

Abstract
Neutrophils, the most abundant type of leukocytes in blood, can form neutrophil extracellular traps (NETs). These are pathogen-trapping structures generated by expulsion of the neutrophil's DNA with associated proteolytic enzymes. NETs produced by infection can promote cancer metastasis. Here, we show that metastatic breast cancer cells can induce neutrophils to form metastasis-supporting NETs in the absence of infection. Using intravital imaging, we observed NET-like structures around metastatic 4T1 cancer cells that had reached the lungs of mice. We also found NETs in clinical samples of triple-negative human breast cancer. The formation of NETs stimulated the invasion and migration of breast cancer cells in vitro. Inhibiting NET formation or digesting NETs with DNase I blocked these processes. Treatment with NET-digesting, DNase I-coated nanoparticles markedly reduced lung metastases in mice. Our data suggest that induction of NETs by cancer cells is a previously unidentified metastasis-promoting tumor-host interaction and a potential therapeutic target.
Introduction
Breast cancer metastasis is associated with very high mortality rates. Cancer cells can acquire the ability to metastasize by expressing metastasis-promoting genes, such as epithelial-to-mesenchymal-transition promoting transcription factors or metalloproteinases (1, 2). However, cancer cells also can recruit and activate leukocytes, including macrophages, to promote metastasis (3). Neutrophils – the most abundant leukocytes in human blood –similarly promote metastasis (4-8), although they can kill disseminated cancer cells under certain conditions (9). Neutrophils and their precursors are sensitive to many chemotherapy regimens, causing dangerously low neutrophil numbers (neutropenia) during the course of treatment. Because neutropenia carries a risk of life-threatening infections, the American Society of Clinical Oncology recommends prophylactic treatment with neutrophil-stimulating factors, including granulocyte colony stimulating factor (G-CSF), for certain chemotherapeutic regimens (10). It is therefore important to determine the conditions under which neutrophils promote metastatic spread.
Neutrophils' normal function is to kill harmful microorganisms in three ways: 1) phagocytosis, a process whereby bacteria or fungi are engulfed and digested, 2) degranulation of cytotoxic enzymes into the extracellular space, and 3) neutrophil extracellular traps (NETs), which are DNA meshes with associated cytotoxic enzymes that are released into the extracellular space where they trap microorganisms (11). NETs form in tissues and have been documented in human pancreatic, liver, and gastric cancer (12-14), but whether they participate in cancer progression remains unclear. NETs can also form intravascularly, and they can damage vascular cells when they form (15). Recently, it was shown that NETs induced within the vasculature by experimentally induced systemic bacterial infection or surgical stress aided in metastatic seeding of cancer cells in the liver (5, 12).
We sought to observe how disseminating cancer cells interacted with neutrophils upon arrival in the lungs, a major site of metastatic colonization in breast cancer. We developed confocal intravital lung imaging (CILI), a modification of a lung-imaging approach used with two-photon microscopy (16). Here, we show that NET-like structures form around disseminated cancer cells in lungs using CILI. We show that cancer cells stimulate neutrophils to form NETs in the absence of pathogens, in vitro. Finally, we show that NETs stimulate cancer cell migration and invasion, and that treatment with NET-digesting DNase-I-coated nanoparticles inhibits metastasis. NET formation is a mechanism by which signals from cancer cells activate host cells to enhance metastasis. Understanding the contribution of neutrophils to metastases has pressing clinical implications, because many cancer patients receiving chemotherapy also receive prophylactic treatment with neutrophil stimulating factors.
Results
Metastatic cancer cells induce formation of NETs
To investigate whether neutrophils play a role in metastasis, we first compared neutrophil infiltration into tumors from orthotopically transplanted 4T1 and 4T07 murine breast cancer cells. These cells originate from the same mammary tumor of a BALB/c mouse, but only the 4T1 cells metastasize (17). We observed significantly (p=0.0009) more neutrophils in primary 4T1 tumors than in 4T07 tumors (Figs. 1A, B). Because the CXCL1 chemokine can recruit neutrophils (18), we measured its mRNA and protein and found higher amounts in 4T1 than in 4T07 cells and in 4T1-derived tumors than in 4T07-derived tumors (Fig. 1C, and fig. S1). Reduction of CXCL1 (and the homologous CXCL2) in 4T1 cells with short hairpin RNAs reduced neutrophil infiltration into tumors and increased primary tumor growth, but had no effect on macrophage infiltration or cancer cell proliferation in vitro (fig. S1). Tumors with reduced expression of CXCL1/2 had an approximately doubled tumor burden, but, despite this, metastatic burden was not increased (fig. S1). Instead, metastatic burden from CXCL1/2 knockdown cells was significantly (p=0.0008) decreased when equal numbers of cancer cells were injected intravenously into mice (Fig. 1D). We speculated that increased tumor burden after CXCL1/2 knockdown was caused by reduced recruitment of tumor-reactive T lymphocytes (19). Consistent with this idea, CXCL1/2 knockdown did not increase primary tumor growth in T cell-deficient, nude mice (Fig. 1E). However, CXCL1/2 knockdown did significantly (p=0.03) reduce spontaneous metastasis in nude mice (Fig. 1F). This suggests that the reduced metastasis is not caused by reduced T cell recruitment, and that neutrophils mediate this effect.
Figure 1. Neutrophil extracellular traps (NETs) form during metastasis of breast cancer.

(A, B) More neutrophils infiltrated metastatic 4T1 tumors than non-metastatic 4T07 tumors (Ly6G immunostaining, mean ± SEM; n=4 mice, t-test). Scale bar: 50 μm.
(C) CXCL1 protein level was higher in 4T1 than in 4T07 tumors (mean ± SEM, n=5 mice, t-test).
(D) Cancer cell-derived CXCL1 promoted metastatic seeding after intravenous injection of luciferase-expressing cells (bioluminescence radiance 19 days after cell injection; individual mice and means indicated; one-way ANOVA p=0.0008, Dunnett's multiple comparison: p<0.01).
(E) CXCL1 secretion by 4T1 cells did not affect primary tumor growth in nude mice (mean ± SEM, n=5 mice).
(F) Knockdown of Cxcl1 in 4T1 cells reduced lung metastasis in nude mice (individual mice and means ± SD indicated; t-test).
(G, H) NET-like structures of extracellular DNA, sensitive to intravenous DNase I, were found around 4T1-cancer cells in LysM-EGFP mice using CILI. Grey scale insert shows DNA channel (shown and quantified 30-60 minutes after cancer cell injection, values from individual mice and mean ± SD are indicated; one-way ANOVA p=0.002, Sidak's multiple comparison: p<0.01). Scale bar: 50 μm.
