

Abstract
Objective
Lipoprotein(a) is causally associated with calcific aortic valve disease (CAVD). Lipoprotein(a) carries pro-inflammatory and pro-calcific oxidized phospholipids (OxPL). We tested whether the CAVD risk is mediated by the content of OxPL on lipoprotein(a).
Approach and Results
A case-control study was performed within the Copenhagen General Population Study (N=87980), including 725 CAVD cases (1977–2013) and 1413 controls free of cardiovascular disease. OxPL carried by apolipoprotein B-100 (OxPL-apoB) or apolipoprotein(a) (OxPL-apo(a)) containing lipoproteins, lipoprotein(a) levels, LPA kringle IV type 2 (KIV-2) repeat and rs10455872 genetic variants were measured. OxPL-apoB and OxPL-apo(a) levels correlated with lipoprotein(a) levels among cases (r=0.75 and r=0.95, both p<0.001) and controls (r=0.65 and r=0.93, both p<0.001). OxPL-apoB levels associated with risk of CAVS with odds ratios of 1.2(95%CI:1.0–1.6) for 34th–66th percentile levels, 1.6(1.2–2.1) for 67th–90th percentile levels, 2.0(1.3–3.0) for 91st–95th percentile levels, and 3.4(2.1–5.5) for levels >95th percentile, versus levels <34th percentile (trend, p<0.001). Corresponding odds ratios for OxPL-apo(a) were 1.2(1.0–1.5), 1.2(0.9–1.6), 2.1(1.4–3.1), and 2.9(1.9–4.5) (trend, p<0.001), and were similar for lipoprotein(a). LPA genotypes associated with OxPL-apoB, OxPL-apo(a), and lipoprotein(a) levels and explained 34%, 46%, and 39%, respectively, of the total variation in levels. LPA genotypes associated with risk of AVS; a doubling in genetically determined OxPL-apoB, OxPL-apo(a), and lipoprotein(a) levels associated with odds ratio of CAVD of 1.18(1.10–1.27), 1.09(1.05–1.13), and 1.09(1.05–1.14), respectively, comparable to the corresponding observational estimates of 1.27(1.16–1.39), 1.13(1.08–1.18), and 1.11(1.06–1.17).
Conclusions
OxPL-apoB and OxPL-apo(a) are novel genetic and potentially causal risk factors for CAVD and may explain the association of lipoprotein(a) with CAVD.
Keywords: lipoprotein(a), oxidized phospholipids, aortic valve disease
Graphical abstract
INTRODUCTION
Recent data, encompassing genome wide association and mendelian randomization studies, have strongly implicated genetic variants in the LPA gene that are associated with elevated lipoprotein(a) plasma levels as risk factors for calcific aortic valve disease (CAVD)1–3. Over the last decade, a large number of studies have shown that lipoprotein(a) is the preferential lipoprotein carrier of phosphocholine (PC) containing oxidized phospholipids (OxPL) and that some of the clinical risk in mediating cardiovascular disease may be due to its content of OxPL4–8. This naturally leads to the hypothesis that the risk of lipoprotein(a) in mediating CAVD may be due to its content of OxPL.
A variety of pathways may exist in mediating CAVD, including inflammation, oxidized lipids, osteoprogenitor cells, osteoblasts, osteoclasts, as well as related proteins and regulatory factors9,10. In particular, PC containing OxPL, such as the products of oxidized PAPC (1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine) have been suggested to play an important role in CAVD11. Although less well studied in vivo, OxPL have been shown in vitro to upregulate alkaline phosphatase activity and induce differentiation of calcifying vascular cells12, suggesting that they are involved in aortic valve calcification13. Many of these phosphocholine containing OxPL are detected by the OxPL-apoB assay, i.e. OxPL measured on apoB-100 lipoproteins using the murine monoclonal antibody E0614. OxPL-apoB detects OxPL on all apoB-100-containing lipoproteins including lipoprotein(a), and we have previously shown most (85–90%) of the OxPL in this assay format reflect OxPL on lipoprotein(a)4,5,14. Recently, a study using this assay and conducted in patients with pre-existing CAVD have demonstrated that patients with elevated OxPL-apoB, OxPL on apolipoprotein(a) (OxPL-apo(a)), and lipoprotein(a) levels have much faster progression of CAVD and more frequent need for aortic valve replacement15.
In the present case-control study, we tested the hypothesis that observationally as well as genetically elevated OxPL-apoB and OxPL-apo(a) are associated with increased risk of CAVD. We used two LPA genetic variants, the kringle IV type 2 repeat polymorphism (KIV-2) determining the number of apolipoprotein(a) kringle structures and the rs10455872 intron single nucleotide polymorphism (SNP), partly tracking the KIV-2 genotype, both associated with CAVD2,16, to conduct Mendelian randomization analyses17 testing whether genetically elevated OxPL-apoB or OxPL-apo(a) associate with increased risk of CAVD indicating causality.
