

Abstract
In the last several decades, research in the field of drug delivery has been challenged with the fabrication of carrier systems engineered to deliver therapeutics to the target site with sustained and controlled release kinetics. Herein, we report the fabrication of microparticles composed of two distinct compartments: i) one compartment containing a pH responsive polymer, acetal-modified dextran, and PLGA (polylactide-co-glycolide), and ii) one compartment composed entirely of PLGA. We demonstrate the complete release of dextran from the microparticles during a 10-hour period in an acidic pH environment and the complete degradation of one compartment in less than 24 h. This is in congruence with the stability of the same microparticles in neutral pH over the 24-hour period. Such microparticles can be used as pH responsive carrier systems for drug delivery applications where their cargo will only be released when the optimum pH window is reached. The feasibility of the microparticle system for such an application was confirmed by encapsulating a cancer therapeutic, irinotecan, in the compartment containing the acetal-modified dextran polymer and the pH dependent release over a 5-day period was studied. It was found that upon pH change to an acidic environment, over 50% of the drug was first released at a rapid rate for 10 h, similar to that observed for the dextran release, before continuing at a more controlled rate for 4 days. As such, these microparticles can play an important role in the fabrication of novel drug delivery systems due to the selective, controlled, and pH responsive release of their encapsulated therapeutics.
Keywords: Drug delivery, Stimuli responsive polymers, Multicompartmental microparticles, Cancer therapeutics, Electrohydrodynamic co-jetting
1. Introduction
Currently, the administration of therapeutics is hampered by the accurate control over bioavailability. These shortcomings lead to patient compliance issues, and, in some cases, severe side effects and drug resistance [1,2]. The use of drug delivery microcarriers can provide an alternative for the administration of therapeutics, [2] because microparticles have the potential to act as a depot, initially protecting the encapsulated therapeutic and releasing the drug in a manner that can ideally be adjusted to the pharmacokinetics of a selected therapeutic [3,4]. In addition, such carriers can be biocompatible and/or biodegradable and intrinsically harmless to the body [5,6]. First formulations have been evaluated in clinical trials and some have already been turned into pharmaceutical products [7,8]. While these early formulations have demonstrated the clinical potential of prolonged release formulations, these carriers are still far from optimal and lack important aspects, such as tailored release kinetics [9]. Another concern is that, as delivery carriers become more efficient in increasing local therapeutic concentrations, the risks associated with the unsolved issue of cellular drug resistance can arguably be elevated [10,11].A promising approach to avoid drug resistance may be the combined use of multiple therapeutics. This delivery will require complex release profiles to accommodate the differences in the pharmacological windows of the respective drugs [12]. Thus, a new generation of drug carriers is needed that can release (i) two or more drugs; (ii) with fully decoupled release profiles. If chosen carefully, such carrier systems would also benefit from possible synergistic effects of the drugs that can further enhance the therapeutic efficacy of the combined delivery [13,14]. Several researchers have proposed possible strategies for the incorporation of complex release profiles in carrier systems, some of which include the use of stimuli responsive material for on-demand and complex release kinetics [9,15–18]. Responsive material can be highly sensitive to external stimuli such as pH, temperature, light, and oxidative stress [18]. The structure and degradation of microparticles composed of stimuli responsive material can then be controlled, and on-demand pharmacokinetics of the encapsulated therapeutics can be achieved [16,18,19]. Beyond simple prolongation of the release, [8] these drug carrier systems can be tailored to the therapeutic window associated with a specific disease [15,17].
The challenge in developing such delivery systems has shifted towards the design and preparation of the ideal carrier particles that can spatioselectively encapsulate separate therapeutics and release them with distinct pharmacokinetics [9]. Current carrier systems that are loaded with multiple therapeutics in a single isotropic particle lack this characteristic and may even promote antagonistic cross-interactions between drugs [9,20–22]. Thus far, several approaches have been developed to fabricate carrier systems that can selectively encapsulate therapeutics in unique environments (composed of different materials) such as capsules [18], LbL (Layer-by-Layer) particles [23,24], templated particles [25,26], core-shell structures [27,28], and dendrimers [29,30]. While these strategies have allowed for the spatioselective encapsulation of therapeutics, few have led to distinct release profiles of two drugs from the same particle [31].
Alternatively, electrohydrodynamic (EHD) co-jetting [32] can result in anisotropic particles with distinct internal geometries [32–37]. The unique multicompartmental architecture allows for the possible incorporation of multiple therapeutics – and polymers – in selected compartments and can result in distinct release profiles for each therapeutic. While most of the work published using EHD co-jetting has focused on the use of similar polymers in different compartments, [35,36,38,39] our more recent work has demonstrated the incorporation of a variety of different polymers, such as hydrogels (e.g., poly(ethylenimine) and poly(ethylene oxide)) [37,40], polysaccharides (dextran) [41], or poly(acrylamide-co-acrylic acid) [42], poly(methyl methacrylate) [37], poly(styrene) [37], and poly(vinyl cinnamate) [37,43,44]. In addition, we have also illustrated the incorporation and release of siRNA from bicompartmental particles with pronounced efficacy in an in vitro model [40]. In this manuscript, we demonstrate the selective incorporation of a stimuli responsive polymer in bicompartmental microparticles via EHD co-jetting and confirm the anisotropic degradation of one compartment only. We further demonstrate the incorporation of a low molecular weight cancer therapeutic selectively in one compartment and display pH dependent release from anisotropic microparticles.
2. Materials and methods
2.1. Materials
Dextran, chloroform, dimethylformamide (DMF), dimethyl sulfoxide (DMSO), pyridinium p-toluenesulfonate, 2-methoxypropane triethylamine, acetic acid, sodium acetate, phosphate buffered saline (PBS), poly[tris(2,5-bis(hexyloxy)-1,4-phenylenevinylene)-alt-(1,3-phenylenevinylene)] (PTDPV) as the green marker for confocal imaging, poly[(m-phenylenevinylene)-alt-(2,5-dihexyloxy-p-phenylenevinylene)] (MEHPV) as the blue marker for confocal imaging, and tween 20 were used as purchased from Sigma Aldrich, USA. Polylactide-co-glycolide (PLGA) with a ratio of 50:50 lactide-to-glycolide and a molecular weight of 44 kDa was purchased from Corbion Purac Corporation. Irinotecan was purchased from Ontario Chemicals.
