

Abstract
ProNGF signaling through p75NTR has been associated with neurodegenerative disorders. Retinitis pigmentosa (RP) comprises a group of inherited retinal dystrophies that causes progressive photoreceptor cell degeneration and death, at a rate dependent on the genetic mutation. There are more than 300 mutations causing RP, and this is a challenge to therapy. Our study was designed to explore a common mechanism for p75NTR in the progression of RP, and assess its potential value as a therapeutic target. The proNGF/p75NTR system is present in the dystrophic retina of the rd10 RP mouse model. Compared with wild-type (WT) retina, the levels of unprocessed proNGF were increased in the rd10 retina at early degenerative stages, before the peak of photoreceptor cell death. Conversely, processed NGF levels were similar in rd10 and WT retinas. ProNGF remained elevated throughout the period of photoreceptor cell loss, correlating with increased expression of α2-macroglobulin, an inhibitor of proNGF processing. The neuroprotective effect of blocking p75NTR was assessed in organotypic retinal cultures from rd10 and RhoP mouse models. Retinal explants treated with p75NTR antagonists showed significantly reduced photoreceptor cell death, as determined by the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay and by preservation of the thickness of the outer nuclear layer (ONL), where photoreceptor nuclei are located. This effect was accompanied by decreased retinal-reactive gliosis and reduced TNFα secretion. Use of p75NTR antagonist THX-B (1,3-diisopropyl-1-[2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-purin-7-yl)-acetyl]-urea) in vivo in the rd10 and RhoP mouse models, by a single intravitreal or subconjunctival injection, afforded neuroprotection to photoreceptor cells, with preservation of the ONL. This study demonstrates a role of the p75NTR/proNGF axis in the progression of RP, and validates these proteins as therapeutic targets in two different RP models, suggesting utility irrespective of etiology.
Retinitis pigmentosa (RP) refers to a group of inherited retinal dystrophies that are clinically similar despite arising from a large set of genetic mutations (http://www.sph.uth.tmc.edu/Retnet/disease.htm). These mutations usually trigger photoreceptor cell degeneration and death, leading to visual function decline and, eventually, blindness.1 While the time of onset and the rate of neurodegeneration are specified by the mutation, most, if not all, forms of RP share molecular and cellular mechanisms that include inflammation, microglial activation and reactive gliosis. These features are shared with other retinal diseases without a pure genetic origin, such as glaucoma, diabetic retinopathy and age-related macular degeneration.2, 3
Various therapeutic strategies for RP, including gene, cell and regenerative therapies, as well as pharmacological treatments, are gradually progressing from the animal models to clinical trials4, 5, 6 and http://clinicaltrials.gov/ct2/results?term=retinitis+pigmentosa. However, there is not yet any approved treatment for the neurodegenerative component of retinal diseases. The genetic complexity in the etiology of RP, comprising more than 300 described mutations in over 50 different genes, calls for the development of treatments targeting common mechanisms independently of the causative mutation. This would entail the detailed characterization of the processes leading to retinal deterioration, as a strategy to discover novel therapeutic targets.
As a monogenic genetic disease of high penetrance, a variety of animal genetic models recapitulate the signs and symptoms of human RP.4 A missense mutation in the Pde6b gene causes blindness in the rd10 mouse model of autosomal recessive RP.7 The RhoP mouse model of autosomal dominant RP carries the mutant human rhodopsin Pro347Ser transgene.8 The course of the disease in these models recapitulate human progression and allows for effective experimental interventions.9, 10, 11, 12, 13, 14, 15, 16 We used these independent models of RP to study the involvement of the p75NTR/proNGF axis in the course of disease.
ProNGF is the precursor of mature NGF. ProNGF and NGF display opposite effects, depending on the receptor-complex stimulated. Both bind p75NTR that mediates a neurotoxic effect. Indeed, p75NTR activation is involved in several neurodegenerative conditions.17, 18 The deleterious signals of p75NTR require an interacting protein sortilin, and are ligand dependent and activated by proNGF (although some signals can also be ligand independent).18 Conversely, NGF also binds the neuroprotective receptor Trk-A that can counterbalance p75NTR. Other proneurotrophins also bind p75NTR, although their functional impact is less characterized than in the case of proNGF.18
In retinal neurodegeneration associated with glaucoma and diabetic retinopathy, proNGF stimulates p75NTR-dependent production and secretion of TNFα and α2-macroglobulin (α2M) by activated glia.19, 20, 21, 22, 23 These are neurotoxic factors that trigger neuronal death in a paracrine manner. In addition, neurotoxic mechanisms are further promoted by α2M interacting with amyloid,24 and by α2M stabilizing proNGF and enhancing p75NTR activation.22
Hence, expression of proNGF, TNFα, α2M, sortilin, as well as neuronal damage and glial activation, serve as markers of p75NTR-mediated neurodegeneration in vivo. Here, we characterize these components in the dystrophic rd10 and RhoP retinas, as well as the use of p75NTR antagonists to reduce neurotoxicity and delay neurodegeneration, both in retinal explants and in vivo. This work validates inhibition of p75NTR as a therapeutic target for treatment of many forms of RP.
Results
Previous findings demonstrate the involvement of the proNGF/p75NTR system in optic nerve axotomy, diabetic retinopathy and glaucoma, diseases primarily affecting the retinal ganglion cell neurons.19, 20 We studied whether this mechanism is functional in a disease primarily affecting photoreceptor neurons, by quantifying the expression of p75NTR and proNGF as well as the downstream effectors TNFα and α2M, and the glial activation during the neurodegenerative process associated with RP.
