

Abstract
Objectives
Bipolar I disorder is a disabling illness affecting 1% of people worldwide. Family and twin studies suggest that psychotic bipolar disorder (BDP) represents a homogenous subgroup with an etiology distinct from non-psychotic bipolar disorder (BDNP) and partially shared with schizophrenia. Studies of auditory electrophysiology [e.g., paired-stimulus and oddball measured with electroencephalography (EEG)] consistently report deviations in psychotic groups (schizophrenia, BDP), yet such studies comparing BDP and BDNP are sparse and, in some cases, conflicting. Auditory EEG responses are significantly reduced in unaffected relatives of psychosis patients, suggesting that they may relate to both psychosis liability and expression.
Methods
While 64-sensor EEGs were recorded, age- and gender-matched samples of 70 BDP, 35 BDNP {20 with a family history of psychosis [BDNP(+)]}, and 70 psychiatrically healthy subjects were presented typical auditory paired-stimuli and auditory oddball paradigms.
Results
Oddball P3b reductions were present and indistinguishable across all patient groups. P2s to paired-stimuli were abnormal only in BDP and BDNP(+). Conversely, N1 reductions to stimuli in both paradigms and P3a reductions were present in both BDP and BDNP(−) groups but were absent in BDNP(+).
Conclusions
While nearly all auditory neural response components studied were abnormal in BDP, BDNP abnormalities at early- and mid-latencies were moderated by family psychosis history. The relationship between psychosis expression, heritable psychosis risk, and neurophysiology within bipolar disorder, therefore, may be complex. Consideration of such clinical disease heterogeneity may be important for future investigations of the pathophysiology of major psychiatric disturbance.
Keywords: biomarker, electroencephalography, gating, N100, oddball, P200, P300, psychosis
Persons with bipolar I disorder experience distressing and disabling affective instability including both manic and depressive symptomology. Approximately 60% of patients with bipolar I disorder experience concurrent psychosis, which carries additional devastating clinical and psychosocial consequences (1, 2). Psychosis breeds true within families with bipolar disorder (3). Patients with bipolar disorder psychosis (BDP) are 2–3 times more likely to have relatives with BDP than bipolar disorder without psychosis (BDNP) (4), and quantitative psychotic symptomology is significantly familial (Spearman’s rho = 0.33) among siblings with bipolar disorder (5) [for a detailed review of its heritability see (3)]. Across the Diagnostic and Statistical Manual of Mental Disorders–IV (DSM-IV) diagnoses, psychosis carries a similar neurophysiological signature (6, 7) and displays shared heritability and genetics (3). Individuals with schizophrenia have increased rates of BDP versus BDNP in their family (8), and twin studies indicate a strong genetic basis for this association (9). Psychosis may capture unique pathophysiological substrates with implications for how bipolar disorder is characterized, studied, and treated.
If BDP and BDNP represent distinct pathophysiological entities, then evidence supporting this distinction should be present in independent biological or cognitive measurements. Most investigations into the biological or cognitive correlates of bipolar disorder have commingled BDP and BDNP (10). Studies separating these groups demonstrate that BDP tend to have more severe disturbances of some, but not all, cognitive functions (11, 12). Ventricular enlargement may be present only in BDP (13, 14), but other neuroanatomical deviations reliably associated with psychotic psychopathology, including gray matter thickness reductions (15), may not differentiate BDP from BDNP (10, 16, 17).
Studies of auditory neurophysiology are informative for identifying psychosis-related biological deviations. Hallucinations are commonly auditory in psychosis, and auditory neurophysiological deviations are state-invariant and appear in unaffected relatives of individuals with psychotic disorders (18–20). The presentation of auditory stimuli elicits a series of event-related potentials [(ERPs) measured with electroencephalography], including the P1 (25–75-msec post-stimulus onset) reflecting stimulus registration in primary auditory cortices (21), the N1 (75–125 msec) reflecting early synchronization between primary and secondary auditory cortices in the superior lateral temporal lobes (22), and the P2 (175–250 msec) reflecting further processing and more widespread integration as auditory cortices are synchronized with tertiary and associative cortical regions [see (22) for a review]. Attentional context (24) and inter-stimulus interval (25) strongly modulate N1 amplitude implying that it indexes the earliest cognitively influenced auditory neural event reliably measureable with EEG.
Classic auditory paired-stimuli paradigms involve presentation of clicks (S1 and S2 separated by 500 msec with long interval between pairs) while ERPs (P1, N1, and P2) to the stimuli are quantified and compared (19). Many studies show smaller differences between ERPs to S1 and S2 from individuals with psychosis compared to healthy subjects, an effect determined by smaller responses to S1 (26, 27) and/or larger responses to S2 (26, 28). The few studies directly comparing BDP and BDNP ERPs have reported abnormalities either: (i) limited to BDP and their relatives (19, 29); (ii) limited only to BDNP (30); or (iii) present in both groups (31, 32). Therefore, the degree to which paired-stimuli ERPs mark psychosis in bipolar disorder is unresolved.
Auditory oddball paradigms involve presenting repeated tones (standard stimuli) interspersed with deviant target stimuli (e.g., at 80/20% frequencies, respectively). The auditory P3 ERP, an event occurring about 300 msec post-targets, is associated with novelty detection (P3a) and/or context updating (p3b) (32), reflects widespread cortical synchronization and temporal orienting (33, 34), and is reliably reduced in schizophrenia and BDP. Earlier ERPs, including N1, P2, and N2 to standard (STD) and target stimuli, also show promise as psychosis markers (35), indicating that fundamental disruption in auditory target differentiation might contribute to P3 reductions in psychosis. Again, comparisons between BDP and BDNP are inconclusive regarding auditory neurophysiological heterogeneity in bipolar disorder (36).
Inconsistency across auditory processing studies fails to support a difference-in-kind taxonomy of BDP versus BDNP as suggested by other data. Of particular importance for addressing this issue may be consideration of psychosis risk rather than just psychosis expression. Auditory electrophysiological responses are significantly heritable (estimated proportion of variance explained by genetic factors equal to 0.4 to 0.7) (37, 38, 39) and may index factors predisposing an individual to psychosis. If so, then one might expect different ERP presentations in BDNP with a family history of psychosis (+) compared to those without such a history (−) regardless of equivalent clinical presentation. For instance, if an auditory ERP component purely marks risk for developing psychosis, BDNP(+) would be expected to deviate from healthy and BDNP(−) but be similar to BDP. Conversely, some ERPs marking affective disturbance could index resilience to psychosis (40) and thus be at least normal in BDNP(+) while being deviant in BDNP(−) and BDP, capturing important etiological variance and predictive power (41). The likely commingling of BDNP (+) and (−) in previous studies could account for inconsistent findings; an effect of family history of psychosis on auditory neurophysiology would implicate previously unrecognized etiological factors.
In contrast to other reports of auditory ERPs that assessed only peak estimates from 1–3 EEG scalp sensors, the present study quantified ERPs across the entire scalp using spatial principal components analysis (PCA) and compared waveforms across the entire recording epoch in temporal bins, making maximal use of the available information. Previous work from the current group and others has established these methods as reliable and sensitive quantifiers of auditory neurophysiology in auditory ERP paradigms (26, 30, 35, 42). In addition to the use of robust methods and a sizable, well-matched sample (n = 175), the current study examined whether family history of psychosis moderated BDNP deviations from BDP and from healthy subjects.
