

Abstract
Salivary biomarkers for disease detection, diagnostic and prognostic assessments have become increasingly well established in recent years. Despite salivary transcriptomic analyses have created a new paradigm in the emerging field for non-invasive molecular diagnosis, other types of RNA, especially non-coding RNAs (ncRNAs), have been the main interest in the scientific community, due to their better stability in a cell-free context than mRNAs. In this chapter, we will overview the development of sensitive and robust technique for the discovery of salivary ncRNAs, such as RNA-sequencing (RNA-seq), technique applicable not only to find disease biomarkers in human saliva, but also to fully describe the landscape of extracellular RNAs (exRNAs) in saliva. RNA is isolated from cell-free saliva (CFS) and quality controls (QCs) have been done to evaluate exRNAs present in saliva from healthy donors. In this chapter we explain the current leading technology that has been used to characterize ncRNAs: HiSeq from Illumina platform for RNA sequencing. Therefore, the chapter is divided into two main sections regarding the type of the library (small and long ncRNA libraries) constructed, from RNA extraction and quantification to cDNA generation and corresponding QCs. Using these invaluable technical tools, one can identify thousands of ncRNA species in saliva. These methods indicate that salivary exRNA provides an efficient medium for biomarker discovery of oral and systemic diseases.
Keywords: Saliva, exRNAs, small and long ncRNA profiling, biomarkers, RNA sequencing
1. Introduction
Extracellular RNA (exRNA) in human saliva is an emerging field for noninvasive diagnostic applications. The discoveries of saliva-derived mRNA in normal and oral cancer patients [1–3] and other forensic applications [4,5] were the first approaches that have opened up a new field for non-invasive molecular diagnosis. Our laboratory has extensively studied microarray-based gene profiling followed by real-time quantitative-PCR (RT-qPCR) for saliva mRNA detection. We have identified certain macromolecules associated with salivary mRNA that were protecting against ribonucleases [6]. Salivary RNA was found in complexes with lipids, proteins, lipoproteins, and phospholipids as well [7,8]. Apoptotic bodies [9] or other vesicular structures in saliva also play protection role. Therefore, RNA in the saliva may not be as fragile as it was previously assumed to be. Despite the numerous studies based on characterizing and finding mRNA diagnostic biomarkers in saliva, the introduction of deep sequencing technologies [10,11] has made revealing a new landscape of salivary exRNA [12]: micro-RNAs (miRNAs), piwi-interacting-RNAs (piRNAs), circular-RNAs (circRNAs) and other non-coding RNAs (ncRNAs). Most of the studies on characterizing ncRNAs in saliva have been recently reviewed [13], pointing out that very few studies have been addressed though RNA-sequencing (RNA-Seq) technologies. However, the group has established a network with several groups to join efforts on characterizing exRNA in several body fluids, including saliva, by using this leading technique [14]. Therefore, in this chapter update, we post the detailed methodology for the RNA extraction, cDNA library construction and quality controls (QCs), and data analysis of sequencing data. Although a variety of platforms are available for RNA-seq, the Illumina® platform is the most used nowadays. Increasing knowledge on salivary composition thanks to this platform will make a difference on understanding the biology of the diagnostic biomarkers found in saliva, either for local and systemic diseases.
The purpose of this chapter update is to provide robust and reliable methods for isolating and profiling of salivary exRNA, dividing it in two main sections regarding the type of the library (small and long ncRNA libraries) constructed. We also describe a protocol for RNA extraction after saliva collection, including detailed explanations of DNase treatment, RNA precipitation for sample concentration and specific QCs for the extracted RNA. The commercial kits for RNA extraction allow high RNA yield but the eluted RNA usually is contained in big volumes and therefore low concentrations, which is not very recommended for subsequent steps, since library preparation starts with little sample volumes in a high concentration rate. Either way, the lower limit of detection of the QCs makes the RNA precipitation crucial for sample concentration, resulting in high reproducibility among samples and accurate RNA and cDNA quantification, which at the same time is translated into good quality of raw read data after sequencing. Thus, our protocols are a guide for RNA-seq at least of salivary exRNA, but also some concepts and methodology may be applied to other type of body fluids.
2. Materials
2.1. Saliva collection and processing
50 mL sterile tube
Laboratory vortex
Refrigerated bench top centrifuge with 50 mL tube adapters
SUPERase-In RNase inhibitor, Cat# AM2694 Ambion
2.2. RNA sequencing of salivary ncRNAs
2.2.1. RNA isolation
TRIzol, Cat# 10296-028 Ambion
Chlorophorm, Cat# AM9732 Ambion
miRNeasy micro Kit (50), Cat# 21708 QIAGEN
Absolute Ethanol
2.2.2. DNase treatment and RNA precipitation
TURBO DNase, Cat# AM1907 Ambion
NaOAc 3M, Cat# AM9740 Ambion
Glycogen, Cat# AM9510 Ambion
Absolute Ethanol
RNase free water, Cat# AM9930 Ambion
2.2.3. RNA quantification and quality controls (QCs)
QuanTi™ RiboGreen RNA assay Kit, Cat# R11490 Invitrogen
96-well half area microplate (black solid plate), Cat# 3694 Corning
Agilent RNA 6000 Pico Kit, Cat# 5067-1513 Agilent Bioanalyzer
2.2.4. cDNA Library preparation
2.2.4.1. Small ncRNA library
NEBNext® Multiplex Small RNA library Prep Set for Illumina®, Cat# E7300 BioLabs
Exiqon Spike-in miRNA kit v2, Cat# 208041 Exiqon
8-tube PCR strip
Thermal Cycler PCR machine
6% Novex® TBE PAGE gel, 1.0 mM 10-well, #EC6265BOX Life Technologies
Gel broker tubes, Cat#, 3388-100 SeqMatic
Corning®, Costar®, Spin-X® Centrifuge Tube Filters, Cat# CLS8162 Sigma Aldrich
QIAquick PCR Purification Kit, Cat# 28104 QIAGEN
2.2.4.2. Long ncRNA library
NEBNext Ultra Directional RNA Library Prep kit for Illumina®, NEB# E7420 Kit
ERCC spike-in, Cat# 4456740 Ambion
8-tube PCR strip
Thermal Cycler PCR machine
DynaMag™-2 Magnet, Cat# 12321D ThermoFisher Scientific
Agentcourt® AMPure® XP Beads, Cat# A63881 Beckman Coulter
2.2.5. cDNA library quantification and QCs
Qubit® dsDNA BR Assay Kit, Cat# Q32853 Invitrogen
96-well half area microplate (black solid plate), Cat# 3694 Corning
Agilent High Sensitivity DNA Kit, Cat# 5067-4626 Agilent Bioanalyzer
2.2.6. RNA sequencing by Illumina ®
EB buffer, Cat# 19086 QIAGEN
Tween™ 20 Surfact-Amps™ Detergent Solution, Cat# 28320 ThermoFisher Scientific
HiSeq2000 Illumina System
2.2.7. De-multiplexing and data processing
Cutadapt
Bowtie mapping 16s rRNA/Microbiome
Bowtie mapping Human Genome
3. Methods
3.1. Saliva collection and processing
Saliva collection from human subjects has to be approved by the Institutional Review Board.