(I, J) Extracellular DNA and neutrophil elastase activity co-localized near 4T1, but not 4T07, cancer cells (shown and quantified 30-60 minutes after cancer cell injection, Fisher's exact test). Scale bar: 50 μm.
(K, L) The number of NETs in the lungs was higher after 4T1 cell injection than in controls (co-localized myeloperoxidase and citrullinated histone H3 immune staining). White arrow: NET; yellow arrow: intact neutrophil (mean ± SEM, n=3 mice). Scale bar: 10 μm.
The assays with CXCL1/2-knockdown cells suggest that neutrophils support metastasis upon their recruitment by cancer cells. To investigate the functions of neutrophils at the metastatic site, we performed confocal intravital lung imaging (CILI) of mCherry-expressing 4T07 and 4T1 cells injected intravenously into LysM-EGFP mice, which express EGFP predominantly in neutrophils (20).Greater numbers of extracellular DNA structures, sensitive to intravenously injected DNase I, were found around 4T1 than around 4T07 cancer cells (Figs. 1G, H, movie S1). To explore whether these structures could be NETs, we next used CILI to determine if the extracellular DNA structures co-localized with the NET-associated protease neutrophil elastase using a probe that produces fluorescent signal after specific cleavage by neutrophil elastase. Neutrophil elastase activity co-localized with extracellular DNA in 14 out of 17 imaged fields of view after injection of the 4T1 cancer cells but not after injection of 4T07 cells (Figs. 1I, J, p=0.0008). To further explore whether NETs are formed after injection of cancer cells, we performed immunofluorescent staining for NETs in tissue sections from lungs of mice injected with 4T1 cancer cells. This analysis confirmed that NETs formed in lungs shortly after tail vein injection of 4T1 cells and that the amounts of NETs were elevated for days after injection (Figs. 1K, 1L, and S2). These data demonstrate that metastatic cancer cells stimulate NET formation at sites of dissemination in the absence of infection.
To determine whether NETs could also be found in metastatic human breast cancer, we performed immunofluorescence staining on a small panel of clinical samples of primary tumor and matched metastatic lung lesions from breast cancer patients (Fig. 2A). We detected NETs in 16 out of 20 primary tumors, and in 13 out of 19 metastatic lung lesions (Fig. 2B). The number of NETs varied between tumors, with the highest numbers in triple-negative tumors (6 of 6 primary tumors and 7 of 7 metastases had detectable NETs), and an absence or very low numbers in luminal breast cancers (Fig. 2C). Thus, we detected NETs in human breast cancer and found the presence of NETs to be associated with an aggressive subtype of breast cancer.
Figure 2. NETs are present in metastatic, triple-negative human breast cancer.

(A) Detection of NETs by immunofluorescence in human breast tumors and lung metastases. White arrows point to NETs (defined as co-localized myeloperoxidase, citrullinated histone H3, and DNA), and yellow arrows point to intact neutrophils. Scale bars: 20 μm.
(B) Number of NETs in matched primary tumors and lung metastases (paired t-test).
(C) Number of NETs in primary tumor of different breast cancer subtypes (ANOVA, Tukey's multiple comparisons test).
NETs promote cancer cell migration and invasion
To determine whether cancer cell-induced NET formation required direct interaction with neutrophils or could be induced over a distance, we performed a transwell chamber assay. Specifically, neutrophils isolated from the mouse bone marrow were plated in the lower wells, and cancer cells were added on top of Matrigel-coated membranes in the upper wells (fig. S3). Neutrophils co-cultured in this manner with metastatic 4T1 cells formed extensive NETs, whereas neutrophils similarly co-cultured with non-metastatic 4T07 cells formed few NETs (Figs. 3A, B).
Figure 3. Formation of NETs by metastatic 4T1 breast cancer cells is associated with cancer cell invasion.

(A, B) 4T1 but not 4T07 cells increased the formation of NETs (immunostaining for histone H3 and neutrophil elastase, mean ± SEM, n=3, t-test). Scale bar: 50 μm.
(C) Neutrophils promoted invasion of 4T1 but not 4T07 cells (mean ± SEM, n=5, t-test).
(D, E) DNase I (1.5 U) digested NETs (mean ± SEM, n=3, t-test). Scale bar: 50 μm.
(F) DNase I treatment inhibited neutrophil-stimulated invasion of 4T1 cells (mean ± SEM, n=5-7, t-test).
(G) Primary C3(1)-Tag cancer cells induced NETs. Scale bar: 50 μm.
(H) DNase I treatment inhibited neutrophil-stimulated migration of C3(1)-Tag cancer cells (mean ± SEM, n=3, t-test).
(I, J) Human BT-549 breast cancer cells promoted NET formation (immunostaining for histone H3 and myeloperoxidase, mean ± SEM, n=3, t-test). Scale bar: 50 μm.
(K) DNase I (1.5 U) treatment blocked neutrophil-stimulated invasion of BT-549 breast cancer cells (mean ± SEM; BT-549 cells only or BT-549 cells with neutrophils and vehicle or DNase I: n=5; BT-549 cells and vehicle or DNase I in 10% FCS: n=2).
Co-culturing with neutrophils increased the invasion of 4T1 cells, but had little effect on the invasion of 4T07 cells (Fig. 3C). To test whether NETs promoted cancer cell invasion, we digested extracellular DNA by adding DNase I to the cultures (Figs. 3D, E). The neutrophils' ability to stimulate invasion of 4T1 cells was lost when the DNA of the NETs was digested, whereas the addition of DNase I had no effect on fetal calf serum (FCS)-stimulated invasion (Fig. 3F).
To test whether other metastatic cancer cells also induce NETs, we isolated primary cancer cells from C3(1)-Tag mice, a genetically engineered mouse model of metastatic basal/triple-negative breast cancer (21). Tumors in this model have high numbers of infiltrating neutrophils and high expression of CXCL1 (22). Like 4T1 cells, C3(1)-Tag primary cancer cells induced NETs when co-cultured with mouse neutrophils (Fig. 3G). C3(1)-Tag cells have a limited ability to invade through Matrigel, but neutrophils significantly (p=0.047) increased the migration of C3(1)-Tag cells across uncoated membranes. This increased migration was blocked by DNase I treatment (Fig. 3H). Extensive NETs were also formed when triple-negative human BT-549 breast cancer cells were cultured with neutrophils from healthy female volunteers (Figs. 3I, J, fig. S3). As observed with 4T1 cells, digesting NETs with DNase I blocked the neutrophils' ability to stimulate BT-549 invasion through Matrigel (Fig. 3K). Thus, both human and mouse triple-negative breast cancer cells can promote the formation of pro-migratory/invasive NETs, whereas treatment with NET-digesting DNase I blocks migration and invasion induced by species-matched neutrophils.
