

ABSTRACT
Understanding the genetic basis of host shifts is a key genomic question for pathogen and parasite biology. The Bacillus cereus group, which encompasses Bacillus thuringiensis and Bacillus anthracis, contains pathogens that can infect insects, nematodes, and vertebrates. Since the target range of the essential virulence factors (Cry toxins) and many isolates is well known, this group presents a powerful system for investigating how pathogens can diversify and adapt to phylogenetically distant hosts. Specialization to exploit insects occurs at the level of the major clade and is associated with substantial changes in the core genome, and host switching between insect orders has occurred repeatedly within subclades. The transfer of plasmids with linked cry genes may account for much of the adaptation to particular insect orders, and network analysis implies that host specialization has produced strong associations between key toxin genes with similar targets. Analysis of the distribution of plasmid minireplicons shows that plasmids with orf156 and orf157, which carry genes encoding toxins against Lepidoptera or Diptera, were contained only by B. thuringiensis in the specialized insect clade (clade 2), indicating that tight genome/plasmid associations have been important in adaptation to invertebrate hosts. Moreover, the accumulation of multiple virulence factors on transposable elements suggests that cotransfer of diverse virulence factors is advantageous in terms of expanding the insecticidal spectrum, overcoming insect resistance, or through gains in pathogenicity via synergistic interactions between toxins.
KEYWORDS: Bacillus thuringiensis, host specialization, invertebrate pathogen, population genomics
IMPORTANCE
Population genomics have provided many new insights into the formation, evolution, and dynamics of bacterial pathogens of humans and other higher animals, but these pathogens usually have very narrow host ranges. As a pathogen of insects and nematodes, Bacillus thuringiensis, which produces toxins showing toxicity to many orders of insects and other invertebrates, can be used as a model to study the evolution of pathogens with wide host ranges. Phylogenomic analysis revealed that host specialization and switching occur at the level of the major clade and subclade, respectively. A toxin gene co-occurrence network indicates that multiple toxins with similar targets were accumulated by the same cell in the whole species. This accumulation may be one of the strategies that B. thuringiensis has used to fight against host resistance. This kind of formation and evolution of pathogens represents a different path used against multiple invertebrate hosts from that used against higher animals.
INTRODUCTION
Many pathogens originate from nonpathogenic ancestors by acquiring virulence genes through horizontal gene transfer (HGT) (1). Virulence factors (VFs) have a range of functions to ensure successful infection and survival in host environments. The evolution of pathogenicity does not occur in a static environment, since hosts are capable of developing resistance or acquiring immunity to prevalent infectious agents (2). In turn, pathogens can evolve new methods to overcome host resistance (3, 4). Mutations that overcome resistance can impair pathogen infectivity in previously susceptible hosts, leading to trade-offs and specialization in particular genotypes (5, 6). In a community with a wide range of possible hosts, coevolutionary dynamics can lead to host switching or “coevolutionary alternation,” as parasites may be favored to specialize in the currently least well-defended host (7). If host switching is rapid, as predicted by coevolutionary theory, and requires relatively few allelic or genomic changes, then closely related pathogens would be expected to be able to exploit a wide taxonomic range of hosts. Conversely, if host switching by pathogens requires multiple new VFs or many allelic changes, then pathogen clades may typically exploit a relatively narrow taxonomic range of hosts.
Comparative genomics has provided many new insights into the formation, evolution, and adaptation of some bacterial pathogens and provided evidence for both of these scenarios. For Salmonella, most serotypes are host specialized, causing diseases in certain hosts, and strains with similar host ranges are clustered together phylogenetically, with different combinations of Salmonella pathogenicity islands (PAIs) and different functional losses, respectively (8, 9). In Campylobacter, different lineages tend to be specialized for different vertebrate taxa, although ecological conditions in the farmed environment are undermining the basis of specialization (10, 11). In Escherichia coli, different combinations of VFs determine different pathotypes that are specialized for different host niches and can cause enteric/diarrheal disease, urinary tract infections, and sepsis/meningitis. Population genomics has shown that E. coli pathotypes are not restricted to specialized phylogenetic groups (12–14).
The Bacillus cereus group contains both pathogenic and nonpathogenic bacteria, and the taxonomic range of hosts susceptible to this group strains includes three phyla (Arthropoda, Nematoda, Chordata) (15, 16), making this group a powerful system for investigating the genomics of host specialization and changes in host range. The group includes Bacillus anthracis, which specializes in grazing ungulate herbivores (17, 18); B. cereus sensu stricto, an opportunistic intestinal pathogen of vertebrates (19); and Bacillus thuringiensis, which is an invertebrate specialist infecting nematodes and three insect orders. B. cereus and B. thuringiensis are currently differentiated on the basis of the production of crystal (Cry) toxin inclusion bodies, the genes for which are located predominantly on plasmids (20). However, previous phylogenetic analysis has identified different major clades in the B. cereus group that are ecologically and genetically distinct. In particular, the two major pathogen clades vary in their propensity to cause serious infections in either vertebrates or invertebrates, while the third clade, composed primarily of Bacillus weihenstephanensis and Bacillus mycoides, has a predominantly saprophytic niche (16, 21–24).
Toxins produced by B. thuringiensis that play a major role in the targeting of insects and other invertebrates include insecticidal Cry proteins, which are obligate VFs, vegetative insecticidal protein (Vip) toxins, and cytotoxin (Cyt) proteins (25, 26). More than 750 Cry proteins from 74 families (Cry1 to Cry74) have been described; each family being composed of members with >45% amino acid identity. More than 150 Vip toxins and 38 Cyt proteins from three families have also been reported (http://btnomenclature.info/). These proteins were reported to be toxic to many orders of insects and other invertebrates, such as nematodes, mites, and protozoa. Different toxins have different targets; Cry1, Cry2, and Vip3 are highly toxic to Lepidoptera; Cry3, Cry8, and Vip1/Vip2 kill mainly Coleoptera; Cry4 and Cyt are active against Diptera insects; and Cry5, Cry6, Cry14, and Cry21 proteins are toxic to nematodes (27).
To examine the formation and evolution of host specialization in the B. cereus group, we built a genome-based phylogeny of B. thuringiensis isolates and studied the distribution and dynamics of toxin genes among these strains. We aimed to test whether adaptations to specialize on particular host taxa are phylogenetically constrained or more widely distributed and how the gain and loss of toxins have contributed to host specialization. Historically, flagellar H serotyping was a useful classification of B. thuringiensis isolates; and the serotypes defined by this system represent a representative sample of the diversity of this species (28). We sequenced and collected the genomes of the antiserum standard strains of all of the serotypes, including multiple strains of several serotypes. Combined with genome sequences of the B. cereus group obtained from GenBank, we used B. thuringiensis as a model to study the formation, evolution, and adaptation of bacterial pathogens.
RESULTS
Population structure of the B. cereus group.
Genome sequences from 83 serovars of B. thuringiensis were used in this study. Ninety-five strains were sequenced by us, including antiserum standard strains from 57 serovars (see Table S1 in the supplemental material). Genome sequences of the remaining 26 serovars were obtained from GenBank. A maximum-likelihood (ML) phylogenetic tree based on the core genome was constructed by PhyML, and population structure was determined by the Bayesian clustering method (Bayesian analysis of population structure [BAPS]). Similar methods were used to analyze the population structure of the whole B. cereus group on the basis of the genomes of the 140 B. thuringiensis and 152 other strains in this group.