Expression of p75NTR in the rd10 and the RhoP retina
In both wild-type (WT) and the dystrophic rd10 and RhoP retinas, p75NTR immunostaining was mostly localized in Müller glia cells, as shown in P21 retinal sections (Figure 1). This observation was confirmed by costaining with the Müller glial cell marker glutamine synthetase (GS; Figure 1). Moreover, comparable p75NTR immunostaining levels were found in both the WT and the dystrophic retinas.
Figure 1.

p75NTR and GS immunostaining in the WT, rd10 and RhoP retinas. Representative images of P21 retinal sections stained for p75NTR (green) and GS (red). Nuclei are stained with DAPI. Scale bar: 20 μm. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer.
Unprocessed proNGF and inflammatory markers are increased in the rd10 retina
We analyzed gene expression of the proNGF/p75NTR system components in the retina of the rd10 mouse, in comparison with WT, starting at stages previous to any observable sign of degeneration (P12) and during the initial period of photoreceptor cell degeneration and death, the time suited for a therapeutical treatment. Quantitative RT-PCR analysis did not show significant differences between WT and rd10 retinas in the mRNA levels of either NGF, the p75NTR and Trk-A receptors, or the p75NTR coreceptor sortilin, at P21, before the peak of photoreceptor cell death but when the degenerative process is well established (Figure 2a; WT expression level=1). Indeed, the mRNA profiles between P12 and P21 showed minor, no significant differences between WT and rd10 retinas (Supplementary Figure 1). In contrast, the mRNA levels of the proinflammatory cytokines IL-1β and TNFα, as well as that of the neurodegeneration-associated, secreted protein, α2M were highly expressed at P21 in the rd10 retina (Figure 2b; WT expression level=1). Moreover, a marked reactive gliosis in the rd10 retina at this age was revealed by the increase of GFAP mRNA, whereas CRALBP, a general Müller glia cell transcript, remained unchanged with respect to WT retina (Figure 2b). GFAP and α2M overexpression was confirmed by immunolabeling of P21 retinal sections. The increased expression of GFAP and α2M was observed in both the rd10 and the RhoP retinas (Figure 2c). GFAP immunoreactivity was associated to Müller gliosis, whereas α2M immunoreactivity was localized in the most external part of the dystrophic retinas, in close contact with the photoreceptor segments, retinal pigmented epithelium and choroids (Figure 2c and Supplementary Figure 2).
Figure 2.

Expression of the NGF system components and inflammation markers. (aand b) RT-qPCR of WT and rd10 retinas at P21. The levels of the different transcripts were normalized to the TBP RNA levels and relativized to WT levels (=1). (c) Representative images of P21 retinal sections from WT, rd10 and RhoP mice, immunostained for glial fibrillary acidic protein (GFAP) (cyan) and α2M (green). Nuclei are stained with DAPI. (d) Representative confocal images at the level of the photoreceptor segments, in P23 WT and rd10 retinas immunostained for CD11b. (e) Magnification of inset from the rd10 retina. (f) Quantification of the number of CD11b-positive cells in the different layers of the WT (gray bars) and rd10 (black bars) retinas. Bars represent the mean±S.E.M.; n≥3; **P<0.01, ***P<0.001. OS, outer segment; ONL, outer nuclear layer; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Scale bars: (c) 30 μm; (d) 50 μm and (e) 20 μm
All these inflammatory signs concur with a marked increase of microglial cells in the rd10 retina, determined at P23 (Figures 2d–f). Particularly relevant was the observed microglial infiltration in the surroundings of the degenerating rd10 photoreceptor cells, including the outer plexiform layer, the ONL and the outer segments of photoreceptors, where α2M immunolabeling was localized.
Next, we examined the persistence of unprocessed proNGF in the rd10 retina (Figure 3). The levels of proNGF, as determined by ELISA, were similar in both the WT and the rd10 retinas at P13, before any detectable photoreceptor degeneration. Strikingly, proNGF protein significantly increased in the rd10 retina at P18, an early degenerative stage close to the beginning of photoreceptor cell death. ProNGF levels remained elevated in the rd10 retina for the rest of the analyzed period, covering the interval of major photoreceptor cell loss, while proNGF in WT retinas persisted at similar, relatively low levels throughout the same time period (Figure 3a). Interestingly, a parallel determination of processed NGF did not show any increase in the rd10 retina (Figure 3b).
Figure 3.

Precursor (proNGF) and processed protein (NGF) levels in the WT and rd10 retinas. (a) ProNGF was quantified by enzyme-linked immunosorbent assay (ELISA) in four retina pools, at the indicated ages. (b) NGF was quantified by ELISA in four retina pools (the contralateral ones, from the same animals than proNGF) at the indicated ages. Two-way analysis of variance (ANOVA) for proNGF, F=12.99, P=0.0227; two-way ANOVA for NGF, F=1.49, P=0.2890
These results suggest that elevated proNGF protein levels, likely activating p75NTR in Müller glia cells, may have a role in the pathogenesis of RP.
Blocking of p75NTR decreases photoreceptor cell death and reactive gliosis in cultured rd10 retinas
We initially tested three small-molecule antagonists of p75NTR 20 in organotypic rd10 retinal explants from P22 mice (Figure 4). Retinal explants recapitulate RP-associated photoreceptor cell death seen in vivo and allow for an easier primary screening before in vivo testing.25 In controls, very scarce photoreceptor cell death is observed in the time frame of the experiment in organotypic retinal explants from P22 WT mice (data not shown).
Figure 4.