2.2. Polymer 1 synthesis and analysis
Acetal-modified dextran (polymer 1) was synthesized as previously described by Fréchet et al. [45]. The polymer was analyzed via IR and NMR spectroscopy to confirm the presence of the appropriate functional groups and polymer purity. The analysis for the degree of protection and cyclic vs. noncyclic protection was done through NMR analysis.
2.3. Electrohydrodynamic Co-jetting
Microparticles were fabricated using the electrohydrodynamic co-jetting procedure as described previously [39]. The jetting solutions were made with a 97:03 ratio of chloroform to DMF and a polymer concentration of 15% w/v. For compartments that contained polymer 1, 75% w/w of the total polymer was polymer 1 while the rest was PLGA. Bicompartmental microparticles containing PLGA in one side and a mixture of PLGA and polymer 1 in the second hemisphere were fabricated using side-by-side 26 gauge needles and a voltage of approximately 6–7 kV. EHD co-jetting was done at a flow rate of 0.4 ml/h, a needle-to-substrate distance of 30 cm, and at room temperature. A metal sheet acted as the counter electrode, onto which the microparticles were deposited and then collected for further processing. The jetted microparticles were imaged via CLSM (Confocal Laser Scanning Microscopy) using an Olympus Confocal Microscope, and an Amray SEM (Scanning Electron Microscopy), at the Microscopy & Imaging Laboratory facilities at the University of Michigan.
2.4. De-protection of polymer 1 and soluble dextran release analysis
Microparticles containing polymer 1 in one compartment were tested for dextran deprotection and release in acidic and neutral conditions at physiological temperatures. The neutral buffer (pH 7.4) used was PBS and the acidic buffer (pH 5) is a mixture of acetic acid and sodium acetate [45]. Both buffers contained 1% Tween 20 (v/v). Different aliquots were tested via the Bicinchoninic Acid (BCA, ThermoScientific) test to determine the amount of dextran released.
2.5. Zeta potential measurements
The pure PLGA microparticles and polymer 1 containing microparticles were dispersed in DI water with 0.01% Tween 20 (v/v) and their zeta potential was measured with a Malvern Zetasizer. Each measurement was done in triplets.
2.6. Microparticle porosity and degradation analysis
Fabricated anisotropic microparticles were suspended in neutral or acidic buffers, filtered through a 10 μm filter mesh, and placed in a 37°C incubator. Samples were taken every 5 h, washed 3–5 times with DI water, and imaged via SEM. Microparticle diameter measurements were done using Image-J software [46–48]. More than 150 microparticles were measured per sample.
2.7. Encapsulation of therapeutics and release data
Drug loaded microparticles were fabricated using the same procedure as described above. Here, the drug, irinotecan, was mixed in the jetting solution containing polymer 1 prior to the jetting. The encapsulation was done as a drug-to-polymer ratio (5:100, 10:100, 25:100, and 50:100). These ratios correspond to 2.4, 4.8, 11.1, and 20% drug loading of the particles, respectively, taking into account both compartments. The drug loading was calculated as the drug mass divided by the combined mass of the polymer and drug.
For release studies, the microparticles were collected from the counter electrode and 20 mg of the microparticles was dispersed in 5 ml at pH 7.4. The solutions were then placed in dialysis membrane tubing before inserting them into containers filled with 35 ml of the same buffer. The dialysis membrane separated the internal solution and limited the diffusion of material at a cut-off range of 100 kDa, thereby allowing the diffusion of the drug, but not the particles, out of the membrane. The samples were incubated at 37°C. Eight samples were prepared for each of the drug-to-polymer ratios. At predetermined intervals, the membranes were switched to a new container with fresh buffer, and the buffer containing the released drugs was measured using a UV spectrometer to determine the amount of drug released according to a calibration curve. The amount released at each stage was added to the previous amounts to determine the total amount of drug released at each fixed point. At 24 h, four out of eight samples for each ratio were centrifuged down and the buffer was fully replaced with a pH 5 buffer. The samples were placed back in the incubator and the release was continued with 4 samples at pH 5 and 4 samples at pH 7.4.
3. Results and discussion
Herein, we report on the fabrication and analysis of drug carriers with distinct compartments; each composed of a chemically distinct polymer (Fig. 1). The polymers employed were polylactide-co-glycolide (PLGA) and acetal-modified dextran (1). PLGA is a biodegradable polymer widely used in the biomaterials field for its versatility and tunability with respect to its molecular weight and lactide-to-glycolide ratio. These factors determine the PLGA degradation rate and its physical characteristics [49,50]. Here, we have used a 40-kDa PLGA polymer with a 50:50 lactide-to-glycolide ratio.
Fig. 1.
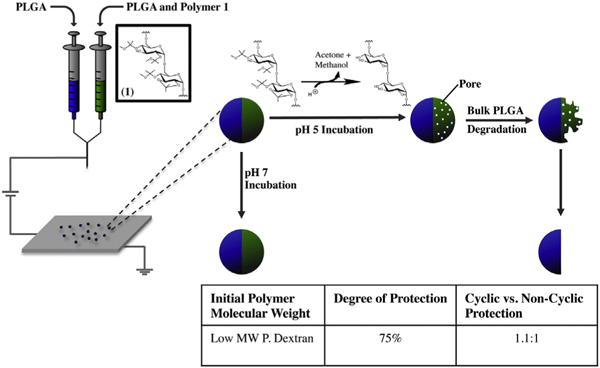
Particle fabrication procedure and hypothesized structure of particles depending on pH of environment. The table contains the polymer characterization information of polymer 1.
Unmodified dextran polymers are water-soluble polysaccharides that have been used extensively in many biomaterial applications [51]. It is this water-solubility, however, that limits the broader use of dextran for drug delivery and tissue engineering, because drug carriers, implants, or scaffolds fabricated from dextran will dissolve in water within a matter of minutes [52]. Masking of the hydroxyl groups on the backbone of the dextran polymer can render this polymer water-insoluble and thereby increase its applicability in this field. Depending on the type of chemistry used, this protection can be pH responsive and, in so doing, a polymer that can be water soluble or insoluble upon demand can be synthesized [45,53,54]. In addition to PLGA, we have employed polymer 1, as a pH responsive polymer that is water-insoluble at physiological pH, but water-soluble in acidic environments [45]. This change in solubility is caused by the hydrogen-catalyzed cleavage of the acetal groups that chemically protect the hydroxyl groups of the unmodified dextran (Fig. 1). Here polymer 1 can potentially be used in the fabrication of pH sensitive carriers that can respond selectively to specific physiological or pathophysiological cues such as the extracellular matrix surrounding tumors and inflamed tissue [16,55].