The following inclusion criteria will be used for normal subject selection: age ≥30 years, and no history of malignancy, immunodeficiency, autoimmune disorders, hepatitis, HIV infection, or smoking. The study population can be composed of males and females, with an average age of 42 years (range from 32 to 55 years).
Un-stimulated saliva samples can be collected between 9 a.m. and 10 a.m. in accordance with published protocols [15]. Briefly, saliva is allowed to accumulate in the floor of the mouth and the subject spits it out into the pre-weighed or graduated test tube every 60 seconds (sec). The subjects should be instructed to rinse the mouth thoroughly with water prior to the collection and to void the mouth of saliva. The subject should be seated comfortably with eyes open, head tilted slightly forward, and (for unstimulated saliva collection) instructed to rest for 5 minutes (min) and to minimize orofacial movements. Five minutes of collection time is usually enough for sufficient amount of saliva (∼5 mL) for analysis. Before collection, subjects were asked to refrain from eating, drinking, smoking, or oral hygiene procedures for at least one hour.
Following collection, saliva samples has to be centrifuged at 2600 × g for 15 min at 4°C. Saliva supernatant will be then separated from the cellular phase. SUPERase-In RNase inhibitor (at a ratio of 1 μL/mL) will be added to 1 mL of cell-free saliva (CFS) supernatant for preserving exRNA degradation. Aliquots of 1 mL CFS will be stored at -80°C for further analysis.
3.2. RNA sequencing of salivary exRNA
3.2.1. RNA isolation
Thaw 4 aliquots of 1 mL of saliva, resting the tubes on ice and not for more than half an hour (see Note 1).
Split the sample in 500 μL of CFS and centrifuge for 5 min at 10.000 × g (see Note 2). Collect the SN to proceed with the step 2, and discard the pellet fraction.
Split 0.5 mL of cell free saliva (CFS) in two tubes −250 μL in each.
Add 750 μL of Qiazol to 250 μL of CFS. Vortex for 30 sec and incubate 5 min at RT
Add 200 μL chloroform and mix by vortex for 30 sec, and then incubate 5 min at RT.
Centrifuge the sample at 12,000 × g for 15 min at 4°C.
Carefully collect 600 μL (at least) of upper aqueous phase and transfer to the new tubes.
Add 900 μL (1.5 Vol) of 100% ethanol and mix thoroughly by pipetting up and down several times. Do not centrifuge. Continue without delay to the next step.
Pipette 700 μL of the sample into an RNeasy MinElute spin column. Centrifuge at 10000 rpm (9300 × g) for 30 sec at RT. Discard the flow-through. Repeat this step using the remainder sample.
Pipet 700 μL buffer RWT into the RNeasy MinElute spin column and centrifuge for 30 sec at 10000 rpm to wash. Discard the tube with flow-through and place the column in a new 2 mL collection tube.
Pipet 700 μL Buffer RPE onto the RNeasy MinElute spin column. Close the lid gently and centrifuge for 30 sec at 10000 rpm to wash the column. Discard the flow-through.
Pipet 300 μL Buffer RPE onto the RNeasy MinElute spin column. Close the lid gently and centrifuge for 30 sec at 10000 rpm to wash the column. Discard the flow-through.
Pipet 500 μL of 80% ethanol onto the RNeasy MinElute spin column (see Note 3). Close the lid and centrifuge for 2 min at ≥ 10000 rpm to wash membrane. Discard the collection tube with the flow-through.
Place the RNeasy MinElute spin column into a new 2 mL collection tube. Centrifuge at full speed for 5 min to dry the membrane. Discard the collection tube.
Place the column in a new 1.5 mL tube. Add 30 μL pre-heated water (∼50°C) directly to the center of the membrane. Close the lid and incubate for 1-2 min at RT, and then centrifuge for 1 min at full speed.
Maintain the column in the same tube. Add 30 μL more (see Note 4) of pre-heated water directly to the center of the membrane. Close the lid and incubate for 1-2 min at RT (see Note 5), and then centrifuge for 1 min at full speed. Proceed directly to DNase treatment and RNA precipitation step (see Note 6).
3.2.2. DNase treatment and RNA precipitation
-
Mix the next components to perform off-column DNase treatment to the eluted RNA of 8 samples at the same time:
- 2μL TURBO DNase - 18 μL (for 8 samples) - 11μL Buffer - 99 μL (for 8 samples) - 27μL H2O (RNAse free) - 243 μL (for 8 samples) Add 40 μL DNase Mix/sample (100 μL final volume= 60 μL RNA + 40 μL DNase Mix).