Effect of p75NTR antagonists in rd10 retinal explants. P22 rd10 retinas were cultured for 24 h in the absence (vehicle) or presence of three different p75NTR antagonists. (a) Representative images of retina explants stained for the TUNEL assay. (b) Quantification of the number of TUNEL-positive cells in the photoreceptor layer. Bars represent the mean±S.E.M.; n≥3; *P<0.05, **P<0.01. Scale bar: 20 μm
THX-B (1,3-diisopropyl-1-[2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydro-purin-7-yl)-acetyl]-urea) was very effective and reduced by 60% the density of TUNEL-positive nuclei in the photoreceptor layer of treated rd10 retinas compared with vehicle-treated rd10 retinas (Figure 4b). THX-A and THX-C (also known as LM11A-2426) are less active small-molecule analogs of THX-B, and exhibited a trend to reducing photoreceptor death. Another p75NTR antagonist LM11A-3126 also partially reduced the density of TUNEL-positive nuclei in the retinal explants (Supplementary Figure 3a).
In these experiments, THX-B had the highest relative potency in decreasing photoreceptor cell death; hence, THX-B was selected for a more detailed characterization of its effect ex vivo and in vivo. THX-B treatment of cultured P22 rd10 retinas attenuated the thickening and enlargement of processes of astrocytes and Müller glia cells visualized by GFAP immunostaining (Figure 5).
Figure 5.
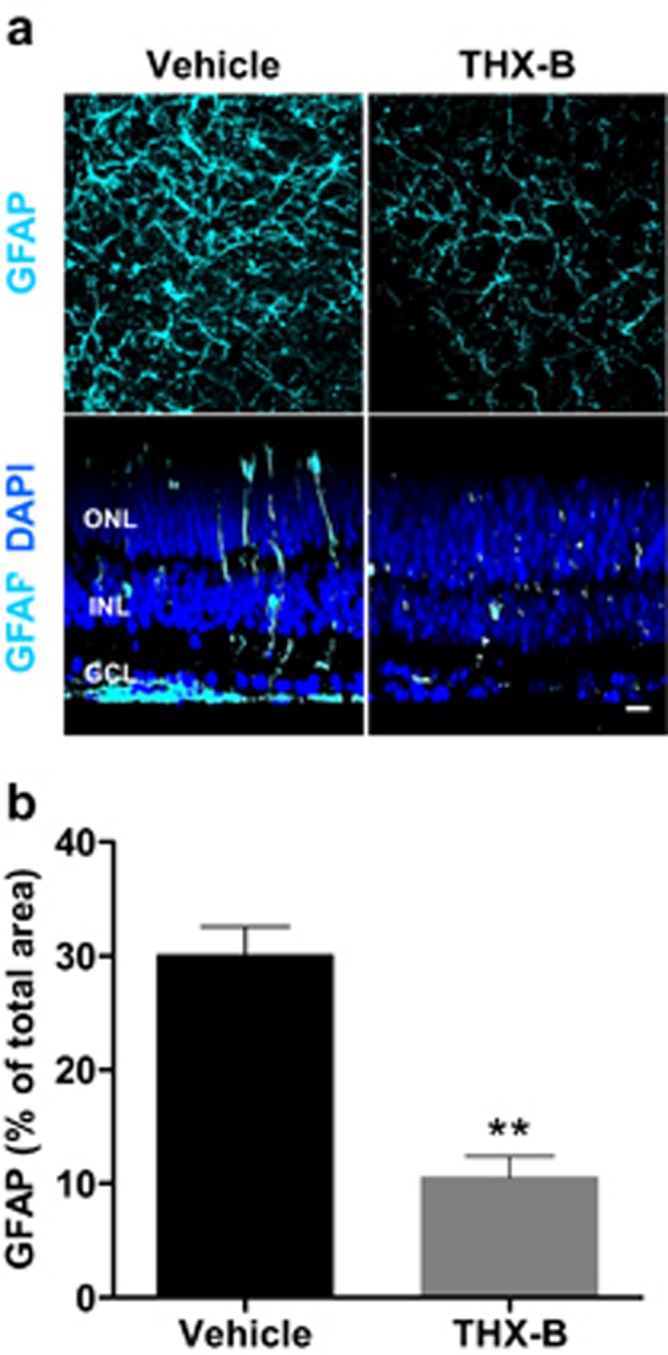
Effect of the p75NTR antagonist THX-B on reactive gliosis in rd10 retinal explants. P22 rd10 retinas were cultured for 24 h in the absence (vehicle) or presence of THX-B. (a) Representative confocal optical sections of retinal whole mounts (xy, upper panels; xz, lower panels) stained for glial fibrillary acidic protein (GFAP) (cyan). Nuclei counterstained with DAPI (blue) are shown in the lower panels. (b) Quantification of the area filled with GFAP staining. Bars represent mean±S.E.M.; n≥3; **P<0.01. Scale bar: 20 μm. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer
In vivo blocking of p75NTR preserves the ONL
The results obtained in organotypic rd10 retinal explants suggest that p75NTR may be a pharmacological target for RP. Hence, we studied the in vivo effect of THX-B in the progression of the retinal degeneration in the murine RP models. A single intravitreal injection of THX-B at P17 in the rd10 mouse eye elicited a neuroprotective effect on photoreceptor cells, observable at P22. The number of photoreceptor rows as well as the ONL/INL ratio were significantly higher in the treated eye than in the vehicle-injected eye from the same animal (Figures 6a–c). This concurred with a significant decrease in the total number of microglial cells in the treated retinas (Figure 6d), as well as a reduction in some of the inflammatory signs, such as the mRNA levels of GFAP, α2M and the proinflammatory cytokines IL-1β and TNFα (Figure 6e; untreated retinas expression level=1). Moreover, these effects concur with the reduced levels of TNFα secreted into the culture medium by LM11A-31-treated rd10 retinal explants (Supplementary Figure 3c), supporting the proinflammatory role of p75NTR in RP.