B. thuringiensis strains used in this study. Download TABLE S1, XLSX file, 0.02 MB (21.1KB, xlsx) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenomic analysis divided the B. cereus group into four clades (Fig. S1). These clades correspond closely to groups previously described on the basis of multilocus sequence typing analysis, which uses partial sequences of seven housekeeping genes for isolate typing. Clades 1 to 3 correspond largely to the clades with those names in previous studies (16, 23, 24), with clade 1 being the “Anthracis” clade and clade 3 being the “Weihenstephanensis” clade. Most typical B. thuringiensis isolates were in clade 2, which can be considered as the “Thuringiensis” clade. We compared the basic genomic features of members of the two major clades (Fig. 1). Strains in clade 2 have significantly larger genomes (most of them are >6 Mbp) than those in clade 1 (usually <6 Mbp; Fig. 1A; P = 3.711 × 10−6 [Wilcoxon-Mann-Whitney test]), which may be due to the carriage of more plasmids in clade 2. On the phylogenetic tree, members of clade 1 had longer branches than those of clade 2, indicating greater genomic diversity within this clade. The results of average nucleotide identity (ANI) analysis support this conclusion (Fig. 1B). The ANIs of isolates in clade 2 are all >95%, significantly higher than those of isolates in clade 1 (between 92 and 95%; P < 2.2 ×10−16 [Wilcoxon-Mann-Whitney test]) and those between the members of the two clades (<92%; P < 2.2×10−16 [Wilcoxon-Mann-Whitney test]).
FIG 1 .

Different features of the two major clades in the B. cereus group. (A) Strains in clade 2 have significantly larger genomes than those in clade 1 (P = 3.711e-6). (B) ANIs between all pairs of strains in clade 1, between all pairs of strains in clade 2, and between strains in clades 1 and those in clade 2 (P < 2.2e-16).
Population structure of the B. cereus group. Four clades are represented by four different background colors of the tree labels. B. thuringiensis strains are red. The binary squares and circles represent the presence and absence of different plasmids. From the innermost circle to the outermost circle, solid squares and circles represent plasmids with orf156/orf157, rep228, rep466, repX, pXO1-14/pXO1-16, repA, ori43, ori44, ori60, CT43_P825207-like, CT43_P851308-like, rep165, and pBMB2062-like minireplicons. Download FIG S1, PDF file, 1.9 MB (2MB, pdf) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Though B. thuringiensis can be found in all clades of the B. cereus group, most of them (132 of 140) were clustered into two major clades (Fig. 2; Fig. S1). On the basis of BAPS analysis, clades 1 and 2 can be divided into two and three lineages, respectively (Fig. 2). We explored the diversity of B. thuringiensis by focusing on the phylogenetic relationships among strains with the same or different serotypes. In most cases, multiple strains of the same serovar were found in the same lineage. For example, all of the strains of serovars thuringiensis, kurstaki, aizawai, and israelensis were clustered together in lineages 2.1, 2.2, 2.3, and 2.3, respectively. In addition, many strains of different serovars clustered tightly on the basis of core gene single nucleotide polymorphism (SNP) analysis, especially in clade 2. In lineage 2.3, for example, strains of serovars kurstaki, galleriae, aizawai, tolworthi, colmeri, amagiensis, and azorensis were on the same branch, with few differences in their core gene SNPs.
FIG 2 .

Population structure of B. thuringiensis. The ML tree shown was inferred by PhyML with the best model (JTT+I+G) on the basis of the alignments of the concatenated protein sequences of single-copy core genes. Bootstrap support values were calculated from 1,000 replicates, and only values of >80% are shown as white circles on the branches of the tree. The antiserum standard strains of all of the H serotypes (serovars); several strains of serotypes H1, H3abc, H5ab, and H7; and some strains with specific features were included in the analysis. The background colors of the labels represent different clades. Branches with different colors represent different lineages obtained by BAPS software. Colors represent isolated areas as follows: blue for Asia, red for Europe, green for North America, and purple for South America. The outermost circle describes the reported targets of these strains as follows: blue for Lepidoptera, purple for Diptera, dark green for nematodes, and red for Coleoptera. Target information for each strain was predicted from the host range of its toxin complement.
Isolates of clade 2 contain more toxin genes than those of clade 1.
As a pathogen of insects and nematodes, B. thuringiensis can produce three types of toxin with lethal activity, Cry, Vip, and Cyt. We focused on toxins with protein sequences >95% identical to described variants, which is the boundary representing the third rank in the nomenclature of Cry toxins. Four hundred fifty-four cry genes from 36 of the 74 families of rank 1 (members of each family with a protein sequence identity of >45%) reported until now were predicted (Fig. 3A). The most abundant family was cry1, making up 40% of the total toxin genes. Most families (23 of the 36) had <5 members of the 140 B. thuringiensis genomes studied here. In addition, 101 vip and 26 cyt genes were predicted (Table S2).
FIG 3 .

Toxin gene complements of the B. thuringiensis strains studied. (A) Composition of cry genes. (B) PAIs of the B. thuringiensis strains studied. Four hundred fifty-four cry genes from 36 rank 1 families were predicted. Red arrows represent toxin genes, and blue and gray arrows represent genes encoding transposases and genes with other functions, respectively.
Details about Cry proteins predicted in B. thuringiensis. Download TABLE S2, DOCX file, 0.02 MB (24.3KB, docx) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We analyzed the distribution of toxin genes and found that B. thuringiensis strains in clade 1 had significantly fewer than those in clade 2 (P < 2.2 × 10−16 [Wilcoxon-Mann-Whitney test]; Fig. S2 and Table S2). Most of the B. thuringiensis strains in clade 1 had only one or two cry genes, and no vip or cyt genes were found in these strains. Conversely, B. thuringiensis strains in clade 2 usually had more than two toxin genes, and many of them had more than five. Serovars containing strains with more than five toxin genes included thuringiensis and tolworthi in lineage 2.1; alesti, toumanoffi, thompsoni, and israelensis in lineage 2.2; and kurstaki, galleriae, aizawai, tolworthi, colmeri, amagiensis, and azorensis in lineage 2.3. Closely related strains could either have the same composition of toxin genes or carry divergent suites of toxin genes (Table S2). In lineage 2.1, among the six strains of thuringiensis, there were five types of toxin gene composition, with four of them containing cry1Aa, cry1Ba, vip1Bb, and vip2Aa. In israelensis of lineage 2.2, 4Q1 contained cry60Aa and cry60Ba and HD-789 had an additional seven toxin genes besides these two. In lineage 2.3, 14 strains of aizawai had seven types of toxin genes, with cry1Da, cry1Ia, cry2Ab, cry9Ea, and vip3Aa shared by all of these strains. Thus, the composition of toxins in B. thuringiensis has a very high level of diversity, implying that the gain and loss of these genes are very frequent in this species.
Differences between the cry gene numbers of the two major groups. Download FIG S2, TIF file, 0.1 MB (146.6KB, tif) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Co-occurrence network of all of the Cry, Cyt, and Vip proteins with known hosts from the B. thuringiensis Toxin Nomenclature database (http://btnomenclature.info/). Only strains with more than two toxin genes displayed in the database were analyzed. Download FIG S3, PDF file, 2.1 MB (2.2MB, pdf) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Multiple HGT events contribute to the accumulation of toxins in the same strain.
Most of the isolates in clade 2 have multiple toxin genes; and most of the genes are harbored by plasmids. We focused on the location relationships of the toxin genes among different genomes to study the dynamics of these toxins. We analyzed the putative PAIs of all of the toxin genes predicted (Fig. 3B). BtPAI-I and its variant BtPAI-II were found to be widespread in lineages 2.1 and 2.3 (Fig. 3B; Table S2). BtPAI-III, BtPAI-IV, and BtPAI-V were predicted for multiple strains of lineage 2.2 (Fig. 3B; Table S2). We suggest that many toxin genes are moved among different strains of B. thuringiensis, as PAIs and several toxins are usually transferred collectively.