Leave it for 15 min at RT. Continue with step 4 for RNA precipitation.
Add 10 μL (0.1 Vol) of sodium acetate 3M pH5.5.
Add 5ug of glycogen – 1 μL – (Glycogen is at 5 μg/μL concentration).
Vortex briefly.
Add 250 μL (2.5Vol) of 100% ETOH (see Note 7).
Vortex briefly.
Incubate at -80°C for overnight (O/N) or at least 1,5 hour incubation at -80°C.
3.2.3. RNA quantification and quality controls (QCs)
3.2.3.1. QuanTi™ RiboGreen RNA assay
Prepare serial dilution of rRNA standards (12.5 ng/mL to 200 ng/mL).
Make 70 μL aliquots of each standard and stock at -80°C for future uses.
Make 5 μL aliquots of fluorescent dye (Component A in RiboGreen kit) and stock them at -80 °C (see Note 8).
Take one set of standards and one Fluorescent Dye aliquot from freezer and thaw them at room temperature (RT) in the dark (important for the Dye) (see Note 9).
Prepare RNA sample dilutions at 1/30 in 1× TE buffer: Mix 1 μL of RNA/sample and 29 μL of 1× TE buffer for each sample.
Prepare enough working solution (WS) for all the experiment at a ratio of 1:200 dilution of Fluorescent Dye: 1× TE into a 15 mL tube (in the darkness).
Plate 15 μL of the standards (multichannel micropipette is recommended for reproducibility) in triplicate, and 15 μL of diluted samples in duplicate.
Add 15 μL of the WS into each well (standard and samples) and incubate the plate for 15 min at RT in the dark with lid.
Read the plate at 480-520 nm in a spectrophotometer (see Note 10).
3.2.3.2. Agilent Bioanalyzer, Eukaryotic RNA Pico Chip
Take out the reagents 30 min prior to run the Chip and allow them to RT in the dark.
Follow the manufacture instructions for preparing the gel-dye-matrix properly, and running the chip (45 min in total).
Criteria of QCs of extracted RNA
-
-
Quant-iT Ribogreen RNA assay: if total RNA amount < 5 ng is not permitted to proceed with the library construction (RNA yield is about 50-80 ng/mL saliva).
-
-
RNA 6000 Pico Chip, Bioanalyzer: detection of intact ribosomal RNA peak will make the exclusion of the sample for further analysis, since will evidence of residual cell contamination (eukaryotic: 18S (1869 nt), 28S rRNA (5070 nt); prokaryotic: 16S (1542 nt), 23S rRNA (2906 nt)).
3.2.4. cDNA Library preparation
3.2.4.1. Small ncRNA library
Prepare the Exiqon Spike-in miRNA kit v2 (Cat# 208041, Exiqon)
Dissolve the miRCURY LNA™ Array Spike-in microRNA Kit v2 in 30 μL/vial of RNase free water (supplied) upon receipt. Vortex to thoroughly dissolve the lyophilized RNA, pulse briefly in a microfuge, and leave the suspension on ice for 30 min to dissolve. Aliquot the dissolved spike-in miRNAs and store at –80°C until use and avoid repeated cycles of freeze/thawing.
1. Ligate the 3′ SR Adaptor
-
Mix the following components in a sterile nuclease-free PCR tube:
Saliva RNA 5.5 μL Exiqon Spike-in 0.5 μL (Green) 3′ SR Adaptor for Illumina 1 μL
Total volume 7 μL Incubate in a preheated thermal cycler for 2 min at 70°C. Transfer tube to ice.
-
Add the following Components:
[RNA+ 3′ SR Adaptor mix 7 μL] (Green) 3′ Ligation Reaction Buffer (2×) 10 μL (Green) 3′ Ligation Enzyme Mix 3 μL
Total volume 20 μL Incubate for 1 hour at 25°C in a thermal cycler.
2. Hybridize the Reverse Transcription Primer
-
Add the following components to the ligation mixture from step 4 and mix well:
[3′ Ligation Reaction mix from step 1 20 μL] Nuclease-Free Water 4.5 μL (Pink) SR RT Primer for Illumina 1 μL
Total volume now should be 25.5 μL Heat samples for 5 min at 75°C. Transfer to 37°C for 15 min, followed by 15 min at 25°C.
3. Ligate the 5′ SR Adaptor
With 5 min remaining, resuspend the (yellow) 5′ SR adaptor in 120 μL of nuclease free water and store at -80°C (This step is only for first time opened the kit).
Aliquot 1.1 μL × N of the (yellow) 5′ SR Adaptor into a separate, 200 μL nuclease-free PCR tube, with N equal to the number of samples being processed for the current experiment.
Incubate the adaptor in the thermal cycler at 70°C for 2 min and then immediately place the tube on ice. Keep the tube on ice and use the denatured adaptor within 30 min of denaturation.
-
Add the following components to the ligation mixture from step 6 and mix well:
[Reaction mix from step 2 25.5 μL] (Yellow) 5′ SR Adaptor for Illumina (denatured) 1 μL (Yellow) 5′ Ligation Reaction Buffer (10×) 1 μL (Yellow) 5′ Ligation Enzyme Mix 2.5 μL
Total volume 30 μL Incubate for 1 hour at 25°C in a thermal cycler.
4. Perform Reverse Transcription
Mix the following components in a sterile, nuclease-free tube:
| [Adaptor Ligated RNA from Step 3 | 30 μL] |
| (Red) First Strand Synthesis Reaction Buffer | 8 μL |
| (Red) Murine RNase Inhibitor | 1 μL |
| (Red) ProtoScript II Reverse Transcriptase | 1 μL |
|
| |
| Total volume | 40 μL |
Incubate for 60 min at 50°C.