Figure 6.

In vivo effect of the p75NTR antagonist THX-B in the rd10 retina. P17 rd10 mice were intravitreally injected with THX-B in one eye and with vehicle in the contralateral eye, and analyzed at P22. (a) Representative sections. Nuclei are stained with DAPI. (b) Comparison of the photoreceptor number per row between the THX-B-treated eye and the control eye. Paired eyes are individually represented. (c) Comparison of the ONL/INL thickness ratio between the THX-B-treated eye and the control eye. Paired eyes are individually represented. (d) Quantification of the microglial (TL-positive) cell density in the THX-B treated retina versus the control retina from the same animal. Paired eyes are individually represented. (e) RT-qPCR of THX-B-treated and control retinas. The levels of the different transcripts were normalized to the TBP RNA levels and relativized to control retina levels. Bars represent mean±S.E.M.; n≥3; *P<0.05, **P<0.01; two-way analysis of variance (ANOVA) for RT-qPCR of treated versus control retinas, F=4.40, P=0.0467. Scale bar: 20 μm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; TL, tomato lectin
Taken together, our observations support the involvement of p75NTR in the molecular and cellular processes occurring in the dystrophic retina and leading to degeneration.
To check the generality of our observations, we treated the RhoP mouse, which presents photoreceptor cell loss with a pace comparable to that of the rd10 mouse,8 as well as similar features concerning, at least, p75NTR, GFAP and α2M expression patterns (Figures 1 and 2c). We first confirmed the effect of THX-B on photoreceptor cell death in P18 organotypic RhoP retinal explants. As in the case of the rd10 retinas, THX-B significantly reduced the number of TUNEL-positive photoreceptor nuclei in comparison with untreated retinas (Figure 7a). We then tested the effect of THX-B in vivo in two different ways. A single THX-B intravitreal injection at P17 resulted, after 5 days, in higher ONL/INL thickness ratio than that of the contralateral vehicle-injected eye (Figures 7b and c), as previously shown for the rd10 mouse (Figures 6a–c). Although intravitreal injection is a common clinical practice in human patients, we tested a less invasive delivery method. The RhoP mice were treated with a single subconjunctival injection of THX-B at P18 and the retinas were analyzed at P22. Again, the eyes treated with THX-B presented significant photoreceptor cell preservation as determined by the observed higher density of photoreceptor nuclei in the THX-B-treated eye than in the control eye (Figures 7d and e).
Figure 7.

Effect of the P75NTR antagonist THX-B, both ex vivo and in vivo, in the RhoP retina. (a) P18 RhoP retinas were cultured for 24 h in the absence (vehicle) or presence of the THX-B. Photoreceptor cell death was visualized by TUNEL and quantified. (b) P17 RhoP mice were intravitreally injected with THX-B in one eye and with vehicle in the contralateral eye, and analyzed at P22. Nuclei in whole-mount retinas were counterstained with DAPI. Representative cryosections. (c) Comparison of the ONL/INL thickness ratio between the THX-B-treated eye and the vehicle control eye. Paired eyes are individually represented. (d) P18 RhoP mice were subconjunctivally injected with THX-B in one eye and with vehicle in the contralateral eye and analyzed at P24. Representative cryosections stained with toluidine. (e) Comparison of the density of nuclei in the ONL between the THX-B-treated eye and the vehicle control eye. Paired eyes are individually represented. Bars represent the mean±S.E.M.; n≥3; **P<0.01; *** P<0.001. Scale bar: 20 μm. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer
Altogether, our results confirm that p75NTR has a relevant role in the RP-associated degenerative process, and constitutes a relevant therapeutic target to attenuate the course of this incurable group of inherited retinal dystrophies.
Discussion
In the present work, we have analyzed the possible involvement of proNGF/p75NTR signaling in the photoreceptor degeneration process associated with RP. Moreover, we have shown the potential therapeutic value of p75NTR antagonists for RP treatment. THX-B administration reduced photoreceptor cell death ex vivo and in vivo, in two different RP models, thus providing an initial proof of concept on a possible therapy for RP. Remarkably, subconjunctival administration arises as a medical plausible treatment for RP, as well as for other retinal dystrophies.27
The proneurotophin–p75NTR axis is emerging as a key player in the progression of several neurodegenerative pathologies.18 In the present study we have seen an increase in the proNGF protein levels in the rd10 retinas before the massive loss of photoreceptors while no significant changes were observed in the processed NGF. Moreover, blocking p75NTR decreased photoreceptor loss. Several studies in models of retinal and brain neurodegenerative damage showed a coordinated upregulation of several components of the proNGF-p75NTR system.28, 29 In this study, we did not observe changes in the gene expression levels of NGF, Trk-A, p75NTR or its coreceptor sortilin. Indeed, the p75NTR immunostaining pattern is similar in the WT, rd10 and RhoP retinas, besides those associated with the degenerative changes. The nature of the damage as well as the age at which degeneration occurs may account for the differential regulation of the proNGF-p75NTR system components in our model. In the rd10 mouse, a model of early onset RP, the increase in proNGF at the protein level may be sufficient to trigger the neurotoxic response through p75NTR. In addition, p75NTR may also bind other proneurotrophins18 (namely pro-BDNF, pro-NT3) in the degenerating rd10 retinas, an alternative that will be addressed in future studies. The mechanistic role of the proNGF-p75NTR signaling in neurodegenerative diseases has been widely attributed to its proapoptotic action. Indeed, p75NTR belongs to the tumor necrosis factor superfamily characterized by the presence of an intracellular ‘death domain’ that interacts with components of the program cell death machinery.30 However, recent studies in retina have shown the contribution of proNGF-p75NTR signaling to neuronal death through a non-cell-autonomous, paracrine mechanism. ProNGF acting on its p75NTR receptor in Müller glial cells is able to induce TNFα production by Müller glial cells, which provokes ganglion cell degeneration.23, 31 We have observed a selective location of p75NTR in Müller glial cells in WT, rd10 and RhoP retinas (Figure 1) at the ages when RP-associated photoreceptor degeneration is taking place.
