

Abstract
AIM
To counteract/reveal celecoxib-induced toxicity and NO system involvement.
METHODS
Celecoxib (1 g/kg b.w. ip) was combined with therapy with stable gastric pentadecapeptide BPC 157 (known to inhibit these lesions, 10 μg/kg, 10 ng/kg, or 1 ng/kg ip) and L-arginine (100 mg/kg ip), as well as NOS blockade [N(G)-nitro-L-arginine methyl ester (L-NAME)] (5 mg/kg ip) given alone and/or combined immediately after celecoxib. Gastrointestinal, liver, and brain lesions and liver enzyme serum values in rats were assessed at 24 h and 48 h thereafter.
RESULTS
This high-dose celecoxib administration, as a result of NO system dysfunction, led to gastric, liver, and brain lesions and increased liver enzyme serum values. The L-NAME-induced aggravation of the lesions was notable for gastric lesions, while in liver and brain lesions the beneficial effect of L-arginine was blunted. L-arginine counteracted gastric, liver and brain lesions. These findings support the NO system mechanism(s), both NO system agonization (L-arginine) and NO system antagonization (L-NAME), that on the whole are behind all of these COX phenomena. An even more complete antagonization was identified with BPC 157 (at both 24 h and 48 h). A beneficial effect was evident on all the increasingly negative effects of celecoxib and L-NAME application and in all the BPC 157 groups (L-arginine + BPC 157; L-NAME + BPC 157; L-NAME + L-arginine + BPC 157). Thus, these findings demonstrated that BPC 157 may equally counteract both COX-2 inhibition (counteracting the noxious effects of celecoxib on all lesions) and additional NOS blockade (equally counteracting the noxious effects of celecoxib + L-NAME).
CONCLUSION
BPC 157 and L-arginine alleviate gastrointestinal, liver and brain lesions, redressing NSAIDs’ post-surgery application and NO system involvement.
Keywords: BPC 157, Celecoxib, L-arginine, N(G)-nitro-L-arginine methyl ester, Rats
Core tip: In rats treated with the COX-2 inhibitor celecoxib, BPC 157 (given intraperitoneally) counteracted lesion development in the stomach, liver and brain. BPC 157 treatment alongside with N(G)-nitro-L-arginine methyl ester (L-NAME) also attenuated any effect of L-NAME that would otherwise have intensified the deleterious regular course. Consistently, with exacerbation (induced by L-NAME administration) and amelioration (due to L-arginine) of gastric, liver and brain lesions, L-arginine amelioration prevailed (i.e., the gastric, liver and brain lesions were attenuated) when given together with L-NAME (L-NAME + L-arginine), an effect further reversed toward a marked beneficial effect by the addition of BPC 157 (L-NAME + L-arginine + BPC 157).
INTRODUCTION
We suggest that celecoxib, a specific COX-2 blocker, causes gastrointestinal, liver and brain lesions in rats when given in high doses, as has already been shown with non-selective nonsteroidal antiinflammatory drugs (NSAIDs)[1]. These results should also be related to the hitherto undetermined interaction with NO system dysfunction as well presenting the role of the NO system in gastrointestinal lesions[2,3], liver damage and hepatic encephalopathy[4]. These findings would correspond to hitherto reported lesions after the use of non-selective NSAIDs[5-8].
To clarify and combat the side effects of celecoxib, we focused on the stable gastric pentadecapeptide BPC 157, which is known to counteract lesions induced by non-selective NSAIDs[5-8] and to interact with the NO system in different models and species[9]. We also explored mechanisms behind the protection induced by the NO synthetase (NOS) substrate L-arginine and the aggravation induced by the NOS blocker N(G)-nitro-L-arginine methyl ester (L-NAME).
It was previously shown that celecoxib improved gastric lesions[10], induced the regression of preneoplastic lesions in the liver[11] and counteracted brain lesions and convulsions[12-14].
However, research into celecoxib-induced damage showed gastric[15-21] or intestinal lesions[22,23] and aggravation of kainic acid-convulsions[24]. Finally, both celecoxib and indomethacin prevented the gastroprotective effects induced by a nitric oxide donor or an inducer of nitric oxide synthesis[25]. These findings reveal more gastrointestinal lesions, more liver toxicity, and more toxicity to the brain as known risks for non-selective NSAIDs[5-8], which are also associated with adverse drug reactions in humans[26,27].
Therefore, these results would define the possible liver and brain lesions in the post-application period during a course of celecoxib-induced damage. In this scenario, the combined application of both of the NO agents L-NAME and L-arginine, i.e., the application of NOS blockade and an NOS substrate, testing the dual significance of NO, would clarify eventual therapeutic attempts and resolve possible controversies. For example, L-NAME was shown to attenuate indomethacin-induced microvascular injuries and leakage while L-arginine worked in conjunction with indomethacin[28].
A likely beneficial effect for the stable gastric pentadecapeptide BPC 157 on potential celecoxib-induced toxicity might be related to NO system dysfunction, which follows from consistent evidence[1,9]. BPC 157 (as an anti-ulcer peptide that is native and stable in human gastric juice) has been designated to be a novel mediator of cytoprotection[29] that has been implemented in inflammatory bowel disease and is in trial now to treat multiple sclerosis[30]. Recent reviews[1,9] cover that BPC 157 counteracts COX-1/COX-2-induced gastric, intestinal, liver and brain lesions[5-8], aspirin-prolonged bleeding and thrombocytopenia[31] and might prevent and rescue adjuvant arthritis[32]. BPC 157 also interacts with the effects of both L-arginine and L-NAME-induced aggravation in different models and species. Thus, we examined these possible therapies and the mechanisms behind them[9].
Consequently, we focused on the disturbances at 24 h and 48 h after celecoxib application, including identifying any gastrointestinal, liver and brain lesions as well as identifying any possible noxious stimuli that could further aggravate the existing conditions, e.g., the NOS blocker L-NAME, which was given alone and combined with celecoxib administration. We also focused on possible therapies, including the stable gastric pentadecapeptide BPC 157 and the NOS substrate L-arginine.