Immediately proceed to PCR amplification.
Safe Stopping Point: If you do not plan to proceed immediately to PCR amplification, then heat inactivate the RT reaction at 70°C for 15 min. Samples can be safely stored at –15°C to – 25°C.
5. Perform PCR Amplification
Add the following components to the RT reaction mix from step 4 and mix well:
| [RT reaction mix from step 4 | 40 μL] |
| (Blue) LongAmp Taq 2× Master Mix | 50 μL |
| (Blue) SR Primer for Illumina | 2.5 μL |
| (Blue) Index Primer | 2.5 μL |
| Nuclease free water | 5 μL |
|
| |
| Total volume now should be | 100 μL |
PCR Cycling Conditions
| Initial Denaturation | 94°C | 30 sec 1 cycle |
| Denaturation | 94°C | 15 sec |
| Annealing | 62°C | 30 sec 15 cycles |
| Extension | 70°C | 15 sec |
| Final Extension | 70°C | 5 min 1 cycle |
| Hold | 4°C |
6. QIAQuick PCR Purification
*Purify the PCR amplified cDNA construct (100 μu) using a QIAQuick PCR Purification Kit.
Add 500 μL Buffer PB to the PCR reaction and mix.
Apply the sample to the QIAquick column and centrifuge at 13000 rpm for 30–60 sec.
Add 750 μL Buffer PE to the QIAquick column and centrifuge at 13000 rpm for 30–60 sec.
Centrifuge the column with the lid of the spin column open for 5 min at 13200 rpm (see Note 11).
Place each QIAquick column in a clean 1.5 mL microcentrifuge tube.
To elute amplified DNA add 26 μL Nuclease-free Water. Let the column stand for 1 min, and then centrifuge 13000 rpm for 1min.
7. Size Selection using 6% Polyacrylamide Gel and purification of size selected library
Prepare 500 mL Running Buffer (100 mL 5× Running Buffer + 400 mL Water), leave 100 mL Running Buffer with 10 μL SYBR Gold stain for later use.
Mix the purified PCR product (25 μL) with 10 μL of Gel Loading Dye, Blue (6×).
Load 5 μL of Quick-Load pBR322 DNA-MspI Digest in a well on the 6%PAGE 10-well gel.
Load two wells with 17 μL each of mixed amplified cDNA and loading dye on the 6% PAGE 10-well gel.
Run the gel for ∼1.5 hour at 90 V. Do not let the blue dye exit the gel.
Remove the gel from the apparatus and stain the gel with SYBR Gold nucleic acid gel stain in a clean container for 2–3 minutes and view the gel on a UV transiluminator. The 140 and 150 nt bands correspond to adapter-ligated constructs derived from the 21 and 30 nt RNA fragments, respectively. For miRNAs, isolate the bands corresponding to ∼140 bp. For piRNAs, isolate the band corresponding to ∼150 bp (see Figure 1).
Place the two gel slices from the same sample in gel breaker tube with a 2 mL tube, then centrifuge at 14,000 × g for 1 min, and then soak in 400 μL DNA Gel Elution buffer (1×).
Rotate in eppendorf shaker for at least 2 hours at RT.
Transfer the eluate and the gel debris to SpinX column with 1 cm diameter Whatman filter.
Centrifuge the filter for 2 min at > 13,200 rpm.
Recover eluate and add 1 μL Linear Acrylamide, 40 μL 3M sodium acetate, pH 5.5 and 500 μL of 100% ethanol, 500 μL of isopropanol. Vortex well.
Precipitate at −20°C for at least 4 hours or -80°C at least 1.5 hours.
Spin > 15,000 × g for 30 min at 4°C.
Remove the supernatant taking care not to disturb the pellet.
Wash the pellet with 500 μL 80% ethanol.
Spin > 15,000 × g for 10 min at 4°C.
Air dry pellet for up to 10 min at RT to remove residual ethanol.
Resuspend pellet in 12 μL EB Buffer (2 of 12 μL will be used for cDNA library quantification in 3.2.5).
Figure 1. Transiluminator view of miRNA and piRNA bands.
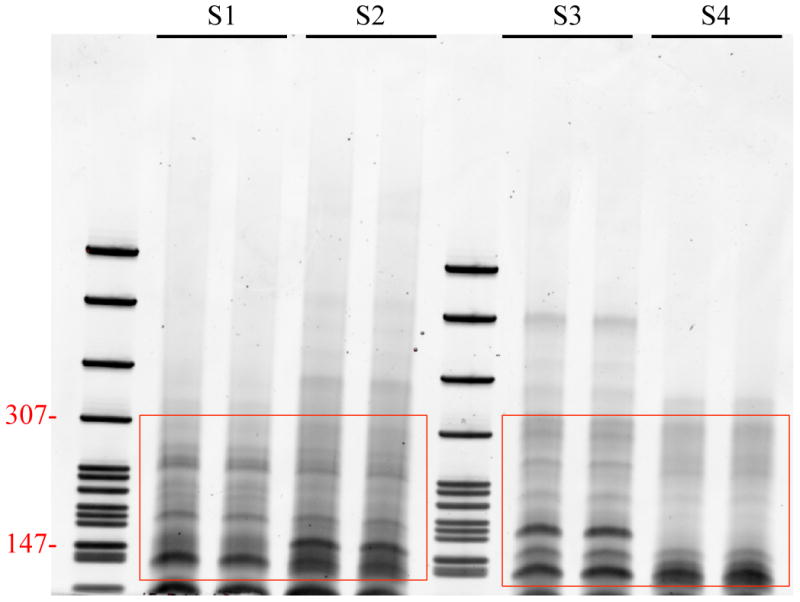
The lanes SI to S4 correspond to 4 different small ncRNA libraries. Each library has been run per duplicate and bands were cut below 140 bp and above 300 bp. miRNAs isolated bands correspond to -140 bp. piRNAs isolated bands correspond to -150 bp.