The PAIs of B. thuringiensis toxins commonly contain multiple transposase genes (Fig. 3B), indicating that transposons have played an important and recent role in the dynamics and horizontal transfer of toxins. We searched for putative transposase genes by using TnpPred (29). We defined gene locations as toxin associated on the basis of the coding sequences in the five upstream and five downstream positions of genes encoding Cry, Vip, or Cyt proteins. Among the 2,252 genes in toxin-associated locations, 360 (15%) encoded transposases. This percentage is much higher than that of genes in all other locations on the genome (i.e., in loci not encoding toxins), 3% of which are associated with transposases (17,584/611,107). Transposases in the IS4, IS6, IS66, IS605, and Tn3 families were particularly associated with toxin genes (Fig. 4).
FIG 4 .
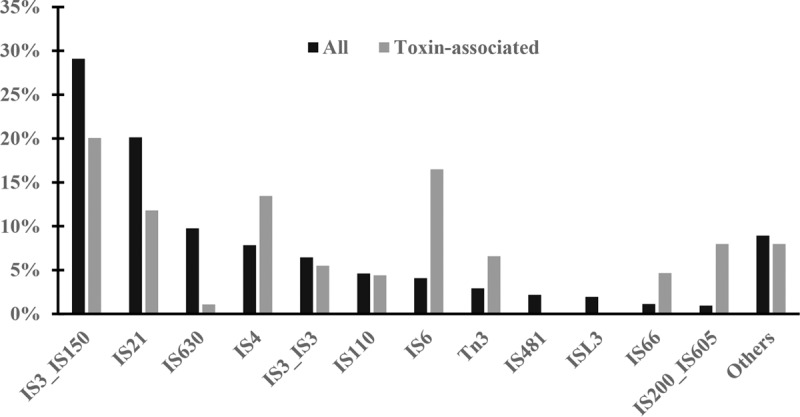
Comparison of transposase families with toxin-associated locations and other locations.
Multiple toxins from the same isolates have similar targets in the whole B. thuringiensis species.
As described above, different toxin genes were frequently contained by the same B. thuringiensis strain. We studied how different toxins coexist in the same strain by constructing a co-occurrence network. Toxin target information was obtained from the literature (27, 30) and the B. thuringiensis toxin specificity database (http://www.glfc.cfs.nrcan.gc.ca/bacillus). In the network, nodes represent Cry, the background color of the node refers to the major target reported for Cry, and edges represent co-occurrence. The edge widths were weighted by the frequency of co-occurrence; some cry genes have multiple copies in the same strain, so many Cry proteins were self-connected. In the four most frequently reported host classes, almost all of the Cry proteins with similar targets were clustered in the same subnetwork, with very few exceptions (Fig. 5).
FIG 5 .

Co-occurrence network of all of the Cry, Cyt, and Vip proteins predicted in this study. Nodes represent different Cry proteins, and node colors represent their targets. Cry proteins that coexist in the same strain are represented by edge, and those that coexist more frequently are connected by thicker lines.
The largest subnetwork contained toxins in the Cry1, Cry2, Cry9, and Vip3 families, and all of these toxins were reported to have high toxicity for insects of the order Lepidoptera. Exceptions (with different host targets) in this subnetwork included Vip1 and Vip2, which are toxic to coleopteran insects, and Cry11Bb, which can kill dipteran insects. The other subnetwork containing Cry proteins toxic to lepidopteran insects was composed of only Cry7Ba. Toxins Cry3, Cry23, Cry37, Vip1, and Vip2 were also clustered into one subnetwork; all of these toxins were toxic to coleopteran insects. In contrast, Cry proteins killing dipteran insects were clustered into six different subnetworks. Cry4 and Cyt2 proteins were found in three of them, respectively, in agreement with experimental work that showed that combinations of these toxins play a crucial role in the targeting of dipteran insects (31). Two small subnetworks had toxins targeting nematodes that contain Cry5, Cry6, and Cry55. Physical linkage can explain some co-occurrence of toxin genes within a network, and we found that all of the toxins encoded by the same PAI have similar targets. Toxins encoded by BtPAI-I, BtPAI-II, and BtPAI-III, which include different members of the Cry1 and Cry2 families and Vip3, are toxic to Lepidoptera and co-occur in the same network. Toxins of the Cry4, Cry10, Cry11, and Cyt families encoded by BtPAI-IV and BtPAI-V can kill Diptera and were also clustered in the same subnetwork.
Plasmids with the orf156/orf157 minireplicon plays a crucial role in the formation and evolution of B. thuringiensis in clade 2.
Previous genomic analysis of B. thuringiensis showed that most toxin genes were on megaplasmids with sizes of >100 kbp (32, 33). We explored the distribution of megaplasmids among all B. thuringiensis strains by studying the minireplicons they contained as described previously (32) and tried to find out the relationship between plasmid distributions and loss and gain of B. thuringiensis toxins (Fig. 6). Of the six types of minireplicons, three (ori44, pXO1-14/pXO1-16, and rep466) had very wide distributions within and between clades of the B. cereus group (Fig. 6; Fig. S1). Moreover, the orf156/orf157 minireplicon, which was first reported to be essential for replication of the large plasmid pBtoxis (34), was contained only by B. thuringiensis in clade 2 (Fig. S1). In lineage 2.1, strains clustered close to serovar thuringiensis contained orf156/orf157 plasmids. In lineage 2.2, strains with this plasmid showed a dispersed distribution on the phylogenetic tree, while in lineage 2.3, strains on the branch containing serovars kurstaki and aizawai had this plasmid (Fig. 6).
FIG 6 .

Distribution of cry genes and plasmids in B. thuringiensis. The tree shown is similar to that in Fig. 1. The innermost line represents target hosts of B. thuringiensis strains, blue for Lepidoptera, purple for Diptera, dark green for nematodes, and red for Coleoptera. In the heat map on the right, shades of green represent the numbers of genes for insecticidal proteins and shades of blue represent the numbers of Rep proteins encoded by genes on plasmids.
We studied the association between the toxin genes and the orf156/orf157 minireplicon by analyzing all of the available B. thuringiensis strains with complete genome sequences. BtPAI-I is contained by plasmids with orf156/orf157, such as pCT281, pIS56-285, pBMB299, and pBMB300 from B. thuringiensis strains CT-43, IS5056, BTK, and YBT-1520, respectively. In HD-789, BtPAI-IV and BtPAI-V and another two toxin genes were all on plasmid pBTHD789-3, which has the orf156/orf157 minireplicon. We also studied the synteny of these plasmids with complete genomes and other putative plasmids with similar minireplicons from their draft assemblies. We found that plasmids harboring similar PAIs usually have similar structures. Compared to pCT281, almost all of the plasmids with BtPAI-I or BtPAI-II have similar structures (Fig. S4A). The situation is the same for plasmids having BtPAI-IV and BtPAI-V in B. thuringiensis of lineage 2.2, such as serovars israelensis and novosibirsk (Fig. S4B).