Materials and methods
Subjects
As part of a large, multi-site data collection project (B-SNIP), 175 subjects were recruited, interviewed, and tested at five sites: University of Illinois (Chicago, IL), Yale University/IOL (Hartford, CT), University of Texas Southwestern (Dallas, TX), Harvard University (Boston, MA), and University of Maryland (Baltimore, MD). Clinically stable participants outside of an acute episode of illness were recruited via community advertisements, linked community facilities and programs, and local NAMI-type organizations. Three age- and gender-matched groups were constructed based on DSM-IV diagnosis and clinical history and blind to brain activity measurements: 70 BDP, 35 BDNP, and 70 healthy persons. Groups were matched on age, gender, and proportion of subjects from each recruitment site (Table 1 and Supplementary Table S1). All subjects provided written informed consent prior to participation. All procedures were approved by the Institutional Review Boards at each recruitment and analysis site and are in accordance with the Helsinki Declaration of 1975. No EEG data in this manuscript have been used in a previous publication.
Table 1.
Demographic and clinical statistics
| Healthy subjects (n = 70) |
BDP (n = 70) |
BDNP(−) (n = 15) |
BDNP(+) (n = 20) |
Statistics | |
|---|---|---|---|---|---|
| Females (%) | 55.7 | 55.7 | 53.3 | 55.0 | χ2(2) = 0.032 p = 0.998 |
| Age, years, mean | 38.4 | 38.2 | 41.9 | 36.7 |
F(3,171) = 0.53 p = 0.65 |
| Paired-stimuli trials accepted, mean (SD) | 140 (13.00) | 141 (8.74) | 143 (8.26) | 142 (10.20) |
F(3,171) = 0.52 p = 0.669 |
| Standard trials accepted, mean (SD) | 535 (45.4) | 542 (45.3) | 549 (24.5) | 535 (44.5) |
F(3,171) = 0.64 p = 0.590 |
| Target trials accepted, mean (SD) | 95.5 (7.05) | 96.4 (5.64) | 97.0 (4.02) | 95.8 (6.44) |
F(3,171) = 0.38 p = 0.769 |
| Percent targets detected, mean (SD) | 95.8 (9.0) | 93.3 (10.5) | 88.7 (16.9) | 91.2 (15.3) |
F(3,150) = 2.0 p = 0.114 |
| GAF score, mean (SD) | – | 60.6 (12.4) (n = 68) |
63.6 (3.6) (n = 14) |
65.3 (10.0) (n = 20) |
F(2,99) = 1.31 p = 0.275 |
| PANSS–Positive score, mean (SD) | – | 12.2 (4.13) (n = 69) |
– | – | – |
| PANSS–Negative score, mean (SD) | – | 12.0 (3.66) (n = 69) |
– | – | – |
| PANSS–General score, mean (SD) | – | 28.1 (7.94) (n = 69) |
– | – | – |
| MADRS score, mean (SD) | – | 10.6 (9.29) (n = 67) |
9.60 (9.17) (n = 15) |
8.00 (9.24) (n = 8) |
F(2,87) = 0.32 p = 0.725 |
| YMRS score, mean (SD) | – | 5.57 (6.00) (n = 67) |
4.87 (5.30) (n = 15) |
4.89 (9.58) (n = 9) |
F(2,88) = 0.11 p = 0.900 |
SD = standard deviation; BDP = bipolar disorder with psychosis; BDNP(−) = bipolar disorder without psychosis without first-degree family history of psychosis; BDNP(+) = bipolar disorder without psychosis with first-degree family history of psychosis; GAF = Global Assessment of Functioning; MADRS = Montgomery-Åsberg Depression Rating Scale; PANSS = Positive and Negative Syndrome Scale; YMRS = Young Mania Rating Scale.
Medical and family history, structured clinical interview for DSM-IV diagnosis [Structured Clinical Interview for DSM-IV (SCID) patient or nonpatient version as appropriate), Positive and Negative Symptom Scale (PANSS) (35), Young Mania Rating Scale (YMRS) (44), Montgomery-Åsberg Depression Rating Scale (MADRS) (45), and Global Assessment of Functioning scale (GAF) (Axis V of DSM-IV) were acquired by trained and experienced clinicians. Presence of serious medical, neuro-opthalmological, or neurological illness (e.g., cancer, seizure disorders, coarse brain-disease), mental retardation, head trauma with > 30 minutes unconsciousness, current substance use ascertained by history as well as urine drug screens on the day of testing (eight-panel screen for amphetamines, barbiturates, cocaine, methadone, opiates, cannabinoids, propoxyphene, and tricyclic antidepressants), abuse in the past three months, and dependence within six months or extensive history of drug dependence (DSM-IV) were criteria for exclusion. Healthy persons were free of any DSM-diagnosis themselves and of any psychosis in a first-degree relatives. The family history of psychotic illnesses was assessed for all participants using Family History Research Diagnostic Criteria (46). Twenty BDNP had first-degree relatives with BDP (n = 17), schizophrenia (n = 7), and/or schizoaffective disorder (n = 8). The remaining 15 BDNP had no first- or second-degree relatives with any psychotic disorder. Additionally, all healthy persons (H) had no first or second-degree relatives with major affective or psychotic diagnoses. All analyses in this manuscript were therefore completed using four groups: H, BDP, BDNP with no first- or second-degree relative with psychosis [BDNP(−)] and BDNP with at least one first-degree relative with a psychotic disorder [BDNP(+)].
All clinical information (including study diagnosis) for each subject were reviewed and confirmed in a best estimate diagnostic meeting including at least one senior psychiatrist/psychologist and the clinician who conducted the structured interview and completed the clinical ratings. Instructions for rating on the SCID diagnostic scale, PANSS, YMRS, and MADRS were carried out at the beginning and updated at six-month intervals during the study, while inter-rater reliability was kept at > 0.85 (intraclass correlation coefficients or Kappa) across all sites (47).
Stimuli
Recording conditions were equivalent and stimulus presentation and recording equipment identical across sites. Seated in a sound and electrically shielded booth (ambient sound = 61–63 dB; luminance = 0.11–0.12 foot-candles) subjects listened to tones delivered by two 8-ohm speakers located 50 cm in front of them. For the paired-stimuli task, subjects passively listened to 150 binaural broadband auditory stimuli pairs (4-msec duration at 75 dB) separated by an average of 9.5 sec (9–10-sec inter-pair interval; rectangular distribution), with 500 msec between stimuli in a pair. For the oddball task, subjects listened to 567 STD (1500 Hz) and 100 target (1000 Hz) tones presented in pseudorandom order (1300-msec inter-trial interval). Subjects were asked to press a button when a target was detected, and the percentage of targets detected was compared between subject groups with a one-way ANOVA. Button press data were unavailable for subjects at the Dallas site. Participants refrained from smoking one hour prior to testing.
Recording
EEG were continuously recorded from 64 Ag/AgCl sensors [impedance < 5 ΚΩ; Quik-Cap (Compumedrics Neuroscan, El Paso, TX, USA)], positioned according to the standard 10–10 EEG system plus mastoids and CP1/2 locations to provide greater sampling below the cantho-meatal line, with nose reference and forehead ground. Recordings were amplified (12,500×) and digitized (1000 Hz) using Neuroscan Acquire and Synamps2 recording systems (Compumedrics Neuroscan).
