

Abstract
The proteolytic activity of Membrane-type 1 Matrix Metalloproteinase (MT1-MMP) is crucial for cancer cell invasion and metastasis. To visualize the protease activity of MT1-MMP with high spatiotemporal resolution at the extracellular plasma membrane surface of live cancer cells, a genetically encoded fluorescent biosensor of MT1-MMP has been developed. Here we describe the design principles of the MT1-MMP biosensor, the characterization of the MT1-MMP biosensor in vitro, and the live-cell imaging protocol used to visualize MT1-MMP activity in mammalian cells. We also provide brief guidelines for observing MT1-MMP subcellular activity by fluorescence resonance energy transfer (FRET) in a cell migration assay.
Keywords: Fluorescent biosensor, FRET, Live-cell imaging, MT1-MMP, Matrix Metalloproteinase, Protease, Cancer cell
1 Introduction
FRET occurs when two fluorophores are in proximity, with the emission spectrum of the donor overlapping the excitation spectrum of the acceptor. Any change of the distance and/or relative orientation between these two fluorophores can affect the efficiency of FRET and therefore the ratio of acceptor to donor emission [1]. Previous studies have shown that fusion proteins with interacting peptide partners sandwiched between two fluorescent protein color variants are capable of monitoring various cellular events in live cells with high spatial and temporal resolution [2 – 10]. A high-efficiency FRET pair, which uses an enhanced cyan fluorescence protein (ECFP) as the donor and a variant of the yellow fluorescence protein (YPet) as the acceptor, enabled the detection of possibly moderate but physiologically important molecular activities, such as vascular endothelial growth factor (VEGF)-induced Src signal in endothelial cells [11]. This, however, still only allowed the visualization of one type of active molecular event in a single live cell. Biosensors with new FRET pairs need to be developed such that multiple active molecular events can be visualized simultaneously within the same live cells. Fluorescence proteins (FPs) with different excitation and emission wavelengths have been recently developed through the directed evolutionary strategy [12]. Among these FPs, mOrange2 and mCherry, with the emission spectrum of mOrange2 overlapping the excitation spectrum of mCherry, appear to have the potential to constitute another new FRET pair with spectra that are distinguishable from those of the CFP and YPet pair. Therefore, mOrange2 and mCherry could be used as a new FRET pair to enable the visualization of a second molecular event in the same cells.
The extracellular matrix (ECM) in surrounding tissue provides a barrier function against cancer growth, invasion and metastasis. One of the matrix metalloproteinase (MMP) family proteins, membrane type 1 MMP (MT1-MMP), has been shown to be crucial for tumor cells to negotiate with and invade human dermis and cross-linked type I collagen gels, which mimic the in vivo situation [13]. In clinical samples, MT1-MMP can also be detected in tumors and their surrounding tissues [14]. Although the exact mechanism is not clear, MT1-MMP may degrade type I collagen through MMP-2-dependent and -independent pathways [13, 15]. MT1-MMP can also directly digest a variety of ECM proteins, including fibronectin, vitronectin, collagen type I, II, III, and other plasma membrane receptors such as CD44 and integrins [16, 17]. Thus, MT1-MMP activity can be representative of the ability of the cancer cells to invade basement membrane or to induce metastasis of the tumor.
Here we describe the design principles of the MT1-MMP protease biosensor with the ECFP/YPet or the mOrange2/mCherry FRET pair. We provide the protocols used to characterize the ECFP/YPet-based MT1-MMP biosensor in vitro and how to visualize the MT1-MMP activity in mammalian cells by live-cell FRET imaging.
2 Materials
2.1 In Vitro Assay
Prepare all solutions using ultrapure water and analytical grade reagents.
LB (Luria Broth) medium: dissolve 25 g of Luria Broth Base (Invitrogen) containing 10 g of peptone, 5 g of yeast extract, and 10 g of sodium chloride into 1 L of water, and autoclave at 120 °C for 30 min.
Ampicillin (Amp) stock solution (100 mg/ml): dissolve 1 g of Amp powder into 10 ml of water, and filter sterilize with a 0.45 μm syringe filter. Keep stock solution in 1 ml aliquots at −20 °C.
LB-Amp agar plates: add 15 g of agar powder per 1 L of LB medium. After autoclaving, cool down the solution to 60 °C and supplement with 100 μg/ml of Amp. Pour 15–20 ml of the solution to each 10 cm-diameter Petri dish and, after solidification at room temperature, seal the plates and store at 4 °C.