Polymer 1 was synthesized according to previously published procedures, [45] and the synthesis was characterized by NMRanalysis. Based on this analysis, approximately 75% of the hydroxyl moieties of the polysaccharide were protected with acetal groups. Among the protected moieties, the ratio of cyclic to noncyclic units was approximately 1.1:1. This degree of protection resulted in a polymer that was water-insoluble in physiological pH, while retaining some of its polar characteristics due to the small amount of free hydroxyl groups. Upon incubation at acidic pH, the acetal groups were cleaved, de-masking the original hydroxyl groups of the polysaccharide and lead to a water soluble polymer [45].
Upon confirmation of the polymer structure and characterization, we focused on the preparation of anisotropic microparticles from these polymer systems. The electrohydrodynamic (EHD) co-jetting process has been developed in our laboratory [32] and has been established as a reliable, reproducible, and versatile method for the fabrication of mono-dispersed anisotropic microparticles and fibers [34–36,39,56]. In the EHD process (Fig. 1), multiple polymer solutions are flown in a laminar regime through syringes tipped with metal needles [38]. The needles are connected to a high voltage source, which is grounded via a metal collector placed at a defined distance from the tip of the syringes. As a DC voltage is applied to the needles, the solutions at their tip form into a Taylor cone [57]. At the end of this Taylor cone, a thin, high-speed jet is formed that travels toward the grounded collector. The jet exiting the tip of the Taylor cone becomes thinner and eventually breaks into small droplets. During this process, the solvents evaporate rapidly, leaving behind solid anisotropic microparticles that are collected on the counter electrode [33]. Due to the rapid evaporation of the solvents and the laminar flow regime used, the polymers do not have sufficient time to mix, and thus result in microparticles with distinct compartments. In these microparticles, the number of compartments is determined by the number of individual needles used for the fluid manipulation [33,35]. Because we were interested in developing environmentally responsive bicompartmental microparticles, we selected polymer 1 and PLGA in one compartment and pure PLGA in the second compartment (Fig. 1). We hypothesized that such microparticles would be anisotropically responsive in low pH environments, where one of the compartments would rapidly degrade and the other would remain stable, while being isotropically responsive in physiological pH environments, where both compartments would remain stable.
First, we fabricated bicompartmental microparticles containing a mixture of polymer 1 and PLGA in the same compartment. For the microparticle fabrication, several different PLGA’s were explored featuring various molecular weights and lactide-to-glycolide ratios, as well as different dextran polymers with varying percentages of acetal protection. Ultimately, the 40 kDa, 50:50 PLGA was used due to its relatively faster degradation period, which might be suitable for drug delivery applications. For the compartment containing polymer 1, we observed that the polymer was more polar than PLGA. In order to create a homogeneous solution for jetting and to ensure that the polymer was still water-insoluble, a protection yield of 75% was found to be optimal for polymer 1.
The bicompartmental microparticles containing polymer 1 were characterized by Confocal Laser Scanning Microscopy (CLSM) and Scanning Electron Microscopy (SEM), as shown in Fig. 2. The CLSM images indeed confirmed the bicompartmental geometry of the microparticles, which contained a green polymeric dye (PTDPV) in the first compartment (PLGA + polymer 1) and a blue polymeric dye (MEHPV) in the PLGA compartment. Fig. 2A and B displays the blue (DAPI) and green (FITC) channels, representing the two separate compartments. Fig. 2C is an overlay image of the two channels confirming that the dyes do not mix and that the microparticles are, therefore, composed of distinct compartments. The three-dimensional shape and size distribution of the microparticles were assessed by SEM (Fig. 2D). Using the Image-J analysis software [46–48], it was found that the as-jetted microparticles are approximately 8.4 ± 0.07 μm in diameter on average, with few, larger microparticles at 10–15 μm that were easily removed via filtration.
Fig. 2.
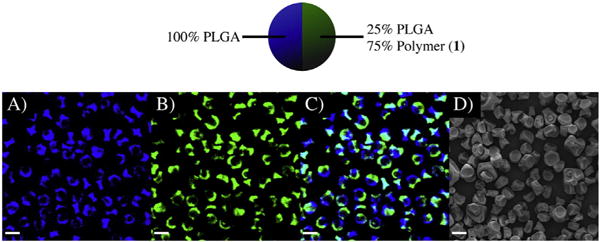
Characterization of microparticles containing polymer 1 + PLGA in one compartment and PLGA in the other compartment. A.–C.) Confocal images of the particles, where A.) is the blue channel (PLGA compartment), B.) is the green channel (PLGA + polymer 1 compartment), and D.) is the overlay of the two images. E.) SEM image of the particles showing their three-dimensional shape and size. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The polymer 1 containing microparticles were stable and similar in properties to PLGA microparticles when incubated in a PBS buffer and kept for 24 h at physiological pH and temperature. The stability of the microparticles in terms of their three-dimensional shape is confirmed in Supplemental Fig. 1. Moreover, unlike PLGA microparticles that require stabilizers such as a polyethylene glycol (PEG) or surfactants to disperse in aqueous solvents [58,59], microparticles containing polymer 1 were well dispersed without the need for further surface modification. This can be attributed to the residual hydroxyl groups of polymer 1. While most hydroxyl groups are protected (75%), the polymer still retains enough free hydroxyl groups to make the surface of the microparticles hydrophilic and keep them dispersed in an aqueous medium. This assumption was further supported by zeta potential measurements, which allowed for approximating the surface charge of the bicompartmental microparticles. The graph in Fig. 3 displays the differences in surface charge between pure PLGA microparticles and polymer 1 containing microparticles. PLGA microparticles have a zeta potential of −4.5 ± 1.2 mV, while polymer 1 containing microparticles have a more negative zeta potential measurement of −15.6 ± 2.1 mV. These results confirm that the polymer 1 containing microparticles are more hydrophilic and polar than PLGA microparticles due to the free hydroxyl groups present on their surface.