Furthermore, we should emphasize that the previous high-dose regimens used to demonstrate the toxicity of all non-selective NSAIDs[5-8] were compared with μg-ng regimens of BPC 157 as a potential antidote to counteract these effects[1] and to examine the safety profile of celecoxib. Thus, we consistently used this selective NSAID at very high doses that consequently markedly exceeded the maximal dose used in patients[10]. Likewise, in rats treated with celecoxib, both BPC 157 regimens, μg and ng, were also validated against the NO-related agents L-NAME and L-arginine, which were given either alone and/or in combination.
MATERIALS AND METHODS
Animals
Male albino Wistar (200 g) rats were used in all the experiments (at least 10 rats per experimental group and interval). Experiments were approved by the local ethics committee and assessed by observers unaware of the given treatment.
Drugs
The medications used, without carrier or peptidase inhibitor, included pentadecapeptide BPC 157 (a partial sequence of the human gastric juice protein BPC that is freely soluble in water at pH 7.0 and in saline). It was prepared as a peptide with 99% (HPLC) purity (1-des-Gly peptide was the main impurity, manufactured by Diagen, Ljubljana, Slovenia, GEPPPGKPADDAGLV, M.W. 1419). Celecoxib, L-NAME, and L-arginine (Sigma, United States) were prepared as previously described[1,9].
Protocol
Celecoxib (1 g/kg) was given intraperitoneally, and then immediately, as medications, we intraperitoneally administered the stable gastric pentadecapeptide BPC 157 (10 μg/kg, 10 ng/kg, 1 ng/kg), L-NAME (5 mg/kg) and L-arginine (100 mg/kg), while controls simultaneously received an equal volume of saline (5 mL/kg) intraperitoneally.
Gastrointestinal assay
Gastric lesions: Injury severity was assessed immediately after sacrifice. The sum of the longest lesion’s diameters was assessed as described previously[5,7,8], and gastric tissues were processed for routine microscopy analysis as described previously[5,7,8].
Liver assays - Bilirubin and enzyme activity: To determine the serum values of aspartate transaminase (AST), alanine transaminase (ALT) (IU/L) and total bilirubin (μmol/L), blood samples were obtained immediately after sacrifice and were centrifuged for 15 min at 3000 rpm. All tests were measured using an Olympus AU2700 analyzer with original test reagents (Olympus Diagnostica, Lismeehan, Ireland)[5-8].
Liver lesions: Liver tissue was immediately placed in 10% neutral buffered formalin for 24 h and subsequently embedded in paraffin. Hematoxylin-eosin-stained sections were analyzed on three high-power fields. The number of nuclei and area of cytoplasm as well as their diameter were measured using the ISSA program (Vamstec, Zagreb, Croatia), and the number of binucleated cells was also counted. Microvesicular steatosis was scored from 1-3: 1, less than 20% of hepatocytes showing microvesicular steatosis; 2, 20%-60% of hepatocytes showing microvesicular steatosis; and 3, over 60% of hepatocytes showing microvesicular steatosis. Parenchymal necrosis, eosinophilic cytoplasm, pyknotic nuclei and conspicuous nucleoli were scored semiquantitatively as follows: 0, showing no changes; 1, minimum; 2, moderate; and 3, maximum changes[5-8].
Brain assay
Brain lesions: Brains were fixed in 10% formalin for two days. Upon fixation, the brain was grossly inspected and cut into consecutive coronal sections. Brain slabs were dehydrated in graded ethanol and embedded in paraffin. Paraffin blocks were cut into 5-μm slices. Paraffin slices were deparaffinated in xylene, rehydrated in graded ethanol and stained with hematoxylin and eosin. The intensity and distribution of brain lesions (balloonized or red neurons), brain edema and cyanosis were described and evaluated semiquantitatively on two scales as follows where 0 generally indicated no changes: 0-3, edema (1, weakly diffuse and/or perifocal; 2, moderate; and 3, strong and generalized); 0-4, balloonized or red neurons (1, 5%; 2, 5%-30%; 3, 30%-50%; and 4, > 50%)[5-8].
Statistical analyses
Statistical analyses of the quantified data were performed by analysis of variance (ANOVA). Post hoc comparisons were appraised using the conservative Bonferroni/Dunn test. Data are presented as the mean ± SD. Non-parametric statistical analyses were performed for categorical data using the Kruskal-Wallis and post hoc Mann-Whitney U test. Values are expressed as min/med/max. Values of P < 0.05 were considered statistically significant.
RESULTS
Gastric lesions
Celecoxib induced severe gastric lesions (histologically, they appeared as mucosal defects ranging from one half of the mucosal thickness to full-blown ulcers; the defect bed was debris-covered, partially by erythrocytes, and it was surrounded by an edematous lamina propria with polymorphonuclear infiltration) showed a gradual increase from 24 h to 48 h after administration, while the liver and brain lesions showed sustained levels (Table 1, Figure 1).
Table 1.
Gastric lesions assessment after celecoxib, BPC 157, L-NAME, L-arginine
| Medication (/kg intraperitoneally) immediately after celecoxib 1 g/kg intraperitoneally |
Sum of longest lesions diameters (means ± SD, mm) assessed rafter celecoxib and mediation application |
|
| 24 h | 48 h | |
| Control, saline 5 mL | 10 ± 2 | 17 ± 3.5 |
| BPC 157 10 μg | 0 ± 0a | 7.5 ± 2.8a |
| BPC 157 10 ng | 2 ± 0.8a | 4 ± 1.2a |
| BPC 157 1 ng | 2.2 ± 0.6a | 4.3 ± 1a |
| L-NAME 5 mg | 13 ± 2 | 30 ± 5a |
| L-arginine 100 mg | 7 ± 1.5 | 5 ± 1a |
| L-NAME 5 mg + L-arginine 100 mg | 8 ± 2 | 10 ± 3a |
| L-NAME 5 mg + BPC 157 10 μg | 2 ± 1a | 4 ± 2a |
| L-NAME 5 mg + BPC 157 10 ng | 3.2 ± 0.8a | 5 ± 0.6a |
| L-arginine 100 mg + BPC 157 10 μg | 1 ± 0.5a | 2.5 ± 0.8a |
| L-arginine 100 mg + BPC 157 10 ng | 2.3 ± 0.4a | 4.5 ± 0.6a |
| L-NAME 5 mg + L-arginine 100 mg + BPC 157 10 μg | 1 ± 0.5a | 2.5 ± 0.6a |
| L-NAME 5 mg + L-arginine 100 mg + BPC 157 10 ng | 2.5 ± 0.4a | 4.7 ± 0.7a |
P < 0.05 vs control.
