMR, ECD and OR with quantum chemical calculation methods. Compound 1 showed cytotoxicity against K562 cell lines with IC50 value of 8.4 μM and antifouling activity against the larvae of the barnacle Balanus albicostatus with EC50 value of 11.9 μg/mL and potent therapeutic index (LC50/EC50 = 3.66). Also the racemic (±)-3 showed cytotoxicities against both HL-60 and K562 cell lines with IC50 values of 2.2 and 3.7 μM, respectively. A semisynthetic trial was performed to validate the proposed biosynthetic hypotheses.
Introduction
Azulene is one of the most important non-benzenoid aromatic compounds1, 2. Especially the azulenes containing nitrogen, mainly including aza-azulenes, N-heterocyclic fused azulene, and other azulene derivatives coupled with nitrogen units, possess special physico-chemical properties and significant pharmacological and therapeutic actions3–5. Although many terrestrial plants and marine corals are the rich resources of natural azulene derivatives with the biogenic fixed guaiazulene (GA) skeleton6–10, their structural variety almost limits to different exocyclic oxygenate patterns or only a few dimmers having the space comfort linkage of C-3–C-3′ or C-2–C-3′. In addition, almost all the azulenes containing nitrogen were obtained from chemical synthesis. However, gorgonian showed some specificity in GA derivation. In 1984, the first GA alkaloid, N,N-dimethylamino-3-guaiazulenyl methane, was isolated from an unidentified blue gorgonian11. A recent study of Anthogorgia species showed an interesting coexistence of GA and indene derivatives10.
Recently we encountered a rare gorgonian Muriceides collaris shaping in fan with unique azure branches, which is distributed only in few regions of South China Sea. To date, apart from our previous preliminary work on M. collaris, resulting in the isolation of cholesterol, batylalcohol, uracil, thymine, (2′-deoxyuridine, 2′-deoxythymidine, and thymidine12. There were few reports on the chemical profiles of genus Muriceides (family Plexauridae) containing more than 20 species. In present study, bioassay-guided isolation of M. collaris yielded three new guaiazulene-type alkaloids and one new bis-sesquiterpene (Fig. 1), named as muriceidines A–C (1–3) and muriceidone A (4), together with the known GA (5) and 3-formyl guaiazulene (6). The muriceidines showed a novel structure architected by guaiazulene coupling with an inner-salt Δ 1-pipecolic acid via a unique sp2 methine-bridged linkage, and the unprecedented bis-sesquiterpene muriceidone A was characterized by a high steric linkage between azulene and indene units through a carbon-carbon σ-bond. All the chiral members 2–4 were initially obtained as racemates and were successfully separated by chiral HPLC methods. Apart from using the extensive spectroscopic analyses including IR, MS and NMR, especially the inner-salt structures in 1–3 and absolute configurations of 2–4 were fully elucidated by calculated 13C NMR, ECD and OR with quantum chemical calculation methods using time-dependent density functional theory (TDDFT). Additionally, cytotoxic and antifouling activities were assayed for all the new compounds 1–6.
Figure 1.
Structures of 1–6 from Gorgonian Muriceides collaris.
Results and Discussion
Structure Elucidation
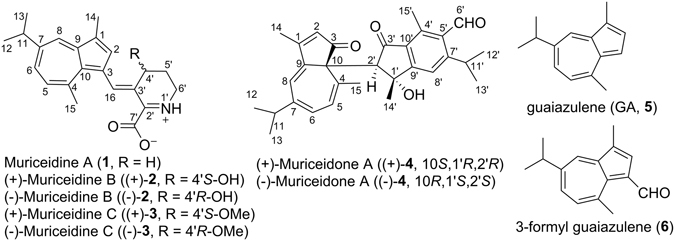
Muriceidine A (1) was obtained as dark red amorphous power. Its molecular formula was determined as C22H25NO2 by HRESIMS (m/z 336.1959 [M + H]+, calcd 336.1958; 358.1776 [M + Na]+, calcd 358.1778), indicating eleven degrees of unsaturation. The IR absorption bands indicated the presence of carbonyl (1701 cm−1) and imino (1649 cm−1) groups. In the 1H NMR spectrum of 1 (Table 1), the characteristic proton signals represented by an aromatic ABX coupling system at δ H 8.17 (1H, d, J = 2.2 Hz, H-8), 7.51 (1H, dd, J = 11.0, 2.2 Hz, H-6), and 7.33 (1H, d, J = 11.0 Hz, H-5), one aromatic singlet proton signal at δ H 7.81 (1H, s, H-2), two olefinic methyl proton signals at δ H 2.58 (3H, s, H3-14) and 3.17 (3H, s, H3-15), as well as one isopropyl proton signals at δ H 3.11 (1H, septet, J = 6.6, 6.6 Hz, H-11) and 1.37 (6H, d, J = 6.6 Hz, H3-12/13), strongly suggested the presence of a C-3 substituted GA moiety in 1 by comparing its NMR data with those of GA derivatives13. The key HMBC correlations from H-12 and H-13 to C-7 (δ C 146.8) and C-11 (δ C 38.2), from H-6 and H-8 to C-11, from H-14 to C-1 (δ C 128.2), C-2 (δ C 139.0) and C-9 (δ C 142.8), and from H-15 to C-4 (δ C 149.3), C-5 (δ C 133.4) and C-10 (δ C 141.0) further confirmed this speculation (Fig. 2A).
Table 1.
NMR Data for muriceidines A–C (1–3) (δ in ppm).