Synteny analysis of plasmids containing the orf156/orf157 minireplicon based on complete and draft genomes. (A) Plasmids having BtPAI-I or BtPAI-II. From the innermost circle to the outermost circle, pCT281; pIS56-285 (thuringiensis); pBMB293 (kurstaki); pBMB29 (kurstaki); and plasmids from different strains of sumiyoshiensis, fukuokaensis, andalousiensis, galleriae, galleriae, tolworthi, tolworthi, tolworthi, toumanoffi, thompsoni, colmeri, thuringiensis, aizawai, aizawai, aizawai, aizawai, aizawai, NBIN-866, and BtPAI-I/II are represented (2). Plasmids of B. thuringiensis are toxic to Diptera. From the innermost circle to the outermost circle, pBtoxis (israelensis); pBTHD789-3 (israelensis); and plasmids from novosibirsk, medellin, and IBL4222 (israelensis-like) are represented. Download FIG S4, PDF file, 0.5 MB (508.2KB, pdf) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We combined the information about B. thuringiensis strain targets, major toxin genes, and megaplasmids to investigate the evolution and dynamics of pathogenicity of B. thuringiensis (Fig. 6). In clade 1, the only toxin gene with activity on Lepidoptera was cry7Ba. In clade 2, strains targeting Lepidoptera insects were mainly in lineages 2.1 and 2.3, which mostly contained the Cry1, Cry2, and Vip3 toxins and had orf156/orf157 plasmids. Diptera-toxic strains were distributed discretely around the phylogenetic tree and contained mainly Cry4 and Cyt proteins, with some strains carrying Cry32. Among them, strains in clade 2 had orf156/orf157-like plasmids. In strains targeting coleopteran insects, those in clade 1 usually had genes encoding Cry8 and those in clade 2 additionally had genes for Vip1 and Vip2. Only four strains were inferred to be toxic to nematodes, and they were not clustered together on the phylogenetic tree. The specific toxin genes that they contained included cry5, cry6, cry21, and cry55. We infer that plasmids with orf156/orf157 minireplicons are the most important vectors of the HGT contributing to the formation and dynamics of pathogenicity of B. thuringiensis in clade 2, especially those toxic to Lepidoptera and Diptera insects.
DISCUSSION
As reported previously, B. thuringiensis was found in all clades of the B. cereus group and cannot be separated from other members of this group by phylogenetic analysis; in other words, it does not form a monophyletic group (24, 35) (Fig. S1). Since this species is defined by the expression of cry genes, a single event, namely, loss of the plasmid carrying cry genes, can result in a change in species designation. A substantial number of strains designated B. thuringiensis in this study carried no cry genes with a match to a protein in the database; subsequent examination by optical microscopy showed that they also lacked crystal inclusions, indicating that they are, in fact, B. cereus (Fig. S5). Misidentification and contamination have inflated the reporting of B. thuringiensis strains in unusual clades (16). A previous study has shown that “B. thuringiensis” serovars bolivia, vazensis, and navarrensis meet the description of B. weihenstephanensis (clade 3), although expression of crystal inclusions was not investigated (36); serovar navarrensis, from the evidence of this study, is not B. thuringiensis (Fig. 6). Although this study found fewer strains with no toxin genes predicted in clade 2 than in other clades, this is, in part, an artifact of the study design, which focused on strains described as B. thuringiensis. In previous studies, B. cereus or Cry null strains were widely distributed in all of the major clades (24), although B. thuringiensis was more common in clade 2, especially within some lineages (16).
Optical microscopy of B. thuringiensis strains with no toxin genes predicted from their genome sequences. Download FIG S5, PDF file, 0.4 MB (419.2KB, pdf) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Phylogenetic analysis and ANI results showed that members of clade 2 represent a good bacterial species, in comparison to the other clades (Fig. 1B and 2). Clade 2 is quite genetically coherent, with an ANI among its strains of >95%; i.e., within the 95 to 96% boundary suggested in reference 37 as a new standard for defining bacterial species. Clade 2 contains many strains with high levels of toxicity to insect pests and which have been used as pesticides, including those from serovars thuringiensis, kurstaki, galleriae, aizawai, tenebrionis, and israelensis (38). The facts that clade 2 contains the strains that are the most effective invertebrate pathogens and contains three lineages dominated by Cry toxin producers (16) indicate that the typical B. thuringiensis strains with high levels of toxicity to insects originated from the same ancestor and evolved into three sublineages, respectively. On the basis of phylogenetic analysis, three subspecies could be proposed, i.e., lineages 2.1, 2.2, and 2.3.
It follows that Cry-producing strains in clades 1 and 3 may not warrant the designation “Thuringiensis”; their genetic background is distinct from invertebrate specialists (Fig. 1), and they appear to be ecologically distinct. Strains in clade 3 are closely related to B. weihenstephanensis, a species that is likely to be primarily saprophytic on the basis of the fact that it is psychrotolerant, is almost exclusively isolated from soil or plant material, and contains relatively few genes encoding VFs (21, 39, 40). In contrast, the vast majority of serious vertebrate infections are caused by isolates in clade 1 or the “Anthracis” clade, and members of this clade have the greatest potential to cause food poisoning in humans (16, 40). It seems likely that any invertebrate-specific toxins carried by strains in clades 1 and 3 have been acquired by recent HGT events; strains in these clades contain relatively few toxins at loci that are relatively unusual, i.e., not associated with orf156/orf157-containing megaplasmids. The fact that a large number of isolates originally designated thuringiensis in clades 1 and 3 appear to contain no cry genes at all also suggests that carriage of mobile elements with these genes might be relatively unstable, although we cannot exclude the role of contamination or misclassification here.
The co-occurrence of multiple toxins with similar invertebrate targets was typical for most B. thuringiensis strains and was particularly common in clade 2. Co-occurring toxins could either have similar protein structures or be derived from very different toxin classes. For instance, many strains in clade 2 with toxicity to Lepidoptera contained Cry1 toxins with a protein sequence homology of >78%, which is the boundary representing the second rank in the nomenclature of Cry toxins (41). Toxins in the same class may have similar receptors and modes of action. Nevertheless, accumulation of similar toxins in the same isolate can improve the speed with which some hosts are killed or expand the insecticidal spectrum among some phylogenetically related insects (42), as has been the practice with currently genetically engineered crops targeting lepidopteran pest complexes (43). In other cases, co-occurring toxins targeting Lepidoptera can be derived from a range of classes, such as Cry2 and Cry9 (with identities to those of Cry1 of <45%) or Vip3, which has no similarity to any Cry proteins. These toxins have different modes of action (44). Strains with putative toxicity to Diptera contained toxins of the Cry4, Cry10, Cry60, and Cyt families, and the identities of their protein sequences are <45%. Strains expressing coleopteran-active toxins usually contained the binary toxins Vip1/Vip2 and some Cry toxins, such as Cry3, Cry23, and Cry37. Combining toxins with different modes of action can lead to synergistic interactions, i.e., increased killing power per unit of protein, and so can provide an immediate selective benefit (45–48). Different toxins with different modes of action on similar insects can reduce the rate of evolution of resistance in targets, while Cyt toxins have the ability to mask genotypic resistance to other toxins (31, 49, 50); thus, combinations of toxins may provide a benefit in the long term or have evolved in response to resistance in some hosts (51).
Thus, synergism, extended host range, or improved resilience to resistance may have contributed to the co-occurrence of multiple toxins with broadly similar phylogenetic targets in single strains. This adds weight to the argument that B. thuringiensis strains, especially those in clade 2, represent specialized pathogens. This co-occurrence implies a continuous evolutionary history in hosts of a particular order. Thus, many strains must have undergone multiple successful cycles of infection in a restricted range of hosts. Some strains, such as B. thuringiensis serovar kurstaki ST8, may have particular adaptations (the ability to colonize plants) that enable them to infect a subset of hosts with greater efficiency (21), in addition to the Cry toxins described here.
Some B. thuringiensis toxins have been reported to have cross-order activity (30). For example, Cry2 toxins have been reported to be highly toxic to both Lepidoptera and Diptera insects. In our network analysis, all of the members of this family were in the subnetwork active against Lepidoptera. We suggest Lepidoptera insects as the original targets of Cry2 and Diptera insects as extended targets. Actually, each toxin with cross-order activity was found in a single subnetwork, which suggests that the alternative targets may have been formed by recent host range extension.