Figure 1.
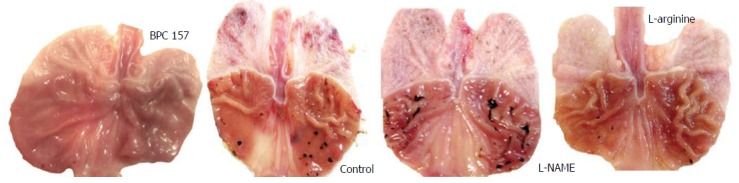
Gross presentation of celecoxib-induced gastric lesions at 48 h.
Only when these lesions became stronger, at the 48 h mark after celecoxib, were they aggravated by L-NAME and attenuated by L-arginine. When combined, the beneficial effect of L-arginine completely overrode the damaging effect of L-NAME. BPC 157 presented a stronger beneficial effect. Illustratively, given immediately after celecoxib, BPC 157 (in either of the regimens) completely alleviated the lesions induced by celecoxib, both at 24 h and 48 h. Likewise, when given together with other agents, BPC 157 consistently demonstrated the same beneficial effect.
Liver lesions
Celecoxib induced marked steatosis, congestion and necrosis at 24 h and at 48 h, along with increased enzyme serum values. The lesions were markedly attenuated by L-arginine at 24 h and at 48 h. Its beneficial effect was preserved at 24 h but decreased to control values at 48 h when combined with L-NAME, though L-NAME by itself could not affect celecoxib-liver lesions. A stronger beneficial effect occurred with BPC 157. BPC 157 alone completely alleviated celecoxib-induced liver lesions, both at 24 h and 48 h. In combination with the other agents, BPC 157 consistently demonstrated the same beneficial effect (Table 2, Figure 2).
Table 2.
Liver lesions assessment after celecoxib, BPC 157, L-NAME, L-arginine
| Medication (/kg intraperitoneally) immediately after celecoxib 1 g/kg intraperitoneally | Time after celecoxib |
Liver lesions assessment |
||||
|
Microscopic assessment, Score (0-3), Min/Med/Max |
Serum enzymes values (IU/L), means ± SD |
|||||
| steatosis | congestion | necrosis | ALT | AST | ||
| Control, saline 5 mL | 24 h | 2/2/3 | 2/2/3 | 2/2/3 | 352 ± 12 | 75 ± 8 |
| 48 h | 2/3/3 | 2/2/3 | 2/2/3 | 356 ± 18 | 84 ± 9 | |
| BPC 157 10 μg | 24 h | 1/1/1a | 1/1/2a | 1/1/1a | 138 ± 14a | 41 ± 11a |
| 48 h | 1/1/1a | 1/1/2a | 1/1/1a | 63 ± 12a | 35 ± 12a | |
| BPC 157 10 ng | 24 h | 1/1/1a | 1/1/2a | 1/1/1a | 154 ± 13a | 45 ± 8a |
| 48 h | 1/1/2a | 1/1/2a | 1/1/1a | 72 ± 19a | 40 ± 7a | |
| BPC 157 1 ng | 24 h | 1/1/1a | 1/1/2a | 1/1/1a | 170 ± 15a | 48 ± 10a |
| 48 h | 1/1/2a | 1/1/2a | 1/1/1a | 80 ± 15a | 43 ± 8a | |
| L-NAME 5 mg | 24 h | 2/3/3 | 2/2/3 | 2/2/3 | 375 ± 11 | 86 ± 12 |
| 48 h | 3/3/3 | 2/3/3 | 2/2/3 | 400 ± 12 | 80 ± 10 | |
| L-arginine 100 mg | 24 h | 1/2/2a | 1/2/2a | 1/2/2a | 315 ± 14a | 68 ± 14a |
| 48 h | 1/2/2a | 1/2/2a | 1/2/2a | 300 ± 21a | 65 ± 10a | |
| L-NAME 5 mg + L-arginine 100 mg | 24 h | 1/2/2a | 1/2/2a | 1/2/2a | 293 ± 17a | 68 ± 9a |
| 48 h | 2/2/2 | 2/2/3 | 2/2/2 | 270 ± 15a | 73 ± 11a | |
| L-NAME 5 mg + BPC 157 10 μg | 24 h | 1/1/1a | 1/2/2a | 1/1/1a | 170 ± 15a | 69 ± 9a |
| 48 h | 1/1/2a | 1/2/2a | 1/1/1a | 150 ± 12a | 55 ± 13a | |
| L-NAME 5 mg + BPC 157 10 ng | 24 h | 1/1/1a | 1/2/2a | 1/1/1a | 210 ± 16a | 70 ± 7a |
| 48 h | 1/1/2a | 1/2/2a | 1/1/1a | 182 ± 14a | 60 ± 8a | |
| L-arginine 100 mg + BPC 157 10 μg | 24 h | 1/1/1a | 1/1/1a | 1/1/1a | 165 ± 25a | 55 ± 8a |
| 48 h | 1/1/2a | 1/1/1a | 1/1/1a | 160 ± 18a | 45 ± 8a | |
| L-arginine 100 mg + BPC 157 10 ng | 24 h | 1/1/1a | 1/1/2a | 1/1/1a | 190 ± 11a | 63 ± 10a |
| 48 h | 1/1/2a | 1/1/2a | 1/1/1a | 170 ± 13a | 55 ± 9a | |
| L-NAME 5 mg + L-arginine 100 mg + BPC 157 10 μg | 24 h | 1/1/2a | 1/1/2a | 1/1/1a | 167 ± 11a | 57 ± 10a |
| 48 h | 1/1/2a | 1/1/2a | 1/1/1a | 176 ± 14a | 57 ± 8a | |
| L-NAME 5 mg + L-arginine 100 mg + BPC 157 10 ng | 24 h | 1/1/2a | 1/1/2a | 1/1/1a | 202 ± 9a | 62 ± 7a |
| 48 h | 1/1/2a | 1/1/2a | 1/1/1a | 190 ± 12a | 65 ± 8a | |
P < 0.05 vs control.