| no. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δ C a | δ H b (mult J in Hz) | δ C c | δ H d (mult J in Hz) | δ C a | δ H b (mult J in Hz) | |
| 1 | 128.2, C | 131.5, C | 128.7, C | |||
| 2 | 139.0, CH | 7.81 (s) | 141.7, CH | 8.51 (s) | 140.0, CH | 7.88 (s) |
| 3 | 123.5, C | 124.2, C | 123.0, C | |||
| 4 | 149.3, C | 149.9, C | 149.3, C | |||
| 5 | 133.4, CH | 7.33 (d, 11.0) | 136.3, CH | 7.58 (d, 10.4) | 133.8, CH | 7.36 (d, 10.4) |
| 6 | 137.1, CH | 7.51 (dd, 11.0, 2.2) | 138.8, CH | 7.75 (d, 10.4) | 137.2, CH | 7.54 (d, 10.4) |
| 7 | 146.8, C | 150.9, C | 147.3, C | |||
| 8 | 134.8, CH | 8.17 (d, 2.2) | 136.2, CH | 8.35 (br s) | 134.8, CH | 8.18 (s) |
| 9 | 142.8, C | 146.5, C | 143.8, C | |||
| 10 | 141.0, C | 143.9, C | 141.5, C | |||
| 11 | 38.2, CH | 3.11 (dq, 6.6, 6.6) | 39.3, CH | 3.21 (dq, 6.6, 6.6) | 38.2, CH | 3.12 (dq, 6.6, 6.6) |
| 12 | 24.5, CH3 | 1.37 (d, 6.6) | 24.6, CH3 | 1.40 (d, 6.6) | 24.6, CH3 | 1.37 (d, 6.6) |
| 13 | 24.5, CH3 | 1.37 (d, 6.6) | 24.6, CH3 | 1.40 (d, 6.6) | 24.6, CH3 | 1.37 (d, 6.6) |
| 14 | 13.3, CH3 | 2.58 (s) | 13.2, CH3 | 2.62 (s) | 13.4, CH3 | 2.59 (s) |
| 15 | 29.7, CH3 | 3.17 (s) | 29.8, CH3 | 3.13 (s) | 29.8, CH3 | 3.18 (s) |
| 16 | 149.4, CH | 9.56 (s) | 151.4, CH | 9.16 (s) | 152.8, CH | 9.90 (s) |
| 2′ | 172.3, C | 173.1, C | 170.5, C | |||
| 3′ | 118.0, C | 117.9, C | 117.5, C | |||
| 4′ | 24.7, CH2 | 2.95 (t, 6.0) | 61.6, CH | 5.05 (s) | 69.7, CH | 4.69 (s) |
| 5′ | 20.4, CH2 | 2.03 (m) | 29.8, CH2 | 2.16 (br d, 13.8), 1.95 (t, 13.8) | 23.5, CH2 | 2.43 (br d, 14.3), 1.82 (m) |
| 6′ | 42.9, CH2 | 3.77 (t, 5.5) | 38.5, CH2 | 3.81 (m), 3.63 (dd, 15.0, 4.8) | 38.2, CH2 | 3.82 (br d, 7.7) |
| 7′ | 163.9, C | 167.2, C | 162.9, C | |||
| OMe | 54.3, CH3 | 3.45 (s) | ||||
aRecorded at 150 MHz in CDCl3.
bRecorded at 600 MHz in CDCl3.
cRecorded at 150 MHz in CD3OD.
dRecorded at 600 MHz in CD3OD.
Figure 2.
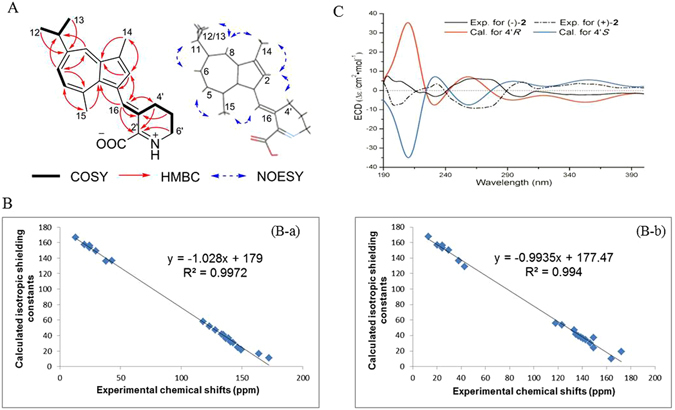
Structure elucidation of muriceidines A-C (1–3). (A) Key COSY, HMBC and NOESY correlations of 1. (B) Correlation of experimental chemical shifts and calculated isotropic shielding constants of compound 1 with the respective inner-salt (B-a) and non-ionized (B-b) structure. (C) Calculated and experimental ECD curves of compounds (+)-2 and (−)-2.
Another key fragment of -CH2-CH2-CH2- in 1 was readily recognized from COSY correlations between H2-4′/H2-5′ and H2-5′/H2-6′. Thus, the HMBC correlations from H2-6′ [δ H 3.77 (2H, t, J = 5.5 Hz)] to C-2′ (δ C 172.3), and from H2-4′ [δ H 2.95 (2H, t, J = 6.0 Hz)] to C-2′, combined with the HRESIMS data could establish the Δ 1-pipecolic acid moiety in 1 (Fig. 2A). This elucidation was further confirmed by comparing the NMR data with those of furpipate derivatives formed from furfural or 5-hydroxy-methylfurfural in the presence of Lysine and those of anthosamines14, 15. The sequential connection between GA moiety and Δ 1-pipecolic acid was achieved by HMBC correlations from the methine-bridged proton of H-16 [δ H 9.56 (1H, s, H-16)] to C-2′, C-3′ (δ C 118.0), C-4′ (δ C 24.7), C-2, C-3 (δ C 123.5) and C-10, and from H2-4′ to C-16 (δ C 149.4) to form the planar structure of 1 (Figs 2A and SS10 in Supporting Information).
The geometry of the double bond ∆3′ 16 in 1 was assigned as E evident from NOESY correlations of H-2/H2-4′ and H-16/H3-15 but lack of correlation of H-16/H2-4′ (Figs 2A and SS11 in Supporting Information), which was supported by a molecular model analysis (the spatial distance of H-2/H2-4′ is <2.5 Å in E geometry but >4.7 Å in Z geometry) on the basis of conformational search in the Spartan 10 package showing that 1 fits in a seemingly planar structure with the C-2–C-3–C-16–C-3′ dihedral angel of 22° (Fig. S2 and Table S2 in Supporting Information). However, there was still a pending issue whether compound 1 could be inner salt or non-ionized structure. Therefore, the quantum-mechanical GIAO calculations for the 13C NMR chemical shifts of 1 were performed using the DFT theory method at the RB3LYP/6-311+g(2d,p) level16. The calculated isotropic shielding constants of inner salt structure of 1 were in good correlation with the experimental 13C NMR chemical shifts (Table 2). After linear regression which gives a reasonable R 2 of 0.9972 and slope of −1.028 (Fig. 2B)16, the mean absolute error with respect to the experimental data was 2.16 ppm for 1. Whereas, the calculation data of non-ionized structure of 1 gave an unacceptable linear relation along with a low R 2 value of 0.9941 and a high mean absolute error of 3.09 ppm (Fig. 2B)16. It was noticeable that the calculated chemical shifts for C-2′ and C-7′ in the two cases showed distinct difference. The resonance of C-2′ in inner salt structure is more deshielded relative to C-7′, whereas it will be opposite in non-ionized structure of 1 (Tables 2 and S5 in Supporting Information). Thus, the structure of 1 was fully elucidated.
Table 2.