BL21(DE3) competent E. Coli (Invitrogen).
Isopropyl β-D-1-thiogalactopyranoside (IPTG) stock solution (1 M): dissolve 2.38 g of IPTG into 10 ml of water and filter sterilize with a 0.45 μm syringe filter. Keep stock solution in 1 ml aliquots at −20 °C.
Phenylmethylsulphonylfluoride (PMSF) stock solution (100 mM): dissolve 0.174 g of PMSF in 10 ml of isoproponal, and store in 1 ml aliquots at −20 °C.
pRSETb and pDisplay vectors (Invitrogen).
Bacterial lysis solution: dissolve half of a protease inhibitor cocktail tablet (Merck) in 10 ml of B-PER protein extraction reagents (Thermo Scientific) and supplement with 100 μM of PMSF.
HisPur Ni-NTA agarose resin (Thermo Scientific).
Affinity purification column (Sigma) for His-tagged protein purification from bacterial lysate.
TBS solution: 50 mM Tris–HCl, pH 7.4, 300 mM NaCl.
Washing buffer: 50 mM Tris–HCl, pH 7.4, 300 mM NaCl, 10 mM imidazole.
Elution buffer: 50 mM Tris–HCl, pH 7.4, 300 mM NaCl, 100 mM imidazole.
Matrix Metalloprotease proteolysis assay buffer: 50 mM HEPES titrated with 1 M NaOH solution to pH 6.8, 10 mM CaCl2, 0.5 mM MgCl2, 50 μM ZnCl2, and 0.01 % Brij-35.
Bio-Rad protein assay kit (Bio-Rad).
Recombinant catalytic domain of human MT1-MMP, MT2-MMP or MT3-MMP, or active human MMP-2, MMP-9 (Calbiochem).
SDS-PAGE gel fixing solution: 50 % methanol and 10 % glacial acetic acid in water.
Coomassie Blue staining solution: 0.1 % Coomassie Brilliant Blue R-250, 50 % methanol (v/v), and 10 % (v/v) acetic acid.
Destaining solution: 50 % (v/v) methanol in water with 10 % (v/v) acetic acid.
2.2 Mammalian Cell Culture and Imaging Process
MiniPrep or MaxiPrep kits (Qiagen).
Mammalian expression vector: PCR3.1 Uni (Invitrogen).
HeLa cells, human breast cancer MDA-MB-231 cells, human HT1080 fibrosarcoma cells, human fibroblasts (ATCC).
Mammalian cell culture medium: Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1 mM sodium pyruvate (Invitrogen).
Phosphate buffered saline (PBS) tablets (Sigma).
35 mm glass-bottom dishes (Cell E&G).
Lipofectamine 2000 (Invitrogen).
Opti-MEM I reduced serum medium (Invitrogen).
Serum-starvation medium: DMEM culture medium containing only 0.5 % FBS.
CO2-independent medium (Invitrogen).
Epithelial growth factor (EGF, Sigma).
MetaFluor 6.2 software (Universal Imaging).
Fibronectin (Sigma).
3 Methods
3.1 Design Principles of MT1-MMP FRET Biosensors
To monitor the protease activity of MT1-MMP, a FRET bio-sensor contains an MT1-MMP specific substrate peptide sandwiched between a FRET donor and an acceptor fluorescent protein (Fig. 1a). The substrate peptide, CPKESC NL FVLKD, is derived from the MT1-MMP cleavage site identified in its substrate molecule, proMMP-2 [18].
A highly efficient FRET pair, ECFP and YPet, are chosen as the donor and acceptor, respectively, in the FRET biosensor [11]. When a biosensor is intact, ECFP and YPet form a dimer which displays a high FRET signal. Upon the cleavage of the substrate peptide by active MT1-MMP, YPet separates from ECFP and diffuses away, which leads to a significant decrease of the FRET signal corresponding to an increase in the ECFP/YPet emission ratio (Fig. 1b).
