

Abstract
The development of an entirely solid-phase peptide synthesis-based synthesis of the quorum sensing signal gelatinase biosynthesis-activating pheromone (GBAP) from Enterococcus faecalis is reported. The method was used to prepare three libraries of analogues to investigate the structure-activity relationships (SARs) of the GBAP signal. The SAR studies revealed new characteristics of the GBAP signal and uncovered the most potent quorum sensing activator in E. faecalis known to date.
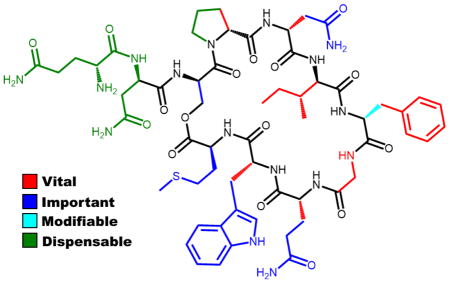
Graphical Abstract
The rapid development of resistance by bacteria to antibiotics has limited the effectiveness of current treatment options, especially in cases where pathogens have acquired multi-drug resistance, including resistance against treatments of last resort.1–3 There is, therefore, an urgent need to develop novel therapeutic strategies against these so called “superbugs”. One such approach is to target processes related to the pathogenicity of the bacteria with the goal of enabling the patient’s own immune system to clear the infection. Because this approach targets non-essential pathways, the selective pressure for resistance is lessened, reducing the risk of antibiotic resistance development.
Quorum sensing (QS) is a process by which bacteria regulate group behaviors through monitoring cell density using diffusible small signal molecules.4–7 These group behaviors include symbiotic (bioluminescence, nodulation) as well as pathogenic (virulence factor production, biofilm formation, competence) phenotypes.4–7 The regulatory role of QS in pathogenicity has led to significant research efforts aimed at targeting QS for the development of anti-infective treatments.5–11
While normally not pathogenic in healthy individuals, in clinical settings, Enterococcus faecalis is responsible for approximately 80% of all enterococcus infections including urinary tract infections, wound infections, bacteremia, and endocarditis.1,2 The fsr QS circuit (Figure 1) has been shown to govern the pathogenicity of E. faecalis by regulating the formation of robust biofilms as well as the production of virulence factors such as gelatinase.12,13 Gelatinase biosynthesis-activating pheromone (GBAP) is a lactone-based macrocyclic auto-inducing peptide (AIP) used by E. faecalis to activate the fsr QS circuitry.4,5,12–14 In this study, we report the design of an entirely solid phase peptide synthetic approach for the preparation of GBAP and GBAP analogues. Previously, GBAP and some analogues have been prepared using a combination of solid-phase and solution-phase synthesis.15–17 Synthesis conducted entirely on resin allows for greater compatibility with the use of automatic peptide synthesizers and reduces the number of intermediate purification steps required as part of the synthesis. We demonstrated the usefulness of our approach by preparing three focused libraries of GBAP analogues and evaluating their ability to modulate QS using a cell-based reporter assay. Our analysis revealed structure-activity relationship (SAR) trends of the GBAP signal and uncovered the most potent QS activator in E. faecalis known to date.
Figure 1.

Diagram of the fsr QS circuit. FsrB processes the 53 residue propeptide into mature GBAP and exports it out of the cell. Upon reaching a threshold concentration, GBAP binds to the histidine kinase receptor FsrC, triggering the phosphorylation of FsrA, which then upregulates the expression of the fsr QS circuitry genes, including the production of more GBAP (auto-induction). Genes involved in pathogenicity are also upregulated by FsrA.