In addition, toxin network analysis may provide a powerful tool for predicting the virulence of newly described toxins with no known host affiliation. For example, in several subnetworks, some Cry proteins, such as Cry29, Cry50, Cry53, Cry68, and Cry70, had no reported target. We hypothesize that these toxins may be toxic to dipteran insects since they are contained within dipteran subnetworks. Actually, Cry29A was reported to synergize the toxicity of Cry11Bb against Aedes aegypti (Diptera: Culicidae), and Cry50Aa, Cry50Ba, Cry54Ba, Cry68Aa, and Cry70Aa were all found in some strains with high toxicity against mosquitoes (52–55). Similarly, in the coleopteran subnetwork, Cry26 and Cry28 had no reported toxicity; we suspect that they may target coleopteran hosts. Two small subnetworks had toxins targeting nematodes; again, the toxin Cry65Aa is likely to be active against this group of hosts, as it belongs to the same network as proteins such as Cry5 with well-described nematode toxicity (56).
Strains with complete genome sequences showed multiple toxins located in different positions on the same plasmid or different plasmids. Ten toxin genes, including seven cry and three cyt genes on three PAIs, were distributed separately on the same plasmid of HD-789 (57). In CT-43 and HD-1, five toxins were encoded by genes from two different plasmids, with four of them on the larger one and the other on a smaller one, respectively (58). In MC28, 11 toxin genes were contained by three different plasmids (53), and in IS5056, nine toxins were encoded by genes on four different plasmids (59). The range of loci encoding toxins implies that multiple toxins in the same genome were gained by multiple independent HGT events.
Plasmids play crucial roles in conferring pathogenicity on many pathogens (60). In the B. cereus group, genes encoding anthrax toxins of B. anthracis and those encoding emetic toxin of some B. cereus were also reported to be on plasmids (61, 62). All of the plasmids carrying these toxin genes have two minireplicons, pXO1-16/pXO1-14 and repX, which belongs to one type of the tubZ/tubR class (32). The former class of minireplicon was carried by most of the strains of the B. cereus group but not by bacteria outside this group (Fig. S1). They are commonly combined with other types of minireplicons to enable the replication of megaplasmids (32, 63). Many megaplasmids carrying B. thuringiensis toxin genes also contained this kind of minireplicon; moreover, they harbored orf156/orf157 belonging to another tubZ/tubR minireplicon. As reported previously, megaplasmids in the B. cereus group were formed by integrating smaller plasmids (32). We infer that the ancestral plasmids containing pXO1-16/pXO1-14 were important for the formation of the B. cereus group, as they integrated with different plasmids to make the host strains with different toxicities. For example, the clade 2 B. thuringiensis strains toxic to Lepidoptera were formed by the integration of one plasmid with orf156/orf157 and another plasmid with pXO1-16/pXO1-14.
The distribution of key virulence genes indicates that repeated interactions with a subset of hosts (e.g., insect orders) led to the accumulation of multiple toxins by successful clones and by successful PAIs. This accumulation may be one of the strategies that B. thuringiensis has used to combat host resistance or to increase its pathogenicity. While B. thuringiensis is not a monophyletic group, the vast majority of isolates were concentrated into a single clade and all of the strains in this clade contained multiple invertebrate VFs, suggesting that a phylogeny-based revision of the nomenclature of the B. cereus group could be valuable. Plasmids play a crucial role in HGT, and notably, plasmids carrying PAIs with multiple invertebrate toxins were restricted to this one clade. If clade 2 contains invertebrate specialists that are defined by their plasmid complement and by their genome, this leads to the question of what the evolutionary and ecological relationships between closely related B. thuringiensis and B. cereus strains within clade 2 are. Potentially, B. cereus clones in clade 2 represent specialist lineages that exploit invertebrate/vertebrate cadavers. Alternatively, B. cereus may persist as a toxin cheater and depend on coinfection with Cry-producing B. thuringiensis to access host resources (64, 65).
MATERIALS AND METHODS
Bacterial strains.
To investigate the genomics of B. thuringiensis, antiserum standard strains of each serovar and some additional strains of serotypes H5ab, H5ac, and H7 were obtained from the Bacillus Genetic Stock Center or the Institute Pasteur. Other strains used in this study were isolated and kept in our laboratory (Table S1). Genomic DNA for sequencing was prepared as described previously (33).
Genome sequencing and assembly.
Samples were sequenced by using multiplexed libraries with Illumina HiSeq 2000, 2500, or 4000 to produce pair-end reads with lengths of 100, 125, and 150 bp. For each sample, reads were assessed with the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and low-quality reads were filtered by Quake (66). All of the assemblies were performed by ABySS with different k-mers (67); the best assembly for each strain, with the largest scaffold N50, was annotated by Prokka (68).
Phylogenetic analysis.
All B. thuringiensis protein sequences were clustered by the MCL algorithm with an inflation value of 2, after an all-against-all BlastP search with an E value of <10−5 (69). Coding sequences of the single-copy core protein in each cluster were aligned by MUSCLE (70), and the alignment was trimmed by TrimAl (71). All alignments were concatenated with an in-house Perl script. The ML phylogenetic tree was constructed with RAxML by using the generalized time reversible (GTR) model and a gamma distribution to model site-specific rate variation (72). Bootstrap support values were calculated from 500 replicates.
For the whole B. cereus group, all of the genome sequences available were obtained from GenBank. Assemblies with a scaffold N50 of <50 kbp were filtered out. The SNPs of the core sequences of all of the genomes, including those sequenced in this study and those downloaded from GenBank, were obtained by the northern Arizona SNP pipeline (https://github.com/TGenNorth/NASP). An ML phylogeny was constructed by FastTree with a GTR+CAT (GTR with per-site rate categories) model of approximation for site rate variation (73). Bootstrap support values were calculated from 500 replicates.
B. thuringiensis population structure was defined with the hierBAPS module of the BAPS software, which delineates the population structure by nested clustering (74). Three independent iterations with upper population sizes of 8, 16, and 24 were used to obtain optimal clustering of the population.
Analysis of toxins and plasmids.
Genes for Cry, Vip, and Cyt proteins were predicted from all of the B. thuringiensis genome sequences used in this study by the second version of BtToxin_scanner (75). It predicted all of the putative toxins on the basis of the homologies to all of the reported toxins and classified them on the basis of their sequence identities to those from the public database by using the nomenclature described by Crickmore et al. (41). Toxin target information was collected from the B. thuringiensis toxin specificity database (http://www.glfc.cfs.nrcan.gc.ca/bacillus) and from literature sources (27, 30). The frequency of co-occurrence of each pair of toxins was explored by using an in-house Perl script. The co-occurrence network was constructed and visualized by the Cytoscape software (76). Toxins of B. thuringiensis strains that were reported to contain two or more toxin genes were collected from the B. thuringiensis toxin database (http://www.btnomenclature.info). Co-occurrence analysis was performed as described above.
Minireplicons that contain the origin of replication and genes encoding replication proteins were used to analyze the distribution and dynamics of plasmids in the B. cereus group as described in our previous work. We focused on putative plasmids with similarity to those mentioned in our previous work (32).
Availability of supporting data.
The data sets supporting the results of this article are included in the supplemental material.
ACKNOWLEDGMENTS
This work was supported by the National Key Research and Development Program of China (2017YFD0201201), the China 948 Program of the Ministry of Agriculture (2016-X21), the National Natural Science Foundation of China (NSFC) (31500003 and 31670085), the China Postdoctoral Science Foundation-funded project (2015M580649 and 2016T90700), and Chinese Fundamental Research Funds for the Central Universities (2662016QD039, 2662015PY123, and 2662017PY094).
