

Abstract
AIM
To investigate the relation of two different mutations to the outcome of partial external biliary diversion (PEBD) in severe bile salt export pump (BSEP) deficiency.
METHODS
Mutations in the gene encoding BSEP leading to severe BSEP deficiency in two unrelated patients were identified by genomic sequencing. Native liver biopsies and transiently transfected human embryonic kidney (HEK) 293 cells expressing either wild-type or mutated BSEP were subjected to immunofluorescence analysis to assess BSEP transporter localization. Bile acid profiles of patient and control bile samples were generated by ultra-performance liquid chromatography-tandem mass spectrometry. Wild-type and mutant BSEP transport of [3H]-labeled taurocholate (TC) and taurochenodeoxycholate (TCDC) was assessed by vesicular transport assays.
RESULTS
A girl (at 2 mo) presented with pruritus, jaundice and elevated serum bile salts (BS). PEBD stabilized liver function and prevented liver transplantation. She was heterozygous for the BSEP deletion p.T919del and the nonsense mutation p.R1235X. At the age of 17 years relative amounts of conjugated BS in her bile were normal, while total BS were less than 3% as compared to controls. An unrelated boy (age 1.5 years) presenting with severe pruritus and elevated serum BS was heterozygous for the same nonsense and another missense mutation, p.G1032R. PEBD failed to alleviate pruritus, eventually necessitating liver transplantation. BS concentration in bile was about 5% of controls. BS were mainly unconjugated with an unusual low amount of chenodeoxycholate derivatives (< 5%). The patients’ native liver biopsies showed canalicular BSEP expression. Both BSEP p.T919del and p.G1032R were localized in the plasma membrane in HEK293 cells. In vitro transport assays showed drastic reduction of transport by both mutations. Using purified recombinant BSEP as quantifiable reference, per-molecule transport rates for TC and TCDC were determined to be 3 and 2 BS molecules per wild-type BSEP transporter per minute, respectively.
CONCLUSION
In summary, our findings suggest that residual function of BSEP as well as substrate specificity influence the therapeutic effectiveness of PEBD in progressive familial intrahepatic cholestasis type 2 (PFIC-2).
Keywords: Familial intrahepatic cholestasis type 2, Partial external biliary diversion, Bile salt export pump, ATP binding cassette transporter, Intrahepatic cholestasis
Core tip: PFIC-2, a severe form of BSEP deficiency, is caused by mutations of the bile salt export pump (BSEP), an ATP binding cassette transporter exclusively expressed in hepatocytes. In this study we determine the functional impact of two distinct BSEP mutations on transporter localization and function in two unrelated PFIC-2 patients showing a drastically different response to partial external biliary diversion (PEBD). Our data demonstrate that residual bile salt transport by BSEP is likely required for a beneficial outcome of PEBD.
INTRODUCTION
Chronic cholestasis is a major cause of severe liver disease in young children. Apart from biliary atresia, four types of progressive familial intrahepatic cholestasis (PFIC) are currently diagnosed. While PFIC-4 is caused by leaky bile canalicular tight junctions due to mutations in the TJP2 gene[1], PFIC-1 to -3 are caused by inherited defects in transporter proteins residing in the canalicular membrane of hepatocytes. Mutations in the ATP8B1 gene encoding a phosphatidylserine flippase (FIC1) cause PFIC-1[2,3]. PFIC-2 is linked to mutations in ABCB11 encoding the bile salt export pump (BSEP)[4]. BSEP is an ATP binding cassette transporter and transports conjugated bile salts (BS), with highest transport capacity for tauro-/glycochenodeoxycholic acid (TCDC/GCDC) and taurocholic acid (TC)[5]. Defects in canalicular BSEP functionality cause severe liver disease by compromising BS excretion into bile, while intracellular and serum BS concentrations rise to toxic levels. Milder forms of BSEP disease have been denoted as benign recurrent intrahepatic cholestasis (BRIC). BRIC is characterised by self-limiting cholestatic episodes with pruritus. BRIC-associated mutations are in general less severe as compared to PFIC-related mutations, which more often affect transmembrane helices[6] and which include nonsense and frame-shift mutations[7].
PFIC-3 is caused by mutations of the multidrug resistance protein 3 (MDR3) encoded by the ABCB4 gene[8-10]. MDR3 transports phosphatidylcholine-type lipids from the inner to the outer canalicular membrane leaflet, from where they are likely extracted by BS to form mixed micelles with cholesterol[11]. The flip of phosphatidylserine from the outer to the inner leaflet by the ATP8B1 gene product compensates this membrane destabilization[12]. In PFIC-1 and PFIC-2 γ-glutamyltransferase (GT) levels are normal, which are attributed to low concentrations of free toxic BS in bile of these patients[13], whereas PFIC-3 is characterized by elevated serum γGT.
Many PFIC patients receive ursodeoxycholic acid (UDCA) as a first-line treatment. UDCA stimulates BSEP expression, inhibits BS synthesis and stimulates BS clearance via urine[14]. When symptoms do not improve sufficiently, partial external biliary diversion (PEBD) may be considered as a surgical intervention to reduce the toxic bile salt pool in PFIC-1 and -2[15]. The continuous removal of bile may alleviate the otherwise intractable pruritus associated with PFIC, improving quality of life as well as prolonging transplant-free survival[15]. While many PFIC-1 and -2 patients improve upon PEBD, some fail to benefit from the procedure. The reasons for the different therapeutic responses are not well understood.