Figure 2.
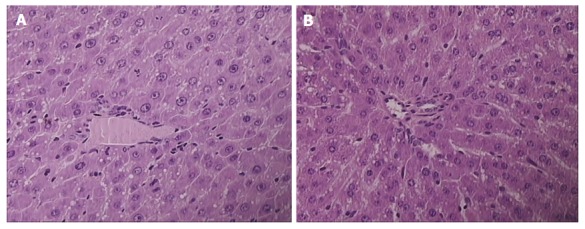
Presentation of celecoxib-induced liver lesions at 48 h. Controls presented with pronounced microvesicullar and macrovesicullar steatosis, dilated sinusoids, and piecemeal necrosis (A); BPC 157 rats presenting with minimal microvesicullar steatosis and no necrosis (B). HE × 40.
Brain lesions
Brain edema was commonly absent, though celecoxib-treated rats presented with damaged (balloonized) red neurons without any inflammation that were markedly expressed particularly in the cerebral cortex and in the Purkinje cells (Table 3, Figure 3). At 48 h, these lesions were attenuated by L-arginine, but when combined with L-NAME, the beneficial effect of L-arginine was decreased to the control values. Treatment with L-NAME by itself, however, did not affect the extent of the celecoxib-brain lesions. Again, BPC 157 presented a stronger beneficial effect; alone, it completely alleviated celecoxib-induced brain lesions, both at 24 h and 48 h. Likewise, given together with other agents, BPC 157 consistently demonstrated the same beneficial effect.
Table 3.
Brain lesions assessment after celecoxib, BPC 157, L-NAME, L-arginine
| Medication (/kg intraperitoneally) immediately after celecoxib 1 g/kg intraperitoneally | Time after celecoxib |
Brain lesions assessment |
||
|
Microscopic assessment, Score (0-4),
Min/Med/Max |
Microscopic assessment, Score (0-3), Min/Med/Max |
|||
| Purkinje cells (red neurons) | Cerebral cortex (red neurons) | Edema | ||
| Control, saline 5 mL | 24 h | 2/2/2 | 2/2/2 | 0/0/0 |
| 48 h | 2/2/3 | 2/2/3 | 0/0/0 | |
| BPC 157 10 μg | 24 h | 0/0/0a | 0/1/1a | 0/0/0 |
| 48 h | 0/1/1a | 0/1/1a | 0/0/0 | |
| BPC 157 10 ng | 24 h | 0/1/1a | 0/1/1a | 0/0/0 |
| 48 h | 0/1/1a | 1/1/1a | 0/0/0 | |
| BPC 157 1 ng | 24 h | 0/1/1a | 0/1/1a | 0/0/0 |
| 48 h | 0/1/1a | 0/1/1a | 0/0/0 | |
| L-NAME 5 mg | 24 h | 2/2/3 | 2/2/3 | 0/0/0 |
| 48 h | 2/3/3 | 2/3/3 | 0/0/0 | |
| L-arginine 100 mg | 24 h | 1/2/2 | 1/2/2 | 0/0/0 |
| 48 h | 1/2/2a | 1/2/2a | 0/0/0 | |
| L-NAME 5 mg + L-arginine 100 mg | 24 h | 2/2/2 | 2/2/2 | 0/0/0 |
| 48 h | 2/2/2 | 2/2/2 | 0/0/0 | |
| L-NAME 5 mg + BPC 157 10 μg | 24 h | 1/1/1a | 1/1/2a | 0/0/0 |
| 48 h | 1/1/2a | 1/1/2a | 0/0/0 | |
| L-NAME 5 mg + BPC 157 10 ng | 24 h | 1/1/1a | 1/1/2a | 0/0/0 |
| 48 h | 1/1/2a | 1/1/2a | 0/0/0 | |
| L-arginine 100 mg+ BPC 157 10 μg | 24 h | 0/1/1a | 0/1/1a | 0/0/0 |
| 48 h | 1/1/2a | 0/1/1a | 0/0/0 | |
| L-arginine 100 mg + BPC 157 10 ng | 24 h | 0/1/1a | 0/1/1a | 0/0/0 |
| 48 h | 1/1/2a | 1/1/1a | 0/0/0 | |
| L-NAME 5 mg + L-arginine 100 mg + BPC 157 10 μg | 24 h | 1/1/2a | 1/1/1a | 0/0/0 |
| 48 h | 1/1/2a | 1/1/1a | 0/0/0 | |
| L-NAME 5 mg + L-arginine 100 mg + BPC 157 10 ng | 24 h | 1/1/2a | 1/1/1a | 0/0/0 |
| 48 h | 1/1/2a | 1/2/1a | 0/0/0 | |
P < 0.05 vs control.
Figure 3.
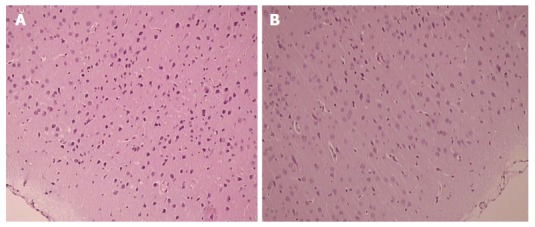
Presentation of celecoxib-induced cerebral cortex lesions at 48 h. Control celecoxib rats presented more damaged (balloonized) red neurons without any inflammation markedly expressed in particular in the cerebral cortex (A), unlike BPC 157 + celecoxib rats (B). HE × 40.
DISCUSSION
This study argues that the celecoxib-induced stomach, liver and brain lesions resulting from extended COX-2 inhibition are a function of NO system dysfunction, which is particularly worsened after a high-dose application. Considering the advanced safety profile of celecoxib[10], lower celecoxib regimens such as 200 mg/kg and 500 mg/kg given intraperitoneally were without notable effect on gastric, liver or brain lesions (thus, these results are not specifically shown). These lesions could be all influenced by the NOS substrate L-arginine and in particular by the stable pentadecapeptide BPC 157, which is an agent known to counteract non-selective NSAID-induced ulcerogenesis as well as the liver and brain lesions that interact with the NO system[1,9]. The additional support comes from the similar therapy effects obtained with the correspondingly high dose range of BPC 157 therapy, which was similar to the dosing used in other studies as well[33,34].