The calculated 13C NMR chemical shifts of compound 1.
| no. | δ exp | δ calc | δ scalc | δ mcalc | δ mscalc | no. | δ exp | δ calc | δ scalc | δ mcalc | δ mscalc |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 128.2 | 135.0 | 128.5 | 132.0 | 127.3 | 12 | 24.5 | 25.9 | 22.4 | 25.9 | 22.0 |
| 2 | 139.0 | 145.1 | 138.3 | 144.5 | 139.7 | 13 | 24.5 | 25.8 | 22.3 | 25.9 | 21.9 |
| 3 | 123.5 | 130.1 | 123.7 | 128.8 | 124.2 | 14 | 13.3 | 15.2 | 11.9 | 14.8 | 10.9 |
| 4 | 149.3 | 160.7 | 153.4 | 158.5 | 153.7 | 15 | 29.7 | 32.7 | 29.0 | 32.1 | 28.0 |
| 5 | 133.4 | 140.4 | 133.7 | 135.3 | 130.6 | 16 | 149.4 | 160.1 | 152.9 | 145.2 | 140.5 |
| 6 | 137.1 | 145.5 | 138.7 | 143.1 | 138.4 | 2′ | 172.3 | 170.9 | 163.4 | 163.1 | 158.2 |
| 7 | 146.8 | 157.2 | 150.0 | 152.1 | 147.3 | 3′ | 118.0 | 123.8 | 117.6 | 126.8 | 122.2 |
| 8 | 134.8 | 140.7 | 134.0 | 140.9 | 136.2 | 4′ | 24.7 | 28.5 | 24.9 | 29.5 | 25.5 |
| 9 | 142.8 | 151.3 | 144.3 | 147.8 | 143.1 | 5′ | 20.4 | 24.4 | 20.9 | 25.5 | 21.6 |
| 10 | 141.0 | 150.3 | 143.3 | 146.2 | 141.4 | 6′ | 42.9 | 45.2 | 41.1 | 53.4 | 49.3 |
| 11 | 38.2 | 46.0 | 41.9 | 46.0 | 41.9 | 7′ | 163.9 | 165.4 | 158.1 | 172.3 | 167.4 |
δ calc: unscaled chemical shifts of inner salt structure relative to TMS at the same level of theory.
δ scalc: calculated chemical shifts of inner salt structure after linear scaling.
δ mcalc: unscaled chemical shifts of non-ionized structure relative to TMS at the same level of theory.
δ mscalc: calculated chemical shifts of non-ionized structure after linear scaling.
(±)-Muriceidine B (2) had molecular formula of C22H25NO3 from its HRESIMS. The UV, IR, and 1D NMR spectra of compound 2 were very similar to those of 1 (Table 1 and Supporting Information), except for an oxygenated methine signal (δ H 5.05, s, H-4′; δ C 61.6, d, C-4′) in 2 instead of one methylene signal in 1, indicating that 2 was a hydroxylated product of 1. The position of the hydroxyl group in 2 was assigned at C-4′ deduced from COSY correlations of H-4′/H2-5′ and H2-5′/H2-6′, as well as HMBC correlations from H-4′ to C-2′ (δ C 173.1), C-3′ (δ C 117.9), C-6′ (δ C 38.5) and C-16 (δ C 151.4). NOESY correlations of H3-15/H-16 and H-2/H-4′ suggested a Z geometry for the double bond ∆3′ 16. The initial speculation of racemic mixture for 2 was caused by the failed observation of optical rotation value and ECD spectrum. Further chiral separation was undertaken on chiral HPLC to yield (+)- and (−)-2 (Fig. S1 in Supporting Information). (+)-2 and (−)-2 gave almost opposite optical rotation values and exhibited mirror-like ECD curves (Fig. 2C). To determine their absolute configurations, the stable conformers of respective (+)- and (−)-2 were studied theoretically by TDDFT/ECD calculations at RB3LYP/DGDZVP level (Supporting Information)17. The experimental ECD spectrum of (+)-2 exhibited three moderate positive Cotton effects (CEs) at 232.7, 302.0 and 347.3 nm and two strong negative CEs at 199.1 and 263.3 nm, which matched well with the calculated ECD data for 4′S configuration (Fig. 2C). On the contrary, calculated ECD of 4′R configuration exhibited mirror-like CEs consistent with the experimental data of (−)-2. Thus, 4′S and 4′R were finally assigned for (+)-2 and (−)-2, respectively.
(±)-Muriceidine C (3) was also obtained as a racemic mixture in initial isolation. Appearance of an extra methoxyl signal (δ H 3.45, s, 3H; δ C 54.3, q) in 3 comparing to 2 indicated that 3 was a methylated derivative of 2, which was supported by HMBC correlations from OMe to C-4′ (δ C 69.7). After chiral separation, the experimental optical rotation values and ECD spectra of (+)-and (−)-3 were consistent with those of respective (+)- and (−)-2 (Fig. 2C), suggesting that they share the same configurations.
(±)-Muriceidone A (4) was obtained as yellow amorphous power. Its HRESIMS (m/z 481.2360 [M+Na]+ (calcd 481.2349)) provided molecular formula as C30H34O4, requiring fourteen degrees of unsaturation. The IR absorption bands indicated the presence of carbonyl (1701, 1697, 1695 cm−1), phenyl (1556, 1457 cm−1), and hydroxyl (3621 cm−1) groups. In accordance with the molecular formula, 30 carbon signals in its 13C NMR spectrum were distinguished as eight methyls, nine methines (five olefinic), thirteen quaternary carbons (one oxygenated, eight olefinic, and three carbonyl carbons) by DEPT and HMQC spectra (Table 3 and Figs SS39 and SS40 in Supporting Information). 1H NMR spectrum of compound 4 showed the presence of two isopropyl groups [(δ H 2.69 (1H, dq, J = 6.6, 7.2 Hz), 3.59 (1H, dq, J = 6.6, 6.6 Hz), 1.30 (3H, d, J = 6.6 Hz), 1.27 (3H, d, J = 6.6 Hz), 1.17 (3H, d, J = 7.2 Hz), 1.13 (3H, d, J = 6.6 Hz)], four olefinic methyl groups [δ H 2.69 (3H, s), 2.31 (3H, s), 1.87 (3H, s), 1.83 (3H, s)], and an aromatic AB system [δ H 6.54 (1H, d, J = 6.6 Hz) and 6.26 (1H, d, J = 6.6 Hz)]. The aforementioned spectral information strongly suggested that compound 4 could be a bis-sesquiterpene with derivative GA or indene unit10. HMBC correlations from H3-12/13 to C-7 and C-11, from H3-14 to C-1, C-2 and C-9, from H3-15 to C-4, C-5 and C-10, from H-2 to C-3 and C-10, from H-8 to C-9, C-10, and C-11, further confirmed the presence of 3-oxo-10-substituted dihydroguaiazulene moiety (Fig. 3A). The presence of 1′-hydroxyl-5′-aldehyde-3′-oxo-indene moiety in 4 was evident from the HMBC correlations from H3-12′/13′ to C-7′ and C-11′, from H3-14 to C-1′, C-2′ and C-9′, from H3-15′ to C-4′, C-5′ and C-10′, from H-8′ to C-1′, C-5′, C-10′ and C-11′, from H-6′ to C-5′, from H-2′ to C-3′, and from the hydroxyl proton (δ H 2.17, br s) to C-2′ and C-9′ (Figs 3A and SS42 in Supporting Information). Finally, the key HMBC correlations from H-2′ to C-3, C-9 and C-10 could establish the planar structure of 4 as shown in Fig. 3A by a carbon-carbon σ-bond between C-10 and C-2′.