We investigated the functional properties of two BSEP missense mutations causing PFIC-2 in two unrelated children that responded fundamentally different to UDCA and PEBD treatment.
MATERIALS AND METHODS
Bile sample collection and analysis
The study was performed according to the guidelines of the declaration of Helsinki and informed written consent was obtained from patients or their parents. Bile samples from both patients were collected by external drainage. Bile samples from six non-cholestatic patients were collected intraoperatively. These patients were operated for oncological reasons and bile was collected from the common bile duct before routine gallbladder removal. Six bile samples from the common bile ducts were collected from patients with obstructive cholestasis (2 × PSC, 2 × choledocholithiasis, 1 × benign and 1 × malignant bile duct obstruction) during endoscopic retrograde cholangiography (ERC).
Solid phase extraction of bile acids and analysis of BS profiles by liquid chromatography-tandem mass spectrometry (LC-MS/MS) was performed as described previously[6].
Sequencing of ABCB11 (BSEP, NM_003742.2), ATP8B1 (FIC1, AF038007) and ABCB4 (MDR3, NM_018849.2) was performed using genomic DNA extracted from EDTA blood and standard sequencing as described recently[16,17]. Exons containing the start codon were defined as the exon one and the adenine of the start codon was counted as “one”.
Immunofluorescence staining of liver biopsies and transfected HEK293 cells was performed as described[17,18]. Liver biopsies were stained using the K24 antiserum for BSEP[19], P3II-26 for MDR3 and a mix of antibodies M2I-4 and M2III-6 for MRP2 (Enzo Life Sciences). Methanol-fixed HEK293 cells were stained using the BSEP antibody F-6 (Santa Cruz Biotechnology) and the Na+/K+-ATPase antibody M7-PB-E9 (Sigma Aldrich). Appropriate fluorophore-labelled secondary antibodies were used.
Cloning of BSEP and mutagenesis for expression in HEK293 cells
For expression of human BSEP, a tag-less version of plasmid pEYFP-N1-OriLeu-BSEP[6] was generated resulting in a BSEP reading frame without any artificial tags or cloning artifacts as determined by gene sequencing. Deletion of the EYFP-tag and introduction of the p.T919del and p.G1032R mutations were performed using the DREAM method[20]. For immunofluorescence studies, HEK293 cells seeded onto coverslips were transfected at sub-confluence with 2 μg of plasmids using polyethyleneimine (MW 25 kDa; Sigma Aldrich). For isolation of plasma membrane vesicles, cells were transfected at a confluence of 30%-40% with 30 μg of each plasmid per 15-cm diameter cell culture dish and cultured for 48 h.
Isolation of human embryonic kidney 293 plasma membrane vesicles
Transfected cells were collected, re-suspended in ice-cold hypotonic buffer (10 mmol/L Tris-HCl pH 8.0, 10 mmol/L KCl, 5 mmol/L EDTA) containing protease inhibitor cocktail (Roche) and incubated for 30 min at 4 °C on a rotator. All subsequent steps were carried out at 4 °C. Cells were disrupted with ten strokes using a tight-fitting Potter-Elvehjem homogenizer. Cellular debris was spun down for 10 min at 400 × g and the supernatant was further centrifuged at 1000 × g for 10 min. The resulting supernatant was centrifuged at 265000 × g in a Ti70 rotor (Beckman). The membrane pellet was re-suspended in 50 mmol/L Hepes-Tris, pH 7.4, 100 mmol/L KNO3, 50 mmol/L sucrose, snap-frozen in liquid nitrogen and stored at -80 °C until further use.
Quantification of BSEP in plasma membrane vesicles
Human BSEP was expressed in Pichia pastoris and purified as described in Ellinger et al[21]. Protein concentrations were determined using the Coomassie Plus Assay (Thermo Scientific). A specified amount of purified BSEP was loaded onto a 7% SDS-PAGE gel together with 20 μg of each plasma membrane vesicle preparation. Samples were incubated in 3% SDS for 30 min at 37 °C, supplemented with 1/4 volume of 5 × reducing Laemmli sample buffer and further incubated for 10 min at 65 °C. SDS-PAGE and Western blotting was performed as described[21]. BSEP was detected by the F-6 antibody and a goat anti-mouse-HRP secondary antibody (Jackson ImmunoResearch), using the Western lightning plus ECL Kit (Perkin Elmer). Images were acquired using the Chemigenius2 detection system (Syngene) according to the manufacturer’s guidelines. The time of exposition was adjusted to prevent signal saturation. Signals were subsequently quantified with the GeneTools analysis software (V.3.06; Syngene) by comparing bands corresponding to known amounts of purified BSEP with bands in the lanes containing vesicle preparations.
Vesicular transport studies were essentially performed as described in Herédi-Szabó et al[22]. Reactions were set up to contain equal BSEP amounts in a final reaction volume of 70 μL and included either 2 μmol/L [3H]-TC or [3H]-TCDC. Transport was started by addition of ATP or transport buffer as background control. Reactions were incubated at 37 °Cfor 5 min or 30 min and stopped with ice-cold washing buffer followed by rapid filtration through 0.22 μm mixed cellulose membrane filters (type GSTF; Millipore). Filters were air-dried and suspended in Ultima GoldTM scintillation liquid in polypropylene vials (Perkin Elmer) for liquid scintillation counting. BSEP-dependent transport was calculated by subtracting the amount of radioactivity in BSEP-containing vesicles obtained in the presence of ATP/Mg2+ from that obtained in the absence of ATP.