Cancer cells induce lytic NETs, and blocking NET formation reduces invasion
Next, we wanted to determine how tumor-induced NETs were formed. G-CSF can prime neutrophils for NET formation and is secreted by 4T1 cells (23). Indeed, anti-G-CSF blocking antibodies significantly (p=0.03) reduced the ability of 4T1 cells to induce NETs in the transwell chamber assay (Fig. 4A). Conversely, human recombinant G-CSF induced NET formation by human neutrophils (fig. S4A). Together, this suggests that G-CSF secreted by cancer cells induces NETs.
Figure 4. Cancer cells induce NET formation through G-CSF, and neutrophil-stimulated invasion requires NADPH oxidase and PAD4 activity.

(A) Blocking anti-G-CSF antibodies (1.6 μg/mL) decreased 4T1-induced NET extension (mean ± SEM, neutrophils with vehicle or anti-G-CSF: n=3; neutrophils with 4T1 cells and vehicle or anti-G-CSF: n=5, t-test).
(B) The NADPH oxidase inhibitor apocynin (10 μM) inhibited NET formation (mean ± SEM, neutrophils and vehicle or NAPDH oxidase inhibitor: n=3, neutrophils with cancer cells and vehicle or apocynin: n=5, t-test).
(C) Apocynin (10 μM) inhibited neutrophil-stimulated invasion (mean ± SEM, 4T1 cells only or 4T1 cells with neutrophils and vehicle or apocynin: n=4; 4T1 cells only and vehicle or apocynin in 10% FCS: n=1, t-test).
(D) PAD4 inhibition (200 μM Cl-amidine) reduced cancer cell-induced NET formation (mean ± SEM; neutrophils and vehicle or PAD4 inhibitor: n=6; neutrophils with 4T1 cells and vehicle or PAD4 inhibitor: n=4, t-test).
(E) PAD4 inhibition (200 μM Cl-amidine) blocked neutrophil-stimulated cancer cell invasion (mean ± SEM; n=3, t-test).
An initial step in the intracellular signaling cascade in neutrophils that results in pathogen-induced NET formation is activation of the phagocytic NADPH oxidase (NOX2) enzyme complex (24). In our transwell assay, apocynin, a pharmacologic inhibitor of NOX2, reduced NET formation and neutrophil-promoted cancer cell invasion (Figs. 4B, C). The phox47 subunit is a critical component of NOX2 in neutrophils. The extension of NETs was greatly reduced for neutrophils isolated from p47phox-/- compared to those isolated from p47phox+/+ mice, and p47phox-/- neutrophils did not promote cancer cell invasion (fig. S4). Unfortunately, the p47phox-/- strain is on the C57BL/6 background, precluding in vivo experiments with the BALB/c-derived 4T1 cells.
Next, we determined the importance of peptidylarginine deiminase type 4 (PAD4), the enzyme responsible for histone modifications required for decondensing neutrophil DNA before expulsion (25). The PAD4 inhibitor Cl-amidine reduced NET formation (Fig. 4D) and blocked the neutrophils' ability to promote invasion (Fig. 4E). Thus, metastatic cancer cells activate signaling pathways that include NADPH oxidase and PAD4 in neutrophils, causing the formation of invasion-promoting NETs.
Several types of NET formation can occur. One requires NADPH oxidase activity and generally results in neutrophil lysis 2-4 hours after activation (24), whereas another process occurs rapidly (5-60 min), independent of NADPH oxidase-activity, and leaves the neutrophil plasma membrane intact (26). Cancer cell-induced NET formation was dependent on NADPH oxidase activity, which suggested that it occurred through a lytic process. To address this directly, we performed electron microscopy. Scanning electron microscopy revealed that extracellular meshes extruded from non-intact neutrophils 3 hours after plating and stimulation with cancer cells, whereas intact neutrophils were rarely discernible (fig. S5). These findings were largely similar to those observed after stimulation with phorbol 12-myristate 13-acetate (PMA) (fig. S5), which is known to induce NETs through a lytic mechanism (24). We next analyzed the integrity of the neutrophils by transmission electron microscopy. Three hours after stimulation with cancer cells, neutrophils typically had decondensed chromatin dispersed in the cytosol and plasma membrane breakdown (fig. S5). This morphology was identical to the morphology of the neutrophils induced to form NETs by PMA. Of >50 evaluated neutrophils, none showed evidence of DNA-containing vesicles budding from the nucleus, the morphology described as typical for non-lytic NET-forming neutrophils (26). Finally, to demonstrate that the extracellular meshes observed by scanning electron microscopy contained DNA, we performed immunogold labeling with anti-DNA antibodies, and detected expelled DNA in the meshes after stimulation with 4T1 cancer cells (fig. S5). Because we did not observe neutrophils with extruded nuclear DNA that were otherwise intact, we conclude that, in our in vitro setting, cancer cells induce NETs through a lytic process.
NETs are defined by the association of neutrophil proteases with the extracellular neutrophilic histone-bound DNA (27). So we speculated that the pro-invasive effects of NETs required NET-associated protease activity. Indeed, inhibition of cathepsin G, a protease associated with NETs, blocked the neutrophils' ability to promote invasion with no effect on the cancer cells' ability to invade towards FCS. Surprisingly, inhibition of cathepsin G also reduced the extension of NETs (Figs. 5A & B). This suggests that cathepsin G is required for NET release. Cleavage of histones by neutrophil elastase is required for chromatin de-condensation and NET release (28, 29), and cathepsin G may have a similar role. The neutrophil elastase inhibitor sivelestat also reduced the extension of cancer cell-induced NETs (Fig. 5C). Although the effect of sivelestat on neutrophil-induced invasion was not significant in assays with murine 4T1 cells (Figs. 5D), inhibition of neutrophil elastase significantly (p=0.02) blocked the neutrophils' ability to stimulate the invasion of human BT-549 breast cancer cells, as did inhibition of NADPH oxidase (Fig. 5E).
Figure 5. NET formation and neutrophil-stimulated invasion require neutrophil protease activity.

(A) Cathepsin G inhibitor I (2 μM) reduced cancer cell-induced NET formation (mean ± SEM, n=4, t-test).
(B) Cathepsin G inhibitor I (2 μM) inhibited neutrophil-stimulated invasion (mean ± SEM; 4T1 cells only, 4T1 cells with neutrophils: n=4; 4T1 cells in 10% FCS: n=3, t-test).
(C) Neutrophil elastase inhibitor sivelestat (10 μM) reduced cancer cell-induced NET formation (mean ± SEM; n=3, t-test).