3.2.4.2. Long ncRNA library
Prepare the ERCC spike in (Cat# 4456740, Ambion)
Dissolve in RNAse free water the lyophilized product making 1:100 dilution stocks. Vortex to thoroughly dissolve the lyophilized RNA, pulse briefly in a microfuge, and leave the suspension on ice for 30 min to dissolve. Aliquot (1-2 μL) the dissolved spike-in RNAs and store at −80°C until use and avoid repeated cycles of freeze/thawing.
Preparation of First Strand Reaction Buffer and Random Primer Mix
| Total Saliva RNA + ERCC spike-in (4.5 μL of RNA + 0.5 μL Spike-in) | 5 μL |
| (Pink) NEBNext First Strand Synthesis Reaction Buffer (5×) | 4 μL |
| (Pink) NEBNext Random Primers | 1 μL |
|
| |
| Total volume | 10 μL |
2. RNA fragmentation
Incubate the samples at 94°C for 2 min.
Transfer the tube on ice
Proceed to First Strand cDNA Synthesis
3. First Strand cDNA Synthesis
*Dilute Actinomycin D stock solution (5 μg/μL) to 0.1 μg/μL in nuclease-free water for immediate use.
| The fragmented and primed mRNA | 10 μL |
| (Pink) Murine RNase Inhibitor | 0.5 μL |
| Actinomycin D (0.1 μg/μL) | 5 μL |
| (Pink) ProtoScript II Reverse Transcriptase | 1 μL |
| Nuclease free water | 3.5 μL |
|
| |
| Final volume | 20 μL |
Incubate the sample in a preheated thermal cycler as follows:
10 min at 25°C
min at 42°C
min at 70°C
Hold at 4°C
4. Second Strand cDNA Synthesis
| The First Strand Synthesis reaction mixes | 20 μL |
| Nuclease free water | 48 μL |
| (Orange) Second Strand Synthesis Reaction Buffer (10×) | 8 μL |
| (Orange) Second Strand Synthesis Enzyme Mix | 4 μL |
|
| |
| Total volume | 80 μL |
Mix thoroughly by gentle pipetting.
Incubate in thermal cycler for 1 hour at 16°C, with heated lid set at ≥40°C.
Purify the Double-stranded cDNA Using 2.5× Agencourt AMPure XP Beads
Vortex AMPure XP beads to resuspend.
Add 200 μL (2.5×) of resuspended AMPure XP beads to the second strand synthesis reaction (≈80 μL). Mix well on a vortex mixer or by pipetting up and down at least 10 times.
Incubate for 5 min at RT.
Quickly spin the tube in a microcentrifuge to collect any sample on the sides of a tube. Place the tube on an appropriate magnetic rack (DynaMag™-2 Magnet) to separate beads from supernatant. After the solution is clear (about 5 min), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets.
Add 200 μL of freshly prepared 80% ethanol to the tube while in the magnetic rack. Incubate at RT for 30 sec, and then carefully remove and discard the supernatant.
Repeat Step 5 once for a total of 2 washing steps.
Air-dry the beads for 10 min while the tube is on the magnetic rack with lid open (recommended hood).
Elute the DNA target from the beads into 60 μL nuclease-free water. Mix well on a vortex mixer or by pipetting up and down. Quickly spin the tube and then place it in the magnetic rack until the solution is clear.
Remove 55.5 μL of the supernatant and transfer to a clean nuclease-free PCR tube.
6. Perform End Repair/dA-tail of cDNA library
| The purified double-stranded cDNA | 55.5 μL |
| (Green) NEBNext End Repair Reaction Buffer (10×) | 6.5 μL |
| (Green) NEBNext End Prep Enzyme Mix | 3 μL |
|
| |
| Total volume | 65 μL |
Incubate the sample in a thermal cycler as follows:
30 min at 20°C
30 min at 65°C
Hold at 4°C
Proceed immediately to Adaptor Ligation
7. Perform Adaptor Ligation
*Dilute the NEBNext Adaptor for Illumina (15 uM) to 1.5 uM with a 10-fold dilution (1:9) with sterile water for immediate use.
| The dA-Tailed cDNA | 65 μL |
| (Red) Blunt/TA Ligase Master Mix | 15 μL |
| (Red) Diluted NEBNext Adaptor * | 1 μL |
| Nuclease-free water | 2.5 μL |
|
| |
| Total volume | 83.5 μL |
Incubate 15 min at 20°C in a thermal cycler.
*The adaptor is provided in NEBBext Singleplex (NEB #E7350) or NEBNext Multiplex (NEB #E7335, #E7500) Oligos for Illumina
8. Purify the ligation Reaction Using AMPure XP Beads
To the ligation reaction (83.5 μL), add 16.5 μL nuclease-free water to bring the reaction volume to 100 μL.
Add 100 μL (1.0×) resuspended AMPure XP beads and mix well on a vortex mixer or by pipetting up and down at least 10 times.
Incubate for 5 min at RT.
Quickly spin the tube in a microcentrifuge and place the tube on an appropriate magnetic rack to separate beads from supernatant. After the solution is clear (about 5 min), discard the supernatant that contain unwanted fragments. (Caution: do not to disturb the beads).
Add 200 μL of freshly prepared 80% ethanol to the tube while in the magnetic rack. Incubate at RT for 30 sec, and then carefully remove and discard the supernatant.
Repeat Step 5 once for a total of 2 washing steps.
Briefly spin the tube, and put the tube back in the magnetic rack.
Completely remove the residual ethanol, and air-dry beads for 10 min while the tube is on the magnetic rack with the lid open (recommended hood).