RESULTS
Two unrelated PFIC-2 patients with different responses to UDCA treatment and PEBD
A two-months-old girl presented with non-obstructive cholestasis und severe pruritus. γGT levels were normal, while serum BS were strongly elevated (150 μmol/L). A liver biopsy at the age of 1 year showed no signs of inflammation but some fibrotic thickening of the portal tracts together with intra- and intercellular cholestasis. Therefore PFIC was suspected and at the age of five PEBD was initiated to avert further cholestatic liver damage. Serum BS decreased and symptoms completely resolved. At the age of 17 years, a moderate deterioration of liver function was observed. Fibroscan was 14.1 kPa reflecting fibrosis[23] or cholestasis[24]. A liver biopsy (for immunostaining), a DNA sample and diverted bile were taken and analysed. Because a low-γGT PFIC was suspected, sequencing of ATP8B1 (FIC1) and ABCB11 (BSEP) was performed. While ATP8B1 only showed two heterozygous intronic variants, sequencing of ABCB11 revealed a heterozygous nonsense mutation (c.3703C>T; p.R1235X) and a heterozygous triplet deletion (c.2756_2758delCCA), causing loss of a single threonine at amino acid position 919 (p.T919del). Genetic analysis of the healthy parents indicated paternal inheritance of the nonsense mutation and maternal inheritance of the deletion. The patient continued to be symptom-free under PEBD without the need for liver transplantation in the continuous presence of UDCA treatment.
An unrelated boy of 18 mo presented with non-obstructive cholestasis and severe pruritus and total serum BS were increased (371 μmol/L). PFIC was suspected, however, UDCA treatment did not alleviate pruritus. PEBD was performed at two years and seven months of age, which led to a drop of serum bile salts from 412 μmol/L (the day before PEBD) to 207 μmol/L (5 d post-OP), 70 μmol/L (7 mo after PEBD) and 93 μmol/L (1 year after PEBD). Symptoms only improved transiently. A biopsy taken during the PEBD showed no signs of fibrosis. Sequencing of ATP8B1 (FIC1) showed the common heterozygous variant p.R952Q and the common homozygous variant p.N168N was found in the ABCB4 gene. ABCB11 sequencing, however, uncovered the same heterozygous nonsense mutation (c.3703C>T; p.R1235X) as found in the first patient along with a heterozygous missense mutation (c.3094G>C) leading to an amino acid change from glycine to arginine at position 1032 (p.G1032R). Genetic testing of both healthy parents showed paternal inheritance of the nonsense and maternal inheritance of the missense mutation. At the age of four, this patient had to be transplanted due to his intractable pruritus despite PEBD. Histology of the explant confirmed absence of cirrhosis.
Bile salt analysis of serum and bile confirm defects in bile salt transport
Total BS concentration in bile of the female patient (p.T919del) was 1531 μmol/L (including 931 μmol/L of UDCA derivatives), which is only 2.5% of total biliary BS in non-cholestatic patients (Table 1 and Figure 1). As in controls and in patients with obstructive cholestasis, we found almost no unconjugated BS in her bile. Notably, the ratio between taurine- and glycine-conjugated bile acids in the patient’s bile was very similar to the ratio in controls (Figure 1A). Likewise, the fraction of CDCA derivatives was comparable to that in controls. The fraction of cholic acid (CA) derivatives was higher in the patient than in controls. This can be attributed to partial disruption of the enterohepatic circulation by PEBD. In healthy individuals, CA and its derivatives are dehydroxylated in the intestine by bacteria to deoxycholic acid (DCA), which is reabsorbed. Indeed, the combined relative amount of DCA and CA derivatives in controls is of the same magnitude as that of CA in the patient. Total serum BS concentration was 150 μmol/L (normal: < 8 μmol/L) including UDCA-derivatives, while relative amounts of single BS species were similar to values in controls. As in bile, DCA and derivatives were absent in the patient’s serum. Taken together, the findings suggest a severe reduction markedly reduced canalicular BSEP function in the presence of the p.T919del mutation despite UDCA treatment, which is known to stabilize BSEP and enhance its expression[14].
Table 1.
Concentrations of bile acids in serum and bile
|
Serum |
Bile |
||||||
| T919del | G1032R | Controls (n = 33) | T919del | G1032R | Controls (n = 6) | Cholestasis (n = 6) | |
| CA | 0.15 ± 0.09 | 2232 | 15 ± 9 | 88 ± 72 | |||
| Tauro-CA | 5.4 | 7.2 | 0.15 ± 0.17 | 85.7 | 549 | 3721 ± 2597 | 754 ± 405 |
| Glyco-CA | 25 | 39 | 0.28 ± 0.31 | 274.5 | 552 | 8189 ± 5978 | 1133 ± 662 |
| CDCA | 0.13 ± 0.12 | 118 | 120 ± 48 | ||||
| Tauro-CDCA | 5.2 | 6.2 | 0.17 ± 0.17 | 54.2 | 3840 ± 2005 | 646 ± 396 | |
| Glyco-CDCA | 26 | 18 | 0.49 ± 0.60 | 185.7 | 8285 ± 8306 | 1100 ± 811 | |
| DCA | 0.25 ± 0.29 | 35 ± 19 | |||||
| Tauro-DCA | 0.11 ± 0.08 | 3028 ± 2562 | 351 ± 328 | ||||
| Glyco-DCA | 0.24 ± 0.25 | 5777 ± 4287 | 537 ± 674 | ||||
| UDCA | 0.01 | 0.11 ± 0.04 | 146 | ||||
| Tauro-UDCA | 7.5 | 77.4 | 357 ± 218 | 384 ± 591 | |||
| Glyco-UDCA | 81 | 0.36 ± 0.18 | 853.2 | 861 ± 635 | 1403 ± 1877 | ||
| total | 150.1 | 70.4 | 1.37 ± 1.61 | 1531 | 3451 | 33603 ± 23354 | 5965 ± 4447 |
| % CDCAderivatives | 21 | 34 | 36 ± 16 | 40 | 3 | 38 ± 8 | 44 ± 5 |
| bile/serum (μmol/L/μmol/L) | 10.2 | 49 | ca. 24500 | ||||
Bile salt derivatives in bile of the female patient (p.T919del) and the male patient (p.G1032R), in six non-cholestatic patients and in six patients with obstructive cholestasis of different causes. Serum control values are from 33 non-cholestatic patients. Values are given in μmol/L. CA: Cholic acid; UDCA: Ursodeoxycholic acid.