Fig. 3.
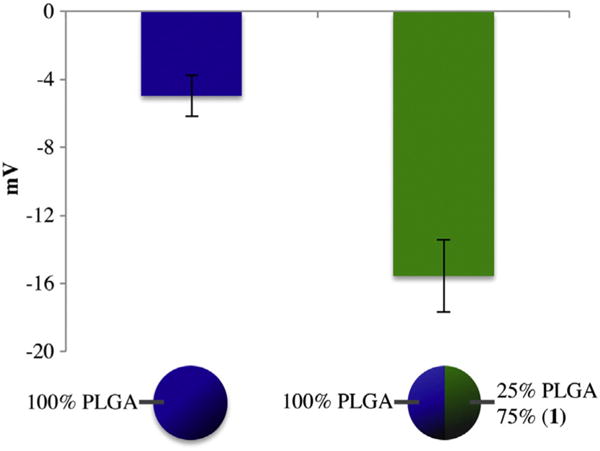
Zeta potential measurements of PLGA and PLGA + polymer 1 containing microparticles.
To verify the anisotropic nature of these microparticles and their ability to selectively respond to environmental changes, microparticles were incubated in two different pH environments, pH 5 and pH 7.4, at physiological temperatures and the release of free dextran and its effect on the structure of the microparticles were quantified. As illustrated in Fig. 4, the release of dextran was observed at acidic pH, while no dextran was released at pH 7.4, indicating that soluble dextran is released only in the acidic environment. The small burst release observed in the pH 7.4 sample is most likely due to a small fraction of polymer 1 with a lower level of protection that can make this polymer water soluble. Overall, these results confirm that a differential release of water-soluble dextran from our microparticles can be achieved.
Fig. 4.
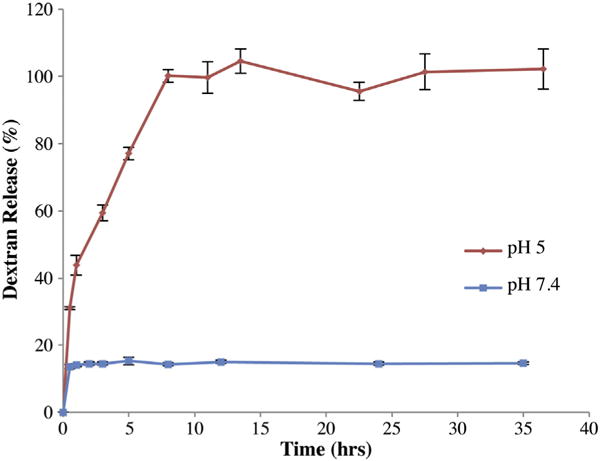
Release of dextran from bicompartmental microparticles. The release at two pH values, pH 5 and 7.4, are displayed.
To quantify the effect of dextran release on the structure of the microparticles, filtered microparticles (to remove a small fraction of larger microparticles) were incubated in two different pH environments (pH 5 and pH 7.4) at physiological temperatures and their degradation kinetics were monitored via SEM imaging (Fig. 5). The initial dextran release studies (Fig. 4) confirmed that the release of dextran was completed within 10 h of incubation. Hence, the appearance of pores due to this release should be observed within that period, followed by rapid bulk degradation of the rest of the compartment (now composed of PLGA) due to the porous remaining network. Pores were observed within the first 10 h of incubation: within 5 h (Fig. 5B) small pores covered one half of the microparticles in comparison to the control at pH 7.4 (Fig. 5F). After 10 h, larger pores were observed (Fig. 5C), as all of the dextran was released. By this time, the size of the microparticles had also decreased and bulk erosion of the PLGA material had begun. After 15 h (Fig. 5D) very few pores could be seen as most of the porous compartment had eroded away. Complete degradation of one side was observed within 20 h (Fig. 5E). In comparison, the same microparticles incubated at pH 7.4 for 20 h (Fig. 5F) remained intact and were approximately twice the size of those incubated for the same period in the acidic environment.
Fig. 5.
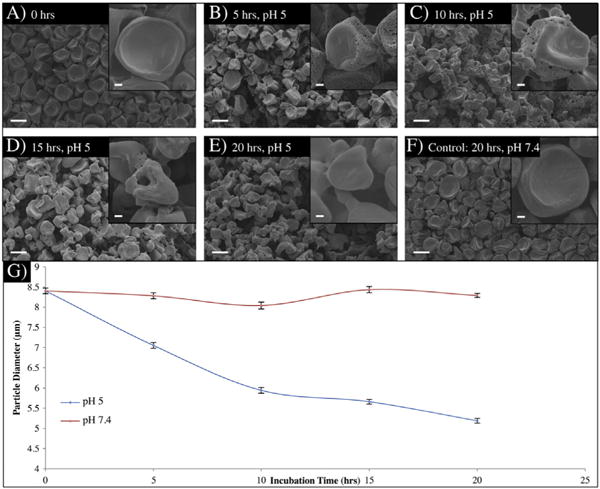
Characterization of polymer 1 containing microparticles in two different pH environments. A.–F.) SEM images of the microparticles overtime in pH 5 and 7.4. G.) Change in the microparticle diameter over time based on Image-J analysis.
In order to quantify the results and confirm the complete and exclusive degradation of one compartment, the size of the microparticles as a function of time was quantified using Image-J software [46–48]. The analysis was done based on SEM images taken of the microparticles at zero, 5, 10, 15, and 20 h for both pH 5 and 7.4 incubations. As seen in Fig. 5G, the pH 7.4 samples were consistent in size 8.29 ± 0.07 μm, while the pH 5 samples decreased to approximately 60% of their original size 5.18 ± 0.06 μm. Based on this analysis, as well as the SEM images, we have demonstrated the fabrication of anisotropic microparticles and control on the selective elimination of one compartment through stimuli responsive chemical degradation.