The source of proNGF may vary according to the type of injury and the affected tissue. It is worth recalling that microglial cells are a potential source of proNGF in the retina during development,32, 33 as well as in models of retinal dystrophies.34 In the rd10 retina, microglia is highly activated and mobilized (Figures 2d–f), and may be the source of proNGF.
ProNGF is efficiently processed in the uninjured adult nervous system and very low levels of the proprotein form are detected under physiological conditions. The intracellular machinery that ensures the efficient conversion of proNGF into mature NGF is not well characterized. Retinal degeneration in the rd10 mouse was accompanied by an increase of α2M, selectively located to the photoreceptor segment (Figures 2b and c and Supplementary Figure 2) and presumptively expressed by Müller glia cells,35 whose role in retinal neurodegeneration has been recently characterized in detail.22, 24 α2M binds to proNGF conferring resistance to processing and increased activity on p75NTR. The marked increase in the α2M in the rd10 retina may account, at least in part, for the accumulation of proNGF. On the other hand, α2M binds to NGF and decreases its activity on the Trk-A receptor.
A hallmark of most forms of RP, despite their genetic origin, is the primary death of rod photoreceptors, in which the mutated gene exerts its function, followed by a secondary loss of cone photoreceptors.5 Photoreceptor degeneration is accompanied and followed by whole retinal remodeling, including retinal pigmented epithelium disorganization, inner neuronal connectivity alterations and vascular disorganization and regression. Concomitant to these alterations an inflammatory process takes place as shown by microglial recruitment, reactive gliosis of Müller glial cells and astrocytes, and a burst of proinflammatory cytokine expression.3 The hierarchies, respective roles and molecular action of microglia, Müller cells and astrocytes during rd10 retinal degeneration remain to be elucidated. Interfering with one or several of these common players may provide a general therapy, despite the broad mutation spectrum of RP. Supporting this approach, suppression of microglial activation by minocycline treatment protected retinal structure and visual function in the rd10 mouse.14 Further, TNFα inhibition by Adalimumab in the rd10 mouse reduces photoreceptor cell death.16
In the present study, we have extended to RP the therapeutic potential of interfering with the proNGF/p75NTR system described for other retinal dystrophies.20, 21, 31
Materials and methods
Animal procedures
The rd10 mouse model of retinal degeneration is a homozygous recessive mutant for phosphodiesterase 6b (Pde6brd10/rd10) on a C57BL/6 J background. The RhoP mouse model of retinal degeneration is a hemizygous dominant mutant that carries the mutant human rhodopsin Pro347Ser transgene on a C57BL/6 J background. WT mice of the same background were used as control. All animals were housed and handled in accordance with the ARVO statement for the Use of Animals in Ophthalmic and Vision Research, and European Union and Canadian guidelines. Mice were bred in the CIB and Lady Davis Institute core facilities.
Intravitreal injections
Rd10 and RhoP mice at P17 were anesthetized with isoflurane. Intravitreal injection was performed under an ophthalmoscope to visualize the retinal fundus. Using a Hamilton syringe (Hamilton AG, Bonaduz, Switzerland; Model 75 RN SYR) with a 33-gauge removable needle, the right eye was injected with 2 μl of the p75NTRantagonist, THX-B (2 μg/μl in 10% (v/v) DMSO in PBS), whereas the left eyes received 2 μl of the vehicle. Animals were killed 5 days after injection, and eyes were processed for cryosectioning as described below. Both male and female mice were used for this study and at least three animals per group were used.
Subconjunctival injection
RhoP at P18 were anesthetized with isoflurane. The conjunctiva was gently pulled from the sclera using tweezers. The conjunctiva of the right eye was injected with 30 μl containing a total of 15 μg THX-B (0.5 μg/μl in 10% (v/v) DMSO in PBS). Half of the total volume of THX-B was delivered into the superior subconjunctival space and the other half into the nasal subconjunctival region using a microsyringe with a 33 G needle. The left eyes received 30 μl of the vehicle. Animals were killed 4 days after injection and processed as above.
p75NTR antagonists synthesis
THX-B and the analogs have been described20 (US20100392647P patent by H Nedev and HU Saragovi). All compounds were purified by HPLC to >99%. The empirical formula of THX-B (F.W. 365) was confirmed by LC/MS, and the 1H NMR spectra (300 MHz) of compounds are consistent with the expected structures.
Retina explant cultures
P20 and P22 rd10 and P18 RhoP mice were killed, and their eyes were enucleated. Retinas were dissected and cultured free floating in M24 multiwell plates for 24 h in 1 ml DMEM/F12 medium containing N2 supplement, except insulin, and in the presence of the indicated p75NTR antagonist THX-A, THX-B, THX-C, LM11A-31 (Tocris, Avonmouth, Bristol, UK), or vehicle. A dose range of 0.2–40 μM drug concentrations were tested (only 20 μM shown). Retinas were subsequently fixed in 4% (wt/vol) paraformaldehyde in phosphate buffer 0.1M, pH 7.4, for 1 h at RT and processed for detection of cell death and immunostaining.