Figure 1.
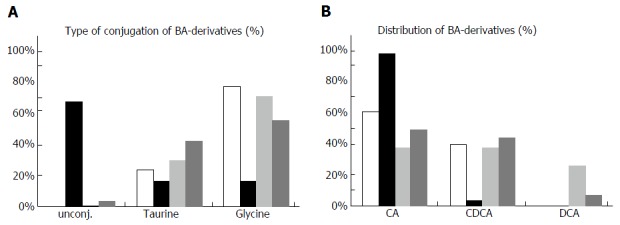
Bile acid derivatives in bile samples of the two patients. Concentrations of different bile acid derivatives were measured in bile samples of two patients possessing the bile salt export pump (BSEP)-T919del and BSEP-G1032R mutation, respectively, as well as in six non-cholestatic and in six cholestatic control samples (Table 1). A: Relative amounts of unconjugated, taurine- or glycine-conjugated bile acids are shown. In the background of the BSEP-G1032R mutation a very high amount ( > 60%) of unconjugated bile salts is found; B: Relative amounts of cholic, chenodeoxycholic and deoxycholic acid (CA, CDCA, DCA) including their corresponding conjugates are shown. In the background of the BSEP-G1032R less than 5% of bile acids are chenodeoxycholic acid and its conjugates (UDCA and conjugates were omitted from these plots). CA: Cholic acid; DCA: Deoxycholic acid; CDCA: Chenodeoxycholic acid.
Bile of the male patient (p.G1032R) showed a total BS amount of 3451 μmol/L (about 5% of controls; Table 1 and Figure 1). Surprisingly, 68% of these were unconjugated bile acids (Figure 1B). Furthermore, there were no conjugates of CDCA and only a minor proportion was unconjugated CDCA. CA and its conjugates amounted to more than 95% of bile acids in the patient’s bile. The absence of CDCA and its conjugates was not caused by defects in conjugation, because tauro- as well as glyco-CDCA was present in elevated amounts with a normal ratio of CDCA derivatives in the serum of the patient (Table 1). While the absolute amount of CA derivatives was 22% of controls, the total amount of CDCA and its derivatives was only 0.4% of controls. These data strongly suggest that the p.G1032R mutation also reduces canalicular BSEP function. The observed shift from CDCA to CA derivatives may be caused by an altered substrate specificity of BSEP, when the mutation p.G1032R is present. The predominance of unconjugated BS may be due to bacterial contamination of the biliary tract after PEBD and subsequent de-conjugation by bacterial enzymes, although the patient had no signs of bacterial cholangitis.
BSEP-T919del and BSEP-G1032R are correctly localized in vivo and and in vitro
Immunofluorescent staining showed normal canalicular expression of MDR3 and MRP2 (bilirubin transporter) in liver biopsies of both patients (Figure 2A). Since BSEP-R1235X lacks the C-terminal 87 amino acid residues containing essential motifs of the second nucleotide binding domain (Walker B, D-and H-loop), this premature stop results in a truncated, non-functional transporter. Using the K24 antiserum raised against the C-terminus of BSEP[19], only the full-length variants carrying the missense mutation or deletion alleles are detected. Both BSEP-T919del and BSEP-G1032R showed normal, canalicular localization. To further confirm this, both mutations were introduced into a mammalian BSEP expression plasmid and transiently transfected into HEK293 cells. Both BSEP-T919del and BSEP-G1032R showed a regular plasma membrane localization comparable to wild-type BSEP (Figure 2B).
Figure 2.