Since MT1-MMP is known to be active at the extracellular surface [19], the MT1-MMP biosensor is subcloned into a pDisplay vector (Invitrogen) for the correct mammalian cell expression and localization (Fig. 1c). The pDisplay vector contains an N-terminal murine Ig κ-chain leader sequence, which directs the biosensor protein to the secretory pathway, and a C-terminal trans-membrane domain of the platelet derived growth factor receptor beta (PDGFR-β) at the C-terminus, which targets the biosensor protein to the plasma membrane. Since both MT1-MMP and PDGFR-β have been demonstrated previously to co-localize at the cell surface [20], the functional domain of the biosensor protrudes outward from the surface of the plasma membrane and is in proximity to the MT1-MMP catalytic domain (see Note 1).
Alternatively, another FRET pair, mOrange2/mCherry, which are spectrally distinguishable from the ECFP/YFP pair, is applied as the donor/acceptor in the MT1-MMP biosensor. Although the mOrange2/mCherry-based MT1-MMP bio-sensor has less dynamic range than the ECFP/YPet version, it allows simultaneous imaging with an ECFP/YFP variant-based biosensor to visualize two kinds of molecular activities simultaneously in a single cell [21].
Fig. 1.
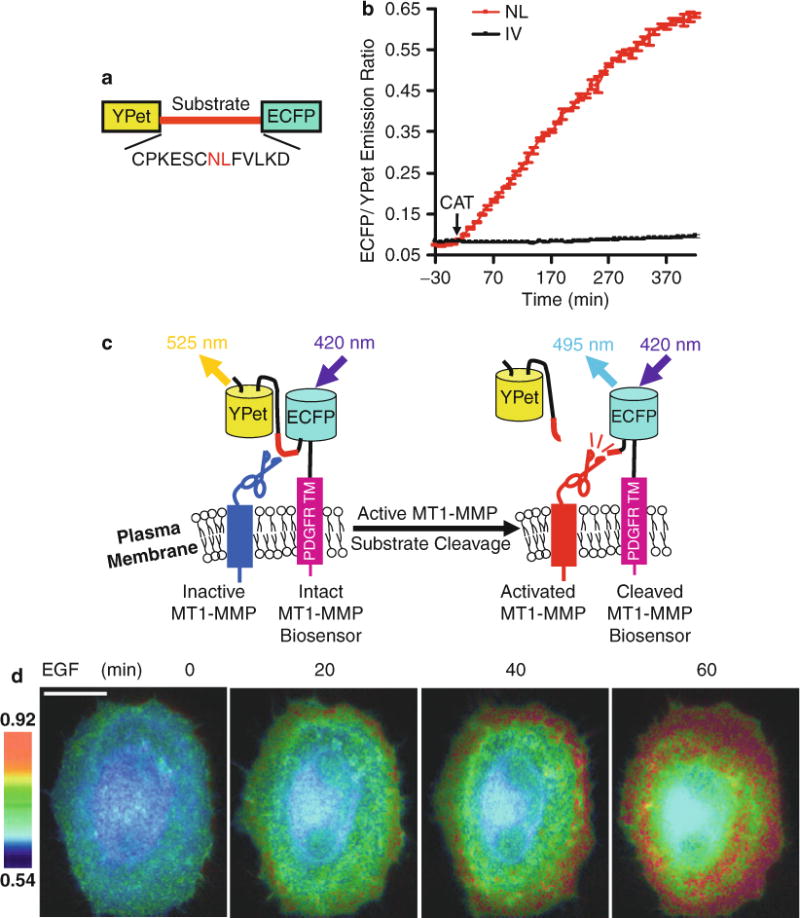
Design strategy of the MT1-MMP biosensor, and its FRET response in vitro and in mammalian cells. (a) Domain structure of MT1-MMP biosensor with YPet and ECFP at its N- and C-termini connected by a substrate peptide of MT1-MMP. (b) The time courses of ECFP/YPet emission ratio (mean ± SD) of wild-type (red line) and mutant (NL to IV mutation; black line) biosensors before and after incubation with the active catalytic domain of MT1-MMP (CAT). (c) The activation mechanism of the cell plasma membrane-tethered MT1-MMP biosensor. The biosensor is fused to the trans-membrane domain of PDGFR to position its sensing element outside of the plasma membrane, making it accessible to MT1-MMP. Active MT1-MMP can cleave the substrate peptide to separate ECFP and YPet, which leads to a decrease in FRET. (d) ECFP/YPet emission ratio images of a HeLa cell co-transfected with the MT1-MMP biosensor and MT1-MMP before and after EGF stimulation. Scale bar: 20 μm. This research was originally published in Journal of Biological Chemistry. Ouyang et al. Visualization of polarized membrane type 1 matrix metalloproteinase activity in live cells by fluorescence resonance energy transfer imaging. J Biol Chem. 2008; 283(25):17740–8. © The American Society for Biochemistry and Molecular Biology
3.2 In Vitro Characterization of MT1-MMP Biosensor
The plasmid encoding the MT1-MMP biosensor with an N-terminal 6x-His tag, which was subcloned into vector pRSETb (Invitrogen) using Bgl II/Hind III sites for bacterial expression, is transformed into BL21(DE3) competent E. Coli. The bacteria are allowed to grow on an LB agar plate supplemented with 100 μg/ml ampicillin at 37 °C overnight (12–16 h).