Data processing
Raw EEG data were inspected for bad sensors and artifacts. Bad sensors were interpolated (< 5% for any subject) using spherical spline interpolation [BESA 5.3 (MEGIS Software, Grafelfing, Germany)]. Data were then converted to an average reference montage and digitally bandpass filtered from 0.5–55 Hz (zero phase filter; rolloff: 6 and 48 dB/octave, respectively). Blink and cardiac artifacts were removed using Independent Components Analysis [EEGLAB 9.0 (48)]. Data were segmented into epochs from 100 msec before to either 550 msec (oddball STDs), 750 msec (oddball targets), or 800 msec after stimulus onset (paired-stimuli S1) based on waveform stabilization and return to baseline (Supplementary Figs. S1–S3). The 100-msec pre-stimulus period was used for baseline adjustment (S1 only for paired-stimuli). Epochs containing activity greater than 75 μV at any sensor were eliminated. The total number of trials used did not differ between groups for any stimulus type (Table 1). Data from good trials were averaged across trial-types within a subject to create 64-sensor ERPs (Butterfly plots available in Supplementary Figs. S1–S3).
PCA data reduction
In order to use EEG data recorded from every sensor and, thus, to most accurately and comprehensively capture the spatial topography of evoked brain responses across time, spatial principal components analysis (PCA) was completed on grand average waveforms acquired from 64-sensor scalp EEG using BESA (MEGIS Software) and Matlab (The Mathworks, Matick, MA, USA). This resulted in component scores that were analyzed instead of single sensors (i.e., as 1–2 virtual sensors), minimizing the number of comparisons and maximizing the signal/noise ratio of the ERP data (49).
For each stimulus type (paired-stimuli, oddball-STD, oddball-target), a PCA with promax (oblique) vector rotation and Kaiser normalization (49) was calculated on the 64 × 64 sensor covariance matrix (time points as observations). Scree tests were used in each case to determine the optimal number of components (50). PCA completed on averaged epochs for the paired-stimuli paradigm revealed a sole component with a frontal-central maximum (FCz) that accounted for 87.9% of the variance in waveforms across sensors (Supplementary Fig. S1). PCA completed on epochs for the oddball paradigm revealed one component with a frontal-central maximum (FCz) for standards accounting for 88.3% of the variance (Supplementary Fig. S3) and two components for target stimuli including a central parietally distributed component accounting for 85.4% of the variance (Pz maximum; with an equivalent timecourse and distribution to the P3b) and one with a frontal-central maximum accounting for 12.2% of the variance (FCz; equivalent to the P3a) (Supplementary Fig. S2). No additional components in any PCA accounted for more than 5% of the variance. When these steps were completed within analysis groups (See Supplementary Figs. S4–S7), the PCA factor weights for both oddball and paired-stimulus did not differ between any analysis group result (all r> 0.90) or between any group and the overall average (all r> 0.95). These factor solutions, along with the substantial equivalence of the results across divergent subject groups, are highly consistent and nearly identical with previous reports from separate (26, 35) and independent samples (30).
Each set of component weights was multiplied by each subject’s grand average data, summed across sensors, and divided by the plus sum of the component weights, reducing waveforms from one for each sensor to one waveform per component for each subject for paired-stimuli, oddball STDs, and oddball targets (four total).
ERP waveform analysis
For each subject waveform data from the entire epoch were grouped into 65–90 separate 10-msec bins and averaged within each bin. For each bin, a one-way ANOVA [F(3,171)] was calculated to determine group differences in waveform amplitude. To control for aberrant significant effects due to a small number of large voltage values within a bin, F-value distributions were created using a bootstrap procedure. For each condition and factor, the same one-way ANOVAs were run 5000 times with group membership randomly shuffled at each step (sampling with replacement). Non-parametric probability estimates (p) of observed F-values were then calculated as the proportion of randomly generated F-values greater than the actual estimate. To control for family-wise error due to multiple comparisons, a clustering method was implemented using Monte Carlo simulations calculated across time-bins using AlphaSim (51, 52). In order to maintain a family-wise alpha of 0.05, three sequential time-bins were required to be significant at p < 0.025.
Post-hoc discriminant analyses
To efficiently summarize variables that uniquely differentiated groups, values from significant time-bin clusters were averaged within clusters for each subject and submitted to a linear discriminant analysis with group as the dependent variable [H, BDP, BDNP(−), BDNP(+)]. Variables which minimized the overall Wilks’ lambda and had individual multiple F-statistics significant at p < 0.05 were entered in a stepwise fashion (53), leaving a parsimonious selection of neurophysiological measures.
Results
The relative distributions of groups across sites did not differ (Supplementary Table S1), and previous reports from our group from larger BSNIP samples demonstrate the lack of significant site or site-by-group effects on auditory ERPs (26, 35). Groups did not differ on number of useable trials for either paradigm or stimulus type and responded equally to targets during the oddball paradigm (Table 1). Spatial PCA reduced 64-sensor ERPs across three stimulus types to a total of four waveforms for each subject for comparisons: paired-stimuli (PS), oddball target component 1 [(TGT1) equivalent to parietal P3b], oddball target component 2 [(TGT2) equivalent to frontal P3a], and oddball STD. Component weights (topographies) are available in Supplementary Figures S1–S3. Time-bin clusters with significant overall group effects are depicted for each waveform are in Figures 1–3. Simple effects from within these clusters are discussed below and means with standard deviations are provided in Table 2.
Fig. 1.
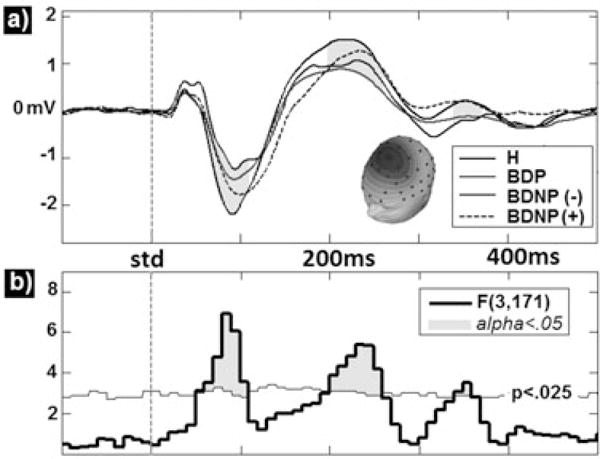
Group comparisons for principal components analysis derived paired-stimuli event-related potentials waveforms (A) averaged within group yield significant effects in the N1–S1 and P2–S1 ranges (shaded regions). F-values for these effects are also presented along with (B) a bootstrapped p < 0.025 probability line (thin horizontal line). Time regions reaching significance at FWalpha< 0.05 (three consecutive bins) are highlighted. H = healthy comparison subjects; BDP = bipolar disorder with psychosis; BDNP(−) = bipolar disorder without psychosis without first-degree family history of psychosis; BDNP(+) = bipolar disorder without psychosis with first-degree family history of psychosis.
Fig. 3.
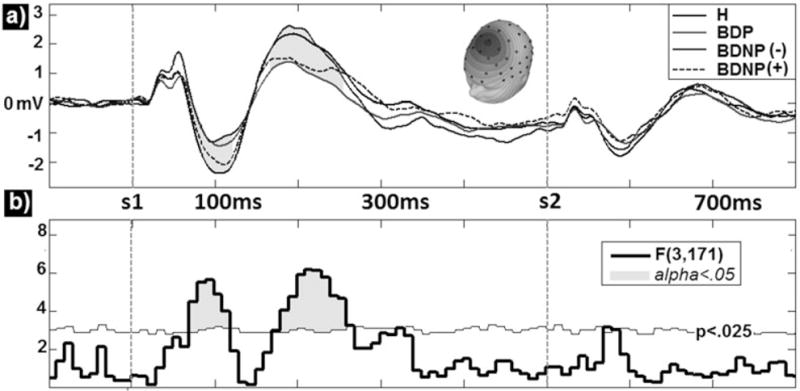
Group comparisons for principal components analysis derived event-related potentials waveforms for oddball standard stimuli (A) averaged within group yield significant effects in the N1, P2, and late N2 time ranges (shaded regions). F-values for these effects are also presented along with (B) a bootstrapped p < 0.025 probability line (thin horizontal line). Time regions reaching significance at FWalpha< 0.05 are highlighted. H = healthy comparison subjects; BDP = bipolar disorder with psychosis; BDNP(−) = bipolar disorder without psychosis without first-degree family history of psychosis; BDNP(+) = bipolar disorder without psychosis with first-degree family history of psychosis.