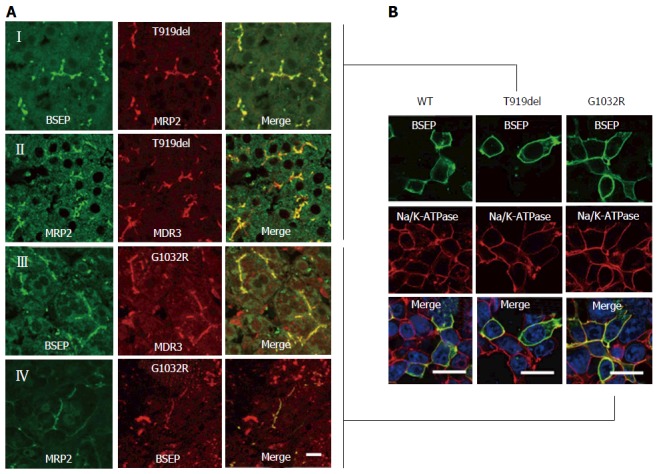
Immunofluorescence studies. A: Liver biopsy from the female patient possessing the BSEP-T919del mutation. (I) Immunoreactivity of BSEP (K24-antibody) was apparently normal as compared to the bilirubin transporter multidrug resistance- associated protein 2 (MRP2; M2I-4- and M2III-6-antibodies). (II) Immunoreactivity of MDR3 (P3II-26-antibody) showed a typical canalicular pattern similar to MRP2 (EAG5-antibody). Liver biopsy from the male patient (p.G1032R mutation) shows an almost regular BSEP expression (K24-antibody) and MDR3 (III) and MRP2 (IV); B: Wild-type BSEP is largely expressed at the cell membrane of human embryonic kidney (HEK293) cells. Likewise, both BSEP mutants, with deletion of threonine at position 919 (p.T919del) and with arginine instead of glycine at amino acid position 1032 (p.G1032R), are localized at the plasma membrane (Bars = 5 μm). BSEP: Bile salt export pump.
Bile salt transport by BSEP-T919del and BSEP-G1032R is impaired in vitro
Plasma membrane vesicles containing wild-type BSEP, BSEP-T919del or BSEP-G1032R were prepared from transiently transfected HEK293 cells. In order to determine a transport rate per BSEP rather than per total protein, we purified recombinant human BSEP from the yeast Pichia pastoris as described in detail in Ellinger et al[21]. Protein purity, as isolated by a two-step purification procedure, was more than 90% (Figure 3A, left panel). Using purified BSEP as quantitative standard, we quantified BSEP amounts in vesicle preparations by immunoblotting (Figure 3A, right panel for an example). Vesicular transport assays (Figure 3B) using equal amounts of each BSEP variant showed a strong reduction of TC and TCDC transport by BSEP-T919del and an even more pronounced reduction in transport for BSEP-G1032R. Notably, the amount of transported TC and TCDC increased from 5 min to 30 min for BSEP-T919del. In contrast, no increase of TC or TCDC accumulation over time was observed for BSEP-G1032R within experimental error.
Figure 3.
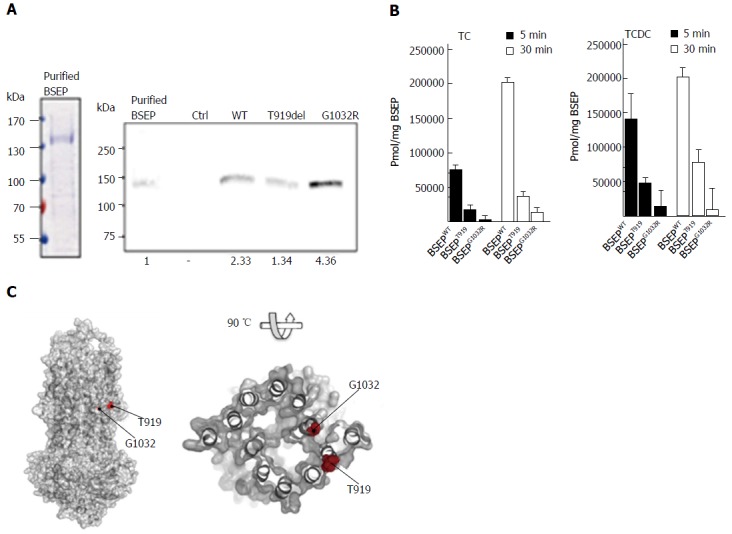
In vitro transport assay of wild-type bile salt export pump, BSEP-T919del and BSEP-G1032R. A: Left, Coomassie brilliant blue (CBB) stained SDS-PAGE gel of purified Bile Salt Export Pump (BSEP) expressed in Pichia pastoris. 9 μg protein was loaded on the lane. Right, Western blot of purified BSEP and membrane preparations of BSEP-T919del, BSEP-G1032R, BSEP wild-type (WT) and empty control (Ctrl). 38.5 ng of purified BSEP or 15 μL of the membrane preparations were subjected to SDS-PAGE. After detection with the monoclonal antibody, the gel was analyzed densitometrically to calculate the amount of wild-type or mutant BSEP or in the membrane preparations (relative densitometric values below panel); B: [3H]-taurocholate (TC) and [3H]-taurochenodeoxycholate (TCDC) transport by wild-type BSEP, BSEP-T919del, and BSEP-G1032R after 5 (black bars) and 30min (white bars). Experimental details are provided in Materials and Methods. The amount of translocated TC (left) or TCDC (right) per mg of BSEP was determined based on the quantification of wild-type BSEP, BSEP-T919del and BSEP-G1032R in the membrane vesicle preparations. All measurements represent the average with standard deviation of triplicate measurements; C: The homology model of BSEP was generated based on the outward-facing crystal structure of the multidrug transporter Sav18662 from S. aureus (pdb entry: 2HYD). The putative localisation of T919 at the outer surface of the transporter molecule and of G1032 close to the potential translocation pore of BSEP is highlighted by red spheres.Pmol/mg BSEP. BSEP: Bile salt export pump.