Based on these results, we hypothesized that we could use this system to deliver cancer therapeutics with on-demand release kinetics. To test this hypothesis, microparticles encapsulating different amounts of irinotecan, a low molecular weight anti-cancer therapeutic, in the compartment containing polymer 1 were fabricated. Microparticles with a range of different drug-to-polymer ratios (5:100 to 50:100, drug: polymer ratio, wt./wt.) could be fabricated using the same polymer formulation and processing parameters. Even for extremely high loading ratios of up to 50:100, microparticles were obtained that maintained similar size and shapes to unloaded microparticles. Next, four different microparticle sets were fabricated with drug-to-polymer ratios of 5:100, 10:100, 25:100, and 50:100. We hypothesized that at physiological pH, there would be a relatively slow release of the drug from our microparticles, while in acidic pH, there would be a rapid release of the majority of the drug. Finally, the rapid release at pH 5 was anticipated to follow a similar pattern (time and rate) as that of the dextran release (Fig. 4).
To test this hypothesis, release studies were conducted with the bicompartmental microparticles. Two sets of samples (4 repetitions each) were used for each drug-to-polymer ratio. For the first 24 h both sets were incubated at pH 7.4 to determine if the release between the sets were consistent, then one set was switched to pH 5 while the other was kept at pH 7.4 as a control. As predicted, the two sets were consistent with a slow release of irinotecan during the initial 24-hour period, after which point the samples were switched to pH 5 and underwent rapid release. Meanwhile, the reference samples, stored at pH 7.4, continued to display a very slow release (Fig. 6B). The detectable, albeit small, burst and the continuous release thereafter match those of the dextran release at the same pH (Fig. 4). We note that the rapid release of irinotecan at pH 5 is consistent with our previous results. While the majority of the compartment was composed of polymer 1, there is still a significant PLGA component in this compartment (25%), which, does not completely degrade within the 10-hour period and retains some of the irinotecan. Thus, irinotecan can be released in two stages: a rapid stage through the release of polymer 1 within a 10-hour period and a slower release phase as the PLGA component degrades.
Fig. 6.
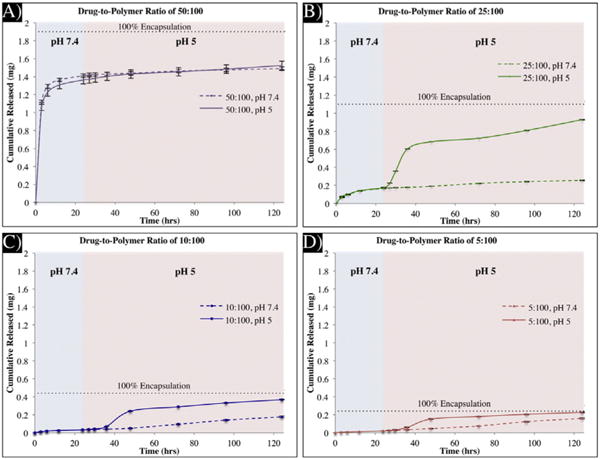
Release of irinotecan from bicompartmental microparticles. A.–D.) Release profiles from microparticles with drug-to-polymer ratios of 50:100 (A.), 25:100 (B.), 10:100 (C.), and 5:100 (D.). The solid lines are of the release in pH 5 and the dotted lines are the release in pH 7.4.
In addition to establishing that pH-triggered, on-demand release of therapeutics can be achieved with bicompartmental microparticles, we were also able to demonstrate that the rate and amount of release can be tuned, depending on the amount of the therapeutic encapsulated in the microparticles. As demonstrated in Fig. 6, as the drug-to-polymer ratio was increased (Fig. 6D to Fig. 6A), the amount of drug and the rate of release were also increased. This is in agreement with previously published results [60] confirming that at higher drug concentrations, the release of the therapeutic is faster due to percolation effects [61,62]. This is particularly evident in the case of the microparticles with a drug-to-polymer ratio of 50:100 (Fig. 6A) where the release of the drug is very fast, irrespective of the pH of the release environment.
Another potentially useful characteristic of bicompartmental carriers containing polymer 1 is their ability to localize high drug content in one compartment without spill over into the second one. This is a feature that was not possible in microparticles containing PLGA. Fig. 7 displays the selective encapsulation of the drug, irinotecan, at high drug-to-polymer concentrations (25:100) in the same compartment containing polymer 1 (Fig. 7A). For comparison, the same drug at the same concentrations was also loaded in bicompartmental microparticles comprised of PLGA in both compartments. For microparticles without polymer 1, diffusion of the drug between the two PLGA compartments was observed (Fig. 7B). In contrast to the microparticles containing polymer 1, irinotecan is distributed throughout the entire microparticle as indicated by the overlay of the blue fluorescence from irinotecan (Fig. 7A3 and B3) and green fluorescence used to label the irinotecan-free compartment (Fig. 7A2 and B2).
Fig. 7.
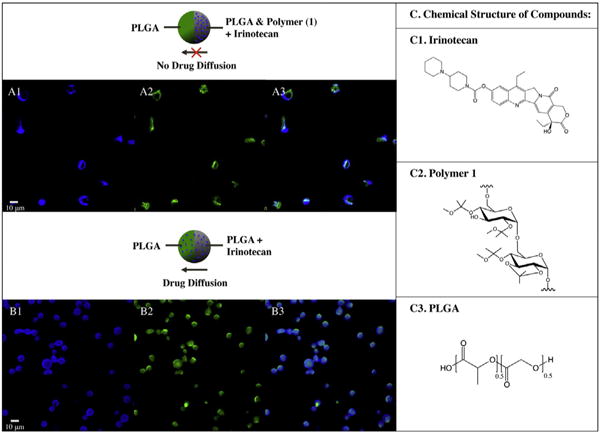
Selective encapsulation of irinotecan in microparticles. A.) In polymer 1 containing microparticles, irinotecan (autofluorescing in blue channel) is compartmentalized in one compartment, as the two dyes do not mix in the overlay image (A3.). B.) In the PLGA containing microparticles, the encapsulated irinotecan is not compartmentalized, as in the overlapped image, B3.), the blue dye is in both compartments. The structure of the drug, irinotecan, and the polymers, polymer 1 and PLGA, are displayed in C1-3.