Table 2.
Significant main effects
| Healthy subjects (n = 70) | BDP (n = 70) | BDNP(−) (n = 15) | BDNP(+) (n = 20) | |
|---|---|---|---|---|
| PS–N1 | −1.94 (1.36) | −1.06 (1.18)a | −0.95 (0.88)b | −1.60 (1.17) |
| PS–P2 | 2.10 (1.66) | 1.07 (1.28)a | 2.23 (2.05) | 1.25 (1.65)c |
| TGT1–P3b | 3.60 (2.54) | 2.35 (1.97)b | 2.46 (1.49) | 2.34 (1.78)c |
| TGT2–N1 | −2.15 (1.17) | −1.46 (1.26)a | −0.92 (0.87)a | −1.59 (0.97) |
| TGT2–P3a | 1.69 (2.53) | 0.66 (1.88)b | 0.02 (2.44)c | 1.27 (2.81) |
| STD–N1 | −1.67 (0.92) | −1.10 (0.97)a | −0.82 (0.71)b | −1.29 (0.88) |
| STD–P2 | 1.36 (0.96) | 0.75 (0.80)a | 0.94 (1.02) | 1.13 (1.20) |
| STD–N2 | 0.16 (0.78) | −0.14 (0.63)c | −0.25 (0.75) | 0.17 (0.58) |
Values presented as mean (standard deviation). BDP = bipolar disorder with psychosis; BDNP(−) = bipolar disorder without psychosis without first-degree family history of psychosis; BDNP(+) = bipolar disorder without psychosis with first-degree family history of psychosis; PS = paired- stimulus waveform; TGT1 = target waveform 1 (parietally distributed); TGT2 = target waveform 2 (frontally distributed); STD = standard waveform.
p< 0.001 (two-tailed t-tests versus healthy subjects).
p< 0.01 (two-tailed t-tests versus healthy subjects).
p< 0.05 (two-tailed t-tests versus healthy subjects).
The PS waveforms for each group (Fig. 1A) and the omnibus F-values compared to the permutated 0.05 probability threshold (Fig. 1B) with significant time-bins shaded are displayed in Figure 1. Two time-bin clusters reached significance. The first was from 70 msec to 120 msec post S1 onset and included the N1, peaking at 95 msec [F(3,171) = 5.68, p < 0.001]. BDP and BDNP(−) groups did not differ but each had significantly lower amplitudes than H [t(138) = 4.08, p < 0.001; t(83) = 2.69, p < 0.01, respectively]. BDNP(+) did not differ from H in the N1 time window. Family history of psychosis therefore appeared to moderate N1 amplitude reductions in BDNP.
The second significant time window lasted from 180 msec to 260 msec post S1 onset and included the P2, peaking at 225 msec [F(3,171) = 6.17, p < 0.001]. BDP and BDNP(+) groups did not differ but each had lower amplitudes than H [t(138) = 4.08, p < 0.001; t(88) = 1.99, p < 0.05, respectively]. BDNP(−) had significantly stronger P2 responses than BDP [t(83) = 2.68, p < 0.01]. Importantly, BDNP(−) did not differ from H. Thus, a family history of psychosis also moderated the paired-stimuli P2 in BDNP, but in a manner opposite to N1; P2 reductions were associated with psychosis risk and not necessarily psychosis expression. This is consistent with a previous report showing that paired-stimuli P2 to S1 is equally reduced in psychotic individuals regardless of DSM diagnostic category (26).
Figure 2 depicts the TGT1 waveforms for each group (Fig. 2A) and associated omnibus F-values (Fig. 2B). A single time-bin cluster lasting from 330 msec to 400 msec after target onset (P3b range) reached significance, peaking at 365 msec [F(3,171) = 5.00, p < 0.01]. BDP [t(138) = 3.25, p < 0.01], and BDNP(+) [t(88) = 2.07, p < 0.05], had significantly smaller amplitude responses than H. All between patient group comparisons, however, were non-significant, and all bipolar subgroup waveforms are highly similar in this time range. This pattern of effects indicates that the P3b is a non-specific marker of psychopathology and is not moderated by psychosis expression or risk.
Fig. 2.
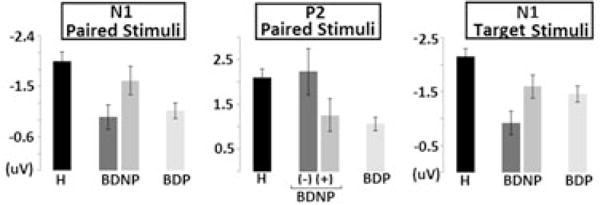
Group comparisons for principal components analysis derived event-related potentials waveforms to oddball target stimuli (A) averaged within group yield significant effects in the P3b range for component 1 (Pz maximum; dark shaded region) and in the N1 and P3a range for component 2 (FCz maximum; light shaded region). F-values for these effects are also presented along with (B) a bootstrapped p < 0.025 probability lines (thin horizontal lines). Time regions reaching significance at FWalpha< 0.05 (three consecutive bins) are highlighted. H = healthy comparison subjects; BDP = bipolar disorder with psychosis; BDNP(−) = bipolar disorder without psychosis without first-degree family history of psychosis; BDNP(+) = bipolar disorder without psychosis with first-degree family history of psychosis.
The TGT2 waveforms and F-values are depicted as thinner lines in Figures 2A and 2B. Two significant time-bin clusters emerged in the omnibus test. The first included the N1 component and lasted from 60 msec to 110 msec post-target onset, peaking at 85 msec [F(3,171) = 6.85, p < 0.001]. Only the BDP and BDNP patient groups had significantly smaller values than H: BDP [t(138) = 3.37, p < 0.001; t(83) = 3.85, p < 0.001, respectively]. Importantly, BDNP(+) had an N1 amplitude significantly larger than BDNP(−) [t(33) = 2.13, p < 0.05]. This family history moderation effect echos the patterns seen in PS-N1. The other significant cluster included the P3a component and lasted from 320 msec to 350 msec, peaking at 325 msec [F(3,171) = 3.57, p < 0.05]. BDP and BDNP(−) groups did not differ but each had smaller amplitudes than H [t(138) = 2.71, p < 0.01; t(83) = 2.33, p < 0.05, respectively]. BDNP(+) did not differ from H in the P3a window for the TGT2 component. A family history of psychosis, therefore, also moderated the P3a in BDNP in a similar manner as it did the paired-stimuli N1 although the overall effect was smaller.