Our synthetic approach for producing GBAP and its analogues utilizes Fluorenylmethyloxycarbonyl (Fmoc)-based solid phase peptide synthesis (SPPS),18–20 and makes use of Asn5 or Gln9 as resin attachment points (Figure 2). By using Rink Amide Methylbenzhydrylamine (Knorr) resin, initial attachment to the resin is accomplished through reaction with either the side chain of Fmoc-protected aspartic acid or glutamic acid that are protected at their C-terminus by an allyloxycarbonyl (alloc) group. Because the attachment to Knorr resin involves the formation of an amide bond, the corresponding asparagine or glutamine is generated upon cleavage from the resin. Two additional protecting groups are important for this approach: the Ser3 side chain is protected with the highly acid-labile trityl protecting group while the N-terminus of Gln1 is protected with a tert-butoxycarbonyl (Boc) group. This combination of protecting groups prevents peptide elongation at the N-terminus and allows for the selective removal of the trityl group. The carboxyl group on Met11 can then be reacted with the deprotected hydroxyl to form the lactone linkage, creating a “branch” in the peptide sequence. The remainder of the peptide sequence can then be extended using standard Fmoc-SPPS methods. Finally, removal of the alloc group makes the C-terminus available for cyclization to achieve the final peptide that can then be cleaved from the resin using standard cleavage conditions.
Figure 2.

The SPPS-based strategy for GBAP synthesis. A) Pathway starting from Asn5; B) Pathway starting from Gln9. In both pathways, the initial amino acid is attached by its side chain to Knorr resin. The peptide is extended to the N-terminus. The acid-labile trityl protecting group is removed and the lactone linkage is made with Ser3 before the remaining amino acids are added. Finally, alloc is removed from the C-terminus, and the peptide is cyclized on the resin before final cleavage.
To aid in optimization of the synthesis, the removal of Fmoc was quantified by UV-Vis spectroscopy for each step in the synthesis (Figures S-1 and S-2). Steps where the apparent loading of the resin dropped precipitously served as major focus points for optimization. General optimizations included changing the coupling reagent (N,N,N′,N′-Tetramethyl-O-(1H-benzotriazol-1-yl)uranium hexafluorophosphate (HBTU) to 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU)), coupling solvents (N,N-dimethylformamide (DMF) to 25% dichloromethane (DCM) in degassed DMF) and reaction time (1 hour to 2 hours). In addition, the initial amino acid loading step to the resin was extended to 16 hours. The steps with the largest drops; coupling of Ser3 and Gln9 for the aspartic acid pathway and Ser3 and Phe7 for the glutamic acid pathway, were improved by conducting double couplings at 50 °C, the first of which was done for 4 hours while the second was done for 16 hours. Lastly, the Met11 coupling was extended to 4 hours and the amount of 4-dimethylaminopyridine (DMAP) used as catalyst was optimized to both minimize the undesired epimerization while still obtaining complete lactone coupling. These combined changes resulted in modest improvements in the Fmoc deprotection assay, but significantly improved yields over the original conditions.
The improvement in bioassay-ready yield was also aided by a change in peptide handling after it was found that the intrinsic instability of many of the peptides resulted in fast and significant degradation of purified peptides. This instability is due to the N-terminal glutamine, which was found to readily cyclize with itself to form pyroglutamic acid.21,22 To minimize this rapid degradation, all peptide-containing fractions were immediately freeze-dried after high performance liquid chromatography (HPLC), and then stored at −80 °C. These precautions greatly reduced the rate at which the peptide degraded and helped boost the obtainable yields. By this method of Fmoc-assay-directed optimization, the bioassay-ready (≥95% pure) yield of GBAP and analogues was improved from less than 0.5% to an acceptable 5–10% range. Nonetheless, significant losses in yield were also seen when more than a single HPLC purification was required (Figure S-3), indicating that further optimization of the HPLC purification step could enhance yields even further.