Inoculate a single bright colony into 50 ml of LB medium supplemented with 100 μg/ml ampicillin, and shake the culture at 250 rpm at 37 °C for 6–8 h. When the OD600 reaches 0.2–0.4, dilute the culture into 200 ml of LB medium with 100 μg/ml ampicillin, add 0.4 mM IPTG to induce the expression of the biosensor protein, and continue shaking at room temperature (around 25 °C) for 12–16 h (see Note 2).
Spin down the bacteria at 5,000 × g at 4 °C for 10 min. Add 10 ml of bacterial lysis solution and gently and completely resuspend the bacterial pellet. Rock the suspension gently at room temperature for 10 min.
Centrifuge the lysate at 20,000 × g at 4 °C for 15 min, and filter the supernatant through a 0.45 μm syringe filter. Add 0.5 ml of a 50 % slurry of Ni-NTA agarose resin and gently rock the mixture in a 15 ml tube at room temperature for 1 h to allow binding (see Note 3).
Assemble the affinity purification column and transfer the biosensor–Ni-NTA resin mixture solution from the binding reaction to the column. Wait until all of the liquid passes through the column. The resin beads bound with biosensor proteins will remain in the column.
Rinse the beads in the column three times with 10 ml of TBS solution (50 mM Tris–HCl, pH 7.4, 300 mM NaCl) before washing the beads five times with 10 ml of washing buffer (see Note 4).
Elute the biosensor proteins from the beads with elution buffer (see Note 5).
Dialyze the biosensor protein solution in matrix metalloprotease proteolysis assay buffer at 4 °C overnight (see Note 6).
Determine the molar concentration of the biosensor protein. One standard method is as follows: (1) calculate the molecular weight of the biosensor protein based on its amino acid sequence; (2) measure the protein mass concentration using the protein assay kit (Bio Rad); (3) the molar concentration of the biosensor is equal to its mass concentration divided by its molecular weight.
Prepare biosensor protein solution (1 μM) in the matrix metal-loprotease proteolysis assay buffer in 96-well plates at 100 μl per well. Measure the biosensor emission profile from 440 to 580 nm with 2 nm intervals following 415 nm excitation using a fluorescence plate reader (TECAN, Sapphire II). Record the emission profile at 10 min intervals at 37 °C for 0.5–1 h and continue for another 6–8 h after adding the recombinant catalytic domain of human MT1-MMP, MT2-MMP or MT3-MMP (2 μg/ml, Mol. Wt. 20 kDa, Calbiochem) or active human MMP-2, MMP-9 (6 μg/ml, Mol. Wt. 66 kDa, Calbiochem). The time course of emission ratios of ECFP/YPet (at 476 nm for ECFP and at 526 nm for YPet) is calculated from the recorded data using Excel (Microsoft) (Fig. 1b) (see Note 7).
The samples with or without the protease treatment are separated by 10 % SDS-PAGE gels followed by gel fixation and Coomassie Blue staining. After de-staining, the biosensor protein is visualized and the image is record using a digital camera.
3.3 Visualizing MT1-MMP Activity in Mammalian Cells by Live-Cell FRET Imaging
For mammalian cell expression, subclone the MT1-MMP bio-sensor into pDisplay (Invitrogen) using the Bgl II/Pst I restriction sites.
The DNA constructs for transfection into mammalian cells should be prepared by commercial MiniPrep or MaxiPrep kits. These constructs include the empty vector (PCR3.1 Uni) and expression vectors encoding wild-type and mutant MT1-MMP biosensors, and human MT1-MMP (see Note 8).