The STD waveforms are depicted in Figure 3 for each group (Fig. 3A) and the associated omnibus F-values (Fig. 3B). Waveform divergences in three time-window clusters achieved between-groups significance. The first window included the N1 component and lasted from 60 msec to 110 msec post-standard onset, peaking at 85 msec [F(3,171) = 6.90, p < 0.001]. Like the PS-N1 and TGT2-N1, BDP, and BDNP(−) groups did not differ but each had lower amplitudes than H [t(138) = 3.57, p < 0.001; t(83) = 3.37, p < 0.001, respectively]. BDNP(+) did not differ from H in the N1 time window. The second window included the P2 component and lasted from 190 msec to 260 msec post-standard onset, peaking at 240 msec [F(3,171) = 4.68, p < 0.01]. Only BDP differed significantly from H with lower amplitude P2s [t(138) = 4.08, p < 0.001]; no other group comparisons reached significance. The third window was in the vicinity of the N2 component and lasted from 340 msec to 370 msec post-standard onset, peaking at 355 msec [F(3,171) = 4.07, p < 0.01]. BDP and BDNP(−) groups did not differ from each other, but BDP had significantly lower amplitude responses than H [t(138) = 2.52, p < 0.05] and BDNP(+) [t(88) = 2.02, p < 0.05]. BDNP(+) did not differ from H in this time period.
Medication effects
As expected, BDP were taking significantly more antipsychotic medications than BDNP [particularly second generation antipsychotics, (see Supplementary Table S2)]. Status for all other medication classes did not differ between patient groups (see Supplementary Table S3 for more details). When sample sizes permitted, t-tests were computed within patient groups and across all patients comparing subjects taking medication and those medication-free within a drug class (antipsychotics, lithium, anticonvulsants, antidepressants, sedatives) on each of the eight effects of interest. In all cases effects were greater than p = 0.10 uncorrected except one; BDNP(+) subjects on anticonvulsant medication had lower P3a amplitudes (mean = − 0.02uV, standard deviation = 2.30) than anticonvulsant-free BDNP(+) (2.87 uV, 2.74). This effect [t(18) = 2.50, p = 0.020] did not exist in any other patient group or in the sample as a whole, and did not survive alpha adjustment for multiple comparisons.
Clinical scores
Young Mania and Montgomery-Åsberg Depression Rating Scale scores were statistically equivalent across patient groups (Table 1), indicating equivalently moderate levels of affective symptomology. GAF scores (DSM-IV-TR Axis V) did not differ between patient groups. None of the eight main ERP effects significantly correlated with any clinical score within or across patient groups.
Linear discriminant analysis
Discriminant analyses indicated that PS-N1, PS-P2, and TGT-N1 each added unique group discrimination variance, and adequately captured the group ERP differences covered by all variables. Results are displayed as bar graphs in Figure 4. The P2 was essentially the only effect out of the eight total effects which included deviations from H for BDP and BDNP(+) but not BDNP(−), suggesting its potential utility in understanding psychosis liability. The overall group discrimination pattern for the PS-N1 and the oddball TGT-N1 were similar such that BDP and BDNP(−) were both significanctly reduced compared with H, while BDNP(+) showed absent or largely attenuated, non-significant deviations from H. This implies that these N1 reductions, though both moderated by familial psychosis history in BDNP patients and correlated at r = 0.49, each carry a degree of unique information across subjects within groups, perhaps related to differences in passive versus active listening contexts.
Fig. 4.
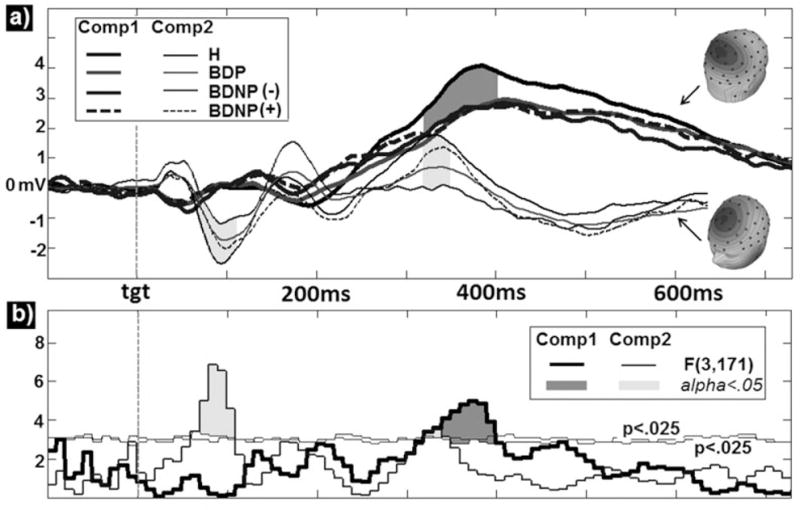
Group averages and standard errors for three main group discriminators determined in the linear discriminant analysis: N1 to paired-stimuli S1, P2 to paired-stimuli S1, and N1 to oddball target stimuli. H = healthy comparison subjects; BDP = bipolar disorder with psychosis; BDNP(−) = bipolar disorder without psychosis without first-degree family history of psychosis; BDNP(+) = bipolar disorder without psychosis with first-degree family history of psychosis.
Discussion
This study investigated whether classically reported auditory neurophysiological biomarkers of psychotic disturbance (paired-stimuli and oddball ERPs) support a unitary representation of bipolar disorder based on psychosis status. The results indicate that auditory paired-stimuli and oddball ERPs do not clearly distinguish bipolar subgroups based on psychosis expression alone (30, 36). When family history of psychosis was considered, however, a pattern emerged that might partially account for inconsistencies and null findings in previous reports. The present results suggest novel interpretations for the pathophysiological meaning of auditory ERP deviations among psychiatric disturbances generally and bipolar disorder variations specifically.
The N1 to auditory stimulus onset was reduced in BDP, replicating previous reports (26, 35). Reduced N1 has been consistently reported in psychotic patients [for a review see (24)]. Some BDNP had N1 reductions at the same levels as BDP, but, importantly, BDNP at high risk for developing psychosis (i.e., with close relatives experiencing psychosis) had normal N1s. This effect was present in both the paired-stimuli and oddball paradigms thus showing replication under three different stimulus-processing conditions in our samples. Studies of N1 amplitudes among relatives of psychosis patients have yielded conflicting results, which may be associated with variable frequencies of comorbid psychiatric conditions across relative samples (37). The present findings are consistent with this thesis, and may implicate alternative means for understanding bipolar disorder’s various etiological substrates. For instance, genes controlling expression of GABA(A) signaling proteins, which influence a critical and ubiquitous receptor in cortical neuronal assemblies playing a role in synchronizing ensembles of pyramidal cells, show association with the N1 (54). Perhaps having intact basic auditory cortical circuitry strengthens the signal/noise ratio in a psychosis-prone bipolar disorder patient’s basic sensory processing system, protecting against inaccuracies in sensory registration contributing to hallucinatory phenomena. Recent clinical neuroscience work has promoted understanding resilience to major psychiatric disturbance (40) and the genetics of individuals relieved from developing such neuropathologies despite being at high-risk thereof (41).
Both BDP and BDNP without a history of psychosis showed N1 reductions, perhaps indicating that N1 marks an indirect relationship to affective psychiatric disturbance while also being associated with psychotic auditory processing abnormalities. Alternatively, BDNP(+) might have hyperexcitable early auditory cortical responses relative to BDNP(−), perhaps indicating constitutional auditory sensory dysregulation in addition to downstream connectivity and/or signal processing deficits marked by reduced longer latency ERPs (e.g., P3b). While requiring additional work to understand its complete neuropathological significance, N1 auditory ERP have promise as specific targets for understanding bipolar disorder’s variable clinical manifestations.