Footnotes
Citation Zheng J, Gao Q, Liu L, Liu H, Wang Y, Peng D, Ruan L, Raymond B, Sun M. 2017. Comparative genomics of Bacillus thuringiensis reveals a path to specialized exploitation of multiple invertebrate hosts. mBio 8:e00822-17. https://doi.org/10.1128/mBio.00822-17.
REFERENCES
- 1.Juhas M. 2015. Horizontal gene transfer in human pathogens. Crit Rev Microbiol 41:101–108. doi: 10.3109/1040841X.2013.804031. [DOI] [PubMed] [Google Scholar]
- 2.Fearon DT, Locksley RM. 1996. The instructive role of innate immunity in the acquired immune response. Science 272:50–53. doi: 10.1126/science.272.5258.50. [DOI] [PubMed] [Google Scholar]
- 3.van der Poll T, Opal SM. 2008. Host-pathogen interactions in sepsis. Lancet Infect Dis 8:32–43. doi: 10.1016/S1473-3099(07)70265-7. [DOI] [PubMed] [Google Scholar]
- 4.Schulte RD, Makus C, Hasert B, Michiels NK, Schulenburg H. 2010. Multiple reciprocal adaptations and rapid genetic change upon experimental coevolution of an animal host and its microbial parasite. Proc Natl Acad Sci U S A 107:7359–7364. doi: 10.1073/pnas.1003113107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lohse K, Gutierrez A, Kaltz O. 2006. Experimental evolution of resistance in Paramecium caudatum against the bacterial parasite Holospora undulata. Evolution 60:1177–1186. doi: 10.1111/j.0014-3820.2006.tb01196.x. [DOI] [PubMed] [Google Scholar]
- 6.Little TJ, Watt K, Ebert D. 2006. Parasite-host specificity: experimental studies on the basis of parasite adaptation. Evolution 60:31–38. doi: 10.1111/j.0014-3820.2006.tb01079.x. [DOI] [PubMed] [Google Scholar]
- 7.Nuismer SL, Thompson JN. 2006. Coevolutionary alternation in antagonistic interactions. Evolution 60:2207–2217. doi: 10.1111/j.0014-3820.2006.tb01858.x. [DOI] [PubMed] [Google Scholar]
- 8.Langridge GC, Fookes M, Connor TR, Feltwell T, Feasey N, Parsons BN, Seth-Smith HMB, Barquist L, Stedman A, Humphrey T, Wigley P, Peters SE, Maskell DJ, Corander J, Chabalgoity JA, Barrow P, Parkhill J, Dougan G, Thomson NR. 2015. Patterns of genome evolution that have accompanied host adaptation in Salmonella. Proc Natl Acad Sci U S A 112:863–868. doi: 10.1073/pnas.1416707112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fookes M, Schroeder GN, Langridge GC, Blondel CJ, Mammina C, Connor TR, Seth-Smith H, Vernikos GS, Robinson KS, Sanders M, Petty NK, Kingsley RA, Bäumler AJ, Nuccio SP, Contreras I, Santiviago CA, Maskell D, Barrow P, Humphrey T, Nastasi A, Roberts M, Frankel G, Parkhill J, Dougan G, Thomson NR. 2011. Salmonella bongori provides insights into the evolution of the Salmonelleae. PLoS Pathog 7:e1002191. doi: 10.1371/journal.ppat.1002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheppard SK, Colles FM, McCarthy ND, Strachan NJC, Ogden ID, Forbes KJ, Dallas JF, Maiden MCJ. 2011. Niche segregation and genetic structure of Campylobacter jejuni populations from wild and agricultural host species. Mol Ecol 20:3484–3490. doi: 10.1111/j.1365-294X.2011.05179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheppard SK, Didelot X, Jolley KA, Darling AE, Pascoe B, Meric G, Kelly DJ, Cody A, Colles FM, Strachan NJC, Ogden ID, Forbes K, French NP, Carter P, Miller WG, McCarthy ND, Owen R, Litrup E, Egholm M, Affourtit JP, Bentley SD, Parkhill J, Maiden MCJ, Falush D. 2013. Progressive genome-wide introgression in agricultural Campylobacter coli. Mol Ecol 22:1051–1064. doi: 10.1111/mec.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salipante SJ, Roach DJ, Kitzman JO, Snyder MW, Stackhouse B, Butler-Wu SM, Lee C, Cookson BT, Shendure J. 2015. Large-scale genomic sequencing of extraintestinal pathogenic Escherichia coli strains. Genome Res 25:119–128. doi: 10.1101/gr.180190.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.von Mentzer A, Connor TR, Wieler LH, Semmler T, Iguchi A, Thomson NR, Rasko DA, Joffre E, Corander J, Pickard D, Wiklund G, Svennerholm AM, Sjöling Å, Dougan G. 2014. Identification of enterotoxigenic Escherichia coli (ETEC) clades with long-term global distribution. Nat Genet 46:1321–1326. doi: 10.1038/ng.3145. [DOI] [PubMed] [Google Scholar]
- 14.Hazen TH, Sahl JW, Fraser CM, Donnenberg MS, Scheutz F, Rasko DA. 2013. Refining the pathovar paradigm via phylogenomics of the attaching and effacing Escherichia coli. Proc Natl Acad Sci U S A 110:12810–12815. doi: 10.1073/pnas.1306836110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruan L, Crickmore N, Peng D, Sun M. 2015. Are nematodes a missing link in the confounded ecology of the entomopathogen Bacillus thuringiensis? Trends Microbiol 23:341–346. doi: 10.1016/j.tim.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 16.Raymond B, Bonsall MB. 2013. Cooperation and the evolutionary ecology of bacterial virulence: the Bacillus cereus group as a novel study system. Bioessays 35:706–716. doi: 10.1002/bies.201300028. [DOI] [PubMed] [Google Scholar]
- 17.Dragon DC, Rennie RP. 1995. The ecology of anthrax spores: tough but not invincible. Can Vet J 36:295–301. [PMC free article] [PubMed] [Google Scholar]
- 18.Muoria PK, Muruthi P, Kariuki WK, Hassan BA, Mijele D, Oguge NO. 2007. Anthrax outbreak among Grevy’s zebra (Equus grevyi) in Samburu, Kenya. Afr J Ecol 45:483–489. doi: 10.1111/j.1365-2028.2007.00758.x. [DOI] [Google Scholar]
- 19.Stenfors Arnesen LP, Fagerlund A, Granum PE. 2008. From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol Rev 32:579–606. doi: 10.1111/j.1574-6976.2008.00112.x. [DOI] [PubMed] [Google Scholar]
- 20.Rasko DA, Altherr MR, Han CS, Ravel J. 2005. Genomics of the Bacillus cereus group of organisms. FEMS Microbiol Rev 29:303–329. [DOI] [PubMed] [Google Scholar]
- 21.Raymond B, Wyres KL, Sheppard SK, Ellis RJ, Bonsall MB. 2010. Environmental factors determining the epidemiology and population genetic structure of the Bacillus cereus group in the field. PLoS Pathog 6:e1000905. doi: 10.1371/journal.ppat.1000905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cardazzo B, Negrisolo E, Carraro L, Alberghini L, Patarnello T, Giaccone V. 2008. Multiple-locus sequence typing and analysis of toxin genes in Bacillus cereus food-borne isolates. Appl Environ Microbiol 74:850–860. doi: 10.1128/AEM.01495-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Didelot X, Barker M, Falush D, Priest FG. 2009. Evolution of pathogenicity in the Bacillus cereus group. Syst Appl Microbiol 32:81–90. doi: 10.1016/j.syapm.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 24.Priest FG, Barker M, Baillie LW, Holmes EC, Maiden MC. 2004. Population structure and evolution of the Bacillus cereus group. J Bacteriol 186:7959–7970. doi: 10.1128/JB.186.23.7959-7970.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schnepf E, Crickmore N, Van Rie J, Lereclus D, Baum J, Feitelson J, Zeigler DR, Dean DH. 1998. Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol Mol Biol Rev 62:775–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakroun M, Banyuls N, Bel Y, Escriche B, Ferré J. 2016. Bacterial vegetative insecticidal proteins (Vip) from entomopathogenic bacteria. Microbiol Mol Biol Rev 80:329–350. doi: 10.1128/MMBR.00060-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Frankenhuyzen K. 2009. Insecticidal activity of Bacillus thuringiensis crystal proteins. J Invertebr Pathol 101:1–16. doi: 10.1016/j.jip.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Lecadet MM, Frachon E, Dumanoir VC, Ripouteau H, Hamon S, Laurent P, Thiéry I. 1999. Updating the H-antigen classification of Bacillus thuringiensis. J Appl Microbiol 86:660–672. doi: 10.1046/j.1365-2672.1999.00710.x. [DOI] [PubMed] [Google Scholar]