HeLa cells, which express only low levels of endogenous MT1-MMP, are used to characterize the MT1-MMP biosensor in mammalian cells.
The day before transfection, pass HeLa cells onto 35 mm glass-bottom dishes and maintain the cells in complete culture medium without antibiotics. The cell density should be around 50–80 % at the time of transfection.
DNA transfection can be carried out with a transfection reagent, such as Lipofectamine 2000, according to the manufacture’s protocol. For co-transfection in the glass-bottom dish, gently and thoroughly mix 1.5 μg of DNA encoding the MT1-MMP biosensor with 1.5 μg of DNA encoding MT1-MMP (or another construct encoding an MT1-MMP mutant) in 200 μl of Opti-MEM I reduced serum medium before adding another 200 μl of Opti-MEM I containing 6–7.5 μl of Lipofectamine 2000. Add the DNA–Lipofectamine 2000 mixture solution to the cells and incubate at 37 °C. After 6–8 h of transfection, replace the culture medium with fresh serum-starvation medium containing only 0.5 % FBS and no antibiotics. Before conducting FRET imaging experiments, transfected cells should be incubated for 36–48 h in the starvation media (see Note 9).
During imaging, maintain the cells in serum-free, CO2-independent medium (Gibco BRL) at 37 °C. Images are collected by a Zeiss axiovert inverted microscope equipped with a cooled charge-coupled device camera (Cascade 512B; Photometrics) using MetaFluor 6.2 software (Universal Imaging). Important imaging parameters, such as dichroic mirrors and excitation and emission filters used for FRET and different fluorescent proteins, are shown in Table 1.
When selecting cells for imaging, choose fluorescent cells that exhibit an intermediate fluorescence intensity so that the FRET change following EGF (50 μg/ml) stimulation can be readily visualized. Collect images at 2 min intervals for half an hour to obtain the basal FRET signal and for another 2–3 h after EGF stimulation to record EGF-induced FRET changes (see Note 10).
To correct for the autofluorescence of the cells under study, the fluorescence intensity of non-transfected cells should be quantified and subtracted from the ECFP and YPet (FRET) signals of transfected cells. The pixel-by-pixel ratio images of ECFP/YPet, representing the FRET efficiency and activation levels of the biosensor, are then calculated directly based on the background-subtracted fluorescence intensity images of ECFP and YPet by the MetaFluor software (Fig. 1d). Finally, emission ratios of ECFP/YPet are averaged within selected regions-of-interest to allow the quantification and statistical analysis by Excel (Microsoft) or MATLAB (The MathWorks).
The specificity of the MT1-MMP biosensor is further charac-terized in HeLa cells co-transfected with different MT1-MMP mutants [22]. Imaging of FRET change can also be achieved in cell lines without co-transfection of MT1-MMP, such as MDA-MB-231 cells, which express substantial levels of endogenous MT1-MMP [22]. FRET levels of the MT1-MMP biosensors in different cell lines can be used to compare their MT1-MMP activity levels, such as among fibroblasts, MT1-MMP-knockout fibroblast, and HT1080 cells [22].
Table 1.
The settings of filters for fluorescence imaging
| Excitation filter (nm) | Dichroic mirror (long pass; nm) | Emission filter (nm) | |
|---|---|---|---|
| CFP YFP (FRET) |
420/20 | 450 | 475/40 535/25 |
| GFP | 495/10 | 515 | 535/25 |
| mCherry | 560/40 | 595 | 653/95 |
3.4 Visualizing MT1-MMP Subcellular Activity During Migration Assay by FRET Imaging
Coat glass-bottom dishes with 10 ng/ml Fibronectin (Fn) at 4 °C overnight (or room temperature for 2 h). By applying micro-fabrication technology, the glass surface on the glass-bottom dishes can be micro-patterned with Fn-coated strips (10–20 μm width) [22]. In this way, cells seeded on the Fn-coated strips can achieve directed migration along the strips (see Note 12). After applying Fn, rinse the coated dishes once with PBS before seeding cells (see Note 11).
Before seeding, HeLa cells co-transfected with the MT1-MMP biosensor and MT1-MMP should be starved in serum-starvation medium containing 0.5 % FBS for 36–48 h in tissue culture dishes. Cells are then passaged onto Fn-coated dishes for 2–6 h before beginning the imaging process (see Note 12).