Like N1, the P2 in the paired-stimuli paradigm was reduced in BDP as previously reported (26). Reductions in paired stimuli P2 in BDNP were also moderated by family psychosis history such that BDNP(+) showed equivalent P2 reductions to BDP, but BDNP(−) showed P2s at healthy levels (in constrast to the N1 effect pattern). Together with a previous study indicating that P2 has a general relationship to psychosis regardless of affective psychopathology (patients with schizophrenia and BDP have equal P2 reductions) (25), this finding extends previous knowledge to indicate that heritable factors related not to psychosis expression but to general psychosis liability are captured by P2 amplitude deviations.
The N1 and P2 demonstrated a pattern of differential effects that have not been directly hypothesized by previous models of psychosis or affective neurophysiology. In addition to occurring later than N1, the P2 shows both superior temporal as well as associative cortical source generators (55), indicating its association with more distributed cortical processing, temporal synchronization, and long range neural communication. An enhanced N1 in BDNP(+), therefore, may represent a functional compensation (not present in BDP) for an inherited cortical network disruption indexed by subsequent decreased signal propagation (reduced P2). While the P2 is commonly conceptualized with the N1 as part of an N1/P2 complex, P2 can vary independently of other auditory ERP components (23), e.g., showing attentional effects substantially different from the N1 (56). This differential relationship to attention could explain why group discriminations differed between passive (PS) and active (OB) listening paradigms for P2 but not for N1. These findings serve as viable clues in future work on the genetics of bipolar disorder specifically and psychosis generally. For example, a set of genes may influence long range connectivity and synchronization (57), correlating with P2 and, theoretically, operating in ways related to, but indeterminate of, psychosis expression. Research on N1 as a resilience factor/marker and P2 as relating to psychosis liability should involve additional quantitative genetic and/or longitudinal approaches. The novelty of this pattern is at once interesting and indicative of the need for replication.
Importantly, a most widely studied ERP, the mesial parietal lobe centered P3b, showed no group specificity, being reduced and nearly equivalent in all bipolar groups. This finding converges with numerous previous reports of equivalent or similar reductions in P3b amplitudes across different psychotic and affective diagnostic categories (35, 58), across different mood, medication, and psychosis states within bipolar disorder (36), and within unaffected family members of bipolar disorder (18). Indeed, P3b abnormalities have been described for multiple behavioral deviations (33, 59, 60), indicating that this brain response may index generalized dysfunction.
The anteriorly distributed P3a was reduced in BDP but additionally showed a relationship to family psychosis history status similar to the N1 [BDNP(+) > BDNP(−)]. Previous reports have implicated the perhaps special importance of the P3a in BDP (61), along with its closer relationship to variations in dopamine-related gene expression than the P3b (62). The P3a is believed to index an orienting responses to novel stimuli (33). A similar effect to N1 and P3a was also present for the later part of the N2 time range to STD stimuli in the oddball task. Responses to oddball STD stimuli in this time range are less commonly reported in the auditory ERP literature than N1 or P3. The N2 response, however, was reduced in both BDP and BDNP(−), but not BDNP(+), again signifying psychosis resilience among a subgroup of patients with bipolar disorder. This effect shared a similar topography and pattern of group discrimination with both N1 and P3a, but across all subjects (n = 175) the STD-N2 correlated poorly with each N1 component (r = −0.06, −0.09, and −0.03 for PS, TGT, and STD, respectively) but significantly with the P3a (r = 0.37). These effects indicate that the psychosis resilience marked by N1 may be separate from that marked by the P3a and STD-N2 components, which may mark novelty-related and/or frontally distributed cognitive processing mediated by dopaminergic mechanisms (33, 62).
Previous work has questioned whether familial (e.g., BDP or schizophrenia with first- or second-degree relatives with psychotic disorders) and sporadic (patients with no family history of psychosis) psychosis represent differentiable clinical or biological subgroups (63–65). The familial versus sporadic distinction may be related, but not equivalent, to the current analysis of psychosis risk versus expression in bipolar disorder. Our sample was not optimized to address the familial versus sporadic issue, but we specifically compared patients with bipolar disorder with familial (n = 16) versus sporadic (n = 51) psychosis (data were not certain for three BDP subjects). Only the paired stimuli N1 approached significance [t(65) = 1.76, p = 0.082], with familial BDP having smaller N1s (mean = −0.33, standard deviation = 1.11) than sporadic BDP (−0.94, 1.23). Interestingly, when compared with BDNP(+), BDP with familial psychosis had significantly smaller PS-N1s [t(34) = 2.73, p < 0.01], further indicating a complex, perhaps additive or protective, relationship of N1 to psychosis expression and risk in bipolar disorder.
The results of the current study mark a crucial step toward understanding how commonly described electrophysiological deviations relate to psychotic and affective psychopathology, and provide information on biomarkers that can be used to guide larger scale efforts to identify and interpret genetic underpinnings of psychiatric disturbance.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (R01s MH077945, MH077862, MH077851, MH078113, and MH085485).
JAS has received grant funding from Janssen; and has served as a consultant for Takeda, Pfizer, and Eli Lilly & Co. MSK has served as an ad-hoc consultant for PureTech Ventures, Eli Lilly & Co., Sunovion, Astellas Pharma, US Inc., and Merck & Co., Inc.; and has served on the advisory board for Intracellular Therapies, Inc.
Footnotes
Disclosures
JPH, LEE, JRS, GDP, CAT, GKT, and BAC do not have any commercial associations that might pose a conflict of interest in connection with this manuscript.