Cell death visualization and counting
Photoreceptor cell death was visualized by DNA fragmentation using the TUNEL assay (DeadEnd Fluorometric TUNEL System; Promega, Madison, WI, USA). Fixed whole-mount retinas were permeated with 2% (wt/vol) Triton X-100 (Merck, Darmstadt, Germany) in PBS for 2 h at RT, followed by incubation in 20 μg/ml proteinase K (Merck) for 15 min at 37 °C and, subsequently, processed for TUNEL staining according to the manufacturer's instructions. After TUNEL labeling, the nuclei were counterstained with 0.05% (wt/vol) propidium iodide (Merck) in PBS, and the retinas were flat mounted in Fluoromount-G (Southern Biotechnology, Birmingham, AL, USA), and analyzed with a laser confocal microscope (TCS SP2; Leica Microsystems, Wetzlar, Germany). Serial optical sections were acquired with the × 63 objective every 1 μm, covering the whole ONL thickness in four central fields around the optic nerve. Direct counting of TUNEL-positive cells was carried out using the FIJI open-source Software.36
Immunolabeling of whole-mount retinas
Fixed whole-mount retinas were permeated with 2% (wt/vol) Triton X-100 in PBS, blocked in BGT (2.5% (wt/vol) BSA, 100 mM glycine, 0.25% (wt/vol) Triton X-100 in PBS) and incubated overnight at 4 °C, with the indicated primary antibodies or 2 h at RT with a biotinylated probe, after washing with TBS. Retinas were then washed with PBS and incubated for 2 h at RT with the secondary antibodies or streptavidin. The nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific, Waltham, MA USA), the retinas were mounted in Fluoromount-G and analyzed with a laser confocal microscope (TCS SP5 and TCS SP2; Leica Microsystems). Antibodies are listed below.
For determining microglia density, serial optical sections were acquired with a × 40 objective every 2.5 μm through the whole retina in four central fields around the optic nerve. To measure the percentage of the area occupied by GFAP staining, serial optical sections were acquired with a × 40 objective every 2 μm, covering the whole ganglion cell layer in 12 fields (four central, four medial and four peripheral) around the optic nerve. Confocal maxima of the ganglion cell layer Z sections were then converted into a black and white image and analyzed with the FIJI Software to calculate the percentage of total area filled with GFAP.
Immunostaining of retinal sections
Animals were killed, their eyes were enucleated, fixed for 1 h in 4% (wt/vol) parafolmaldehyde in phosphate buffer 0.1 M, pH 7.4, and then cryoprotected by incubation in increasing concentrations of sucrose (15–50% in PBS). The eyes were then embedded in Tissue-Tek (Sakura, Leiden, The Netherlands), and frozen on dry ice. Cryostat sections (12 μm) were mounted on Superfrost slides (Thermo Fisher Scientific) and dried at room temperature. For p75NTR immunostaining, retinal sections were rehydrated in PBS, incubated with NaBH4 5 mg/ml in PBS for 5 min, followed by 0.1% (wt/vol) Triton X-100 and 5% (v/v) normal goat serum in PBS for 1 h. For immunostaining with the rest of the antibodies, retinal sections were permeated for 1 h with 0.2% (wt/vol) Triton X-100, and blocked with BGT for another hour. Following the incubation with the primary antibody overnight at 4 °C, the sections were washed with PBS and incubated for 1 h with secondary antibodies and DAPI. Then, sections were washed with PBS and coverslipped with Fluoromont-G. Antibodies are listed below.
To quantify the retinal structure using techniques unbiased to retinal location (i.e. the central regions of the retina are thicker than the peripheral regions), we compared the thickness of the ONL (containing primarily photoreceptors) and the corresponding INL (containing bipolar, horizontal and amacrine neuron as well as Müller glia cell bodies). Both ONL and INL were measured, and the ONL/INL ratios were calculated in treated eyes versus vehicle-injected contralateral eyes. Five sections per retina were analyzed. For each section, one photograph was taken for each of four defined retinal zones. In each photo three measurements of the ONL and INL thickness were performed in triplicate to obtain an average value per retinal zone per section. The measurements were carried out with the FIJI Software (using the ‘freehand line’ and ‘measure’ tools). Alternatively, the number of nucleus rows or the nuclear density in a determined retinal area were quantified in the same way to corroborate the neuroprotective effect and the accuracy of the ONL/INL ratio determination.

ProNGF, NGF and TNFα levels determination
ProNGF and NGF levels were measured in retinal extracts from the same animals by ELISA assay (MyBioSource Inc., San Diego, CA, USA). For proNGF determination, four retinas per age were homogenized in PBS at 100 mg wet weight per ml and stored overnight at −20 °C. After two freeze–thaw cycles, the homogenates were centrifuged for 5000xg at 4 °C for 5 min. The supernatant was removed and assayed immediately following the manufacturer’s instructions. For NGF determination, the four contralateral retinas per age of the same animal used for proNGF determination were homogenized in PBS at 100 mg wet weight per ml and centrifuged for 1500xgfor 15 min. The supernatant was removed and assayed immediately. TNFα concentration was measured in culture supernatant from retinal explants by ELISA assay (BioLegend, San Diego, CA, USA) following the manufacturer’s instructions.