Elute DNA target from the beads with 50 μL nuclease-free water. Mix well on a vortex mixer or by pipetting up and down, and put the tube in the magnetic rack until the solution is clear.
Transfer the 50 μL supernatant to a clean PCR tube. Discard the beads.
To the 50 μL supernatant, add 50 μL (1.0×) of the resuspended AMPure XP beads and mix well on a vortex or by pipetting up and down at least 10 times.
Incubate for 5 min at RT.
Quickly spin the tube in a microcentrifuge and place the tube on an appropriate magnetic rack to separate beads from the supernatant. After the solution is clear (about 5 min), discard the supernatant that contains unwanted fragments (Caution: do not discard the beads).
Add 200 μL of freshly prepared 80% ethanol to the tube while in the magnetic rack. Incubate at RT for 30 sec, and then carefully remove and discard the supernatant.
Repeat Step 14 once for a total of 2 washing steps.
Briefly spin the tube, and put the tube back in the magnetic rack.
Completely remove the residual ethanol, and air-dry beads for 10 min while the tube is on the magnetic rack with the lid open (recommended hood).
Elute DNA target from the bead with 25 μL nuclease-free water. Mix well on a vortex mixer or by pipetting up and down, and put the tube in the magnetic rack until the solution is clear.
-
Without disturbing the bead pellet, transfer 20 μL of the supernatant to a clean PCR tube and proceed to PCR enrichment (see Note 12).
Optional stopping point: at this point cDNA library can be stored at -20°C.
9. Perform USER Excision and PCR Library Enrichment
*The Universal PCR primer and Index (X) Primer are contained in the NEBNext SinglePlex (NEB #E7350) or NEBNext Multiplex (NEB #E7335 or NEB #E7500) Oligos for Illumina.
| The size selected cDNA | 20 μL |
| (Blue) NEBNext USER Enzyme | 3 μL |
| (Blue) NEBNext High-Fidelity PCR Master Mix, 2× | 25 μL |
| (Blue) Universal PCR Primer (25 uM) | 1 μL |
| (Blue) Index (X) Primer (25 uM)* | 1 μL |
|
| |
| Total volume | 50 μL |
PCR Cycling Conditions
| User Digestion | 37°C | 15 min 1 cycle |
| Initial Denaturation | 98°C | 30 sec 1 cycle |
| Denaturation | 98°C | 10 sec |
| Annealing | 65°C | 30 sec 15 cycle |
| Extension | 72°C | 30 sec |
| Final Extension | 72°C | 5 min 1 cycle |
| Hold | 4°C |
10.Purify the PCR Reaction using AMPure XP Beads
Vortex AMPure XP beads to resuspend.
Add 50 μL (1.0×) of resuspended AMPure XP beads to the PCR reaction (≈50 μL). Mix well on a vortex mixer or by pipetting up and down at least 10 times.
Incubate for 5 min at RT.
Quickly spin the tube in a microcentrifuge and place the tube on an appropriate magnetic rack to separate beads from supernatant. After the solution is clear (about 5 min), carefully remove and discard the supernatant. Be careful not to disturb the beads that contain DNA targets.
Add 200 μL of freshly prepared 80% ethanol to the tube while in the magnetic rack. Incubate at RT for 30 sec, and then carefully remove and discard the supernatant.
Repeat Step 5 once for a total of 2 washing steps.
Air-dry the beads for 5 min while the tube is on the magnetic rack with the lid open.
Elute the DNA target from the beads into 23 μL nuclease-free water. Mix well on a vortex mixer or by pipetting up and down, quickly spin the tube in a microcentrifuge and place it in the magnetic rack until the solution is clear.
Transfer 20 μL of the supernatant to a clean PCR tube, proceed with the QCs and quantification step and/or store at -20°C.
3.2.5. cDNA library quantification and QCs
3.2.5.1. Qubit® dsDNA BR Assay
Prepare serial dilution of DNA standards. (1 ng/μL to 20 ng/μL)
Make 500 μL aliquots of each standard and stock at -4°C for future uses (use within a month).
Make 10 μL aliquots of fluorescent dye in Qubit DNA kit and stock at -80 °C (see Note 13).
Remove the high concentrated STD curve from 4°C and one Dye aliquot from -80°C, and bring them to RT in the dark (important for the Dye) (see Note 9).
Prepare enough WS for all the experiment at a ratio of 1:200 (means of 1 μL of Dye/Reagent per 200 μL of Buffer (see Note 14).
Dilute the DNA library samples with 1:60 against WS.
Prepare the low concentrated STD curve (1/10 ratio of high concentrated STD, see Table1).
Plate 30 μL of the low concentration standard curve (multichannel micropipette is recommended for reproducibility) in triplicate, and 30 μL of sample in duplicate.
Incubate the plate for 15 min at RT in the dark (cover well the plate to avoid DMSO evaporations and therefore, variation in the lecture).
Read the plate at 485-530 nm in a spectrophotometer.
Table 1. Qubit DNA standards.
| Initial High Concentration [ng/uL] | Volume of diluted DNA standard | Volume of WS | Final Low Concentration [ng/ul] |
|---|---|---|---|
| 20 | 10ul of 20ng/ul | 90 uL | 2 |
| 10 | 10ul of 10ng/ul | 90 uL | 1 |
| 5 | 10ul of 5ng/ul | 90 uL | 0.5 |
| 2 | 10ul of 2ng/ul | 90 uL | 0.2 |
| 1 | 10ul of 1ng/ul | 90 uL | 0.1 |
| 0 | 10ul of 0ng/ul | 90 uL | 0 |
3.2.5.2. Agilent Bioanalyzer, High Sensitivity DNA Chip
Take out the reagents 30 min prior to run the Chip and allow them to RT in the dark.