Particles containing polymer 1 can play an important role in the delivery of cancer therapeutics due to their on-demand and tunable characteristics. Such particles can be used for the local delivery of cancer therapeutics by direct injection into the tumor site in cases where the resection of the tumor is not possible without major damage to the surrounding organs (i.e. intracerebra, pancreatic, glioblastoma, and gastric unresectable tumors) [63–65], or for prolonged release of therapeutics in the resected areas to avoid the growth of recurrent tumors, similar to already available products such as the Gliadel® wafers [66–68]. Microparticles have a potential advantage in such cases over their nano-sized counterparts due to their higher drug loadings, prolonged release of the loaded therapeutics, and slower clearance by macrophages due to their larger size (optimal size for macrophage uptake is 2–4 μm) [69]. In addition to these advantages, direct injection into the tumor site provides additional benefits including lower side effects, one-time administration and higher patient compliance, and direct access to the cancer sites in difficult to target areas such as the brain due to the blood-brain barrier [68].
In this study, we have demonstrated the fabrication of environmentally responsive anisotropic microparticles for the delivery of the anti-cancer drug irinotecan. Further, we have characterized their on-demand degradation kinetics and have established their application as pH responsive drug delivery vehicles. Future directions of this work include the incorporation of a second therapeutic or an imaging agent in the other compartment. In addition, the fabrication of nanoparticles and surface modification of such particles for the inclusion of targeting moieties will be investigated.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jconrel.2013.08.017.
Supplementary Material
{kind=link}
Acknowledgments
The authors would like to acknowledge funding from the Multidisciplinary University Research Initiative of the Department of Defense and the Army Research Office (W911NF-10-1-0518), American Cancer Society (RSG-080284-01-CDD), the DOD through an Idea award (W81XWH-11-1-0111) and the Tissue Engineering and Regenerative Medicine Training Grant (DE00007057-36) for their support. We would also like to thank Jaewon Yoon, Asish Misra, and Aftin Ross for their insightful discussions.
References
- 1.Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 2.Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 3.Desai N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 2012;14:282–295. doi: 10.1208/s12248-012-9339-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi DL, Bedford NM, Cho HS. Engineered multifunctional nanocarriers for cancer diagnosis and therapeutics. Small. 2011;7:2549–2567. doi: 10.1002/smll.201100436. [DOI] [PubMed] [Google Scholar]
- 5.Nicolas J, Mura S, Brambilla D, Mackiewicz N, Couvreur P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem Soc Rev. 2013;42:1147–1235. doi: 10.1039/c2cs35265f. [DOI] [PubMed] [Google Scholar]
- 6.Anderson JM, Shive MS. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 2012;64:72–82. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 7.Heidel JD, Davis ME. Clinical developments in nanotechnology for cancer therapy. Pharm Res-Dordr. 2011;28:187–199. doi: 10.1007/s11095-010-0178-7. [DOI] [PubMed] [Google Scholar]
- 8.Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 9.Soundararajan V, Warnock K, Sasisekharan R. Multifunctional nanoscale platforms for targeting of the cancer cell immortality spectrum. Macromol Rapid Commun. 2010;31:202–216. doi: 10.1002/marc.200900596. [DOI] [PubMed] [Google Scholar]
- 10.Ling V. Multidrug resistance: molecular mechanisms and clinical relevance. Cancer Chemother Pharmacol. 1997;40:S3–S8. doi: 10.1007/s002800051053. [DOI] [PubMed] [Google Scholar]
- 11.Livingston DM, Silver DP. Cancer — crossing over to drug resistance. Nature. 2008;451:1066–1067. doi: 10.1038/4511066a. [DOI] [PubMed] [Google Scholar]
- 12.Devalapally H, Duan ZF, Seiden MV, Amiji MM. Modulation of drug resistance in ovarian adenocarcinoma by enhancing intracellular ceramide using tamoxifen-loaded biodegradable polymeric nanoparticles. Clin Cancer Res. 2008;14:3193–3203. doi: 10.1158/1078-0432.CCR-07-4973. [DOI] [PubMed] [Google Scholar]
- 13.Zimmermann GR, Lehar J, Keith CT. Multi-target therapeutics: when the whole is greater than the sum of the parts. Drug Discov Today. 2007;12:34–42. doi: 10.1016/j.drudis.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 14.Tran MA, Smith CD, Kester M, Robertson GP. Combining nanoliposomal ceramide with sorafenib synergistically inhibits melanoma and breast cancer cell survival to decrease tumor development. Clin Cancer Res. 2008;14:3571–3581. doi: 10.1158/1078-0432.CCR-07-4881. [DOI] [PubMed] [Google Scholar]
- 15.Colson YL, Grinstaff MW. Biologically responsive polymeric nanoparticles for drug delivery. Adv Mater. 2012;24:3878–3886. doi: 10.1002/adma.201200420. [DOI] [PubMed] [Google Scholar]
- 16.Gao WW, Chan JM, Farokhzad OC. pH-responsive nanoparticles for drug delivery. Mol Pharm. 2010;7:1913–1920. doi: 10.1021/mp100253e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kost J, Langer R. Responsive polymeric delivery systems. Adv Drug Deliv Rev. 2001;46:125–148. doi: 10.1016/s0169-409x(00)00136-8. [DOI] [PubMed] [Google Scholar]
- 18.Esser-Kahn AP, Odom SA, Sottos NR, White SR, Moore JS. Triggered release from polymer capsules. Macromolecules. 2011;44:5539–5553. [Google Scholar]
- 19.Motornov M, Roiter Y, Tokarev I, Minko S. Stimuli-responsive nanoparticles, nanogels and capsules for integrated multifunctional intelligent systems. Prog Polym Sci. 2010;35:174–211. [Google Scholar]