(D) Neutrophil elastase inhibitor sivelestat (10 μM) weakly reduced neutrophil-stimulated invasion of 4T1 cells (mean ± SEM; 4T1 cells only or 4T1 cells with neutrophils and vehicle or sivelestat: n=5; 4T1 cells and vehicle or sivelestat in 10% FCS: n=2).
(E) NADPH oxidase or neutrophil elastase inhibition (10 μM apocynin or sivelestat, respectively) blocked neutrophil-stimulated invasion of human breast cancer cells (mean ± SEM, BT-549 cells only or BT-549 cells with neutrophils and vehicle or apocynin or sivelestat: n=4, BT-549 cells and apocynin or sivelestat in 10% FCS: n=2).
(F) CM from neutrophils induced to form NETs by culturing with cancer cells promoted invasion, but not when NET induction occurred in the presence of NADPH oxidase inhibition (10 μM apocynin, mean ± SEM, n=3-6, t-tests).
The pro-invasive effects of NET-forming neutrophils in transwell chamber assays suggested that a factor (or factors) generated during NET formation acted on cancer cells. To test this possibility, we generated conditioned medium (CM) from unstimulated neutrophils, from neutrophils induced to form NETs by co-culturing with cancer cells, and from neutrophils co-cultured with cancer cells in the presence of the NADPH oxidase inhibitor apocynin to prevent the formation of NETs. CM from cultures induced to form NETs effectively stimulated cancer cell invasion, whereas CM from cultures where NET formation was inhibited with apocynin did not (Fig. 5F). To exclude the possibility that invasion was stimulated by factors secreted by cancer cells into the CM, we also used CM from neutrophils that formed NETs in response to PMA stimulation. CM from PMA-induced, NET-forming neutrophils also stimulated cancer cell invasion. Conditioned medium from both cancer cell-induced and PMA-induced NET-forming neutrophil cultures lost the ability to stimulate invasion if neutrophils were cultured in the presence of DNase I (fig. S6). In addition, the CM lost its ability to stimulate invasion if DNase I was added after collection or upon heat denaturing (fig. S6). This suggests that the pro-invasive factor is not simply extracellular DNA, but protein factors associated with the DNA mesh.
DNase I treatment prevents lung metastasis in mice
Digesting bacterially induced NETs with systemically administered DNase I can reduce experimental metastasis in a mouse model of metastatic lung cancer, but the effects are relatively modest (5). Because we found that DNase I prevented NET-mediated invasion and migration in vitro with three different sources of cancer cells (Figs. 3F, H, K), we speculated that the modest in vivo effects of DNase I were due to its short half-life in blood (30). Immobilization of an enzyme on the surface of nanoparticles can increase enzyme stability (31), and we therefore developed DNase I-coated nanoparticles. In vitro, DNase I-coated nanoparticles digested NETs and blocked invasion as effectively as free DNase I (Fig. 6A). In vivo, a higher concentration of DNase I was found in plasma when mice were treated with DNase I-coated nanoparticles than with free DNase I (Fig. 6B). Daily intraperitoneal injection of DNase I-coated nanoparticles significantly (p=0.002) reduced metastatic burden after intravenous injection of 4T1 cells (Figs. 6C, D). Three out of nine mice treated with DNase-I-coated nanoparticles had no detectable metastasis by histology, whereas all 10 mice treated with control nanoparticles had macroscopic or microscopic metastases. A detailed analysis of the tissue revealed that there was a significant reduction in the number of detectable, individual metastatic foci (Fig. 6E, p=0.0003) and a reduction in the average size of foci (Fig. 6F, p=0.01) after treatment with DNase I-coated nanoparticles. This suggests that NETs are critical for metastatic colonization. NETs that form intravascularly in response to infection can promote extravasation of disseminated cancer cells (5). We therefore examined whether DNA digestion with DNase I-coated nanoparticles or inhibition of NET formation with the PAD4 inhibitor Cl-amidine influenced the number of cancer cells that made it out of the vasculature in our metastasis model, which does not involve infection. Despite a significant (p=0.02) reduction in the number of NETs using either approach (fig. S7A), the number of cells that extravasated into the lung tissue 24 hours after injection was not altered (fig. S7B).
Figure 6. Targeting NETs in vivo reduces metastasis.
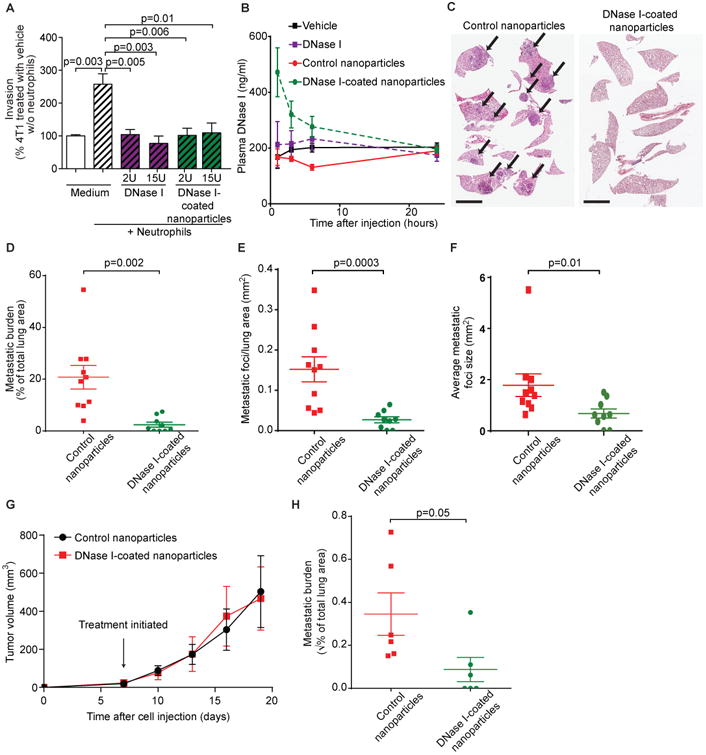
(A) DNase I-coated nanoparticles reduced neutrophil-stimulated cancer cell invasion in vitro (mean ± SEM, n=4, t-test).
(B) Injection of DNase I-coated nanoparticles results in higher plasma nuclease activity than injection of free DNase I (mice injected with 75 U free DNase I, 75 U nanoparticle bound DNase I, or equivalent vehicle, mean ± SEM; n=3 mice).
(C, D) DNase I-coated nanoparticles (75 U/mouse) reduced experimental lung metastasis of 4T1 cells (mean ± SEM; n=9-10 mice; t-test). Arrows point to metastases. Scale bars: 4 mm.