Dilute the DNA samples 1/10 with RNase-free water (Only for the Long RNA libraries).
Follow the manufacture instructions for preparing the gel-dye-matrix properly, and running the chip (45min in total) (see Figure 2).
Figure 2. Agilent Bioanalyzer: Eukaryotic RNA Pico Chip and High Sensitivity DNA Chip.
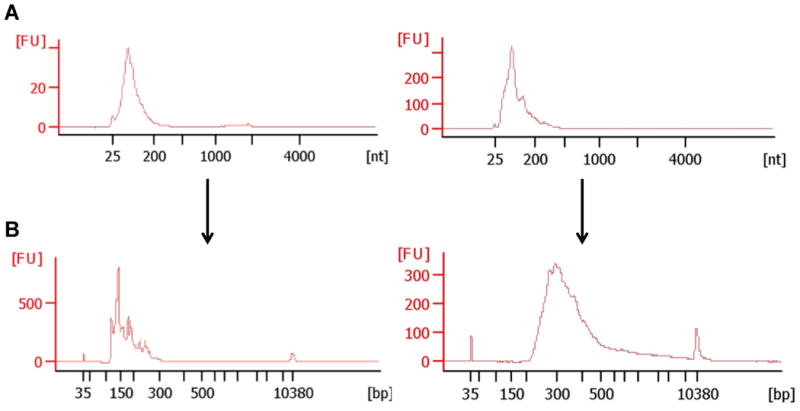
A) RNA profile of salivary exRNA after RNA precipitation. The average length of salivary exRNA is 25-200 nt. B) cDNA library of small ncRNAs (left) and long ncRNAs (right) after size selection and cDNA library purification.
Criteria of QCs of constructed library
-
-
Qubit dsDNA BR assay: concentration > 10 nM
-
-
High Sensitivity DNA Chip, Bioanalyzer: Small RNA library should have a major peak of 140-200 bp; Long RNA library should have a major peak of 300-400 bp.
3.2.6. RNA sequencing by Illumina ®
Sample pooling: 8 libraries of small ncRNA are pooled at 10 nM in total, per lane; 4 libraries of long ncRNA are pooled at 10 nM in total, per lane (see Note 15). Samples are pooled with QIAGEN EB Buffer 0.1% Tween 20 in a total volume of at least 20 μL (preferable).
Sequencing: samples are sequenced by HiSeq Illumina system, and stranded and Single-End 50 base paired (SE50) was used for the procedure (5 days long).
3.2.7. Barcode de-multiplexing and data processing
Raw data: one FastQ file is obtained per lane (8 lanes/flow cell).
De-multiplexing: Each lane needs to be de-multiplexed following the indexing codes of the samples (1-48 index in one set).
Adaptor Trimmed reads: raw data of each individual sample is submitted to Cutadapt software to remove the adaptor sequences from RNA-Seq raw data.
Quality control: QC on adaptor-trimmed reads follows the next aspects: (i) Per Base Sequence quality; (ii) Per Read quality; (iii) per Base N content and (iv) Adaptor content.
-
Mapping:
-
-
Small RNA libraries: Bowtie mapping and Human Genome are used to align the reads to the human genome. Then, RNA read counts are measured using mapping results and RNA annotation (see Table 2).
-
-
Long RNA libraries: Bowtie mapping to 16S rRNA/microbial genome is used before mapping to the Human Genome, and RNA read counts are measured using mapping results and RNA annotation (see Table 2).
-
-
Table 2. Output data of Small and Long ncRNA libraries.
| Small ncRNA libraries | Long ncRNA libraries | ||
|---|---|---|---|
| Average number of RNA detected | Average number of genes detected | ||
| miRNA | 386 | RefSeq genes | 3050 |
| piRNA | 99 | lncRNA genes | 1419 |
| Other ncRNAs* | 145 | ||
Includes snoRNAs, tRNAs, snRNA, and others
4. Notes
Each aliquot allows two RNA extractions – starting volume of 500 μL CFS.
This step gets rid of residual bacteria and cell debris.
Adding a washing step with ethanol 80% step to the commercial protocol improves washing salts and concentrates the RNA in the silica-gel membrane.
Two times of elution may reduce the concentration of RNA but will translate into higher yield after RNA precipitation since more volume for eluting the RNA allows better recovery of the RNA trapped in the silica-gel membrane.
Waiting time (1-2 min) for the membrane to get well soaked as well as using pre-heated water facilitates the elution of all RNA content.
RNA precipitation after DNase treatment will clean the protein content in the sample regarding the DNase enzyme and will result in a high RNA concentration, suitable for starting cDNA library construction.
The use ethanol instead of isopropanol is because the precipitated pellet is firmer and adheres more strongly to the tube wall with ethanol than isopropanol. Ethanol is more volatile, which facilitates removal and less salt will co-precipitate with ethanol than with isopropanol.
Avoiding freezing and thawing of the rRNA standard and fluorescent dye improves notably the reproducibility of the Ribogreen quantification assay.
Little changes in temperature will affect the fluorescence lecture, so it is important to avoid heating the tubes containing Dye before plate lecture.
Both standard curve and samples will end up in ½ diluted when mixed with the Dye → STD curve points: 100, 50, 25, 12.5, 6.25 and 0 ng/mL, RNA samples: 1/60 dilution).
Centrifugation with the lid open ensures that no ethanol remains during DNA elution. Residual ethanol may interfere with the correct loading of the sample on the PAGE gel.
Be sure not to transfer any beads. Trace amounts of bead carry over may affect the optimal performance of the polymerase used in the NEBNext High-Fidelity 2× PCR Master Mix in the subsequent PCR step.
Avoiding freezing and thawing of the fluorescent Dye improves notably the reproducibility of the Qubit DNA quantification assay.
WS already contains the dye so, needs to be prepared freshly and protected from light.