DISCUSSION
Both patients reported here suffered from severe cholestasis due to deficient canalicular transport of BS. It has been shown in earlier studies that absence of BSEP from the canalicular membrane is a diagnostic feature of PFIC-2/BSEP-disease[18,25,26], which is found in up to 72% of affected patients[25]. Reduced or absent canalicular BSEP expression can for example be caused by splicing defects[7,27,28], premature stop codons or by impaired targeting to the canalicular membrane[5,16]. In our patients canalicular BSEP expression was well detectable in liver biopsies and targeting was apparently normal as shown by transient expression of mutated BSEP in HEK293 cells (Figure 2). Instead, the transporter function itself is compromised. While BSEP-T919del showed a residual transport of both BS species (22% and 34% transport of TC and TCDC as compared to wild-type BSEP), the p.G1032R mutation essentially abolished transport of the conjugated bile salts TC and TCDC in vitro. Taking into account that the decrease of the bile-to-serum ratio for BS is less pronounced in the presence of BSEP-G1032R than of BSEP-T919del, it must be assumed that decreased transport capacity of BSEP-G1032R is at least partially compensated by a higher expression level. Nevertheless, the outcome after PEBD-treatment is worse in the boy carrying the p.G1032R mutation, which is otherwise a well-established therapy in severe BSEP-disease[29,30]. Interestingly, determination of BS species in the bile samples of the two patients showed a marked shift in the composition of single bile salts in the boy’s sample. Notably, there are less than 5% of conjugated bile salts and CA outbalances CDCA. The latter finding is in contrast with the known substrate specificity of human BSEP[5], indicating a significant shift in specificity of the mutated transporter.
The presence of unconjugated BS may theoretically be due to de-conjugation within the biliary tract, although clinically there was no evidence of cholangitis or bacterial contamination of bile ducts and gallbladder. The lack of CDCA and its conjugates is not due to a defect in BS synthesis, since CDCA species were found in the serum in a normal distribution. Bile analysis of the BSEP-G1032R affected patient revealed that more unconjugated than conjugated cholate is transported into bile. Considering that the intracellular amount of unconjugated cholate in general is much lower than that of taurine- or glycine-conjugated cholate, it must be assumed that unconjugated cholate is transported much better than conjugated cholate by BSEP-G1032R. This may explain why in our transport assay TC and TCDC transport was almost absent, although the bile-to-serum ratio of all bile acids was approximately 50. Unfortunately, [3H]-labelled unconjugated cholate was not available for use in our assay to test our hypothesis.
Using purified BSEP as a quantitative reference, the amount of wild-type BSEP and both mutations could be determined (Figure 3B). Thus it was possible to obtain a per-molecule transport rate for human BSEP. Assuming that all BSEP molecules within the membrane vesicles are in an uptake-competent orientation (NBD facing the exterior) and generally transport-active, the number of BS molecules transported per BSEP unit per minute is 3 for TCDC and 2 for TC, respectively, under the conditions of the assay (5 min duration). These two values clearly represent the lowest possible estimate for TC and TCDC transport. Calibration of the Western blot signal likely results in values, which are too high since our BSEP preparation displayed homogeneity of approximately 90%. Additionally, not all BSEP-containing vesicles adopt the inside-out orientation (i.e., NBDs facing the exterior) during preparation. Under the experimental conditions, BS transport appears to be rather slow. However, it should be noted that the plasma membrane environment of HEK293 cells is not identical to the apical membrane of hepatocytes, lacking its unique membrane asymmetry and structure and that one cannot ensure that the linear rate of transport is still maintained. Nevertheless, this is, to the best of our knowledge, the first time that a per-transporter translocation rate of TC and TCDC has been established for human BSEP.
To obtain molecular insights into a structure-function relation of these two mutations, we employed a homology model of BSEP (Figure 3C), which is based on the outward-facing crystal structure of Sav18662[31]. Based on this model, T919 is located at the membrane-exposed side of the transporter, while G1032 lines up the putative translocation pore of BSEP. A deletion of threonine at position 919 would result in a shift within the corresponding helix, which obviously interferes with the function of BSEP. Replacement of a small glycine residue at position 1032 against a bulky and charged arginine residue within the pore of the transporter might either sterically block substrate translocation or prevent the switch from inward- to outward-facing conformation, because the large side chain would reduce the flexibility of this region. This conformational switch is absolutely required for transport. In addition, the critical localization of this amino acid might well explain a shift in transporter specificity. In PFIC-2 patients who do not respond to PEBD sufficiently, low residual transporter activity as well as disruption of transporter specificity should be considered by determining BS profiles in serum and bile.
Taking the limitations of individual observations into account, the results suggest that both mutations reported here differently interfere with the function of BSEP, which provides a rational explanation for the individual course of cholestatic liver disease of the two patients.
ACKNOWLEDGMENTS
The authors would like to acknowledge expert technical assistance by Paulina Philippski and Nathalie Walter.
COMMENTS
Background
Genetic defects of the bile salt export pump (BSEP), the major transporter for bile salts at the apical membrane of hepatocytes, may result in severe liver disease, termed progressive familial intrahepatic cholestasis type 2 (PFIC-2). Biliary diversion is a therapeutic option for the treatment of PFIC-2. However, only some patients benefit from this surgical intervention. The reason for variable treatment response is not well understood.
Research frontiers
The molecular understanding of pathophysiological consequences of single mutations or clusters of mutations of BSEP is presently evolving.
Innovations and breakthroughs
The study provides detailed analysis of two BSEP mutations, which have opposite effects on patient’s outcome in terms of treatment response. Most importantly, the results strongly indicate that a single amino acid substitution of BSEP (G1032R) may severely alter its substrate specificity.