Table 3.
1H and 13C NMR data for muriceidone A (4) in CDCl3.
| Position | δ C a | δ H b (J in Hz) | Position | δ C a | δ H b (J in Hz) |
|---|---|---|---|---|---|
| 1 | 167.3, C | 1′ | 76.7, C | ||
| 2 | 132.1, CH | 6.19 (s) | 2′ | 59.5, CH | 3.03 (s) |
| 3 | 206.4, C | 3′ | 202.3, C | ||
| 4 | 133.4, C | 4′ | 140.0, C | ||
| 5 | 124.3, CH | 6.26 (d, 6.6) | 5′ | 134.4, C | |
| 6 | 126.3, CH | 6.54 (d, 6.6) | 6′ | 194.6, CH | 10.61 (s) |
| 7 | 147.5, C | 7′ | 157.4, C | ||
| 8 | 117.1, CH | 6.44 (s) | 8′ | 118.7, CH | 7.49 (s) |
| 9 | 138.3, C | 9′ | 162.4, C | ||
| 10 | 55.7, C | 10′ | 129.3, C | ||
| 11 | 36.0, CH | 2.69 (dq, 6.6, 7.2) | 11′ | 29.6, CH | 3.59 (dq, 6.6, 6.6) |
| 12 | 23.0, CH3 | 1.13 (d, 6.6) | 12′ | 23.9, CH3 | 1.27 (d, 6.6) |
| 13 | 23.5, CH3 | 1.17 (d, 7.2) | 13′ | 24.0, CH3 | 1.30 (d, 6.6) |
| 14 | 15.1, CH3 | 2.31 (s) | 14′ | 28.4, CH3 | 1.87 (s) |
| 15 | 24.1, CH3 | 1.83 (s) | 15′ | 14.3, CH3 | 2.69 (s) |
| OH-3′ | 2.17 (br s) |
aRecorded at 150 MHz. bRecorded at 600 MHz.
Figure 3.
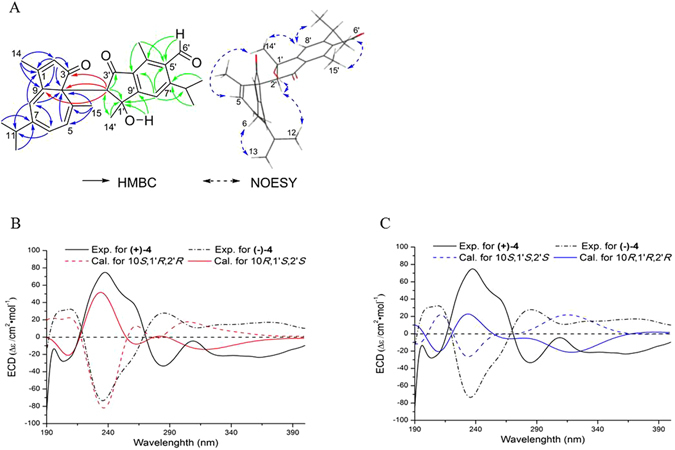
Structure elucidation of muriceidone A (4). (A) Key HMBC and NOESY correlations of 4. (B) Calculated and experimental ECD curves of compounds (+)-4 and (−)-4 in 10S,1′R,2′R/10R,1′S,2′S configuration. (C) Calculated and experimental ECD curves of compounds (+)-4 and (−)-4 in 10S,1′S,2′S/10R,1′R,2′R configuration.
In NOESY spectrum of 4, the NOE correlation of hydroxyl proton with H3-14′ (Figs 3A and SS44 in Supporting Information) and lack of NOE correlation of H3-14′ with H-2′ indicated that H-2′ and the hydroxyl group was located on the same side of the indene plane. The initial optical rotation value near to be zero suggested its enantisomeric feature, and a successful chiral separation on HPLC for 4 yielded optical pure compounds (+)-4 and (−)-4, showing opposite optical rotation values of 308.7 and −316.7, respectively. To determine the absolute configurations of 4, the stable conformers of structural candidates of 10R,1′S,2′S, 10S,1′R,2′R, 10R,1′R,2′R, and 10S,1′S,2′S for 4 were studied theoretically by TDDFT/ECD calculations at RB3LYP/DGDZVP level17. It was evident that whether the main CE at about 230 nm could be positive or negative depends on the C-10 configuration, while the configurations of C-2′ and C-3′ were sensitive to the intensity of the CE (Figs 3B and S6–S9 Supporting Information). Accordingly, the calculated ECD for 10S,1′R,2′R configuration matched well with the experimental ECD spectrum of (+)-4 with a strong negative CE at 235.4 nm and a relatively weak positive CE at 368.4 nm, which was opposite to the experimental ECD spectrum of (−)-4 and calculated ECD for 10R,1′S,2′S configuration (Fig. 3B). The result was further supported by the computed optical rotation (OR) for the optimal B3LYP/6-31G(d,p)-optimized geometries of the four structural candidates (Supporting Information). The calculated OR values for 4a1 and 4b1 with respective 10S,2′R,3′R and 10R,2′S,3′S absolute configurations were correspondingly 276.4 and −278.5 at the B3LYP/6-311++G(2d,p) level17, which were close to the experimental data of 308.7 and −316.7 and differed greatly from the calculated 693.4 and −746.2 for the respective 10S,1′S,2′S and 10R,1′R,2′R candidates (4c1 and 4d1). It indicated that the C-10 configuration played a decisive role in the nature of OR, while C-1′ and C-2′ configurations together reflected their degrees. And this trend was still consistent in those OR of all the dominant conformers and in the combined ones after Boltzmann weighting (Table S18 in Supporting Information). Therefore, the absolute configurations of (+)-4 and (−)-4 were determined as 10S,2′R,3′R and 10R,2′S,3′S, respectively.