- 20.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 21.Pelicano H, Carew JS, McQueen TJ, Plunkett M, Keating MJ, Huang P. Targeting Hsp90 by 17-AAG in leukemia cells: mechanisms for synergistic and antagonistic drug combinations with arsenic trioxide and Ara-C. Leukemia. 2006;20:610–619. doi: 10.1038/sj.leu.2404140. [DOI] [PubMed] [Google Scholar]
- 22.Jia J, Zhu F, Ma XH, Cao ZWW, Li YXX, Chen YZ. Mechanisms of drug combinations: interaction and network perspectives. Nat Rev Drug Discov. 2009;8:111–128. doi: 10.1038/nrd2683. [DOI] [PubMed] [Google Scholar]
- 23.Delcea M, Mohwald H, Skirtach AG. Stimuli-responsive LbL capsules and nanoshells for drug delivery. Adv Drug Deliv Rev. 2011;63:730–747. doi: 10.1016/j.addr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 24.Yan Y, Such GK, Johnston APR, Lomas H, Caruso F. Toward therapeutic delivery with layer-by-layer engineered particles. ACS Nano. 2011;5:4252–4257. doi: 10.1021/nn201793f. [DOI] [PubMed] [Google Scholar]
- 25.Perry JL, Herlihy KP, Napier ME, Desimone JM. PRINT: a novel platform toward shape and size specific nanoparticle theranostics. Acc Chem Res. 2011;44:990–998. doi: 10.1021/ar2000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cui JW, Yan Y, Wang YJ, Caruso F. Templated assembly of pH-labile polymer-drug particles for intracellular drug delivery. Adv Funct Mater. 2012;22:4718–4723. [Google Scholar]
- 27.Siegwart DJ, Whitehead KA, Nuhn L, Sahay G, Cheng H, Jiang S, Ma ML, Lytton-Jean A, Vegas A, Fenton P, Levins CG, Love KT, Lee H, Cortez C, Collins SP, Li YF, Jang J, Querbes W, Zurenko C, Novobrantseva T, Langer R, Anderson DG. Combinatorial synthesis of chemically diverse core-shell nanoparticles for intracellular delivery. Proc Natl Acad Sci USA. 2011;108:12996–13001. doi: 10.1073/pnas.1106379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teng ZG, Zhu XG, Zheng GF, Zhang F, Deng YH, Xiu LC, Li W, Yang Q, Zhao DY. Ligand exchange triggered controlled-release targeted drug delivery system based on core-shell superparamagnetic mesoporous microspheres capped with nanoparticles. J Mater Chem. 2012;22:17677–17684. [Google Scholar]
- 29.Kojima C. Design of stimuli-responsive dendrimers. Expert Opin Drug Deliv. 2010;7:307–319. doi: 10.1517/17425240903530651. [DOI] [PubMed] [Google Scholar]
- 30.Gillies ER, Frechet JMJ. Dendrimers and dendritic polymers in drug delivery. Drug Discov Today. 2005;10:35–43. doi: 10.1016/S1359-6446(04)03276-3. [DOI] [PubMed] [Google Scholar]
- 31.Sengupta S, Eavarone D, Capila I, Zhao GL, Watson N, Kiziltepe T, Sasisekharan R. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature. 2005;436:568–572. doi: 10.1038/nature03794. [DOI] [PubMed] [Google Scholar]
- 32.Roh KH, Martin DC, Lahann J. Biphasic Janus particles with nanoscale anisotropy. Nat Mater. 2005;4:759–763. doi: 10.1038/nmat1486. [DOI] [PubMed] [Google Scholar]
- 33.Roh KH, Martin DC, Lahann J. Triphasic nanocolloids. J Am Chem Soc. 2006;128:6796–6797. doi: 10.1021/ja060836n. [DOI] [PubMed] [Google Scholar]
- 34.Bhaskar S, Hitt J, Chang SWL, Lahann J. Multicompartmental microcylinders. Angew Chem Int Ed. 2009;48:4589–4593. doi: 10.1002/anie.200806241. [DOI] [PubMed] [Google Scholar]
- 35.Bhaskar S, Lahann J. Microstructured materials based on multicompartmental fibers. J Am Chem Soc. 2009;131:6650–+. doi: 10.1021/ja900354b. [DOI] [PubMed] [Google Scholar]
- 36.Bhaskar S, Pollock KM, Yoshida M, Lahann J. Towards designer microparticles: simultaneous control of anisotropy, Shape, Size. Small. 2010;6:404–411. doi: 10.1002/smll.200901306. [DOI] [PubMed] [Google Scholar]
- 37.Lee KJ, Yoon J, Rahmani S, Hwang S, Bhaskar S, Mitragotri S, Lahann J. Spontaneous shape reconfigurations in multicompartmental microcylinders. Proc Natl Acad Sci U S A. 2012;109:16057–16062. doi: 10.1073/pnas.1213669109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhaskar S, Gibson CT, Yoshida M, Nandivada H, Deng XP, Voelcker NH, Lahann J. Engineering, characterization and directional self-assembly of anisotropically modified nanocolloids. Small. 2011;7:812–819. doi: 10.1002/smll.201001695. [DOI] [PubMed] [Google Scholar]
- 39.Bhaskar S, Roh KH, Jiang XW, Baker GL, Lahann J. Spatioselective modification of bicompartmental polymer particles and fibers via huisgen 1,3-dipolar cycloaddition. Macromol Rapid Commun. 2008;29:1655–1660. [Google Scholar]
- 40.Misra AC, Bhaskar S, Clay N, Lahann J. Multicompartmental particles for combined imaging and siRNA delivery. Adv Mater. 2012;24:3850–3856. doi: 10.1002/adma.201200372. [DOI] [PubMed] [Google Scholar]
- 41.Park TH, Eyster TW, Lumley JM, Hwang S, Lee KJ, Misra A, Rahmani S, Lahann J. Photoswitchable particles for on-demand degradation and triggered release. Small. 2013 doi: 10.1002/smll.201201921. http://dx.doi.org/10.1002/smll.201201921. [DOI] [PMC free article] [PubMed]
- 42.Hwang S, Lahann J. Differentially degradable janus particles for controlled release applications. Macromol Rapid Commun. 2012;33:1178–1183. doi: 10.1002/marc.201200054. [DOI] [PubMed] [Google Scholar]
- 43.Lee KJ, Hwang S, Yoon J, Bhaskar S, Park TH, Lahann J. Compartmentalized photoreactions within compositionally anisotropic Janus microstructures. Macromol Rapid Commun. 2011;32:431–437. doi: 10.1002/marc.201000558. [DOI] [PubMed] [Google Scholar]
- 44.Lee KJ, Park TH, Hwang S, Yoon J, Lahann J. Janus-core and shell microfibers. Langmuir. 2013;29:6181–6186. doi: 10.1021/la4009416. [DOI] [PubMed] [Google Scholar]
- 45.Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Frechet JMJ. Acetal-derivatized dextran: an acid-responsive biodegradable material for therapeutic applications. J Am Chem Soc. 2008;130:10494–+. doi: 10.1021/ja803947s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y, Bajaj N, Xu P, Ohn K, Tsifansky MD, Yeo Y. Development of highly porous large PLGA microparticles for pulmonary drug delivery. Biomaterials. 2009;30:1947–1953. doi: 10.1016/j.biomaterials.2008.12.044. [DOI] [PubMed] [Google Scholar]