Applications
The findings of their study imply that in BSEP-associated liver diseases evaluation of bile salt profiles (blood and bile) should be considered in addition to genetic analysis. This will improve the choice for the best treatment and will advance the prognostic assessment.
Terminology
Partial external biliary diversion (PEBD) is a surgical procedure for drainage of bile to the outside of the body. Bile is drained from bile ducts via the gallbladder and a short interposed segment from the small intestine to the surface of the abdomen.
Peer-review
The authors reported two patients with different mutations of the bile salt export pump which affect its residual function and substrate specificity. This is a well-written paper. The article is innovative. The article suggests that residual function of BSEP as well as substrate specificity influence the therapeutic effectiveness of PEBD in PFIC-2.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Germany
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B, B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: The study was reviewed and approved by the Institutional Review board, the “Ethikkommission an der Medizinischen Fakultät der Heinrich-Heine-Universität Düsseldorf” (Study number 2875).
Conflict-of-interest statement: The authors declare no conflict of interest.
Data sharing statement: No additional data are available.
Peer-review started: January 5, 2017
First decision: January 19, 2017
Article in press: June 12, 2017
P- Reviewer: Garcia-Olmo D, Koksal AS, Mansilla-Vivar R, Sira AM, Sun JH, S- Editor: Qi Y L- Editor: A E- Editor: Li D
References
- 1.Sambrotta M, Strautnieks S, Papouli E, Rushton P, Clark BE, Parry DA, Logan CV, Newbury LJ, Kamath BM, Ling S, et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet. 2014;46:326–328. doi: 10.1038/ng.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, Klomp LW, Lomri N, Berger R, Scharschmidt BF, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18:219–224. doi: 10.1038/ng0398-219. [DOI] [PubMed] [Google Scholar]
- 3.Paulusma CC, Oude Elferink RP. Diseases of intramembranous lipid transport. FEBS Lett. 2006;580:5500–5509. doi: 10.1016/j.febslet.2006.06.067. [DOI] [PubMed] [Google Scholar]
- 4.Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, Sokal E, Dahan K, Childs S, Ling V, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20:233–238. doi: 10.1038/3034. [DOI] [PubMed] [Google Scholar]
- 5.Hayashi H, Takada T, Suzuki H, Onuki R, Hofmann AF, Sugiyama Y. Transport by vesicles of glycine- and taurine-conjugated bile salts and taurolithocholate 3-sulfate: a comparison of human BSEP with rat Bsep. Biochim Biophys Acta. 2005;1738:54–62. doi: 10.1016/j.bbalip.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 6.Stindt J, Ellinger P, Weissenberger K, Dröge C, Herebian D, Mayatepek E, Homey B, Braun S, Schulte am Esch J, Horacek M, et al. A novel mutation within a transmembrane helix of the bile salt export pump (BSEP, ABCB11) with delayed development of cirrhosis. Liver Int. 2013;33:1527–1535. doi: 10.1111/liv.12217. [DOI] [PubMed] [Google Scholar]
- 7.Kubitz R, Dröge C, Stindt J, Weissenberger K, Häussinger D. The bile salt export pump (BSEP) in health and disease. Clin Res Hepatol Gastroenterol. 2012;36:536–553. doi: 10.1016/j.clinre.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Deleuze JF, Jacquemin E, Dubuisson C, Cresteil D, Dumont M, Erlinger S, Bernard O, Hadchouel M. Defect of multidrug-resistance 3 gene expression in a subtype of progressive familial intrahepatic cholestasis. Hepatology. 1996;23:904–908. doi: 10.1002/hep.510230435. [DOI] [PubMed] [Google Scholar]
- 9.Ruetz S, Gros P. Phosphatidylcholine translocase: a physiological role for the mdr2 gene. Cell. 1994;77:1071–1081. doi: 10.1016/0092-8674(94)90446-4. [DOI] [PubMed] [Google Scholar]
- 10.van Helvoort A, Smith AJ, Sprong H, Fritzsche I, Schinkel AH, Borst P, van Meer G. MDR1 P-glycoprotein is a lipid translocase of broad specificity, while MDR3 P-glycoprotein specifically translocates phosphatidylcholine. Cell. 1996;87:507–517. doi: 10.1016/s0092-8674(00)81370-7. [DOI] [PubMed] [Google Scholar]
- 11.Small DM. Role of ABC transporters in secretion of cholesterol from liver into bile. Proc Natl Acad Sci USA. 2003;100:4–6. doi: 10.1073/pnas.0237205100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Groen A, Romero MR, Kunne C, Hoosdally SJ, Dixon PH, Wooding C, Williamson C, Seppen J, Van den Oever K, Mok KS, et al. Complementary functions of the flippase ATP8B1 and the floppase ABCB4 in maintaining canalicular membrane integrity. Gastroenterology. 2011;141:1927–1937.e1-4. doi: 10.1053/j.gastro.2011.07.042. [DOI] [PubMed] [Google Scholar]
- 13.Jacquemin E. Role of multidrug resistance 3 deficiency in pediatric and adult liver disease: one gene for three diseases. Semin Liver Dis. 2001;21:551–562. doi: 10.1055/s-2001-19033. [DOI] [PubMed] [Google Scholar]