To demonstrate the utility of our synthetic approach, we synthesized three focused libraries of GBAP analogues (overall 25 analogues excluding native GBAP) and tested them for their ability to modulate QS in E. faecalis using cell-based reporter assays. Previous investigations into the SAR of GBAP included mainly alanine scanning.17 These studies revealed that Phe7 and Trp10 were important residues for binding to the FsrC receptor.16 It was also found that the side chains of the first two amino acids of GBAP composing its “tail” region were not necessary for either binding or agonistic activity.17 Unfortunately, the phenotypic assay utilized in these studies prevented quantification of agonistic activity, thus in-depth analysis of the role each side-chain residue plays in the overall activity was not feasible. We wanted to expand on these results and quantitatively assess the contribution of each side chain and chiral center to the overall activity. Thus, we conducted full alanine and D-amino acid scans of the GBAP signal. To evaluate the activity of each analogue, we utilized an E. faecalis β-galactosidase QS reporter strain.13 In this strain, a plasmid containing pGelE-lacZ was incorporated. Thus, upon activation of the fsr QS circuitry, FsrA will bind pGelE and transcribe lacZ, allowing for quantification of QS activation by measuring β-galactosidase activity.
Initial bioassays revealed that GBAP activates FsrC with a half maximal effective concentration (EC50) value of 1.15 nM. To our knowledge, this is the first report quantifying the potency of GBAP or any other analogue. The results of the ala-nine scan largely recapitulated what had been observed previously.17 However, additional insights were made using the wide range of concentrations necessary to determine the EC50 values of the analogues. With the exception of I6A, F7A and G8A, all the alanine analogues were capable of fully activating the fsr QS circuitry at high concentration (10 μM). Further determination of the EC50 values of these analogues revealed that the side chains of the exocyclic tail (residues Gln1 and Asn2) are dispensable as alanine substitution in these positions resulted in analogues with similar EC50 values to GBAP (Table 1). Within the macrocyclic region, with the exception of Pro4, all the side chain residues were found to be important for activity as all the alanine modifications resulted in significant reduction in potency (>100-fold, Table 1). Interestingly, the replacement of Pro4 with alanine resulted in an analog with improved potency (~4-fold, Table 1). Since proline is known to induce conformational restriction, the improved potency is likely due to the increased flexibility conferred by the alanine replacement. The complete loss of activity in the G8A analogue further supports this hypothesis and highlights the importance of conformational flexibility in the macrocyclic region.
Table 1.
EC50 values for the alanine scan, D-amino acid scan, and tail modification analogues of GBAP against the FsrC receptora
| peptide name | EC50b [95% CI]c (nM) |
|---|---|
| GBAP | 1.15 [0.825–1.59] |
| GBAP-Q1A | 0.549 [0.299–1.01] |
| GBAP-N2A | 0.704 [0.522–0.898] |
| GBAP-P4A | 0.270 [0.202–0.360] |
| GBAP-N5A | 341 [200–581] |
| GBAP-I6Af | > 1,000 |
| GBAP-F7Af | > 1,000 |
| GBAP-G8A | > 10,000 |
| GBAP-Q9A | 312 [222–437] |
| GBAP-W10A | 246 [117–520] |
| GBAP-M11A | 164 [93.8–288] |
| GBAP-q1d | 0.211 [0.0798–0.599] |
| GBAP-n2 | 0.304 [0.168–0.552] |
| GBAP-s3 | 45.1 [17.0–120] |
| GBAP-p4 | > 1,000 |
| GBAP-n5 | > 1,000 |
| GBAP-i6 | > 1,000 |
| GBAP-f7 | 4.97 [3.12–7.77] |
| GBAP-q9 | > 1,000 |
| GBAP-w10 | > 10,000 |
| GBAP-m11 | 231 [129–414] |
| Ac-GBAP | 1.50 [1.31–1.73] |
| GBAP-DesQ1 | 1.26 [1.15–1.39] |
| Ac-GBAP-DesQ1 | 1.44 [0.855–2.44] |
| GBAP-DesQ1N2 lactame,f | > 10,000 |
| Ac-GBAP-DesQ1N2 | 1.01 [0.496–2.06] |
See Supporting Information for details of reporter strain and methods. See Supporting Information for plots of agonism dose response curves. All assays performed in triplicate. The full sequences for the peptides can be found in Table S-5.
EC50 values determined by testing peptides over a range of concentrations.
95% confidence interval.
lower case letters indicate residues replaced with the D-enantiomer.
Lactam (head-to-tail) cyclization.