Biological Activity
It was reported that GA derivatives generally possess antioxidant, antiallergic, and anti-inflammatory activities, and have wide application as cosmetic color additive and anti-ulcer drug13, 18, 19. However, the novel muriceidines showed significant cytotoxic and antifouling bioactivities. In cytotoxic assay against HeLa, K562, HL-60, and A549 human tumor cell lines using the MTT method (Table 4)20, the racemic (±)-3 showed strong cytotoxicities against both HL-60 and K562 cell lines with IC50 values of 2.19 and 3.68 μM, respectively, and compound 1 showed moderate cytotoxicity against K562 cell lines with IC50 value of 8.37 μM, neither racemic (±)-2 nor the optically pure enantiomers, (+)-2 and (−)-2, as well as (±)-4, (+)-4 and (−)-4, were active against the selected tumor cell lines. The optically pure enantiomer (+)-3 was also inactive to the selected four tumor cell lines while (−)-3 showed moderate cytotoxic activity with IC50 value of 5.08 μM against HL-60 cell line. Furthermore, compound 1 also had antifouling activity against the larvae of the barnacle Balanus albicostatus with EC50 value of 11.9 μg/mL with high therapeutic index (LC50/EC50 = 3.66), stronger than the positive control (Cu2+, EC50 and LC50 = 1.0 mg/mL, LC50/EC50 = 1.0)21. Interestingly, 3-formyl guaiazulene (6) showed a lower EC50 value of 2.39 μg/mL but lower therapeutic ratio of LC50/EC50 = 2.60.
Table 4.
Cytotoxic activities of compounds 1–4 (IC50 μM) with adramycin as positive control.
| compounds | A549 | HeLa | K562 | HL-60 |
|---|---|---|---|---|
| 1 | >100 | 17.9 | 8.4 | >100 |
| 2 | >100 | >100 | >100 | >100 |
| (+)-2 | >100 | >100 | >100 | >100 |
| (−)-2 | >100 | >100 | >100 | >100 |
| 3 | 82.7 | >100 | 3.7 | 2.2 |
| (+)-3 | >100 | 53.7 | 47.2 | 22.7 |
| (−)-3 | >100 | 53.2 | 53.2 | 5.1 |
| 4 | >100 | >100 | >100 | >100 |
| (+)-4 | >100 | 42.7 | 88.4 | 54.2 |
| (−)-4 | >100 | 40.0 | >100 | >100 |
| Adramycin | 0.6 | 0.6 | 0.2 | 0.3 |
Proposed Biogenesis
Chemically, GA is known as having rich-electronic property and high reactivity, especially the diverse oxygenation has been proved to be a normal chemical transformation in many synthetic and natural products10, 22, 23. In the present study, the GA showed a unusual derivative pattern featured by coupling to Δ1-pipecolic acid (3,4,5,6-tetrahydropyridine-2-carboxylic acid) and indene moiety. From a biogenic view, Δ1-pipecolic acid was considered to be generated from L-Lys24, and the important precursor molecules as a part of the new skeleton, the co-isolated 3-formyl guaiazulene (6), could be derived from the co-isolated GA (5) through oxygenations. Δ1-pipecolic acid and 6 were speculated to form muriceidines A by an aldol-condensation-like step. Muriceidines B and C were regarded to be the products of oxidation and following methylation in allylic position of muriceidine A. Therefore, a plausible biogenetic pathway for these unusual guaiazulene derivatives (1–3) was proposed as shown in Fig. 4A.
Figure 4.
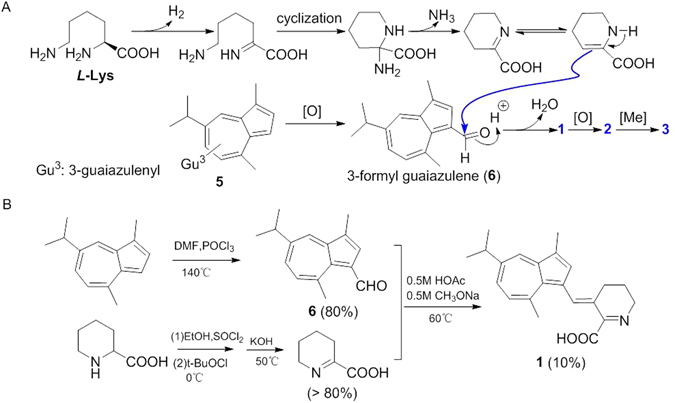
Biosynthetic pathway and semisynthetic procedure of the new compounds 1–3. (A) Proposed Biosynthetic Pathway for Compounds 1–3. (B) Semisynthetic procedure of muriceidine A (1).
Semisynthesis of muriceidines A and B
In order to validate the proposed hypotheses, brief investigations were carried out using a precursor-directed approach (Fig. 4B). Firstly, the Δ1-pipecolic acid was synthesized by L-pipecolinic acid after esterification, chlorination and dehydrochlorination25, 26. The other precusor 3-formyl guaiazulene could be easily prepared by Vilsmeier-Hacck reaction starting with guaiazulene in 80% yield27. Under the presence of sodium methoxide and acetic acid, muriceidine A could be obtained by an aldol-condensation-like reaction between the Δ1-pipecolic acid and 3-formyl guaiazulene in 8% yield28, which not only proved the rationality of the proposed biogenetic pathway but also afforded an available reference for large-scale preparation of the family molecules. Furthermore, the further oxidation, either direct exposure to air, even using relative severe oxylation conditions of SeO2, H2O2 or bromination-hydrolysis with NBS-NaHCO3, in allylic position of muriceidine A to get muriceidines B and C failed. These efforts also indirectly confirmed the natural occurrence of (±)-Muriceidine B and (±)-Muriceidine C.
Experimental Section
General Experimental Procedures
Optical rotations were measured on a Jasco P-1020 digital polarimeter. UV spectra were recorded on a Beckman DU640 spectrophotometer. CD spectra were obtained on a Jasco J-810 spectropolarimeter. IR spectra were taken on a Nicolet NEXUS 470 spectrophotometer in KBr discs. NMR spectra were measured by JEOL JNMECP 600 and Bruker AVANCE III 600 spectrometers. The 7.2600 and 3.3100 ppm resonances of residual CDCl3 and CD3OD, and 77.16 and 49.00 ppm resonances of CDCl3 and CD3OD were used as internal references for 1H and 13C NMR spectra, respectively. HRESIMS spectra were measured on a Micromass Q-Tof Ultima GLOBAL GAA076 LC and Thermo Scientific LTQ orbitrap XL mass spectrometers. Semi-preparative HPLC utilized an ODS column [YMC-Pack ODS-A, 10 × 250 mm, 5 μm, 1.5 mL/min]. Chiral HPLC utilized chiral analytical columns [Daicel Chiralpack IB and IC: 5 μm, 4.6 mm × 250 mm]. Silica gel (200–300 mesh, Qingdao, China) was used for column chromatography, and pre-coated Silica gel plates (GF254, Qingdao, China) were used for TLC, and spots visualized by heating SiO2 plates sprayed with 5% H2SO4 in EtOH.