This study argues that the celecoxib-induced stomach, liver and brain lesions presenting after COX-2 inhibition demonstrate a particular NO system dysfunction. These effects could be all influenced by the NOS substrate L-arginine, particularly by the stable pentadecapeptide BPC 157, which is an agent known to counteract non-selective NSAID-induced ulcerogenesis as well as liver and brain lesions and particularly interacts with the NO system[1,9].
In support of this argument, the evidence for non-selective NSAIDs[5-8] shows that they cause more gastrointestinal lesions, more liver toxicity, and more toxicity in the brain due to the known COX-1/COX-2 relationship[5-8].
On the other hand, after celecoxib treatment, the deleterious course that characterizes COX-2 inhibition should be more complex. This course includes initial stomach lesions and further progressing lesions that deteriorate over time as well as extensive liver and brain lesions that are already sustainably present at the early 24 h period. Further, these lesions are reciprocally potentiated by L-NAME and opposed by L-arginine in a particular way.
In so doing and in substantiating the particularities for the initial lesions in the stomach at 24 h, it is likely that celecoxib provides COX-2 inhibition; at that time, in the stomach, there is no effect of L-arginine or L-NAME. However, the liver and brain lesions likely appear due to COX-2 inhibition and NOS dysfunction, the latter of which is counteracted by L-arginine.
Next, in addition to this opposing effect of L-arginine, NOS blockade specifically contributes NOS dysfunction to COX-2 inhibition as verified over the 24-48 h period. It is only in competition with the progressive stomach lesions that the beneficial effect of L-arginine occurs. Similarly, L-NAME aggravation occurs in stomach lesions, while the liver and brain lesions cannot not be worsened further. Therefore, maximal NOS dysfunction with COX-2 inhibition is present in the brain more than in the liver. Namely, unlike stomach lesions (where L-arginine nullifies L-NAME aggravation) and liver lesions, in brain lesions, L-arginine could not work to counteract an L-NAME-induced NOS blockade.
Thus, there is threefold confirmation of the NO relationship: L-NAME (NOS blockade) in stomach lesions; L-arginine (NOS substrate) counteracts stomach, liver, brain lesions; and L-arginine and L-NAME could specifically affect each other’s response in the stomach, liver and brain.
Finally, for all these lesions in the stomach, liver and brain, celecoxib- and/or L-NAME-induced lesions were completely inhibited by BPC 157 administration. As mentioned, BPC 157, due to its counteraction of NSAID-induced lesions[1] and interaction with NO agents[1,9], is assumed to be more prone to counteracting both COX-1/COX-2 and NOS inhibition than L-arginine as an NOS substrate. In addition, this result is also true for COX-2/NOS inhibition. Further, BPC 157 induces NO release from the gastric mucosa supernatant, similarly to L-arginine[35], but it also functions under conditions where L-arginine does not work[36]. This mechanism assumes a persistent, beneficial effect versus increasing dysfunction of the nitrergic pathway (for instance, heavier loss of endothelium cells from the vascular wall could lead to less NO production ability[37]), making COX-2/NOS inhibition worse as the tissue integrity is damaged further. Likely as a result of this relationship, the same effectiveness is observed in all increasingly damaged circumstances following celecoxib and L-NAME application. This process specifically involves all BPC 157 groups when administered with celecoxib alone and/or with NO agents (L-arginine + BPC 157; L-NAME + BPC 157; L-NAME + L-arginine + BPC 157) in the stomach, liver and brain. BPC 157 may thereby equally counteract both COX-2 inhibition (counteracting the noxious effects of celecoxib on all lesions) and additional NOS blockade (equally counteracting the noxious effects of celecoxib + L-NAME)[1,9]. This outcome occurs equally with either μg- or ng dosing regimens. In addition, BPC 157 affects eNOS gene function as well[38] as that of other genes[38-42].
In the latter cases, in the healing process, BPC-157 regulates the phosphorylation level of extracellular signal-regulated kinases 1 and 2 (ERK1/2) as well as their downstream targets, including c-Fos, c-Jun, and egr-1, key molecules involved in cell growth, migration, and angiogenesis[38-42].
In conclusion, the therapy and syndrome in rats treated with celecoxib described herein (the gastrointestinal tract, liver and brain lesions presented rapidly in rats) overlaps with the definitive follow-up of previous non-selective NSAID studies[1,5-8]. With respect to the administration of pentadecapeptide BPC 157, these results extend and generalize the observed acute and long-term therapeutic effects[5-8]. Then, over longer periods, there is greater deterioration from celecoxib, the COX-2-inhibitor, and the NO synthetase (NOS)-blocker L-NAME when combined, along with prominent rescue by the stable pentadecapeptide BPC 157 and less prominent attenuation with the NOS substrate L-arginine. These findings should likely reveal an aggravating parallelism between COX-2 and NOS inhibition[43], including the role of the NO system in gastrointestinal[2,3] and liver damage and hepatic encephalopathy[4] as well as celecoxib syndrome in particular NO system pathways.
Conclusively, L-arginine, but more so BPC 157, may provide a particular therapy that may alleviate likely gastrointestinal, liver and brain lesions and redress NSAIDs’ post-surgery application and NO system involvement. However, the particular point remains that a single large overdose challenge differs considerably from the lower regular patient regimens throughout a markedly more prolonged treatment duration.
COMMENTS
Background
Non-selective nonsteroidal antiinflammatory drugs (NSAIDs) induce gastrointestinal, liver and brain toxicity in rats, while celecoxib, a COX-2 inhibitor, is considered less toxic.