References
- 1.Keck PE, McElroy SL, Havens JR, et al. Psychosis in bipolar disorder: phenomenology and impact on morbidity and course of illness. Comprehensive Psychiatry. 2003;44:263–269. doi: 10.1016/S0010-440X(03)00089-0. [DOI] [PubMed] [Google Scholar]
- 2.Goodwin FK, Jamison KR. Manic-Depressive Illness: Bipolar Disorders and Recurrent Depression. Vol. 1. Oxford University Press; 2007. p. 1262. [Google Scholar]
- 3.Goes FS, Sanders LLO, Potash JB. The genetics of psychotic bipolar disorder. Current Psychiatry Reports. 2008;10:178–189. doi: 10.1007/s11920-008-0030-5. [DOI] [PubMed] [Google Scholar]
- 4.Potash JB, Toolan J, Steele J, et al. The bipolar disorder phenome database: a resource for genetic studies. Am J Psychiatry. 2007;164:1229–1237. doi: 10.1176/appi.ajp.2007.06122045. [DOI] [PubMed] [Google Scholar]
- 5.O’Mahony E, Corvin A, O’Connell R, et al. Sibling pairs with affective disorders: resemblance of demographic and clinical features. Psychological Med. 2002;32:55–61. doi: 10.1017/s0033291701004986. [DOI] [PubMed] [Google Scholar]
- 6.Thaker GK. Neurophysiological endophenotypes across bipolar and schizophrenia psychosis. Schizophr Bull. 2008;34:760–773. doi: 10.1093/schbul/sbn049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearlson GD, Wong DF, Tune LE, et al. In vivo D2 dopamine receptor density in psychotic and nonpsychotic patients with bipolar disorder. Arch Gen Psychiatry. 1995;52:47471–7. doi: 10.1001/archpsyc.1995.03950180057008. [DOI] [PubMed] [Google Scholar]
- 8.Kendler KS, Gruenberg AM, Tsuang MT. Psychiatric illness in first-degree relatives of schizophrenic and surgical control patients. A family study using DSM-III criteria. Arch Gen Psychiatry. 1985;42:770–779. doi: 10.1001/archpsyc.1985.01790310032004. [DOI] [PubMed] [Google Scholar]
- 9.Cardno AG, Rijsdijk FV, Sham PC, Murray RM, McGuffin P. A twin study of genetic relationships between psychotic symptoms. Am J Psychiatry. 2002;159:539–545. doi: 10.1176/appi.ajp.159.4.539. [DOI] [PubMed] [Google Scholar]
- 10.Emsell L, McDonald C. The structural neuroimaging of bipolar disorder. Int Rev Psychiatry. 2009;21:297–313. doi: 10.1080/09540260902962081. [DOI] [PubMed] [Google Scholar]
- 11.Glahn DC, Bearden CE, Barguil M, et al. The neurocognitive signature of psychotic bipolar disorder. Biol Psychiatry. 2007;62:910–916. doi: 10.1016/j.biopsych.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Weiser M, Reichenberg A, Kravitz E, et al. Subtle cognitive dysfunction in nonaffected siblings of individuals affected by nonpsychotic disorders. Biol Psychiatry. 2008;63:602–608. doi: 10.1016/j.biopsych.2007.05.029. [DOI] [PubMed] [Google Scholar]
- 13.Strasser HC, Lilyestrom J, Ashby ER, et al. Hippocampal and ventricular volumes in psychotic and nonpsychotic bipolar patients compared with schizophrenia patients and community control subjects: a pilot study. Biol Psychiatry. 2005;57:633–639. doi: 10.1016/j.biopsych.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 14.Byne W, Tatusov A, Yiannoulos G, Vong GS, Marcus S. Effects of mental illness and aging in two thalamic nuclei. Schizophr Res. 2008;106:172–181. doi: 10.1016/j.schres.2008.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gur RE, Keshavan MS, Lawrie SM. Deconstructing psychosis with human brain imaging. Schizophr Bull. 2007;33:921–931. doi: 10.1093/schbul/sbm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Javadapour A, Malhi GS, Ivanovski B, Chen X, Wen W, Sachdev P. Hippocampal volumes in adults with bipolar disorder. J Neuropsychiatry Clin Neurosci. 2010;22:55–62. doi: 10.1176/jnp.2010.22.1.55. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi T, Malhi GS, Wood SJ, et al. Gray matter reduction of the superior temporal gyrus in patients with established bipolar I disorder. J Affect Disord. 2010;123:276–282. doi: 10.1016/j.jad.2009.08.022. [DOI] [PubMed] [Google Scholar]
- 18.Hall M-H, Schulze K, Rijsdijk F, et al. Are auditory P300 and duration MMN heritable and putative endophenotypes of psychotic bipolar disorder? A Maudsley Bipolar Twin and Family Study. Psychological Med. 2009;39:1277–1287. doi: 10.1017/S0033291709005261. [DOI] [PubMed] [Google Scholar]
- 19.Schulze KK, Hall M-H, McDonald C, et al. P50 auditory evoked potential suppression in bipolar disorder patients with psychotic features and their unaffected relatives. Biol Psychiatry. 2007;62:121–128. doi: 10.1016/j.biopsych.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Hall M-H, Taylor G, Salisbury DF, Levy DL. Sensory gating event-related potentials and oscillations in schizophrenia patients and their unaffected relatives. Schizophr Bull. 2011;37:1187–1199. doi: 10.1093/schbul/sbq027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yvert B, Crouzeix A, Bertrand O, Seither-Preisler A, Pantev C. Multiple supratemporal sources of magnetic and electric auditory evoked middle latency components in humans. Cerebral Cortex. 2001;11:411–423. doi: 10.1093/cercor/11.5.411. [DOI] [PubMed] [Google Scholar]
- 22.Yvert B, Fischer C, Bertrand O, Pernier J. Localization of human supratemporal auditory areas from intracerebral auditory evoked potentials using distributed source models. Neuroimage. 2005;28:140–153. doi: 10.1016/j.neuroimage.2005.05.056. [DOI] [PubMed] [Google Scholar]
- 23.Crowley KE, Colrain IM. A review of the evidence for P2 being an independent component process: age, sleep and modality. Clin Neurophysiology. 2004;115:732–744. doi: 10.1016/j.clinph.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 24.Hillyard SA, Hink RF, Schwent VL, Picton TW. Electrical signs of selective attention in the human brain. Science. 1973;182:177–180. doi: 10.1126/science.182.4108.177. [DOI] [PubMed] [Google Scholar]
- 25.Rosburg T, Boutros NN, Ford JM. Reduced auditory evoked potential component N100 in schizophrenia—a critical review. Psychiatry Res. 2008;161:259–274. doi: 10.1016/j.psychres.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 26.Hamm JP, Ethridge LE, Shapiro JR, et al. Spatiotemporal and frequency domain analysis of auditory paired stimuli processing in schizophrenia and bipolar disorder with psychosis. Psychophysiology. 2012;49:522–530. doi: 10.1111/j.1469-8986.2011.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clementz BA, Dzau JR, Blumenfeld LD, Matthews S, Kissler J. Ear of stimulation determines schizophrenia-normal brain activity differences in an auditory paired-stimuli paradigm. Neurosci. 2003;18:2853–2858. doi: 10.1111/j.1460-9568.2003.03027.x. [DOI] [PubMed] [Google Scholar]
- 28.Sánchez-Morla EM, García-Jiménez MA, Barabash A, et al. P50 sensory gating deficit is a common marker of vulnerability to bipolar disorder and schizophrenia. Acta Psychiatrica Scandinavica. 2008;117:313–318. doi: 10.1111/j.1600-0447.2007.01141.x. [DOI] [PubMed] [Google Scholar]
- 29.Olincy A, Martin L. Diminished suppression of the P50 auditory evoked potential in bipolar disorder subjects with a history of psychosis. Am J Psychiatry. 2005;162:43–49. doi: 10.1176/appi.ajp.162.1.43. [DOI] [PubMed] [Google Scholar]
- 30.Carroll CA, Kieffaber PD, Vohs JL, O’Donnell BF, Shekhar A, Hetrick WP. Contributions of spectral frequency analyses to the study of P50 ERP amplitude and suppression in bipolar disorder with or without a history of psychosis. Bipolar Disord. 2008;10:776–787. doi: 10.1111/j.1399-5618.2008.00622.x. [DOI] [PubMed] [Google Scholar]
- 31.Patterson JV, Sandman CA, Ring A, Jin Y, Bunney WE. An initial report of a new biological marker for bipolar disorder: P85 evoked brain potential. Bipolar Disord. 2009;11:596–609. doi: 10.1111/j.1399-5618.2009.00734.x. [DOI] [PubMed] [Google Scholar]