Animal Material
The marine gorgonian Muriceides collaris was collected off the coast of Weizhou Island of South China Sea South Sea in April 2010, and was frozen immediately after collection. The specimen was identified by Prof. Lin-Ren Zou, South China Sea Institute of Oceanology, Chinese Academy of Sciences. The voucher specimen (No. WZD-2010-04) was deposited at State Key Laboratory of Marine Drugs, Ocean University of China, P. R. China.
Extraction and isolation
A frozen specimen of Muriceides collaris (9.6 kg, wet weight) was homogenized and then extracted with MeOH four times (each time, 3 days) at room temperature. The combined solutions were concentrated in vacuo and the concentrated extract was subsequently desalted by redissolving with MeOH to yield a residue (221 g). The crude extract was subjected to silica gel vacuum liquid chromatography (VLC), eluting with a gradient of petroleum/acetone (from 10:0 to 1:1, v:v) and subsequently CH2Cl2/MeOH (from 20:1 to 0:1, v-v) to obtain nine fractions (Fr.A−Fr.I). Each fraction was detected by TLC, and was tested for their cytotoxicity against HeLa and K562 cell lines at 50 μg/mL. The unusual spots with natural pink color and strong ultraviolet absorption (254 nm) on UV analyzer were observed on TLC of the most bioactive fraction Fr.G (CH2Cl2/MeOH 20:1) with inhibition rations of 89.0% and 92.8% against HeLa and K562 tumor cell lines, respectively. Fr.G (3.5 g) was then subjected to a Sephadex LH-20 column eluted with CH2Cl2/MeOH (1:1, v/v) to give three subfractions Fr.G-1 and Fr.G-4. Fr.G-3 (1.018 g) was separated by silica gel CC (CH2Cl2/MeOH, 30:1, v-v) to give six subfractions (Fr.G-3-1 to Fr.G-3-6). Fr.G-3-2 (223 mg) was purified by ODS CC with a gradient of MeOH/H2O (from 20% to 100%) to yield four subfractions (Fr.G-3-2-1 to Fr.G-3-2-4). Fr.G-3-2-3 (25.0 mg) was purified by semi-preparative HPLC (ODS, 5 μm, 250 × 10 mm; MeOH/H2O, 65:35, v/v; 1.5 mL/min) to afford muriceidines A (1, 10.3 mg) and B (2, 9.6 mg). Fr.G-3-4 (137 mg) was purified by ODS column chromatograph with a gradient of MeOH/H2O (from 20% to 100%) to yield four subfractions (Fr.G-3-4-1 to Fr.G-3-4-4). And Fr.G-3-4-1 (9.6 mg) was further purified by semi-preparative HPLC (ODS, 5 μm, 250 × 10 mm; MeOH/H2O, 60:40, v/v; 1.5 mL/min) to afford muriceidine C (3, 4.6 mg). Similarly, muriceidone A (4, 3.7 mg) and the co-isolated known 3-formylguaiazulene (6, 9.1 mg) was obtained in subfraction Fr. D. (4.9 g) with medium inhibition ratios of 69.4% and 51.9% against HeLa and K562 cell lines, repectively. And GA (5, 1.5 mg) was obtained in subfraction Fr. A (0.24 g) with only low inhibition ratio of 31.4% against HeLa cell line. Chiral seperations of partial 2 [Daicel Chiralpack IB, 5 μm, 4.6 mm × 250 mm], 3 [Daicel Chiralpack IC, 5 μm, 4.6 mm × 250 mm; MeOH, 1.5 mL/min], and 4 [Daicel Chiralpack IC, 5 μm, 4.6 mm × 250 mm; Hexane/isopropyl alcohol 97:3, 1.5 mL/min] were performed on Agilent analytical HPLC system to afford optically pure (+)-2 (0.9 mg), (−)-2 (1.2 mg), (+)-3 (2.5 mg), (−)-3 (2.2 mg), (+)-4 (1.5 mg), and (−)-4 (1.6 mg) (Fig. S1).
Muriceidine A (1). dark red amorphous power; UV (MeOH) (log ε) λ max 205 (3.23), 235 (3.38), 305 (3.04), 351 (3.11); IR (KBr)ν max 2957, 2863, 1701, 1669, 1649, 1640, 1621, 1557, 1539, 1511, 1493, 1391, 1337, 1260, 1189, 1169, 1034, 859, 761 cm−1; 1H and 13C data, see Table 1; ESIMS m/z 292.2 [M − CO2 + H]+, 336.1 [M + H]+, m/z 358.2 [M + Na]+ ; HRESIMS m/z 336.1959 [M + H]+ (calcd for C22H26NO2, 336.1958), m/z 358.1776 [M + Na]+ (calcd for C22H25NO2Na, 358.1778).
(±)-Muriceidine B (2). dark red amorphous power; UV (MeOH) (log ε) λ max 207 (2.70), 237 (2.76), 308 (2.45), 354 (2.48); IR (KBr)ν max 3747,2958, 2926, 1689, 1682, 1649, 1556, 1539, 1509, 1520, 1493, 1458, 1390, 1337, 1263, 1171, 1097, 1933, 794, 666 cm−1; 1H and 13C data (Table 1); ESIMS m/z 308.2 [M - CO2 + H]+, 352.2 [M + H]+; HRESIMS m/z 352.1912 [M + H]+ (calcd for C22H26O3N, 352.1907), m/z 374.1732 [M + Na]+ (calcd for C22H25NO3Na, 374.1727). (+)-(2): [α]25 D 501.0 (c 0.017, MeOH); CD (c 0.0008 M, MeOH) λ max (Δε) 199.1 (−7.8), 232.8 (3.4), 263.3 (−9.3), 302.0 (4.3), 347.3 (3.6) nm; (−)-(2): [α]25 D −586.7 (c 0.038, MeOH); CD (c 0.08 M, MeOH) λ max (Δε) 193.3 (7.5), 231.8 (−3.1), 263.6 (6.1), 300.8 (−3.3), 348.8 (−2.3) nm.
(±)-Muriceidine C (3). dark red amorphous powder; UV (MeOH) (log ε) λ max 207 (3.00), 236 (3.11), 293 (2.94), 352 (2.82); IR (KBr)ν max 2957, 2927, 1700, 1682, 1650, 1557, 1539, 1509, 1458, 1433, 1419, 1391, 1366, 1337, 1264, 1169, 691, 669 cm−1; 1H and 13C data (Table 1); ESIMS m/z 322.2 [M - CO2 + H]+, 366.1 [M + H]+, m/z 388.2 [M + Na]+; HRESIMS m/z 388.1890 [M + Na]+ (calcd for C23H27NO3Na, 388.1883). (+)-(3): [α]25 D 662.0 (c 0. 025, CDCl3); CD (c 0.0008 M, MeOH) λ max (Δε)) 199.1 (−2.3), 232.2 (2.6), 262.8 (−3.7), 304.7 (0.6), 341.6 (1.7) nm; (−)-(3): [α]25 D −506.6 (c 0. 053, CDCl3); CD (c 0.0007 M, MeOH) λ max (Δε) 197.4 (2.3), 221.0 (−2.2), 267.4 (3.5), 300.9 (−1.1), 335.7 (−2.3) nm.