(E, F) DNase I-coated nanoparticles (75 U/mouse) reduced the number and the size of metastatic foci arising after injection of 4T1 cells (mean ± SEM; n=9-10 mice; t-test. The data were transformed by taking the square root before performing the t-test due to significantly [p=0.0003] different variances [untransformed data graphed]).
(G) DNase I-coated nanoparticles did not affect primary tumor growth. Nanoparticle treatment was initiated 7 days after tumor cell transplantation (mean ± SEM; n=6 mice).
(H) DNase I-coated nanoparticles (75 U/mouse) reduced spontaneous metastasis of 4T1 cells (mean ± SEM; n=6 mice; t-test. The data were transformed by taking the square root before performing the t-test due to significantly [p=0.007] different variances).
To determine whether DNase I-coated nanoparticles also reduced spontaneous metastasis, 4T1 cells were injected into both inguinal mammary glands of female mice and, after seven days, daily treatment with DNase-I coated nanoparticles began. The DNase I-coated nanoparticles had no effect on primary tumor growth (Fig. 6G), but the lung metastatic burden was reduced: all mice treated with control nanoparticles had micrometastases, but 3 out of 6 mice treated with DNase I-coated nanoparticles had no detectable metastases by histology (Fig. 6H). Together, these data suggest that therapeutic targeting of NETs can prevent lung metastasis.
Discussion
Here, we show that cancer cells can hijack neutrophils so that the neutrophils' ability to eradicate pathogens through formation of NETs aids metastatic spread instead. By imaging in live mice, we observed that breast cancer cells can induce neutrophils to form NETs after they arrive in the lungs. We also documented the presence of NETs in the aggressive triple-negative subtype of human breast cancer. Using in vitro models, we further demonstrated that inhibiting the signaling pathways that promote NET formation or digesting NETs with DNase I blocks invasion. Our data also suggest that cathepsin G is not just an effector of NET functions but participates in the release of NETs, similar to what is reported for neutrophil elastase (28). Finally, we demonstrate that treatment with DNase I-coated nanoparticles effectively reduces metastasis in vivo. Unexpectedly, cancer cell-induced NETs did not promote extravasation but did influence the number of histologically detectable metastatic foci. This suggests that NETs mediate the expansion of disseminated cells. Our findings, together with a previous report that NETs induced by infection aid in metastatic seeding of the liver (5), raise the exciting possibility of targeting NETs to prevent metastasis.
Neutrophils and their precursors are sensitive to chemotherapy, and cancer patients can thus develop life-threatening neutropenia. Notably, long-term survival of breast cancer patients receiving chemotherapy is higher for patients with mild chemotherapy-induced neutropenia (32). It has been proposed that this is because neutropenia represents a surrogate marker of adequate chemotherapy dosing for a given patient (33). However, our findings suggest an alternative hypothesis: that mild neutropenia is associated with better survival because neutrophils play a functional role in metastasis. It could therefore be important to develop approaches to prevent or dissolve pro-metastatic NETs while simultaneously leaving the life-saving de-granulating and phagocytic activities of neutrophils intact. Blocking antibodies against G-CSF reduced cancer cell-induced NET formation in our study and can reduce metastases in mice (34, 35). However, G-CSF is used clinically to prevent mortality associated with neutropenic infections and cannot be easily targeted. Inhibitors against NOX2 and neutrophil proteases also blocked cancer cell-induced NET formation. Unfortunately, NOX2 and the neutrophil proteases are also not good targets because NOX2 is critical for bacterial killing and neutrophil proteases are required to eliminate pathogens by phagocytosis and de-granulation. However, the extracellular DNA mesh of NETs is an exciting and feasible target. Treatment with nucleases was reported to reduce metastasis decades ago, but with no insights as to why (36). DNase I treatment is approved by FDA for the treatment of cystic fibrosis, for which it is used to decrease mucus viscosity resulting from NET accumulation triggered by persistent infections. Our results with DNase I-coated nanoparticles serve as proof-of-principle that NETs are a drug target to reduce metastasis. Another strategy may be to prevent NETs from forming, for example by targeting PAD4. Although we were able to prevent NET formation using PAD4 inhibitors in vivo in short-term assays, we could not test the effect on metastasis with the long term treatment needed. Commercially available PAD4 inhibitors have serum half-lives of ∼15 min-4 h (37, 38). It was therefore impossible to compare NET digestion by DNase I-coated nanoparticles to inhibition of NET formation using PAD4 inhibition and establish which approach is the best strategy for preventing metastasis.
Our experimental metastasis assays suggest that NETs play a role during the establishment of metastases. They may do so through multiple mechanisms. The DNA mesh can trap circulating cancer cells at the site of dissemination (5). In addition, intravascular NETs can increase local vascular permeability (15), which would permit cancer cells to extravasate more easily. This role has been proposed for systemically formed NETs. However, our data do not support a role for cancer cell-induced NETs in enhancing extravasation. Instead, our data suggest that NETs may stimulate the further invasion of the cells into the tissue and the expansion of the colonizing cells. The DNA mesh of NETs may promote invasion by acting to concentrate NET-associated proteases or protecting proteases from the endogenous inhibitors abundant in blood. We found that conditioned medium from NET-forming cultures can promote invasion, but that both DNase I digestion and heat denaturing can abolish this effect. This suggests that the DNA mesh is important for mediating the effect, but because denaturing does not degrade DNA, it is not simply free DNA that stimulates invasion. Possibly, a chemotactic factor(s) is associated with NETs. HMGB1 released during NET formation may drive proliferation and migration of cancer cells (12). Because we found NETs in primary human tumors as well as metastases, another important future direction will be to elucidate whether NETs also participate in cancer cell migration and invasion in the primary tumor.
Here, we show that three different types of cancer cells can induce pro-migratory or pro-invasive NETs, but cancer cells can acquire the ability to invade and migrate through many other means. Therefore, assays to identify patients who may benefit from NET-targeted treatments are needed. Detection of NETs in tumor biopsies may be the most accurate method of identifying patients who might benefit from NET-targeting approaches. However, because we identified G-CSF as a critical factor in the induction of NETs by cancer cells, it is possible that cancer cell expression of G-CSF could identify patients at risk of developing NET-promoted metastasis. Both 4T1 cells and C3(1)-Tag tumors express high levels of G-CSF (22, 35), and they are both models of triple-negative breast cancer. Higher amounts of G-CSF are found in human patient samples of triple-negative breast cancers than of other subtypes (34). Our analysis of clinical samples also shows that NETs are found in highest amounts in this breast cancer subtype.
In conclusion, we demonstrated that cancer cells can hijack the physiological processes of microorganism killing through NET formation to promote metastasis. This concept for how cancer cells exploit host cells represents a therapeutic opportunity to prevent metastasis, which accounts for the great majority of cancer-associated deaths.