- 32.Cabranes JA, Ancin I, Santos JL, et al. P50 sensory gating is a trait marker of the bipolar spectrum. Eur Neuropsychopharmacol. 2013;23:721–727. doi: 10.1016/j.euroneuro.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 33.Linden DEJ. The p300: where in the brain is it produced and what does it tell us? Neuroscientist. 2005;11:563–576. doi: 10.1177/1073858405280524. [DOI] [PubMed] [Google Scholar]
- 34.Mulert C, Jager L, Schmitt R, et al. Integration of fMRI and simultaneous EEG: towards a comprehensive understanding of localization and time-course of brain activity in target detection. Neuroimage. 2004;22:83–94. doi: 10.1016/j.neuroimage.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 35.Ethridge LE, Hamm JP, Shapiro JR, et al. Neural activations during auditory oddball processing discriminating schizophrenia and psychotic bipolar disorder. Biol Psychiatry. 2012;72:766–774. doi: 10.1016/j.biopsych.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fridberg DJ, Hetrick WP, Brenner CA, et al. Relationships between auditory event-related potentials and mood state, medication, and comorbid psychiatric illness in patients with bipolar disorder. Bipolar Disord. 2009;11:857–866. doi: 10.1111/j.1399-5618.2009.00758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turetsky BI, Greenwood TA, Olincy A, et al. Abnormal auditory N100 amplitude: a heritable endophenotype in first-degree relatives of schizophrenia probands. Biol Psychiatry. 2008;64:1051–1059. doi: 10.1016/j.biopsych.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahveninen J, Jääskeläinen IP, Osipova D, et al. Inherited auditory-cortical dysfunction in twin pairs discordant for schizophrenia. Biol Psychiatry. 2006;60:612–620. doi: 10.1016/j.biopsych.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 39.Van Beijsterveldt CE, Van Baal GC, Molenaar PC, Boomsma DI, De Geus EJ. Stability of genetic and environmental influences on P300 amplitude: a longitudinal study in adolescent twins. Behav Geneti. 2001;31:533–543. doi: 10.1023/a:1013389226795. [DOI] [PubMed] [Google Scholar]
- 40.Frangou S. Brain structural and functional correlates of resilience to bipolar disorder. Frontiers Hum Neurosci. 2011;5:184. doi: 10.3389/fnhum.2011.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 42.Clementz BA, Blumenfeld LD. Multichannel electroencephalographic assessment of auditory evoked response suppression in schizophrenia. Exper Brain Rese. 2001;139:377–390. doi: 10.1007/s002210100744. [DOI] [PubMed] [Google Scholar]
- 43.Lançon C, Auquier P, Nayt G, Reine G. Stability of the five-factor structure of the Positive and Negative Syndrome Scale (PANSS) Schizophr Res. 2000;42:231–239. doi: 10.1016/s0920-9964(99)00129-2. [DOI] [PubMed] [Google Scholar]
- 44.Young RC, Biggs JT, Ziegler VE, Meyer DA. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133:429–435. doi: 10.1192/bjp.133.5.429. [DOI] [PubMed] [Google Scholar]
- 45.Montgomery SA, Åsberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–389. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- 46.Andreasen NC, Endicott J, Spitzer RL, Winokur G. The Family History Method using diagnostic criteria: reliability and validity. Arch Gen Psychiatry. 1977;34:1229–1235. doi: 10.1001/archpsyc.1977.01770220111013. [DOI] [PubMed] [Google Scholar]
- 47.Tamminga CA, Iveleva EI, Keshavan MS, et al. Clinical phenotypes of psychosis in the Bipolar and Schizophrenia Network on Intermediate Phenotpes (B-SNIP) Am J Psychiatry. 2013 doi: 10.1176/appi.ajp.2013.12101339. in press. [DOI] [PubMed] [Google Scholar]
- 48.Delorme A, Makeig S. EEGLAB: an open source toolbox for analysis of single-trial EEG dynamics including independent component analysis. J Neurosci Meth. 2004;134:9–21. doi: 10.1016/j.jneumeth.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 49.Dien J, Khoe W, Mangun GR. Evaluation of PCA and ICA of simulated ERPs: Promax vs. Infomax rotations. Human Brain Mapping. 2007;28:742–763. doi: 10.1002/hbm.20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cattell RB. The Scree Test for the number of factors. Multivariate behavioral research. Psychology Press. 1966;1:245–276. doi: 10.1207/s15327906mbr0102_10. [DOI] [PubMed] [Google Scholar]
- 51.Forman SD, Cohen JD, Fitzgerald M, Eddy WF, Mintun MA, Noll DC. Improved assessment of significant activation in functional magnetic resonance imaging (fMRI): use of a cluster-size threshold. Magnetic Resonance Med. 1995;33:636–647. doi: 10.1002/mrm.1910330508. [DOI] [PubMed] [Google Scholar]
- 52.Cox LA., Jr Reassessing benzene risks using internal doses and Monte-Carlo uncertainty analysis. Environ Health Perspect. 1996;104(Suppl. 6):1413–1429. doi: 10.1289/ehp.961041413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mardia K, Kent J, Bibby J. Multivariate Analysis. Academic Press; 1980. [Google Scholar]
- 54.Porjesz B, Begleiter H, Wang K, et al. Linkage and linkage disequilibrium mapping of ERP and EEG phenotypes. Biol Psychology. 2002;61:229–248. doi: 10.1016/s0301-0511(02)00060-1. [DOI] [PubMed] [Google Scholar]
- 55.Godey B, Schwartz D, De Graaf JB, Chauvel P, Liégeois-Chauvel C. Neuromagnetic source localization of auditory evoked fields and intracerebral evoked potentials: a comparison of data in the same patients. Clin Neurophysiology. 2001;112:1850–1859. doi: 10.1016/s1388-2457(01)00636-8. [DOI] [PubMed] [Google Scholar]
- 56.Näätänen R. The role of attention in auditory information processing as revealed by event- related potentials and other brain measures of cognitive function. Behav Brain Sci. 1990;13:201–233. [Google Scholar]
- 57.Linke J, Witt SH, King AV, et al. Genome-wide supported risk variant for bipolar disorder alters anatomical connectivity in the human brain. Neuroimage. 2012;59:3288–3296. doi: 10.1016/j.neuroimage.2011.10.083. [DOI] [PubMed] [Google Scholar]
- 58.Blackwood DH, Fordyce A, Walker MT, St Clair DM, Porteous DJ, Muir WJ. Schizophrenia and affective disorders—cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet. 2001;69:428–433. doi: 10.1086/321969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bruder GE. P300 findings for depressive and anxiety disorders. Ann NY Acad Sci. 1992;658:205–222. doi: 10.1111/j.1749-6632.1992.tb22846.x. [DOI] [PubMed] [Google Scholar]
- 60.Johannesen JK, O’Donnell BF, Shekhar A, McGrew JH, Hetrick WP. Diagnostic specificity of neurophysiological endophenotypes in schizophrenia and bipolar disorder. Schizophr Bull. 2012 doi: 10.1093/schbul/sbs093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salisbury DF, Shenton ME, McCarley RW. P300 topography differs in schizophrenia and manic psychosis. Biol Psychiatry. 1999;45:98–106. doi: 10.1016/s0006-3223(98)00208-x. [DOI] [PubMed] [Google Scholar]
- 62.Marco-Pallarés J, Nager W, Kramer UM, et al. Neurophysiological markers of novelty processing are modulated by COMT and DRD4 genotypes. Neuroimage. 2010;53:962–969. doi: 10.1016/j.neuroimage.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 63.Roy MA, Crowe RR. Validity of the familial and sporadic subtypes of schizophrenia. Am J Psychiatry. 1994;151:805–814. doi: 10.1176/ajp.151.6.805. [DOI] [PubMed] [Google Scholar]
- 64.Frommann I, Brinkmeyer J, Ruhrmann S, et al. Auditory P300 in individuals clinically at risk for psychosis. Int J Psychophysiology. 2008;70:192–205. doi: 10.1016/j.ijpsycho.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 65.Malaspina D, Friedman JH, Kaufmann C, et al. Psychobiological heterogeneity of familial and sporadic schizophrenia. Biol Psychiatry. 1998;43:489–496. doi: 10.1016/s0006-3223(97)00527-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.