RNA isolation and quantitative PCR
Total RNA from individual retinas was extracted using the TRIzol reagent, and 2.5 μg of RNA were typically reverse transcribed (RT) using the Superscript III Kit and random primers (all from Thermo Fisher Scientific). Quantitative PCR (qPCR) was performed with the ABI Prism 7900HT Sequence Detection System using TaqMan Universal PCR Master Mix, No-AmpErase UNG and the Taqman assays (listed below) for detection (all from Thermo Fisher).

Statistical analysis
Data size was estimated in accordance with previous literature, following the Reduction principle for animal research. At least three animals were analyzed per experimental point. Data points in graphs represent individual mice, and bars in all panels represent the mean and the standard error of the mean (S.E.M.).
Data were checked for normality using both D’Agostino–Pearson omnibus and Kolmogorov–Smirnov normality tests sequentially. Data were considered to fit a normal distribution only if they passed simultaneously both tests. For normal data, Fisher’s test was used to determine whether the variance of the samples analyzed was comparable (homoscedasticity). Normal data were compared using a two-way ANOVA with Bonferroni's multiple comparison test or an unpaired Student’s T-test, applying Welch’s correction in cases of non-homoscedasticity. In cases of non-normal samples, populations were compared using the Mann–Whitney nonparametric U-test. Outliers were detected by Grubbs’ outlier test, and excluded from further analysis. If previous literature allowed us to make a prediction about the result of the experiment, then one-sided test were applied. Otherwise, tests were two sided. All analyses were performed at a fixed 95% confidence interval, using the GraphPad Prism version 5.01 for Windows (GraphPad Software, San Diego, CA, USA; http://www.graphpad.com). Statistically significant differences are indicated as follows: *P<0.05; **P<0.01; ***P<0.001.
Acknowledgments
This work was supported by the Ministerio de Economía y Competitividad, Spain (Grants SAF2013-41059- R and SAF2016-75681- R to EJdlR), Ministerio de Educación, Cultura y Deporte, Spain (Travel grant to EJdlR), and by CIHR grants (Pharmacology and Proof of Principle) and by the Foundation Fighting Blindness to HUS. MPC has a fellowship from the Ministerio de Economía y Competitividad, Spain. We thank laboratory members for continuous encouragement and ideas, and Cayetana Murillo, José Luis Martínez and the staff of the CIB animal house and microscopy facilities for technical support.
Footnotes
Supplementary Information accompanies this paper on Cell Death and Disease website (http://www.nature.com/cddis)
Edited by A Yaron
McGill University has filed patents for the compounds used in this project (HUS inventor).
Supplementary Material
References
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet 2006; 368: 1795–1809. [DOI] [PubMed] [Google Scholar]
- Chinskey ND, Besirli CG, Zacks DN. Retinal cell death and current strategies in retinal neuroprotection. Curr Opin Ophthalmol 2014; 25: 228–233. [DOI] [PubMed] [Google Scholar]
- Cuenca N, Fernandez-Sanchez L, Campello L, Maneu V, De la Villa P, Lax P et al. Cellular responses following retinal injuries and therapeutic approaches for neurodegenerative diseases. Progr Retin Eye Res 2014; 43: 17–75. [DOI] [PubMed] [Google Scholar]
- Rivas MA, Vecino E. Animal models and different therapies for treatment of retinitis pigmentosa. Histol Histopathol 2009; 24: 1295–1322. [DOI] [PubMed] [Google Scholar]
- Bramall AN, Wright AF, Jacobson SG, McInnes RR. The genomic, biochemical, and cellular responses of the retina in inherited photoreceptor degenerations and prospects for the treatment of these disorders. Annu Rev Neurosci 2010; 33: 441–472. [DOI] [PubMed] [Google Scholar]
- Sacchetti M, Mantelli F, Merlo D, Lambiase A. Systematic review of randomized clinical trials on safety and efficacy of pharmacological and nonpharmacological treatments for retinitis pigmentosa. J Ophthalmol 2015; 2015: 737053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B, Hawes NL, Pardue MT, German AM, Hurd RE, Davisson MT et al. Two mouse retinal degenerations caused by missense mutations in the beta-subunit of rod cGMP phosphodiesterase gene. Vision Res 2007; 47: 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Snyder WK, Olsson JE, Dryja TP. Transgenic mice carrying the dominant rhodopsin mutation P347S: evidence for defective vectorial transport of rhodopsin to the outer segments. Proc Natl Acad Sci USA 1996; 93: 14176–14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrochano S, Barhoum R, Boya P, Arroba AI, Rodriguez-Muela N, Gomez-Vicente V et al. Attenuation of vision loss and delay in apoptosis of photoreceptors induced by proinsulin in a mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci 2008; 49: 4188–4194. [DOI] [PubMed] [Google Scholar]
- Chadderton N, Millington-Ward S, Palfi A, O'Reilly M, Tuohy G, Humphries MM et al. Improved retinal function in a mouse model of dominant retinitis pigmentosa following AAV-delivered gene therapy. Mol Ther 2009; 17: 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramall AN, Szego MJ, Pacione LR, Chang I, Diez E, D'Orleans-Juste P et al. Endothelin-2-mediated protection of mutant photoreceptors in inherited photoreceptor degeneration. PLoS ONE 2013; 8: e58023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone I, Novelli E, Strettoi E. Long-term preservation of cone photoreceptors and visual acuity in rd10 mutant mice exposed to continuous environmental enrichment. Mol Vis 2014; 20: 1545–1556. [PMC free article] [PubMed] [Google Scholar]