- 14.Marschall HU, Wagner M, Zollner G, Fickert P, Diczfalusy U, Gumhold J, Silbert D, Fuchsbichler A, Benthin L, Grundström R, et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology. 2005;129:476–485. doi: 10.1016/j.gastro.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 15.Whitington PF, Whitington GL. Partial external diversion of bile for the treatment of intractable pruritus associated with intrahepatic cholestasis. Gastroenterology. 1988;95:130–136. doi: 10.1016/0016-5085(88)90301-0. [DOI] [PubMed] [Google Scholar]
- 16.Keitel V, Burdelski M, Vojnisek Z, Schmitt L, Häussinger D, Kubitz R. De novo bile salt transporter antibodies as a possible cause of recurrent graft failure after liver transplantation: a novel mechanism of cholestasis. Hepatology. 2009;50:510–517. doi: 10.1002/hep.23083. [DOI] [PubMed] [Google Scholar]
- 17.Kubitz R, Keitel V, Scheuring S, Köhrer K, Häussinger D. Benign recurrent intrahepatic cholestasis associated with mutations of the bile salt export pump. J Clin Gastroenterol. 2006;40:171–175. doi: 10.1097/01.mcg.0000196406.15110.60. [DOI] [PubMed] [Google Scholar]
- 18.Keitel V, Burdelski M, Warskulat U, Kühlkamp T, Keppler D, Häussinger D, Kubitz R. Expression and localization of hepatobiliary transport proteins in progressive familial intrahepatic cholestasis. Hepatology. 2005;41:1160–1172. doi: 10.1002/hep.20682. [DOI] [PubMed] [Google Scholar]
- 19.Noé J, Stieger B, Meier PJ. Functional expression of the canalicular bile salt export pump of human liver. Gastroenterology. 2002;123:1659–1666. doi: 10.1053/gast.2002.36587. [DOI] [PubMed] [Google Scholar]
- 20.Stindt J, Ellinger P, Stross C, Keitel V, Häussinger D, Smits SH, Kubitz R, Schmitt L. Heterologous overexpression and mutagenesis of the human bile salt export pump (ABCB11) using DREAM (Directed REcombination-Assisted Mutagenesis) PLoS One. 2011;6:e20562. doi: 10.1371/journal.pone.0020562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellinger P, Kluth M, Stindt J, Smits SH, Schmitt L. Detergent screening and purification of the human liver ABC transporters BSEP (ABCB11) and MDR3 (ABCB4) expressed in the yeast Pichia pastoris. PLoS One. 2013;8:e60620. doi: 10.1371/journal.pone.0060620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herédi-Szabó K, Kis E, Krajcsi P. The vesicular transport assay: validated in vitro methods to study drug-mediated inhibition of canalicular efflux transporters ABCB11/BSEP and ABCC2/MRP2 2012; Chapter 23: Unit 23.4. Curr Protoc Toxicol. 2012;Chapter 23:Unit 23.4. doi: 10.1002/0471140856.tx2304s54. [DOI] [PubMed] [Google Scholar]
- 23.Göbel T, Schadewaldt-Tümmers J, Greiner L, Poremba C, Häussinger D, Erhardt A. Transient elastography improves detection of liver cirrhosis compared to routine screening tests. World J Gastroenterol. 2015;21:953–960. doi: 10.3748/wjg.v21.i3.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Millonig G, Reimann FM, Friedrich S, Fonouni H, Mehrabi A, Büchler MW, Seitz HK, Mueller S. Extrahepatic cholestasis increases liver stiffness (FibroScan) irrespective of fibrosis. Hepatology. 2008;48:1718–1723. doi: 10.1002/hep.22577. [DOI] [PubMed] [Google Scholar]
- 25.Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerová D, Rayner A, Dutton L, Meier Y, Antoniou A, Stieger B, Arnell H, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134:1203–1214. doi: 10.1053/j.gastro.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 26.Thompson R, Strautnieks S. BSEP: function and role in progressive familial intrahepatic cholestasis. Semin Liver Dis. 2001;21:545–550. doi: 10.1055/s-2001-19038. [DOI] [PubMed] [Google Scholar]
- 27.Byrne JA, Strautnieks SS, Ihrke G, Pagani F, Knisely AS, Linton KJ, Mieli-Vergani G, Thompson RJ. Missense mutations and single nucleotide polymorphisms in ABCB11 impair bile salt export pump processing and function or disrupt pre-messenger RNA splicing. Hepatology. 2009;49:553–567. doi: 10.1002/hep.22683. [DOI] [PubMed] [Google Scholar]
- 28.Dröge C, Schaal H, Engelmann G, Wenning D, Häussinger D, Kubitz R. Exon-skipping and mRNA decay in human liver tissue: molecular consequences of pathogenic bile salt export pump mutations. Sci Rep. 2016;6:24827. doi: 10.1038/srep24827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang KS, Tiao G, Bass LM, Hertel PM, Mogul D, Kerkar N, Clifton M, Azen C, Bull L, Rosenthal P, Stewart D, Superina R, Arnon R, Bozic M, Brandt ML, Dillon PA, Fecteau A, Iyer K, Kamath B, Karpen S, Karrer F, Loomes KM, Mack C, Mattei P, Miethke A, Soltys K, Turmelle YP, West K, Zagory J, Goodhue C, Shneider BL; Childhood Liver Disease Research Network (ChiLDReN) Analysis of surgical interruption of the enterohepatic circulation as a treatment for pediatric cholestasis. Hepatology. 2017;65:1645–1654. doi: 10.1002/hep.29019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Czubkowski P, Jankowska I, Pawlowska J. Successful pregnancy after ileal exclusion in progressive familial intrahepatic cholestasis type 2. Ann Hepatol. 2015;14:550–552. [PubMed] [Google Scholar]
- 31.Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]