Follow the same procedures described in Subheading 3.3 (steps 6 – 8) for imaging processing and FRET quantification.
Acknowledgments
This work is supported by grants from NIH HL098472, CA139272, NS063405, NSF CBET0846429 (Y.W., S. L.), and the Wallace H. Coulter Foundation and Beckman Laser Institute, Inc. (Y.W.). The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
The transmembrane domain of PDGFR-β, which targets the biosensor protein to the plasma membrane, is located at the C-terminus of the biosensor. In order to keep the FRET donor, ECFP, on the membrane after MT1-MMP cleavage, ECFP must be incorporated at the C-terminal side of the substrate peptide, while YPet is incorporated at the N-terminal side.
Due to leaky expression from the T7 promotor, BL21(DE3) bacteria usually express a certain level of biosensor protein after growing overnight on the LB plate. Before inoculating a colony into liquid culture, the fluorescence intensity of the colonies can be easily checked under the 488 nm excitation light with a microscope or under UV light. Try to pick a colony with bright fluorescence for efficient biosensor expression in liquid culture.
To protect the biosensor protein from photobleaching by light, wrap the 15 ml tube containing the mixture solution with aluminum foil during the rocking incubation step. Likewise, avoid any strong light illumination of the biosensor protein during the entirety of purification procedures.
The purpose of the washing steps is to remove nonspecific binding proteins from the beads and biosensor proteins. Most of the time, the washing solution can pass through the column smoothly by gravity flow. If the flow is too slow on some occasions, a small amount of air pressure can be applied to the column to facilitate the flow speed.
During the elution step, the concentration of the biosensor protein is highest in the first 0.5–1 ml of elution solution coming out from the column and gradually becomes lower thereafter. To obtain concentrated biosensor, try to collect the elution into separate tubes at different elution times. However, if the biosensor concentration is too high (for example, above 100 μM), it can become partially precipitated during storage, so try to maintain the biosensor protein at an appropriate concentration, such as 10–50 μM, during storage at 4 °C.
The purpose of the dialysis is to replace the elution buffer with the matrix metalloprotease proteolysis assay buffer. The dialysis can be done in a 2-L beaker with 1.5–1.8 L of matrix metal-loprotease proteolysis assay solution outside the dialysis sacks in a cold room (4 °C) or refrigerator. Keep stirring the dialysis solution slowly with a magnetic stirrer to facilitate the dialysis process. To avoid the direct collision of the stir bar and the dialysis sacks, a small amount of air can be left in the dialysis sacks to keep them afloat during the dialysis process.
Control wells with biosensor protein but without any MMP enzyme are required to measure the biosensor stability during the experimental process. At least three parallel samples for each condition are required during the measurement. In order to avoid significant photobleaching of the biosensor, time intervals during the measurement should not be too short. To fairly compare the cleavage speed of different MMP enzymes, add all enzymes to the biosensor solution at almost the same time. Because it takes several hours to complete the measurement on the plate reader at 37 °C, water evaporation from the solution should be controlled by adding water into the empty wells in the plate and sealing the plate boundary with parafilm.
In order to achieve efficient and controllable expression of the exogenous proteins in mammalian cells after transfection, high DNA quality from the plasmid extraction step is required. Run a DNA agarose gel to check whether the plasmid is intact or contaminated with other DNA.
To achieve high efficiency in co-transfection of two kinds of DNAs in the same cells, it is necessary to mix them thoroughly before further mixing with transfection reagent, such as Lipofectamine 2000. The expression level of the MT1-MMP biosensor in mammalian cells usually peaks 36–48 h after transfection and decreases quickly after 72 h. Therefore, the imaging process should be conducted within 36–72 h after transfection. To increase the dynamic range of MT1-MMP activation and hence FRET changes of the biosensor upon EGF stimulation, it is helpful to maintain the cells in serum starvation medium with 0.5 % FBS to reduce the basal MT1-MMP activity.
It is important to ensure that the imaged cells have appropriate expression levels of each biosensor. Weak expression leads to low signal–noise ratio, while overly high expression can cause abnormal cellular functions and/or intermolecular FRET. Usually weak expression of the biosensors can be detected if the fluorescence image of the cell was not clearly distinguishable from the background. On the other hand, cells with overly high expression of biosensors have bright fluorescent signals and can sometimes shrink quickly during imaging.