Research frontiers
This study argues that the celecoxib-induced stomach, liver and brain lesions resulting from extended COX-2 inhibition are a function of NO system dysfunction, which is particularly worsened after a high-dose application. These lesions could be all influenced by the NO synthetase (NOS) substrate L-arginine and in particular by the stable pentadecapeptide BPC 157, which is an agent known to counteract non-selective NSAID-induced ulcerogenesis as well as the liver and brain lesions that interact with the NO system. The additional support comes from the similar therapy effects obtained with the correspondingly high dose range of BPC 157 therapy, which was similar to the dosing used in other studies as well.
Innovations and breakthroughs
With respect to the administration of pentadecapeptide BPC 157, these results extend and generalize the observed acute and long-term therapeutic effects. Then, over longer periods, there is greater deterioration from celecoxib, the COX-2-inhibitor, and the NOS-blocker L-NAME when combined, along with prominent rescue by the stable pentadecapeptide BPC 157 and less prominent attenuation with the NOS substrate L-arginine. These findings should likely reveal an aggravating parallelism between COX-2 and NOS inhibition, including the role of the NO system in gastrointestinal and liver damage and hepatic encephalopathy as well as celecoxib syndrome in particular NO system pathways.
Applications
Conclusively, L-arginine, but more so BPC 157, may provide a particular therapy that may alleviate likely gastrointestinal, liver and brain lesions and redress NSAIDs’ post-surgery application and NO system involvement.
Peer-review
The manuscript is well written and reports a potentially interesting data.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Croatia
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): 0
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: The study was reviewed and approved by the Department of Veterinary, Ministry of Agriculture, Croatia, No. UP/I 322-01/07-01/210.
Conflict-of-interest statement: The authors state that they have no conflicts of interest.
Data sharing statement: No additional data are available.
Peer-review started: February 19, 2017
First decision: April 5, 2017
Article in press: July 4, 2017
P- Reviewer: Swierczynski JT S- Editor: Gong ZM L- Editor: A E- Editor: Li D
References
- 1.Sikiric P, Seiwerth S, Rucman R, Turkovic B, Rokotov DS, Brcic L, Sever M, Klicek R, Radic B, Drmic D, et al. Toxicity by NSAIDs. Counteraction by stable gastric pentadecapeptide BPC 157. Curr Pharm Des. 2013;19:76–83. doi: 10.2174/13816128130111. [DOI] [PubMed] [Google Scholar]
- 2.Whittle BJ. Nitric oxide-modulating agents for gastrointestinal disorders. Expert Opin Investig Drugs. 2005;14:1347–1358. doi: 10.1517/13543784.14.11.1347. [DOI] [PubMed] [Google Scholar]
- 3.Evans SM, Whittle BJ. Interactive roles of superoxide and inducible nitric oxide synthase in rat intestinal injury provoked by non-steroidal anti-inflammatory drugs. Eur J Pharmacol. 2001;429:287–296. doi: 10.1016/s0014-2999(01)01327-9. [DOI] [PubMed] [Google Scholar]
- 4.Huang HC, Wang SS, Lee FY, Chan CY, Chang FY, Lin HC, Chu CJ, Chen YC, Lee SD. Simvastatin for rats with thioacetamide-induced liver failure and encephalopathy. J Gastroenterol Hepatol. 2008;23:e236–e242. doi: 10.1111/j.1440-1746.2007.04988.x. [DOI] [PubMed] [Google Scholar]
- 5.Ilic S, Drmic D, Franjic S, Kolenc D, Coric M, Brcic L, Klicek R, Radic B, Sever M, Djuzel V, et al. Pentadecapeptide BPC 157 and its effects on a NSAID toxicity model: diclofenac-induced gastrointestinal, liver, and encephalopathy lesions. Life Sci. 2011;88:535–542. doi: 10.1016/j.lfs.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 6.Ilic S, Drmic D, Zarkovic K, Kolenc D, Coric M, Brcic L, Klicek R, Radic B, Sever M, Djuzel V, et al. High hepatotoxic dose of paracetamol produces generalized convulsions and brain damage in rats. A counteraction with the stable gastric pentadecapeptide BPC 157 (PL 14736) J Physiol Pharmacol. 2010;61:241–250. [PubMed] [Google Scholar]
- 7.Ilic S, Drmic D, Zarkovic K, Kolenc D, Brcic L, Radic B, Djuzel V, Blagaic AB, Romic Z, Dzidic S, et al. Ibuprofen hepatic encephalopathy, hepatomegaly, gastric lesion and gastric pentadecapeptide BPC 157 in rats. Eur J Pharmacol. 2011;667:322–329. doi: 10.1016/j.ejphar.2011.05.038. [DOI] [PubMed] [Google Scholar]
- 8.Lojo N, Rasic Z, Zenko Sever A, Kolenc D, Vukusic D, Drmic D, Zoricic I, Sever M, Seiwerth S, Sikiric P. Effects of Diclofenac, L-NAME, L-Arginine, and Pentadecapeptide BPC 157 on Gastrointestinal, Liver, and Brain Lesions, Failed Anastomosis, and Intestinal Adaptation Deterioration in 24 Hour-Short-Bowel Rats. PLoS One. 2016;11:e0162590. doi: 10.1371/journal.pone.0162590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sikiric P, Seiwerth S, Rucman R, Turkovic B, Rokotov DS, Brcic L, Sever M, Klicek R, Radic B, Drmic D, et al. Stable gastric pentadecapeptide BPC 157-NO-system relation. Curr Pharm Des. 2014;20:1126–1135. doi: 10.2174/13816128113190990411. [DOI] [PubMed] [Google Scholar]