Materials and Methods
Study design
The objective of this research was to test the hypothesis that cancer cell-induced NETs promote invasion and metastasis in in vitro and in vivo models of cancer. Animals were excluded from the study if the initial intravenous injection of the cancer cells missed the tail vein. Endpoints for animal experiments were selected before the conduction of the experiments as the time when the first animals in any experimental group had a weight loss of >10%. The number of replicates for specific experiments is listed in the figure legends. The experiments testing the effects of DNase I on invasion in vitro were repeated by three different co-authors. The investigators who assessed, measured, or quantified the results of invasion through Matrigel, NET-like structures by CILI, or metastatic burden after treatment with DNase I-coated nanoparticles were blinded to the specific intervention.
Animals
BALB/c mice were purchased from Charles River Laboratory, p47phox-/- (39) and ACTB-ECFP mice (40) mice from Jackson Laboratory, and C3(1)-Tag mice (21) from the NIH mouse repository. LysM-EGFP mice (20) were provided by Dr. Mark Looney (UCSF). All procedures were approved by the Cold Spring Harbor Laboratory (CSHL) Institutional Animal Care and Use Committee and were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
In vitro invasion, migration, and NET formation assays
Neutrophils (2.5×105 cells) in 750 μL serum-free medium were seeded on poly-L-lysine coated coverslips (BD Biosciences) in 24-well plates for 15-30 min before adding anti-G-CSF (R&D Systems), human recombinant G-CSF (R&D System), apocynin (Abcam), cathepsin G inhibitor I (EMD Millipore), Cl-amidine (Cayman Chemical), sivelestat (Tocris Bioscience), DNase I (Invitrogen, AM2222 or Roche, 10104159001), or DNase I-coated nanoparticles. Cancer cells (1×105 4T1, 4T07, or BT-549 cells) in serum-free medium were then added to rehydrated Matrigel inserts (BD Biosciences). After 22 hours at 37 °C, the non-invading cancer cells from the upper surface of the membrane were wiped off, and cells on the bottom side of the membrane were fixed with 4% PFA and stained with hematoxylin. The number of invading cells was counted under a light microscope in six fields of view, and the average number of cells was calculated.
CM was prepared from neutrophil cultures grown alone or with cancer cells (4T1 or BT549) present in the upper chamber of the transwells, the latter with or without DNase I or apocynin added to the bottom well. The CM from co-cultures of neutrophils and cancer cells was also heat denatured (65 °C for 15 min or 95 °C for 5 min) or DNase I was added to the collected CM. The CM was added to the lower chamber, and cancer cells were seeded in serum-free medium in the upper chamber of Matrigel-coated cell culture inserts. CM was also prepared from neutrophil cultures treated with 20 nM phorbol 12-myristate 13-acetate (PMA) (Sigma) with or without DNase I (Invitrogen). The CM was added to the lower chamber, and 4T1 cells were seeded in serum-free medium in the upper chamber of Matrigel-coated FluoroBlock cell culture inserts. After 22 hours, the cells were stained with 1 μM SYTO13 (Molecular Probes), and the number of invading cells was counted in ten random fields of view using a fluorescence microscope.
To test the effect of NETs on C3(1)-Tag primary cancer cell migration, tumors (∼10-12 mm in diameter) were resected from C3(1)-Tag mice intercrossed with ACTB-ECFP, and cancer cells were isolated as described (42). After growth to subconfluency, 5×104 cells in DMEM containing 0.5% FCS were added to a FluoroBlock cell culture insert with 8 μm pore size (Corning). The plate was incubated for 2 hours to allow cells to adhere, and then neutrophils (2.5×105 cells) were seeded on poly-L-lysine-coated coverslips (BD Biosciences) in the lower chamber in serum-free DMEM. The cancer cell medium was then replaced by serum-free DMEM. After 22 hours, the number of migrating cells was quantified as for the experiments performed with neutrophil CM.
Neutrophils grown on coverslips in the lower chamber were fixed with 4% PFA, permeabilized with 0.5% Triton X-100 in PBS, and incubated in 1× blocking buffer (5% goat serum, 2.5% BSA in PBS) for 1 hour. Cells were stained with primary antibodies in blocking buffer using anti-neutrophil elastase (1:100 dilution, Abcam, rabbit pAb, ab68672) and either anti-histone H1 (1:30 dilution, Abcam, ab62884) or anti-histone H3 (1:50 dilution, Cell Signaling Technology, #3680). After washing with PBS, the cells were stained with fluorochrome-conjugated secondary antibodies (1:150 dilution, Invitrogen) in 0.5× blocking buffer and finally stained with DAPI (Invitrogen, 0.05 mg/mL). NET formation was determined as the percentage of the field of view positive for a histone signal.
In vivo treatment with DNase I-coated nanoparticles
To test the effect of DNase I-coated nanoparticles on experimental metastasis, female BALB/c mice were treated with DNase I-coated nanoparticles (75 U/mouse) or control, uncoated nanoparticles intraperitoneally. Two hours later, 1×105 4T1-luciferase (exp. 1) or 4T1 cells (exp. 2) were injected intravenously. Daily nanoparticle treatment was continued for two weeks, and the mice were euthanized when the first mouse showed >10% weight loss (33 and 26 days after cancer cell injection for exp. 1 and 2, respectively). Metastatic burden was quantified from lung sections as described above, and results from the two experiments were pooled. Of note, nanoparticles were not given intravenously because the repeated injections would damage the tail veins, making it difficult to continue treatment through the entire period. The bio-distribution of intraperitoneally and intravenously administrated nanoparticles is similar (48).
To test the effect of DNase I-coated nanoparticles on spontaneous lung metastasis, 4T1 cells (2.5×105) were injected into both inguinal mammary glands of female BALB/c mice. One week later, daily intraperitoneal injections with DNase I-coated nanoparticles (75 U/mouse) or uncoated nanoparticles were initiated and continued until 19 days after cancer cell transplantation. Tumor growth and lung metastatic burden were quantified as described above.
Statistical analysis
All statistical analyses were performed using GraphPad Prism software, versions 5 and 6. Data were analyzed using two-sided t-tests or one-way ANOVA, as indicated in the figure legends with alpha 0.05. For statistical analyses of the metastatic burden from orthotopically growing tumors after treatment with DNase I-coated nanoparticles and of the effect of DNase I-coated nanoparticles on the size of metastatic foci, the data were first transformed by taking the square root because the variance of the untransformed data was significantly (p=0.007 and p=0.0003, respectively) different between the two groups. The number of sampled units, N, is indicated in the figure legends.
Supplementary Material
Supplementary Figure S1. Effects of CXCL1 on cancer progression.