- Jiang K, Wright KL, Zhu P, Szego MJ, Bramall AN, Hauswirth WW et al. STAT3 promotes survival of mutant photoreceptors in inherited photoreceptor degeneration models. Proc Natl Acad Sci USA 2014; 111: E5716–E5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng B, Xiao J, Wang K, So KF, Tipoe GL, Lin B. Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J Neurosci 2014; 34: 8139–8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Muela N, Hernandez-Pinto AM, Serrano-Puebla A, Garcia-Ledo L, Latorre SH, de la Rosa EJ et al. Lysosomal membrane permeabilization and autophagy blockade contribute to photoreceptor cell death in a mouse model of retinitis pigmentosa. Cell Death Differ 2015; 22: 476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Fernandez de la Camara C, Hernandez-Pinto AM, Olivares-Gonzalez L, Cuevas-Martin C, Sanchez-Arago M, Hervas D et al. Adalimumab reduces photoreceptor cell death in a mouse model of retinal degeneration. Sci Rep 2015; 5: 11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez CF, Simi A. p75 neurotrophin receptor signaling in nervous system injury and degeneration: paradox and opportunity. Trends Neurosci 2012; 35: 431–440. [DOI] [PubMed] [Google Scholar]
- Hempstead BL. Deciphering proneurotrophin actions. Handbook Exp Pharmacol 2014; 220: 17–32. [DOI] [PubMed] [Google Scholar]
- Lebrun-Julien F, Morquette B, Douillette A, Saragovi HU, Di Polo A. Inhibition of p75(NTR) in glia potentiates TrkA-mediated survival of injured retinal ganglion cells. Mol Cell Neurosci 2009; 40: 410–420. [DOI] [PubMed] [Google Scholar]
- Bai Y, Dergham P, Nedev H, Xu J, Galan A, Rivera JC et al. Chronic and acute models of retinal neurodegeneration TrkA activity are neuroprotective whereas p75NTR activity is neurotoxic through a paracrine mechanism. J Biol Chem 2010; 285: 39392–39400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysona BA, Al-Gayyar MM, Matragoon S, Abdelsaid MA, El-Azab MF, Saragovi HU et al. Modulation of p75(NTR) prevents diabetes- and proNGF-induced retinal inflammation and blood-retina barrier breakdown in mice and rats. Diabetologia 2013; 56: 2329–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcelona PF, Saragovi HU. A pro-nerve growth factor (proNGF) and NGF binding protein, alpha2-macroglobulin, differentially regulates p75 and TRKa receptors and is relevant to neurodegeneration ex vivo and in vivo. Mol Cell Biol 2015; 35: 3396–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcelona PF, Sitaras N, Galan A, Esquiva G, Jmaeff S, Jian Y et al. p75NTR and its ligand proNGF activate paracrine mechanisms etiological to the vascular, inflammatory, and neurodegenerative pathologies of diabetic retinopathy. J Neurosci 2016; 36: 8826–8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizi C, Businaro R, Lauro GM, Fumagalli L. Role of alpha2-macroglobulin in regulating amyloid beta-protein neurotoxicity: protective or detrimental factor? J Neurochem 2001; 78: 406–412. [DOI] [PubMed] [Google Scholar]
- Arroba AI, Alvarez-Lindo N, van Rooijen N, de la Rosa EJ. Microglia-mediated IGF-I neuroprotection in the rd10 mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci 2011; 52: 9124–9130. [DOI] [PubMed] [Google Scholar]
- Yang T, Knowles JK, Lu Q, Zhang H, Arancio O, Moore LA et al. Small molecule, non-peptide p75 ligands inhibit Abeta-induced neurodegeneration and synaptic impairment. PLoS ONE 2008; 3: e3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan A, Barcelona PF, Nedev H, Sarunic MV, Jian Y, Saragovi HU. Subconjunctival delivery of p75NTR antagonists reduces the inflammatory, vascular, and neurodegenerative pathologies of diabetic retinopathy. Invest Ophthalmol Vis Sci 2017; 58: 2852–2862. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Harada C, Okumura A, Namekata K, Mitamura Y, Yoshida K et al. Effect of p75NTR on the regulation of photoreceptor apoptosis in the rd mouse. Mol Vis 2005; 11: 1229–1235. [PubMed] [Google Scholar]
- Al-Shawi R, Hafner A, Chun S, Raza S, Crutcher K, Thrasivoulou C et al. ProNGF, sortilin, and age-related neurodegeneration. Ann NY Acad Sci 2007; 1119: 208–215. [DOI] [PubMed] [Google Scholar]
- Liepinsh E, Ilag LL, Otting G, Ibanez CF. NMR structure of the death domain of the p75 neurotrophin receptor. EMBO J 1997; 16: 4999–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun-Julien F, Bertrand MJ, De Backer O, Stellwagen D, Morales CR, Di Polo A et al. ProNGF induces TNFalpha-dependent death of retinal ganglion cells through a p75NTR non-cell-autonomous signaling pathway. Proc Natl Acad Sci USA 2010; 107: 3817–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frade JM, Rodriguez-Tebar A, Barde YA. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 1996; 383: 166–168. [DOI] [PubMed] [Google Scholar]
- Frade JM, Barde YA. Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron 1998; 20: 35–41. [DOI] [PubMed] [Google Scholar]
- Srinivasan B, Roque CH, Hempstead BL, Al-Ubaidi MR, Roque RS. Microglia-derived pronerve growth factor promotes photoreceptor cell death via p75 neurotrophin receptor. J Biol Chem 2004; 279: 41839–41845. [DOI] [PubMed] [Google Scholar]
- Shi Z, Rudzinski M, Meerovitch K, Lebrun-Julien F, Birman E, Di Polo A et al. Alpha2-macroglobulin is a mediator of retinal ganglion cell death in glaucoma. J Biol Chem 2008; 283: 29156–29165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9: 676–682D. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.