- 10.Tsuji S, Miyoshi H, Tomita T, Nakase T, Hamada M, Oomae T, Tsumoto C, Hirata Y, Iguchi M, Edogawa S, et al. Celecoxib, a cyclooxygenase-2 inhibitor, improved upper gastrointestinal lesions in rheumatoid arthritis patients as assessed by endoscopic evaluation. Mod Rheumatol. 2012;22:353–362. doi: 10.1007/s10165-011-0524-6. [DOI] [PubMed] [Google Scholar]
- 11.Arellanes-Robledo J, Márquez-Rosado L, Pérez-Carreón JI, Fattel-Fazenda S, Aguirre-García J, Villa-Treviño S. Celecoxib induces regression of putative preneoplastic lesions in rat liver. Anticancer Res. 2006;26:1271–1280. [PubMed] [Google Scholar]
- 12.Gupta A, Dhir A, Kumar A, Kulkarni SK. Protective effect of cyclooxygenase (COX)-inhibitors against drug-induced catatonia and MPTP-induced striatal lesions in rats. Pharmacol Biochem Behav. 2009;94:219–226. doi: 10.1016/j.pbb.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 13.Abd El-Aal SA, El-Sawalhi MM, Seif-El-Nasr M, Kenawy SA. Effect of celecoxib and L-NAME on global ischemia-reperfusion injury in the rat hippocampus. Drug Chem Toxicol. 2013;36:385–395. doi: 10.3109/01480545.2012.749270. [DOI] [PubMed] [Google Scholar]
- 14.Zandieh A, Maleki F, Hajimirzabeigi A, Zandieh B, Khalilzadeh O, Dehpour AR. Anticonvulsant effect of celecoxib on pentylenetetrazole-induced convulsion: Modulation by NO pathway. Acta Neurobiol Exp (Wars) 2010;70:390–397. doi: 10.55782/ane-2010-1811. [DOI] [PubMed] [Google Scholar]
- 15.Berenguer B, Alarcón De La Lastra C, Motilva V, La Casa C, Herrerias JM, Pozo D, Calero MJ. Effects of celecoxib on acid-challenged gastric mucosa of rats: comparison with metamizol and piroxicam. Dig Dis Sci. 2004;49:937–947. doi: 10.1023/b:ddas.0000034552.20917.5e. [DOI] [PubMed] [Google Scholar]
- 16.Yamakawa N, Suzuki K, Yamashita Y, Katsu T, Hanaya K, Shoji M, Sugai T, Mizushima T. Structure-activity relationship of celecoxib and rofecoxib for the membrane permeabilizing activity. Bioorg Med Chem. 2014;22:2529–2534. doi: 10.1016/j.bmc.2014.02.032. [DOI] [PubMed] [Google Scholar]
- 17.Ishihara T, Tanaka K, Tashiro S, Yoshida K, Mizushima T. Protective effect of rebamipide against celecoxib-induced gastric mucosal cell apoptosis. Biochem Pharmacol. 2010;79:1622–1633. doi: 10.1016/j.bcp.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 18.Ma J, Yuan G, Chen MH. [Non-steroidal anti-inflammatory drug induced gastropathy and preventive effects of teprenone on the gastropathy in rats] Zhonghua Yi Xue Za Zhi. 2006;86:2868–2873. [PubMed] [Google Scholar]
- 19.Pajdo R, Brzozowski T, Szlachcic A, Konturek PC, Ptak-Belowska A, Drozdowicz D, Targosz A, Konturek SJ, Pawlik WW. Lipoxins, the novel mediators of gastroprotection and gastric adaptation to ulcerogenic action of aspirin. Curr Pharm Des. 2011;17:1541–1551. doi: 10.2174/138161211796197043. [DOI] [PubMed] [Google Scholar]
- 20.Toller IM, Hitzler I, Sayi A, Mueller A. Prostaglandin E2 prevents Helicobacter-induced gastric preneoplasia and facilitates persistent infection in a mouse model. Gastroenterology. 2010;138:1455–1467, 1467.e1-1467.e4. doi: 10.1053/j.gastro.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 21.Junqueira-Júnior J, Junqueira AF, Medeiros JV, Barbosa SH, Nogueira AC, Mota JM, Santana AP, Brito GA, Ribeiro RA, Lima-Júnior RC, et al. Role of capsaicin-sensitive primary afferent neurons and non-protein sulphydryl groups on gastroprotective effect of amifostine against ethanol-induced gastric damage in rats. Dig Dis Sci. 2011;56:314–322. doi: 10.1007/s10620-010-1300-8. [DOI] [PubMed] [Google Scholar]
- 22.Sendur P, Ceranowicz P, Sendur R, Cieszkowski J, Warzecha Z, Dembiński A. [Involvement of endogenous tachykinins in the development of jejunal mucosa injury induced by on-steroidal anti-inflammatory drugs] Przegl Lek. 2013;70:48–52. [PubMed] [Google Scholar]
- 23.Zwolinska-Wcislo M, Krzysiek-Maczka G, Ptak-Belowska A, Karczewska E, Pajdo R, Sliwowski Z, Urbanczyk K, Drozdowicz D, Konturek SJ, Pawlik WW, et al. Antibiotic treatment with ampicillin accelerates the healing of colonic damage impaired by aspirin and coxib in the experimental colitis. Importance of intestinal bacteria, colonic microcirculation and proinflammatory cytokines. J Physiol Pharmacol. 2011;62:357–368. [PubMed] [Google Scholar]
- 24.Baik EJ, Kim EJ, Lee SH, Moon C. Cyclooxygenase-2 selective inhibitors aggravate kainic acid induced seizure and neuronal cell death in the hippocampus. Brain Res. 1999;843:118–129. doi: 10.1016/s0006-8993(99)01797-7. [DOI] [PubMed] [Google Scholar]
- 25.Coppelli G, Guaita E, Spaggiari S, Coruzzi G. Gastric effects of the selective cyclooxygenase-2 inhibitor, celecoxib, in the rat. Dig Liver Dis. 2004;36:265–270. doi: 10.1016/j.dld.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 26.Auriel E, Regev K, Korczyn AD. Nonsteroidal anti-inflammatory drugs exposure and the central nervous system. Handb Clin Neurol. 2014;119:577–584. doi: 10.1016/B978-0-7020-4086-3.00038-2. [DOI] [PubMed] [Google Scholar]