Supplementary Figure S2. NET formation in lungs of mice injected with 4T1 cancer cells.
Supplementary Figure S3. Isolation of neutrophils for in vitro assay.
Supplementary Figure S4. The effects of G-CSF and p47phox on NET formation and cancer cell invasion.
Supplementary Figure S5. Breast cancer cells induce the formation of lytic NETs.
Supplementary Figure S6. Conditioned medium from neutrophils induced to undergo NET formation promotes invasion.
Supplementary Figure S7. Cancer cell-induced NETs do not promote extravasation.
Supplementary Movie S1. In vivo formation of NET-like structures around 4T1 cancer cells.
Acknowledgments
We thank Mark Looney (UCSF) for helping to establish CILI and for LysM-EGFP mice, Robert Eifert (CSHL) for designing the thoracic windows, George Peeters (Solamere Technologies) for modification of our microscope for CILI, Marty Brown for obtaining clinical samples, Scott Lowe (Memorial Sloan Kettering Cancer Center/HHMI) for the MSCV-miR30-PGK-NeoR-IRES-GFP (LMN) vector, M. Monestier (Temple University) for antibodies to nucleosomes, and Michelle Long and Vishal Vaghela for technical assistance.
Funding: This work was supported by the CSHL Cancer Center Support Grant 5P30CA045508 and funds to M.E. from the Department of Defense (W81XWH-14-1-0078), the Long Island 2-Day Walk to Fight Breast Cancer, and the Joni Gladowsky Breast Cancer Foundation. M.E., J.S., and A.F.S. were supported by funds from NIH (5U01CA180944-02) and the Hope Foundation. M.S.G. was supported by the Cancer Research Institute CLIP grant; R.W.W. was supported by an NIHGM MSTP Training Award (T32-GM008444); Z.A. was supported by Aid for Cancer Research; L.M. is a George A. & Marjorie H. Anderson fellow and is supported by a fellowship from the Boehringer Ingelheim Fonds; N.P. was supported by a Formación de Profesorado Universitario (FPU) fellowship (AP2010-2197); K.K. was supported by the National Cancer Institute (K99 CA181490); and V.K. was supported by the DFG research fellowship (KU 3264/1-1).
Footnotes
Author contributions: J.P., R.W.W., L.M., and M.E. designed the experiments. J.P. and Y.K. performed the co-culture experiments; R.W.W. and L.M. isolated human neutrophils. R.W.W., J.P., L.M., E.S.N., and J.Q. performed the animal experiments; R.W.W. and M.R.F. performed CILI. Z.A. and M.S.G. designed and engineered DNase I-coated nanoparticles. A.S.A. and N.P. performed FACS; J.P., L.M., R.W.W., and K.K. analyzed NET formation in tissues. J.S., A.F.S., Y.L, and N.H.W. collected breast cancer samples and were responsible for obtaining clinical information on the patients. S.A.H. performed electron microscopy. V.K. designed experiments using conditioned medium. J.P., R.W.W., L.M., and M.E. wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
References and Notes
- 1.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 2.Sethi N, Kang Y. Unravelling the complexity of metastasis - molecular understanding and targeted therapies. Nat Rev Cancer. 2011;11:735–748. doi: 10.1038/nrc3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 4.Tazawa H, Okada F, Kobayashi T, Tada M, Mori Y, Une Y, Sendo F, Kobayashi M, Hosokawa M. Infiltration of neutrophils is required for acquisition of metastatic phenotype of benign murine fibrosarcoma cells: implication of inflammation-associated carcinogenesis and tumor progression. Am J Pathol. 2003;163:2221–2232. doi: 10.1016/S0002-9440(10)63580-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, Bourdeau F, Kubes P, Ferri L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013;123:3446–3458. doi: 10.1172/JCI67484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJ, Ciampricotti M, Hawinkels LJ, Jonkers J, de Visser KE. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345–348. doi: 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015;528:413–417. doi: 10.1038/nature16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spiegel A, Brooks MW, Houshyar S, Reinhardt F, Ardolino M, Fessler E, Chen MB, Krall JA, DeCock J, Zervantonakis IK, Iannello A, Iwamoto Y, Cortez-Retamozo V, Kamm RD, Pittet MJ, Raulet DH, Weinberg RA. Neutrophils suppress intraluminal NK-mediated tumor cell clearance and enhance extravasation of disseminated carcinoma cells. Cancer Discov. 2016 Apr 12; doi: 10.1158/2159-8290.CD-15-1157. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. 2011;20:300–314. doi: 10.1016/j.ccr.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith TJ, Bohlke K, Lyman GH, Carson KR, Crawford J, Cross SJ, Goldberg JM, Khatcheressian JL, Leighl NB, Perkins CL, Somlo G, Wade JL, Wozniak AJ, Armitage JO. Recommendations for the Use of WBC Growth Factors: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol. 2015;33:3199–3212. doi: 10.1200/JCO.2015.62.3488. [DOI] [PubMed] [Google Scholar]
- 11.Branzk N, Papayannopoulos V. Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol. 2013;35:513–530. doi: 10.1007/s00281-013-0384-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, Wang Y, Simmons RL, Huang H, Tsung A. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res. 2016;76:1367–1380. doi: 10.1158/0008-5472.CAN-15-1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merza M, Hartman H, Rahman M, Hwaiz R, Zhang E, Renstrom E, Luo L, Morgelin M, Regner S, Thorlacius H. Neutrophil Extracellular Traps Induce Trypsin Activation, Inflammation, and Tissue Damage in Mice with Severe Acute Pancreatitis. Gastroenterology. 2015;149:1920–1931. doi: 10.1053/j.gastro.2015.08.026. [DOI] [PubMed] [Google Scholar]
- 14.Yang C, Sun W, Cui W, Li X, Yao J, Jia X, Li C, Wu H, Hu Z, Zou X. Procoagulant role of neutrophil extracellular traps in patients with gastric cancer. Int J Clin Exp Pathol. 2015;8:14075–14086. [PMC free article] [PubMed] [Google Scholar]
- 15.Kolaczkowska E, Jenne CN, Surewaard BG, Thanabalasuriar A, Lee WY, Sanz MJ, Mowen K, Opdenakker G, Kubes P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673. doi: 10.1038/ncomms7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Looney MR, Thornton EE, Sen D, Lamm WJ, Glenny RW, Krummel MF. Stabilized imaging of immune surveillance in the mouse lung. Nat Methods. 2011;8:91–96. doi: 10.1038/nmeth.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52:1399–1405. [PubMed] [Google Scholar]
- 18.Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- 19.Owen JL, Criscitiello MF, Libreros S, Garcia-Areas R, Guthrie K, Torroella-Kouri M, Iragavarapu-Charyulu V. Expression of the inflammatory chemokines CCL2, CCL5 and CXCL2 and the receptors CCR1-3 and CXCR2 in T lymphocytes from mammary tumor-bearing mice. Cell Immunol. 2011;270:172–182. doi: 10.1016/j.cellimm.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faust N, Varas F, Kelly LM, Heck S, Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000;96:719–726. [PubMed] [Google Scholar]