Weak inhibitor.
Moving to the D-amino acid scan, unsurprisingly, the “tail” of GBAP was found to be permissive to inversions in chirality at each of its two amino acids (Table 1). Intriguingly, both inversions enhanced the potency, resulting in the identification of the most potent analogues reported to date, along with GBAP P4A (4–5-fold increase in potency). Looking at the macrocycle region, it appears that chirality alterations are less tolerated than alanine substitutions, as the vast majority of analogues resulted in almost complete loss of activity (>1,000-fold decrease, Table 1). Surprisingly, the D-substitution of Phe7, a residue that was found to be critical for activity in the alanine scan, resulted in a potent analogue with only slight reduction in potency (~4-fold, Table 1). This result suggests that for Phe7 the identity of the side chain is more important than the orientation of that side chain, possibly due to a relatively promiscuous binding pocket that only requires a bulky hydrophobic residue. Alternatively, this result can be explained by the adjacency of Phe7 to glycine, which provides little steric constraint on the phenyl side chain, thus allowing it to adopt an active conformation even with the opposite chirality. Lastly, changing the chirality of Ser3, the macrocycle bridging residue, resulted in decreased potency compared to GBAP, but to a much lesser extent than the other macrocycle residues (only ~40-fold decrease, Table 1). This result implies that conformational changes within the scaffold of the macro-cycle are better tolerated than changes in the conformation of the macrocycle side chain residues.
Our alanine and D-amino acid scans revealed that all exocyclic tail modifications are tolerated, suggesting that the tail residues, in their current form, do not contribute to bioactivity. To test this hypothesis, we constructed a focused library of tail modifications. This library consisted of five analogues where the tail residues were sequentially removed and/or acetylated. The results from the tail modification library confirmed that the two amino acids composing the tail in GBAP are not required for activity. A head-to-tail (lactam) cyclization, forming the macrocycle through the reaction of the N-terminus of serine with the C-terminus, was inactive. This loss of activity can be the result of a change in the orientation of the macrocyclic linkage, and/or a change in the macrocycle ring size (28 to 27 atoms), and/or a change in the macrocycle bridge chemistry (lactone to lactam).
Our SAR analysis revealed several analogues that exhibited little to no QS activation, even at high concentration (10 μM). To determine whether these analogues are unable to bind the FsrC receptor or fail to activate it, we tested these analogues for their ability to competitively inhibit GBAP-mediated QS activation (Figure S-5). From the lack of inhibition activity, we concluded that these analogues lost their ability to bind the FsrC receptor.
In conclusion, we developed and optimized an entirely SPPS approach to the preparation of GBAP and demonstrated its applicability by constructing three libraries of analogues. Our optimized synthetic conditions resulted in acceptable yields (5–10%), however, yields can be further improved through the optimization of the HPLC methods used during peptide purification. Our SAR analysis of GBAP revealed several key positions that are required for receptor binding. Furthermore, we showed that the exocyclic tail region, in its current form, is dispensable. Nonetheless, the enhanced activity of GBAP-q1 and GBAP-n2 suggests that this region can contribute to bio-activity, if it is altered correctly. Thus, the exocyclic tail, in its original form, can either be removed or altered to enhance bioactivity, increase solubility, or incorporate different tags that will facilitate mechanistic studies of the fsr QS circuitry. Lastly, we have identified the most potent FsrC activators known with activities in the picomolar range. These analogues can be used as leads to the development of chemical probes with desired activity profiles that would be applied to study interspecies communications. Furthermore, the combination of efficient synthetic approach with in-depth understanding of the SAR trends of the GBAP molecule will facilitate the development of potent QS modulators that could be utilized to regulate pathogenicity in E. faecalis. Such studies are ongoing in our laboratory, along with structural analyses of the GBAP molecule, and will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported by the Nevada INBRE through a grant from the National Institute of General Medical Sciences (GM103440) from the National Institutes of Health (NIH). The E. faecalis reporter strain, TX5274, was kindly provided by B. E. Murray (University of Texas Health Science Center, Houston).