- 27.Bessone F. Non-steroidal anti-inflammatory drugs: What is the actual risk of liver damage? World J Gastroenterol. 2010;16:5651–5661. doi: 10.3748/wjg.v16.i45.5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whittle BJ, László F, Evans SM, Moncada S. Induction of nitric oxide synthase and microvascular injury in the rat jejunum provoked by indomethacin. Br J Pharmacol. 1995;116:2286–2290. doi: 10.1111/j.1476-5381.1995.tb15066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sikiric P, Seiwerth S, Brcic L, Sever M, Klicek R, Radic B, Drmic D, Ilic S, Kolenc D. Revised Robert’s cytoprotection and adaptive cytoprotection and stable gastric pentadecapeptide BPC 157. Possible significance and implications for novel mediator. Curr Pharm Des. 2010;16:1224–1234. doi: 10.2174/138161210790945977. [DOI] [PubMed] [Google Scholar]
- 30.Klicek R, Kolenc D, Suran J, Drmic D, Brcic L, Aralica G, Sever M, Holjevac J, Radic B, Turudic T, et al. Stable gastric pentadecapeptide BPC 157 heals cysteamine-colitis and colon-colon-anastomosis and counteracts cuprizone brain injuries and motor disability. J Physiol Pharmacol. 2013;64:597–612. [PubMed] [Google Scholar]
- 31.Stupnisek M, Franjic S, Drmic D, Hrelec M, Kolenc D, Radic B, Bojic D, Vcev A, Seiwerth S, Sikiric P. Pentadecapeptide BPC 157 reduces bleeding time and thrombocytopenia after amputation in rats treated with heparin, warfarin or aspirin. Thromb Res. 2012;129:652–659. doi: 10.1016/j.thromres.2011.07.035. [DOI] [PubMed] [Google Scholar]
- 32.Sikiric P, Seiwerth S, Grabarevic Z, Rucman R, Petek M, Jagic V, Turkovic B, Rotkvic I, Mise S, Zoricic I, et al. Pentadecapeptide BPC 157 positively affects both non-steroidal anti-inflammatory agent-induced gastrointestinal lesions and adjuvant arthritis in rats. J Physiol Paris. 1997;91:113–122. doi: 10.1016/s0928-4257(97)89474-0. [DOI] [PubMed] [Google Scholar]
- 33.Jelovac N, Sikiric P, Rucman R, Petek M, Marovic A, Perovic D, Seiwerth S, Mise S, Turkovic B, Dodig G, et al. Pentadecapeptide BPC 157 attenuates disturbances induced by neuroleptics: the effect on catalepsy and gastric ulcers in mice and rats. Eur J Pharmacol. 1999;379:19–31. doi: 10.1016/s0014-2999(99)00486-0. [DOI] [PubMed] [Google Scholar]
- 34.Duplancic B, Stambolija V, Holjevac J, Zemba M, Balenovic I, Drmic D, Suran J, Radic B, Filipovic M, Blagaic AB, et al. Pentadecapeptide BPC 157 and anaphylactoid reaction in rats and mice after intravenous dextran and white egg administration. Eur J Pharmacol. 2014;727:75–79. doi: 10.1016/j.ejphar.2014.01.046. [DOI] [PubMed] [Google Scholar]
- 35.Sikirić P, Seiwerth S, Grabarević Z, Rucman R, Petek M, Jagić V, Turković B, Rotkvić I, Mise S, Zoricić I, et al. The influence of a novel pentadecapeptide, BPC 157, on N(G)-nitro-L-arginine methylester and L-arginine effects on stomach mucosa integrity and blood pressure. Eur J Pharmacol. 1997;332:23–33. doi: 10.1016/s0014-2999(97)01033-9. [DOI] [PubMed] [Google Scholar]
- 36.Turkovic B, Sikiric P, Seiwerth S, Mise S, Anic T, Petek M, Rucman R. Stable gastric pentadecapeptide BPC 157 studied for inflammatory bowel disease (PLD116, PL14736, Pliva) induces nitric oxide synthesis. Gastroenterology. 2004;126:A287–A287. [Google Scholar]
- 37.Berra-Romani R, Avelino-Cruz JE, Raqeeb A, Della Corte A, Cinelli M, Montagnani S, Guerra G, Moccia F, Tanzi F. Ca²+-dependent nitric oxide release in the injured endothelium of excised rat aorta: a promising mechanism applying in vascular prosthetic devices in aging patients. BMC Surg. 2013;13 Suppl 2:S40. doi: 10.1186/1471-2482-13-S2-S40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cesarec V, Becejac T, Misic M, Djakovic Z, Olujic D, Drmic D, Brcic L, Rokotov DS, Seiwerth S, Sikiric P. Pentadecapeptide BPC 157 and the esophagocutaneous fistula healing therapy. Eur J Pharmacol. 2013;701:203–212. doi: 10.1016/j.ejphar.2012.11.055. [DOI] [PubMed] [Google Scholar]
- 39.Chang CH, Tsai WC, Lin MS, Hsu YH, Pang JH. The promoting effect of pentadecapeptide BPC 157 on tendon healing involves tendon outgrowth, cell survival, and cell migration. J Appl Physiol (1985) 2011;110:774–780. doi: 10.1152/japplphysiol.00945.2010. [DOI] [PubMed] [Google Scholar]
- 40.Chang CH, Tsai WC, Hsu YH, Pang JH. Pentadecapeptide BPC 157 enhances the growth hormone receptor expression in tendon fibroblasts. Molecules. 2014;19:19066–19077. doi: 10.3390/molecules191119066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang T, Zhang K, Sun L, Xue X, Zhang C, Shu Z, Mu N, Gu J, Zhang W, Wang Y, et al. Body protective compound-157 enhances alkali-burn wound healing in vivo and promotes proliferation, migration, and angiogenesis in vitro. Drug Des Devel Ther. 2015;9:2485–2499. doi: 10.2147/DDDT.S82030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tkalcević VI, Cuzić S, Brajsa K, Mildner B, Bokulić A, Situm K, Perović D, Glojnarić I, Parnham MJ. Enhancement by PL 14736 of granulation and collagen organization in healing wounds and the potential role of egr-1 expression. Eur J Pharmacol. 2007;570:212–221. doi: 10.1016/j.ejphar.2007.05.072. [DOI] [PubMed] [Google Scholar]
- 43.Tetsuka T, Daphna-Iken D, Miller BW, Guan Z, Baier LD, Morrison AR. Nitric oxide amplifies interleukin 1-induced cyclooxygenase-2 expression in rat mesangial cells. J Clin Invest. 1996;97:2051–2056. doi: 10.1172/JCI118641. [DOI] [PMC free article] [PubMed] [Google Scholar]