- 21.Maroulakou IG, Anver M, Garrett L, Green JE. Prostate and mammary adenocarcinoma in transgenic mice carrying a rat C3(1) simian virus 40 large tumor antigen fusion gene. Proc Natl Acad Sci U S A. 1994;91:11236–11240. doi: 10.1073/pnas.91.23.11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park JH, Rasch MG, Qiu J, Lund IK, Egeblad M. Presence of insulin-like growth factor binding proteins correlates with tumor-promoting effects of matrix metalloproteinase 9 in breast cancer. Neoplasia. 2015;17:421–433. doi: 10.1016/j.neo.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, Scadden DT, Wagner DD. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A. 2012;109:13076–13081. doi: 10.1073/pnas.1200419109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176:231–241. doi: 10.1083/jcb.200606027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207:1853–1862. doi: 10.1084/jem.20100239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, Bowden MG, Hussain M, Zhang K, Kubes P. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. 2010;185:7413–7425. doi: 10.4049/jimmunol.1000675. [DOI] [PubMed] [Google Scholar]
- 27.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 28.Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191:677–691. doi: 10.1083/jcb.201006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014;8:883–896. doi: 10.1016/j.celrep.2014.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prince WS, Baker DL, Dodge AH, Ahmed AE, Chestnut RW, Sinicropi DV. Pharmacodynamics of recombinant human DNase I in serum. Clin Exp Immunol. 1998;113:289–296. doi: 10.1046/j.1365-2249.1998.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 32.Han Y, Yu Z, Wen S, Zhang B, Cao X, Wang X. Prognostic value of chemotherapy-induced neutropenia in early-stage breast cancer. Breast Cancer Res Treat. 2012;131:483–490. doi: 10.1007/s10549-011-1799-1. [DOI] [PubMed] [Google Scholar]
- 33.Shitara K, Matsuo K, Oze I, Mizota A, Kondo C, Nomura M, Yokota T, Takahari D, Ura T, Muro K. Meta-analysis of neutropenia or leukopenia as a prognostic factor in patients with malignant disease undergoing chemotherapy. Cancer Chemother Pharmacol. 2011;68:301–307. doi: 10.1007/s00280-010-1487-6. [DOI] [PubMed] [Google Scholar]
- 34.Hollmen M, Karaman S, Schwager S, Lisibach A, Christiansen AJ, Maksimow M, Varga Z, Jalkanen S, Detmar M. G-CSF regulates macrophage phenotype and associates with poor overall survival in human triple-negative breast cancer. Oncoimmunology. 2016;5:e1115177. doi: 10.1080/2162402X.2015.1115177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, Yu L, Ross J, Korsisaari N, Cao T, Bou-Reslan H, Kallop D, Weimer R, Ludlam MJ, Kaminker JS, Modrusan Z, van Bruggen N, Peale FV, Carano R, Meng YG, Ferrara N. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A. 2010;107:21248–21255. doi: 10.1073/pnas.1015855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sugihara S, Yamamoto T, Tsuruta J, Tanaka J, Kambara T, Hiraoka T, Miyauchi Y. Serine protease-induced enhancement of blood-borne metastasis of rat ascites tumour cells and its prevention with deoxyribonuclease. Br J Cancer. 1990;62:607–613. doi: 10.1038/bjc.1990.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knight JS, Subramanian V, O'Dell AA, Yalavarthi S, Zhao W, Smith CK, Hodgin JB, Thompson PR, Kaplan MJ. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis. 2015;74:2199–2206. doi: 10.1136/annrheumdis-2014-205365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.in http://www.thesgc.org/chemical-probes/GSK484.
- 39.Jackson SH, Gallin JI, Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med. 1995;182:751–758. doi: 10.1084/jem.182.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hadjantonakis AK, Macmaster S, Nagy A. Embryonic stem cells and mice expressing different GFP variants for multiple non-invasive reporter usage within a single animal. BMC Biotechnol. 2002;2:11. doi: 10.1186/1472-6750-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE. 2009;4:e6529. doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J, Park J, Sinha P, Bissell MJ, Frengen E, Werb Z, Egeblad M. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21:488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z, Looney MR. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122:2661–2671. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 45.Thornton EE, Krummel MF, Looney MR. Live imaging of the lung. Curr Protoc Cytom. 2012;Chapter 12 doi: 10.1002/0471142956.cy1228s60. Unit12 28. [DOI] [PubMed] [Google Scholar]
- 46.Ewald AJ, Werb Z, Egeblad M. Dynamic, long-term in vivo imaging of tumor-stroma interactions in mouse models of breast cancer using spinning-disk confocal microscopy. Cold Spring Harb Protoc. 2011;2011 doi: 10.1101/pdb.top97. pdb top. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amoozgar Z, Park J, Lin Q, Weidle JH, 3rd, Yeo Y. Development of quinic acid-conjugated nanoparticles as a drug carrier to solid tumors. Biomacromolecules. 2013;14:2389–2395. doi: 10.1021/bm400512g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arvizo RR, Miranda OR, Moyano DF, Walden CA, Giri K, Bhattacharya R, Robertson JD, Rotello VM, Reid JM, Mukherjee P. Modulating pharmacokinetics, tumor uptake and biodistribution by engineered nanoparticles. PLoS One. 2011;6:e24374. doi: 10.1371/journal.pone.0024374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Macanovic M, Lachmann PJ. Measurement of deoxyribonuclease I (DNase) in the serum and urine of systemic lupus erythematosus (SLE)-prone NZB/NZW mice by a new radial enzyme diffusion assay. Clin Exp Immunol. 1997;108:220–226. doi: 10.1046/j.1365-2249.1997.3571249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Effects of CXCL1 on cancer progression.
Supplementary Figure S2. NET formation in lungs of mice injected with 4T1 cancer cells.
Supplementary Figure S3. Isolation of neutrophils for in vitro assay.
Supplementary Figure S4. The effects of G-CSF and p47phox on NET formation and cancer cell invasion.
Supplementary Figure S5. Breast cancer cells induce the formation of lytic NETs.
Supplementary Figure S6. Conditioned medium from neutrophils induced to undergo NET formation promotes invasion.
Supplementary Figure S7. Cancer cell-induced NETs do not promote extravasation.
Supplementary Movie S1. In vivo formation of NET-like structures around 4T1 cancer cells.