- 29.Riadi G, Medina-Moenne C, Holmes DS. 2012. TnpPred: a web service for the robust prediction of prokaryotic transposases. Comp Funct Genomics 2012:678761. doi: 10.1155/2012/678761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Frankenhuyzen K. 2013. Cross-order and cross-phylum activity of Bacillus thuringiensis pesticidal proteins. J Invertebr Pathol 114:76–85. doi: 10.1016/j.jip.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 31.Wirth MC, Georghiou GP, Federici BA. 1997. CytA enables CryIV endotoxins of Bacillus thuringiensis to overcome high levels of CryIV resistance in the mosquito, Culex quinquefasciatus. Proc Natl Acad Sci U S A 94:10536–10540. doi: 10.1073/pnas.94.20.10536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zheng J, Peng D, Ruan L, Sun M. 2013. Evolution and dynamics of megaplasmids with genome sizes larger than 100 kb in the Bacillus cereus group. BMC Evol Biol 13:262. doi: 10.1186/1471-2148-13-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu L, Peng D, Wang Y, Ye W, Zheng J, Zhao C, Han D, Geng C, Ruan L, He J, Yu Z, Sun M. 2015. Genomic and transcriptomic insights into the efficient entomopathogenicity of Bacillus thuringiensis. Sci Rep 5:14129. doi: 10.1038/srep14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang MJ, Bideshi DK, Park HW, Federici BA. 2006. Minireplicon from pBtoxis of Bacillus thuringiensis subsp. israelensis. Appl Environ Microbiol 72:6948–6954. doi: 10.1128/AEM.00976-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zwick ME, Joseph SJ, Didelot X, Chen PE, Bishop-Lilly KA, Stewart AC, Willner K, Nolan N, Lentz S, Thomason MK, Sozhamannan S, Mateczun AJ, Du L, Read TD. 2012. Genomic characterization of the Bacillus cereus sensu lato species: backdrop to the evolution of Bacillus anthracis. Genome Res 22:1512–1524. doi: 10.1101/gr.134437.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soufiane B, Côté JC. 2010. Bacillus thuringiensis serovars Bolivia, vazensis and navarrensis meet the description of Bacillus weihenstephanensis. Curr Microbiol 60:343–349. doi: 10.1007/s00284-009-9547-z. [DOI] [PubMed] [Google Scholar]
- 37.Richter M, Rosselló-Móra R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roh JY, Choi JY, Li MS, Jin BR, Je YH. 2007. Bacillus thuringiensis as a specific, safe, and effective tool for insect pest control. J Microbiol Biotechnol 17:547–559. [PubMed] [Google Scholar]
- 39.Lechner S, Mayr R, Francis KP, Prüss BM, Kaplan T, Wiessner-Gunkel E, Stewart GS, Scherer S. 1998. Bacillus weihenstephanensis sp. nov. is a new psychrotolerant species of the Bacillus cereus group. Int J Syst Bacteriol 48:1373–1382. doi: 10.1099/00207713-48-4-1373. [DOI] [PubMed] [Google Scholar]
- 40.Guinebretière MH, Velge P, Couvert O, Carlin F, Debuyser ML, Nguyen-The C. 2010. Ability of Bacillus cereus group strains to cause food poisoning varies according to phylogenetic affiliation (groups I to VII) rather than species affiliation. J Clin Microbiol 48:3388–3391. doi: 10.1128/JCM.00921-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crickmore N, Zeigler DR, Feitelson J, Schnepf E, Van Rie J, Lereclus D, Baum J, Dean DH. 1998. Revision of the nomenclature for the Bacillus thuringiensis pesticidal crystal proteins. Microbiol Mol Biol Rev 62:807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sauka DH, Sánchez J, Bravo A, Benintende GB. 2007. Toxicity of Bacillus thuringiensis delta-endotoxins against bean shoot borer (Epinotia aporema Wals.) larvae, a major soybean pest in Argentina. J Invertebr Pathol 94:125–129. doi: 10.1016/j.jip.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 43.Carrière Y, Crickmore N, Tabashnik BE. 2015. Optimizing pyramided transgenic Bt crops for sustainable pest management. Nat Biotechnol 33:161–168. doi: 10.1038/nbt.3099. [DOI] [PubMed] [Google Scholar]
- 44.Bravo A, Gill SS, Soberón M. 2007. Mode of action of Bacillus thuringiensis Cry and Cyt toxins and their potential for insect control. Toxicon 49:423–435. doi: 10.1016/j.toxicon.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raymond B, Wright DJ, Crickmore N, Bonsall MB. 2013. The impact of strain diversity and mixed infections on the evolution of resistance to Bacillus thuringiensis. Proc Biol Sci 280:20131497. doi: 10.1098/rspb.2013.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cantón PE, Zanicthe Reyes EZ, Ruiz de Escudero I, Bravo A, Soberón M. 2011. Binding of Bacillus thuringiensis subsp. israelensis Cry4Ba to Cyt1Aa has an important role in synergism. Peptides 32:595–600. doi: 10.1016/j.peptides.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hernández-Soto A, Del Rincón-Castro MC, Espinoza AM, Ibarra JE. 2009. Parasporal body formation via overexpression of the Cry10Aa toxin of Bacillus thuringiensis subsp. israelensis, and Cry10Aa-Cyt1Aa synergism. Appl Environ Microbiol 75:4661–4667. doi: 10.1128/AEM.00409-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu D, Johnson JJ, Federici BA. 1994. Synergism of mosquitocidal toxicity between CytA and CrylVD proteins using inclusions produced from cloned genes of Bacillus thuringiensis. Mol Microbiol 13:965–972. doi: 10.1111/j.1365-2958.1994.tb00488.x. [DOI] [PubMed] [Google Scholar]
- 49.Zhao JZ, Cao J, Li YX, Collins HL, Roush RT, Earle ED, Shelton AM. 2003. Transgenic plants expressing two Bacillus thuringiensis toxins delay insect resistance evolution. Nat Biotechnol 21:1493–1497. doi: 10.1038/nbt907. [DOI] [PubMed] [Google Scholar]
- 50.Georghiou GP, Wirth MC. 1997. Influence of exposure to single versus multiple toxins of Bacillus thuringiensis subsp. israelensis on development of resistance in the mosquito Culex quinquefasciatus (Diptera: Culicidae). Appl Environ Microbiol 63:1095–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tabashnik BE, Brévault T, Carrière Y. 2013. Insect resistance to Bt crops: lessons from the first billion acres. Nat Biotechnol 31:510–521. doi: 10.1038/nbt.2597. [DOI] [PubMed] [Google Scholar]