Each pooled library contributes to 1.25nM and 2.5nM of the total lane, for small and long ncRNA libraries respectively.
Acknowledgments
This work was supported by National Institutes of Health grant (UH2/UH3 TR000923). We thank Kai Kao, Leo lee and Hui Zhou for technical suggestions.
Footnotes
Conflicts of Interest: David Wong is co-founder of RNAmeTRIX Inc., a molecular diagnostic company. He holds equity in RNAmeTRIX, and serves as a company Director and Scientific Advisor. The University of California also holds equity in RNAmeTRIX. Intellectual property that David Wong invented and which was patented by the University of California has been licensed to RNAmeTRIX. Additionally, he is a consultant to PeriRx.
References
- 1.Hu Z, Zimmermann BG, Zhou H, Wang J, Henson BS, Yu W, Elashoff D, Krupp G, Wong DT. Exon-level expression profiling: a comprehensive transcriptome analysis of oral fluids. Clin Chem. 2008;54:824–32. doi: 10.1373/clinchem.2007.096164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Y, Zhou X, St John MAR, Wong DTW. RNA profiling of cell-free saliva using microarray technology. J Dent Res. 2004;83:199–203. doi: 10.1177/154405910408300303. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, St John MAR, Zhou X, Kim Y, Sinha U, Jordan RCK, Eisele D, Abemayor E, Elashoff D, Park NH, Wong DT. Salivary transcriptome diagnostics for oral cancer detection. Clin Cancer Res. 2004;10:8442–50. doi: 10.1158/1078-0432.CCR-04-1167. [DOI] [PubMed] [Google Scholar]
- 4.Juusola J, Ballantyne J. Multiplex mRNA profiling for the identification of body fluids. Forensic Sci Int. 2005;152:1–12. doi: 10.1016/j.forsciint.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 5.Juusola J, Ballantyne J. mRNA profiling for body fluid identification by multiplex quantitative RT-PCR. Journal of Forensic Sciences. 2007;52:1252–1262. doi: 10.1111/j.1556-4029.2007.00550.x. [DOI] [PubMed] [Google Scholar]
- 6.Park NJ, Li Y, Yu T, Brinkman BMN, Wong DT. Characterization of RNA in saliva. Clin Chem. 2006;52:988–94. doi: 10.1373/clinchem.2005.063206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosi A, Guidoni L, Luciani AM, Mariutti G, Viti V. RNA-lipid complexes released from the plasma membrane of human colon carcinoma cells. Cancer Lett. 1988;39:153–160. doi: 10.1016/0304-3835(88)90100-0. [DOI] [PubMed] [Google Scholar]
- 8.Whitelegge JP, Zabrouskov V, Halgand F, Souda P, Bassilian S, Yan W, Wolinsky L, Loo JA, Wong DTW, Faull KF. Protein-Sequence Polymorphisms and Post-translational Modifications in Proteins from Human Saliva using Top-Down Fourier-transform Ion Cyclotron Resonance Mass Spectrometry. Int J Mass Spectrom. 2007;268:190–197. doi: 10.1016/j.ijms.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halicka HD, Bedner E, Darzynkiewicz Z. Segregation of RNA and separate packaging of DNA and RNA in apoptotic bodies during apoptosis. Exp Cell Res. 2000;260:248–256. doi: 10.1006/excr.2000.5027. [DOI] [PubMed] [Google Scholar]
- 10.Park NJ, Zhou X, Yu T, Brinkman BMN, Zimmermann BG, Palanisamy V, Wong DT. Characterization of salivary RNA by cDNA library analysis. Arch Oral Biol. 2007;52:30–5. doi: 10.1016/j.archoralbio.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spielmann N, Ilsley D, Gu J, Lea K, Brockman J, Heater S, Setterquist R, Wong DTW. The human salivary RNA transcriptome revealed by massively parallel sequencing. Clin Chem. 2012;58:1314–21. doi: 10.1373/clinchem.2011.176941. [DOI] [PubMed] [Google Scholar]
- 12.Bahn JH, Zhang Q, Li F, Chan TM, Lin X, Kim Y, Wong DTW, Xiao X. The Landscape of MicroRNA, Piwi-Interacting RNA, and Circular RNA in Human Saliva. Clin Chem. 2014 doi: 10.1373/clinchem.2014.230433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Majem B, Rigau M, Reventós J, Wong DT. Non-coding RNAs in saliva: emerging biomarkers for molecular diagnostics. Int J Mol Sci. 2015;16:8676–98. doi: 10.3390/ijms16048676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laurent LC, Abdel-Mageed AB, Adelson PD, Arango J, Balaj L, Breakefield X, Carlson E, Carter BS, Majem B, Chen CC, Cocucci E, Danielson K, Courtright A, Das S, Abd Elmageed ZY, Enderle D, Ezrin A, Ferrer M, Freedman J, Galas D, Gandhi R, Huentelman MJ, Van Keuren-Jensen K, Kalani Y, Kim Y, Krichevsky AM, Lai C, Lal-Nag M, Laurent CD, Leonardo T, Li F, Malenica I, Mondal D, Nejad P, Patel T, Raffai RL, Rubio R, Skog J, Spetzler R, Sun J, Tanriverdi K, Vickers K, Wang L, Wang Y, Wei Z, Weiner HL, Wong D, Yan IK, Yeri A, Gould S. Meeting report: discussions and preliminary findings on extracellular RNA measurement methods from laboratories in the NIH Extracellular RNA Communication Consortium. J Extracell vesicles. 2015;4:26533. doi: 10.3402/jev.v4.26533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Navazesh M. Methods for collecting saliva. Ann N Y Acad Sci. 1993;694:72–77. doi: 10.1111/j.1749-6632.1993.tb18343.x. [DOI] [PubMed] [Google Scholar]