(±)-Muriceidone A (4). dark red amorphous powder; UV (MeOH) (log ε) λ max 217 (3.49), 253 (3.24); IR (KBr)ν max 3621, 2958, 1701, 1697, 1695, 1649, 1556, 1539, 1520, 1509, 1457, 1432, 1420, 1337, 1232, 1107 cm−1; 1H and 13C data (Table 3); ESIMS m/z 481.2 [M + Na]+; HRESIMS m/z 481.2360 [M + Na]+ (calcd for C30H34O4Na, 481.2349). (+)-(4): [α]25 D 308.7 (c 0.14, MeOH); CD (c 0.0013 M, MeOH) λ max (Δε) 210.2 (31.4), 235.4 (−80.2), 281.9 (28.5), 368.4 (15.8) nm; (−)-(4): [α]25 D −316.7 (c 0.12, MeOH); CD (c 0.0008 M, MeOH) λ max (Δε) 193.8 (−27.0), 241.6 (26.8), 284.9 (−18.6), 365.6 (−10.9) nm.
Computational Section
The quantum chemical calculations were performed by using the density functional theory (DFT) as carried out in the Gaussian 0929. Conformational search was performed by Spartan 10 software using MMFF force filed, and conformers occurring within a 10 kcal/mol energy window from the global minimum were chosen for geometry optimization in the gas phase with the DFT method at the B3LYP/6-31G(d,p) and B3LYP/DGDZVP levels. The stable conformers for 1 were used in 13C NMR shifts calculations using DFT-GIAO model at RB3LYP/6-311+G(2d,p) level in chloroform by using the SCRF/PCM model in agreement with the experiment condition16. The experimental, calculated 13C NMR shifts (relative to TMS-resonance calculated at the same level of DFT) combined after Boltzmann weighting. The spin-allowed excitation energies and rotatory (Rn) and oscillator strengths (fn) of the lowest excited states of stable conformers were calculated for ECD spectra using TD-DFT method with the basis set RB3LYP/DGDZVP17. Solvent effects of methanol solution were evaluated at the same DFT level by using the SCRF/PCM method. Electronic transitions were expanded as Gaussian curves with a FQHM (full width at half maximum) for each peak of 0.32 eV. The ECD spectra were combined after Boltzmann weighting according to their population contribution. And the optical rotation values of the dominant B3LYP/6-31G(d,p)-optimized geometries were calculated at RB3LYP-SCRF(PCM, methanol)/6-311+G(2d,p) level (Figs S6–S9 and Table S18 in Supporting Information)17.
Cytotoxicity assay
The cytotoxicity of Fr. A–I against K562 and HeLa cell lines and compounds 1–4 against K562, HeLa, A-549, and HL-60 human tumor cell lines were determined by MTT method with Adriamycin as a positive control20. The IC50 value of each compound was calculated by Reed and Muench’s method. Experiments were repeated three times and carried out in triplicate.
Antifouling assay
The antifouling activity of compounds 1–6 against against larval of the barnacle Balanus albicostatus Pilsbry were determined according to the literature21. EC50 was calculated as the concentration where 50% of the larval population was inhibited to settle as compared to the control while LC50 was calculated as the concentration where 50% of the larval population was dead. Experiments were repeated three times and carried out in triplicate.
Electronic supplementary material
Acknowledgements
This work was supported by National Natural Science Foundation of China (Nos 41522605, 41476107, 41376142, 21572210), AoShan Talents Program Supported by Qingdao National Laboratory for Marine Science and Technology (No. 2015ASTP), and the Shandong Natural Science Fund for Distinguished Young Scholars (JQ201606). Special thanks are given to Professor J. Li (Ocean University of China, Qingdao, China) for the cytotoxicity tests, Dr. D. Q. Feng (Xiamen University, Xiamen, China) for the antifouling tests, and Professor R. L. Zou (South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China) for the species identification.
Author Contributions
P.L., X.L. and H.Z. contributed equally to this study. P.L. wrote the main manuscript text. Y.L. and G.L. designed the project. H.Z., X.S. and X.T. isolated and elucidated the structures. X.L. did the synthetic experiment. P.L. did the calculations. All authors reviewed the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Pinglin Li, Xiaoling Liu and Hongyan Zhu contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-08100-z
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yonghong Liu, Email: yonghongliu@scsio.ac.cn.
Guoqiang Li, Email: liguoqiang@ouc.edu.cn.