- 52.Zhang WF, Crickmore N, George Z, Xie L, He YQ, Li YZ, Tang JL, Tian L, Wang X, Fang XJ. 2012. Characterization of a new highly mosquitocidal isolate of Bacillus thuringiensis—an alternative to Bti? J Invertebr Pathol 109:217–222. doi: 10.1016/j.jip.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 53.Guan P, Ai P, Dai XJ, Zhang J, Xu LZ, Zhu J, Li Q, Deng QM, Li SC, Wang SQ, Liu HN, Wang LX, Li P, Zheng AP. 2012. Complete genome sequence of Bacillus thuringiensis serovar Sichuansis strain MC28. J Bacteriol 194:6975–6975. doi: 10.1128/JB.01861-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohgushi A, Wasano N, Shisa N, Saitoh H, Mizuki E, Maeda M, Ohba M. 2003. Characterization of a mosquitocidal Bacillus thuringiensis serovar sotto strain isolated from Okinawa, Japan. J Appl Microbiol 95:982–989. doi: 10.1046/j.1365-2672.2003.02068.x. [DOI] [PubMed] [Google Scholar]
- 55.Juárez-Pérez V, Porcar M, Orduz S, Delécluse A. 2003. Cry29A and Cry30A: two novel delta-endotoxins isolated from Bacillus thuringiensis serovar medellin. Syst Appl Microbiol 26:502–504. doi: 10.1078/072320203770865783. [DOI] [PubMed] [Google Scholar]
- 56.Wei JZ, Hale K, Carta L, Platzer E, Wong C, Fang SC, Aroian RV. 2003. Bacillus thuringiensis crystal proteins that target nematodes. Proc Natl Acad Sci U S A 100:2760–2765. doi: 10.1073/pnas.0538072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Doggett NA, Stubben CJ, Chertkov O, Bruce DC, Detter JC, Johnson SL, Han CS. 2013. Complete genome sequence of Bacillus thuringiensis serovar Israelensis strain HD-789. Genome Announc 1:e01023-13. doi: 10.1128/genomeA.01023-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.He J, Wang J, Yin W, Shao X, Zheng H, Li M, Zhao Y, Sun M, Wang S, Yu Z. 2011. Complete genome sequence of Bacillus thuringiensis subsp. chinensis strain CT-43. J Bacteriol 193:3407–3408. doi: 10.1128/JB.05085-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murawska E, Fiedoruk K, Bideshi DK, Swiecicka I. 2013. Complete genome sequence of Bacillus thuringiensis subsp. thuringiensis strain IS5056, an isolate highly toxic to Trichoplusia ni. Genome Announc 1:e0010813. doi: 10.1128/genomeA.00108-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith J. 2001. The social evolution of bacterial pathogenesis. Proc Biol Sci 268:61–69. doi: 10.1098/rspb.2000.1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Okinaka RT, Cloud K, Hampton O, Hoffmaster AR, Hill KK, Keim P, Koehler TM, Lamke G, Kumano S, Mahillon J, Manter D, Martinez Y, Ricke D, Svensson R, Jackson PJ. 1999. Sequence and organization of pXO1, the large Bacillus anthracis plasmid harboring the anthrax toxin genes. J Bacteriol 181:6509–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ehling-Schulz M, Fricker M, Grallert H, Rieck P, Wagner M, Scherer S. 2006. Cereulide synthetase gene cluster from emetic Bacillus cereus: structure and location on a mega virulence plasmid related to Bacillus anthracis toxin plasmid pXO1. BMC Microbiol 6:20. doi: 10.1186/1471-2180-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pomerantsev AP, Camp A, Leppla SH. 2009. A new minimal replicon of Bacillus anthracis plasmid pXO1. J Bacteriol 191:5134–5146. doi: 10.1128/JB.00422-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raymond B, West SA, Griffin AS, Bonsall MB. 2012. The dynamics of cooperative bacterial virulence in the field. Science 337:85–88. doi: 10.1126/science.1218196. [DOI] [PubMed] [Google Scholar]
- 65.Raymond B, Davis D, Bonsall MB. 2007. Competition and reproduction in mixed infections of pathogenic and non-pathogenic Bacillus spp. J Invertebr Pathol 96:151–155. doi: 10.1016/j.jip.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 66.Kelley DR, Schatz MC, Salzberg SL. 2010. Quake: quality-aware detection and correction of sequencing errors. Genome Biol 11:R116. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res 19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 69.Li L, Stoeckert CJ Jr., Roos DS. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cheng L, Connor TR, Sirén J, Aanensen DM, Corander J. 2013. Hierarchical and spatially explicit clustering of DNA sequences with BAPS software. Mol Biol Evol 30:1224–1228. doi: 10.1093/molbev/mst028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ye W, Zhu L, Liu Y, Crickmore N, Peng D, Ruan L, Sun M. 2012. Mining new crystal protein genes from Bacillus thuringiensis on the basis of mixed plasmid-enriched genome sequencing and a computational pipeline. Appl Environ Microbiol 78:4795–4801. doi: 10.1128/AEM.00340-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Demchak B, Hull T, Reich M, Liefeld T, Smoot M, Ideker T, Mesirov JP. 2014. Cytoscape: the network visualization tool for GenomeSpace workflows. F1000Res 3:151. doi: 10.12688/f1000research.4492.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
B. thuringiensis strains used in this study. Download TABLE S1, XLSX file, 0.02 MB (21.1KB, xlsx) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Population structure of the B. cereus group. Four clades are represented by four different background colors of the tree labels. B. thuringiensis strains are red. The binary squares and circles represent the presence and absence of different plasmids. From the innermost circle to the outermost circle, solid squares and circles represent plasmids with orf156/orf157, rep228, rep466, repX, pXO1-14/pXO1-16, repA, ori43, ori44, ori60, CT43_P825207-like, CT43_P851308-like, rep165, and pBMB2062-like minireplicons. Download FIG S1, PDF file, 1.9 MB (2MB, pdf) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Details about Cry proteins predicted in B. thuringiensis. Download TABLE S2, DOCX file, 0.02 MB (24.3KB, docx) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differences between the cry gene numbers of the two major groups. Download FIG S2, TIF file, 0.1 MB (146.6KB, tif) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Co-occurrence network of all of the Cry, Cyt, and Vip proteins with known hosts from the B. thuringiensis Toxin Nomenclature database (http://btnomenclature.info/). Only strains with more than two toxin genes displayed in the database were analyzed. Download FIG S3, PDF file, 2.1 MB (2.2MB, pdf) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Synteny analysis of plasmids containing the orf156/orf157 minireplicon based on complete and draft genomes. (A) Plasmids having BtPAI-I or BtPAI-II. From the innermost circle to the outermost circle, pCT281; pIS56-285 (thuringiensis); pBMB293 (kurstaki); pBMB29 (kurstaki); and plasmids from different strains of sumiyoshiensis, fukuokaensis, andalousiensis, galleriae, galleriae, tolworthi, tolworthi, tolworthi, toumanoffi, thompsoni, colmeri, thuringiensis, aizawai, aizawai, aizawai, aizawai, aizawai, NBIN-866, and BtPAI-I/II are represented (2). Plasmids of B. thuringiensis are toxic to Diptera. From the innermost circle to the outermost circle, pBtoxis (israelensis); pBTHD789-3 (israelensis); and plasmids from novosibirsk, medellin, and IBL4222 (israelensis-like) are represented. Download FIG S4, PDF file, 0.5 MB (508.2KB, pdf) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Optical microscopy of B. thuringiensis strains with no toxin genes predicted from their genome sequences. Download FIG S5, PDF file, 0.4 MB (419.2KB, pdf) .
Copyright © 2017 Zheng et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.