- 47.Impellitteri NA, Toepke MW, Levengood SKL, Murphy WL. Specific VEGF sequestering and release using peptide-functionalized hydrogel microspheres. Biomaterials. 2012;33:3475–3484. doi: 10.1016/j.biomaterials.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duncanson WJ, Figa MA, Hallock K, Zalipsky S, Hamilton JA, Wong JY. Targeted binding of PLA microparticles with lipid-PEG-tethered ligands. Biomaterials. 2007;28:4991–4999. doi: 10.1016/j.biomaterials.2007.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park TG. Degradation of poly(lactic-co-glycolic acid) microspheres — effect of co-polymer composition. Biomaterials. 1995;16:1123–1130. doi: 10.1016/0142-9612(95)93575-x. [DOI] [PubMed] [Google Scholar]
- 50.Kim K, Yu M, Zong XH, Chiu J, Fang DF, Seo YS, Hsiao BS, Chu B, Hadjiargyrou M. Control of degradation rate and hydrophilicity in electrospun non-woven poly(D, L-lactide) nanofiber scaffolds for biomedical applications. Biomaterials. 2003;24:4977–4985. doi: 10.1016/s0142-9612(03)00407-1. [DOI] [PubMed] [Google Scholar]
- 51.Montdargent B, Letourneur D. Toward new biomaterials. Infect Control Hosp Epidemiol. 2000;21:404–410. doi: 10.1086/501782. [DOI] [PubMed] [Google Scholar]
- 52.Baldwin AD, Kiick KL. Polysaccharide-modified synthetic polymeric biomaterials. Biopolymers. 2010;94:128–140. doi: 10.1002/bip.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cohen JA, Beaudette TT, Cohen JL, Brooders KE, Bachelder EM, Frechet JMJ. Acetal-modified dextran microparticles with controlled degradation kinetics and surface functionality for gene delivery in phagocytic and non-phagocytic cells. Adv Mater. 2010;22:3593–+. doi: 10.1002/adma.201000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Broaders KE, Cohen JA, Beaudette TT, Bachelder EM, Frechet JMJ. Acetalated dextran is a chemically and biologically tunable material for particulate immunotherapy. Proc Natl Acad Sci U S A. 2009;106:5497–5502. doi: 10.1073/pnas.0901592106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mahmoud EA, Sankaranarayanan J, Morachis JM, Kim G, Almutairi A. Inflammation responsive logic gate nanoparticles for the delivery of proteins. Bioconjug Chem. 2011;22:1416–1421. doi: 10.1021/bc200141h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lahann J. Recent progress in nano-biotechnology: compartmentalized micro- and nanoparticles via electrohydrodynamic co-jetting. Small. 2011;7:1149–1156. doi: 10.1002/smll.201002002. [DOI] [PubMed] [Google Scholar]
- 57.Taylor G. Disintegration of water drops in electric field. Proc R Soc Lond A. 1964;280:383–+. [Google Scholar]
- 58.Cheng FY, Wang SPH, Su CH, Tsai TL, Wu PC, Shieh DB, Chen JH, Hsieh PCH, Yeh CS. Stabilizer-free poly(lactide-co-glycolide) nanoparticles for multimodal biomedical probes. Biomaterials. 2008;29:2104–2112. doi: 10.1016/j.biomaterials.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 59.Cu Y, Saltzman WM. Controlled surface modification with poly(ethylene)glycol enhances diffusion of plga nanoparticles in human cervical mucus. Mol Pharm. 2009;6:173–181. doi: 10.1021/mp8001254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saltzman WM, Langer R. Transport rates of proteins in porous materials with known microgeometry. Biophys J. 1989;55:163–171. doi: 10.1016/S0006-3495(89)82788-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rothstein SN, Little SR. A “tool box” for rational design of degradable controlled release formulations. J Mater Chem. 2011;21:29–39. [Google Scholar]
- 62.Siegel RA, Kost J, Langer R. Mechanistic studies of macromolecular drug release from macroporous polymers. 1. Experiments and preliminary theory concerning completeness of drug release. J Control Release. 1989;8:223–236. [Google Scholar]
- 63.Li KW, Dang WB, Tyler BM, Troiano G, Tihan T, Brem H, Walter KA. Polilactofate microspheres for paclitaxel delivery to central nervous system malignancies. Clin Cancer Res. 2003;9:3441–3447. [PubMed] [Google Scholar]
- 64.Pradilla G, Wang PP, Gabikian P, Li K, Magee CA, Walter KA, Brem H. Local intracerebral administration of paclitaxel with the Paclimer (R) delivery system: toxicity study in a canine model. J Neuro-Oncol. 2006;76:131–138. doi: 10.1007/s11060-005-5531-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harper E, Dang WB, Lapidus RG, Garver RI. Enhanced efficacy of a novel controlled release paclitaxel formulation (PACLIMER Delivery System) for local-regional therapy of lung cancer tumor nodules in mice. Clin Cancer Res. 1999;5:4242–4248. [PubMed] [Google Scholar]
- 66.Westphal M, Hilt DC, Bortey E, Delavault P, Olivares R, Warnke PC, Whittle IR, Jaaskelainen J, Ram Z. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5:79–88. doi: 10.1215/S1522-8517-02-00023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Valtonen S, Timonen U, Toivanen P, Kalimo H, Kivipelto L, Heiskanen O, Unsgaard G, Kuurne T. Interstitial chemotherapy with carmustine-loaded polymers for high-grade gliomas: a randomized double-blind study. Neurosurgery. 1997;41:44–48. doi: 10.1097/00006123-199707000-00011. [DOI] [PubMed] [Google Scholar]
- 68.Wolinsky JB, Colson YL, Grinstaff MW. Local drug delivery strategies for cancer treatment: gels, nanoparticles, polymeric films, rods, and wafers. J Control Release. 2012;159:14–26. doi: 10.1016/j.jconrel.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Champion JA, Walker A, Mitragotri S. Role of particle size in phagocytosis of polymeric microspheres. Pharm Res-Dordr. 2008;25:1815–1821. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.