References
- 1.Gordon M. The Azulenes. Chem. Rev. 1952;50:127–200. doi: 10.1021/cr60155a004. [DOI] [Google Scholar]
- 2.Ito S, Shoji T, Morita N. Recent Advances in the Development of Methods for the Preparation of Functionalized Azulenes for Electrochromic Applications. Synlett. 2011;16:2279–2298. doi: 10.1055/s-0030-1260316. [DOI] [Google Scholar]
- 3.Kimura M. The chemistry of aza-azulenes. J. Syn. Org. Chem. Jpn. 1981;39:690–700. doi: 10.5059/yukigoseikyokaishi.39.690. [DOI] [Google Scholar]
- 4.Fischer G. Azulenes fused to heterocycles. Adv. Heterocycl. Chem. 2009;97:131–218. doi: 10.1016/S0065-2725(08)00203-1. [DOI] [Google Scholar]
- 5.Barybin MV. Nonbenzenoid aromatic isocyanides: New coordination building blocks for organometallic and surface chemistry. Coord. Chem. Rev. 2010;254:1240–1252. doi: 10.1016/j.ccr.2009.11.002. [DOI] [Google Scholar]
- 6.Yang X-L, Luo D-Q, Dong Z-J, Liu J-K. Two New Pigments from the Fruiting Bodies of the Basidiomycete Lactarius deliciosus. Helv. Chim. Acta. 2006;89:988–990. doi: 10.1002/hlca.200690103. [DOI] [Google Scholar]
- 7.Randau KP, Sproll S, Lerche H, Bracher F. Pernambucone, a new tropone derivative from Croton argyroglossum. Die Pharmazie. 2009;64:350–351. [PubMed] [Google Scholar]
- 8.Zhang C, Liang H, Tu G, Zhao Y. A new natural azulene-type pigment from Oreocnide frutescens. Fitoterapia. 2010;81:849–851. doi: 10.1016/j.fitote.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 9.Zheng J-J, et al. Ochracenoids A and B, Guaiazulene-Based Analogues from Gorgonian Anthogorgia ochracea Collected from the South China Sea. Mar. Drugs. 2014;12:1569–1579. doi: 10.3390/md12031569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen D, Yu S, van Ofwegen L, Proksch P, Lin W. Anthogorgienes A−O, New Guaiazulene-Derived Terpenoids from a Chinese Gorgonian Anthogorgia Species, and Their Antifouling and Antibiotic Activities. J. Agr. Food Chem. 2012;60:112–123. doi: 10.1021/jf2040862. [DOI] [PubMed] [Google Scholar]
- 11.Li MKW, Scheuer PJ. N,N-dimethylamino-3-guaiazulenylmethane from a deep sea gorgonian. Tetrahedron Lett. 1984;25:4707–4708. doi: 10.1016/S0040-4039(01)81498-2. [DOI] [Google Scholar]
- 12.Shi XF, Tang XL, Li GQ, Wang CY, Guan HS. Studies on chemical constituents of the South China Sea Gorgonian Muriceides collaris. ChongguoHaiyangYaowu. 2009;28:18–21. [Google Scholar]
- 13.Zhao L, Bruneau C, Doucet H. A straightforward access to guaiazulene derivatives using palladium-catalysed sp 2 or sp 3 C–H bond functionalization. Chem. Commun. 2013;49:5598–5600. doi: 10.1039/c3cc42226g. [DOI] [PubMed] [Google Scholar]
- 14.TotsuKa H, ToKuzen K, Ono H, Murata M. A novel yellow compound and furpipate derivatives formed from furfural or 5-hydroxymethylfurfural in the presence of lysine. Food Sci. Technol. Res. 2009;15:45–50. doi: 10.3136/fstr.15.45. [DOI] [Google Scholar]
- 15.Tsukamoto S, Kato H, Hirota H, Fusetani N. Pipecolate derivatives, anthosamines A and B, inducers of larval metamorphosis in ascidians, from a marine sponge Anthosigmella aff. Raromicrosclera. Tetrahedron. 1995;51:6687–6694. doi: 10.1016/0040-4020(95)00322-Y. [DOI] [Google Scholar]
- 16.Lodewyk MW, Siebert MR, Tantillo DJ. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic, and synthetic organic chemistry. Chem. Rev. 2012;112:1839–1862. doi: 10.1021/cr200106v. [DOI] [PubMed] [Google Scholar]
- 17.McCann DM, Stephens PJ. Determination of absolute configuration using density functional theory calculations of optical rotation and electronic circular dichroism: chiral alkenes. J. Org. Chem. 2006;71:6074–6098. doi: 10.1021/jo060755+. [DOI] [PubMed] [Google Scholar]
- 18.Takaaki, M., Koji, F. & Yoshihide, O. First components of hair deformation agents containing guaiazulene sulfonates, and hair deformation agents. JP Patent No. 2011/168499.
- 19.Hwang, U. I. et al. Antiwrinkle cosmetics containing guaiane compounds. KR Patent 2008/040356.
- 20.Alley MC, et al. Feasibility of Drug Screening with Panels of Human Tumor Cell Lines Using a Microculture Tetrazolium Assay. Cancer Res. 1984;48:589–601. [PubMed] [Google Scholar]
- 21.Qian PY, Xu Y, Fusetani N. Natural products as antifouling compounds: recent progress and future perspectives. Biofouling. 2010;26:223–234. doi: 10.1080/08927010903470815. [DOI] [PubMed] [Google Scholar]
- 22.Matsubara Y, Takekuma S, Yamamoto H, Nozoe T. 6-(3-Guaiazulenyl)-5-isopropyl-3, 8-dimethyl-1 (6 H)-azulenone and its norcaradiene-isomer. Key intermediates for novel intermolecular one-carbon transfer in autoxidation of guaiazulene. 6-(3-Guaiazulenyl)-5-isopropyl-3,8-dimethyl-1 (6 H)-azulenone and its norcaradiene-isomer. Key intermediates for novel intermolecular one-carbon transfer in autoxidation of guaiazulene. Chem. Lett. 1987;3:455–458. [Google Scholar]
- 23.Takekuma S, Matsubara Y, Yamamoto H, Nozoe T. Autoxidation of solid guaiazulene and of the solution in DMF in the presence of base or acid: A comparative study of the product distribution. Autoxidation of solid guaiazulene and of the solution in DMF in the presence of base or acid: A comparative study of the product distribution. Bull. Chem. Soc. Jpn. 1988;61:475–481. doi: 10.1246/bcsj.61.475. [DOI] [Google Scholar]
- 24.Gatto GJ, Boyne MT, Kelleher NL, Walsh CT. Biosynthesis of pipecolic acid by RapL, a lysine cyclodeaminase encoded in the rapamycin gene cluster. J. Am. Chem. Soc. 2006;128:3838–3847. doi: 10.1021/ja0587603. [DOI] [PubMed] [Google Scholar]
- 25.Juli C, et al. Pipecolic acid derivatives as small-molecule inhibitors of the Legionella MIP protein. J. Med. Chem. 2011;54:277–283. doi: 10.1021/jm101156y. [DOI] [PubMed] [Google Scholar]
- 26.Chen X-M, Chen G, Chen H, Zhang Y, Kitts DD. Elucidation of the Chemical Structure and Determination of the Production Conditions for a Bioactive Maillard Reaction Product,[5-(5,6-Dihydro-4 H-pyridin-3-ylidenemethyl) furan-2-yl] methanol, Isolated from a Glucose–Lysine Heated Mixture. J. Agric. Food Chem. 2015;63:1739–1746. doi: 10.1021/jf505579m. [DOI] [PubMed] [Google Scholar]
- 27.Takekuma S, Matsuoka H, Minematsu T, Takekuma H. Preparation, crystal structure, and spectroscopic, chemical, and electrochemical properties of (2E, 4E)-4-[4-(dimethylamino) phenyl]-1-(3-guaiazulenyl)-1,3-butadiene compared with those of (E)-2-[4-(dimethylamino) phenyl]-1-(3-guaiazulenyl) ethylene. Tetrahedron. 2010;66:3004–3015. doi: 10.1016/j.tet.2010.02.064. [DOI] [Google Scholar]
- 28.Ikeda, I., Utsunomiya, T., Sadamitsu, M., Ozoe, Y. & Mochida, K. J. Pestic. Sci. 31, 417–419 (2006).
- 29.Frisch, M. J. et al. Gaussian 09, Revision A.1; Gaussian, Inc., Wallingford CT (2009).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.