Successfully coating the glass surface with Fn is critical for migration assay of transfected cells. An easy way to confirm successful Fn coating is to put a drop of PBS solution on the surface. If the drop spreads out evenly along the surface, Fn coating is likely successful. If the drop maintains a round shape without spreading, it suggests that the glass surface is hydrophobic and that Fn coating has failed.
The time period after the cells are seeded on the Fn-coated glass needs to be well controlled. Long seeding times will reduce the mobility of cells during the migration assay.
References
- 1.Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 2.Kunkel MT, et al. Spatio-temporal dynamics of protein kinase B/Akt signaling revealed by a genetically encoded fluorescent reporter. J Biol Chem. 2005;280(7):5581–5587. doi: 10.1074/jbc.M411534200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miyawaki A, et al. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388(6645):882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- 4.Mochizuki N, et al. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature. 2001;411(6841):1065–1068. doi: 10.1038/35082594. [DOI] [PubMed] [Google Scholar]
- 5.Pertz O, et al. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440(7087):1069–1072. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- 6.Ting AY, et al. Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc Natl Acad Sci U S A. 2001;98(26):15003–15008. doi: 10.1073/pnas.211564598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Violin JD, et al. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol. 2003;161(5):899–909. doi: 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, et al. Visualizing the mechanical activation of Src. Nature. 2005;434(7036):1040–1045. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- 9.Zhang J, et al. Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature. 2005;437(7058):569–573. doi: 10.1038/nature04140. [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, et al. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc Natl Acad Sci U S A. 2001;98(26):14997–15002. doi: 10.1073/pnas.211566798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ouyang M, et al. Determination of hierarchical relationship of Src and Rac at subcellular locations with FRET biosensors. Proc Natl Acad Sci U S A. 2008;105(38):14353–14358. doi: 10.1073/pnas.0807537105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaner NC, et al. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22(12):1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 13.Sabeh F, et al. Tumor cell traffic through the extracellular matrix is controlled by the membrane-anchored collagenase MT1-MMP. J Cell Biol. 2004;167(4):769–781. doi: 10.1083/jcb.200408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itoh Y, Seiki M. MT1-MMP: a potent modifier of pericellular microenvironment. J Cell Physiol. 2006;206(1):1–8. doi: 10.1002/jcp.20431. [DOI] [PubMed] [Google Scholar]
- 15.Deryugina EI, et al. MT1-MMP initiates activation of pro-MMP-2 and integrin alphavbeta3 promotes maturation of MMP-2 in breast carcinoma cells. Exp Cell Res. 2001;263(2):209–223. doi: 10.1006/excr.2000.5118. [DOI] [PubMed] [Google Scholar]
- 16.Seiki M. Membrane-type 1 matrix metalloproteinase: a key enzyme for tumor invasion. Cancer Lett. 2003;194(1):1–11. doi: 10.1016/s0304-3835(02)00699-7. [DOI] [PubMed] [Google Scholar]
- 17.Seiki M, Yana I. Roles of pericellular proteolysis by membrane type-1 matrix metal-loproteinase in cancer invasion and angiogenesis. Cancer Sci. 2003;94(7):569–574. doi: 10.1111/j.1349-7006.2003.tb01484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kinoshita T, et al. Processing of a precursor of 72-kilodalton type IV collagenase/gelatinase A by a recombinant membrane-type 1 matrix metalloproteinase. Cancer Res. 1996;56(11):2535–2538. [PubMed] [Google Scholar]
- 19.Sato H, et al. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370(6484):61–65. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 20.Lehti K, et al. An MT1-MMP-PDGF receptor-beta axis regulates mural cell investment of the microvasculature. Genes Dev. 2005;19(8):979–991. doi: 10.1101/gad.1294605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ouyang M, et al. Simultaneous visualization of protumorigenic Src and MT1-MMP activities with fluorescence resonance energy transfer. Cancer Res. 2010;70(6):2204–2212. doi: 10.1158/0008-5472.CAN-09-3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ouyang M, et al. Visualization of polarized membrane type 1 matrix metalloproteinase activity in live cells by fluorescence resonance energy transfer imaging. J Biol Chem. 2008;283(25):17740–17748. doi: 10.1074/jbc.M709872200. [DOI] [PMC free article] [PubMed] [Google Scholar]