Footnotes
Note
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website.
Information includes: full details of experimental procedures, GBAP synthesis optimization, peptide characterization, and dose response curves for GBAP analogues (PDF)
References
- 1.Huycke M, Sahm D, Gilmore MS. Emerg Infect Dis. 1998;4:239. doi: 10.3201/eid0402.980211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arias CA, Murray BE. Nat Rev Microbiol. 2012;10:266. doi: 10.1038/nrmicro2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garland M, Loscher S, Bogyo M. Chem Rev. 2017;117:4422. doi: 10.1021/acs.chemrev.6b00676. [DOI] [PubMed] [Google Scholar]
- 4.Cook LC, Federle MJ. Fems Microbiol Rev. 2014;38:473. doi: 10.1111/1574-6976.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rutherford ST, Bassler BL. Cold Spring Harb Perspect Med. 2012;2:a012427. doi: 10.1101/cshperspect.a012427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turan NB, Chormey DS, Büyükpınar Ç, Engin GO, Bakirdere S. TrAC, Trends Anal Chem. 2017;91:1. [Google Scholar]
- 7.Papenfort K, Bassler BL. Nat Rev Microbiol. 2016;14:576. doi: 10.1038/nrmicro.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerdt JP, Blackwell HE. ACS Chem Biol. 2014;9:2291. doi: 10.1021/cb5004288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson BK, Abramovitch RB. Trends Pharmacol Sci. 2017;38:339. doi: 10.1016/j.tips.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen F, Gao Y, Chen X, Yu Z, Li X. Int J Mol Sci. 2013;14:17477. doi: 10.3390/ijms140917477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang K, Zhang XH. Mar Drugs. 2014;12:3245. doi: 10.3390/md12063245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin X, Singh KV, Weinstock GM, Murray BE. Infect Immun. 2000;68:2579. doi: 10.1128/iai.68.5.2579-2586.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin X, Singh KV, Weinstock GM, Murray BE. J Bacteriol. 2001;183:3372. doi: 10.1128/JB.183.11.3372-3382.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakayama J, Cao Y, Horii T, Sakuda S, Akkermans ADL, de Vos WM, Nagasawa H. Mol Microbiol. 2001;41:145. doi: 10.1046/j.1365-2958.2001.02486.x. [DOI] [PubMed] [Google Scholar]
- 15.Nakayama J, Cao Y, Horii T, Sakuda S, Nagasawa H. Biosci Biotechnol Biochem. 2001;65:2322. doi: 10.1271/bbb.65.2322. [DOI] [PubMed] [Google Scholar]
- 16.Nakayama J, Yokohata R, Sato M, Suzuki T, Matsufuji T, Nishiguchi K, Kawai T, Yamanaka Y, Nagata K, Tanokura M, Sonomoto K. ACS Chem Biol. 2013;8:804. doi: 10.1021/cb300717f. [DOI] [PubMed] [Google Scholar]
- 17.Nishiguchi K, Nagata K, Tanokura M, Sonomoto K, Nakayama J. J Bacteriol. 2009;191:641. doi: 10.1128/JB.01029-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan WC, White PD, editors. Fmoc solid phase peptide synthesis: a practical approach. Oxford University Press; New York: 2000. [Google Scholar]
- 19.Behrendt R, White P, Offer J. J Pept Sci. 2016;22:4. doi: 10.1002/psc.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chantell CA, Onaiyekan MA, Menakuru M. J Pept Sci. 2012;18:88. doi: 10.1002/psc.1419. [DOI] [PubMed] [Google Scholar]
- 21.Chelius D, Jing K, Lueras A, Rehder DS, Dillon TM, Vizel A, Rajan RS, Li T, Treuheit MJ, Bondarenko PV. Anal Chem. 2006;78:2370. doi: 10.1021/ac051827k. [DOI] [PubMed] [Google Scholar]
- 22.Povoledo D, Vallentyne JR. Geochim Cosmochim Acta. 1964;28:731. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.