MATERIAL AND METHODS
A case-control study was designed from among 87980 participants in the Copenhagen General Population Study (CGPS), a general population study initiated in 200318. For the present study, we included all CGPS participants diagnosed with CAVD from 1977 until 2013, and additionally for each CAVD case participant, two matched control participants free of cardiovascular disease, dependent on available blood samples (N=2138). The detailed Materials and Methods section is available in the online-only Supplemental Material.
RESULTS
Baseline characteristics of the study participants
Baseline characteristics of the 2138 participants selected from 87980 CGPS participants and stratified by CAVD case-control status and are shown in Table 1 (and stratified by LPA genotypes shown in Supplemental Table I). Patients with CAVD had more diabetes, lower levels of total cholesterol (and were more often on lipid lowering therapy), higher body mass index, higher levels of high-sensitivity C-reactive protein (hs-CRP), higher levels of lipoprotein(a), OxPL-apoB, and OxPL-apo(a), smaller numbers of LPA KIV-2 repeats, and higher prevalence of LPA rs10455872. Overall, 25% and 24% of cases had OxPL-apoB levels ≥5 nmol/L and lipoprotein(a) levels ≥50 mg/dL, with corresponding percentages in controls of 15% and 13%.
Table 1.
Baseline characteristics of participants from the Copenhagen General Population Study.
| Participants with CAVD | Participants free of cardiovascular disease | P | Total Population | |
|---|---|---|---|---|
| No. individuals | 725 | 1413 | 87980* | |
| Women, % | 37 | 37 | matched | 55 |
| Age, years | 74(67–80) | 74(67–79) | matched | 58(48–67) |
| Total cholesterol, mmol/L | 5.4(4.6–6.2) | 5.7(5.0–6.3) | <0.001 | 5.6(4.9–6.3) |
| HDL cholesterol, mmol/L | 1.6(1.2–2.0) | 1.6(1.3–2.0) | 0.11 | 1.6(1.2–1.9) |
| Systolic BP, mmHg | 146(130–160) | 147(133–162) | 0.39 | 136(123–150) |
| BMI, kg/m2 | 27(24–30) | 26(24–28) | <0.001 | 26(23–29) |
| eGFR, mL/min | 69(56–80) | 69(60–80) | 0.05 | 80(69–91) |
| Hs–CRP, mg/L | 2.0(1.2–3.9) | 1.6(1.1–3.0) | <0.001 | 1.4(1.0–2.3) |
| Smoking, % | 19 | 18 | 0.54 | 19 |
| Diabetes, % | 13 | 6 | <0.001 | 4 |
| Lipid lowering therapy, % | 40 | 12 | <0.001 | 12 |
| Lipoprotein(a), mg/dL | 12(4–48) | 8(4–24) | <0.001 | 12(6–31) |
| OxPL-apoB, nmol/L | 2.6(1.5–4.9) | 2.1(1.3–3.5) | <0.001 | NA |
| OxPL-apo(a), nmol/L | 5.0(1.7–29.7) | 3.5(1.4–12.0) | <0.001 | NA |
| LPA rs10455872, % carriers | 22 | 14 | <0.001 | 14 |
| LPA KIV-2, No. repeats in both alleles combined | 33(28–39) | 34(29–39) | 0.04 | 34(29–39) |
Continuous covariates are reported as median (interquartile range). P values were obtained from Kruskall-Wallis tests for continous variables and from chi-square tests for categorical values. Abbreviations: HDL, high density lipoprotein; BP, blood pressure; BMI, body mass index; eGFR, estimated glomerular filtration rate; hs-CRP, high-sensitivity C-reactive protein; OxPL-apoB, oxidized phospholipids bound to apolipoprotein B; OxPL-apo(a), oxidized phospholipids bound to apolipoprotein(a); KIV-2, kringle IV type 2.
Number of participants for individual covariates may vary dependent on availability of covariates.
Association of OxPL-apoB and OxPL-apo(a) with lipoprotein(a) levels
The concentration distributions for OxPL-apoB (and OxPL-apo(a)) and lipoprotein(a) levels were similarly skewed in both controls and cases (Figure 1 and Supplemental Figure I). For both controls and cases, OxPL-apoB and lipoprotein(a) levels were highly correlated with correlation coefficients of 0.65 (p<0.001) and 0.75 (p<0.001) (Figure 2). OxPL-apo(a) and lipoprotein(a) levels were likewise highly correlated (Supplemental Figure II), with corresponding correlation coefficients of 0.93 (p<0.001) and 0.95 (p<0.001). Figures 1 and 2 and Supplemental Figures I and II thus demonstrate the close relationship between lipoprotein(a) measurements and measurements of OxPL-apoB and OxPL-apo(a).
Figure 1.
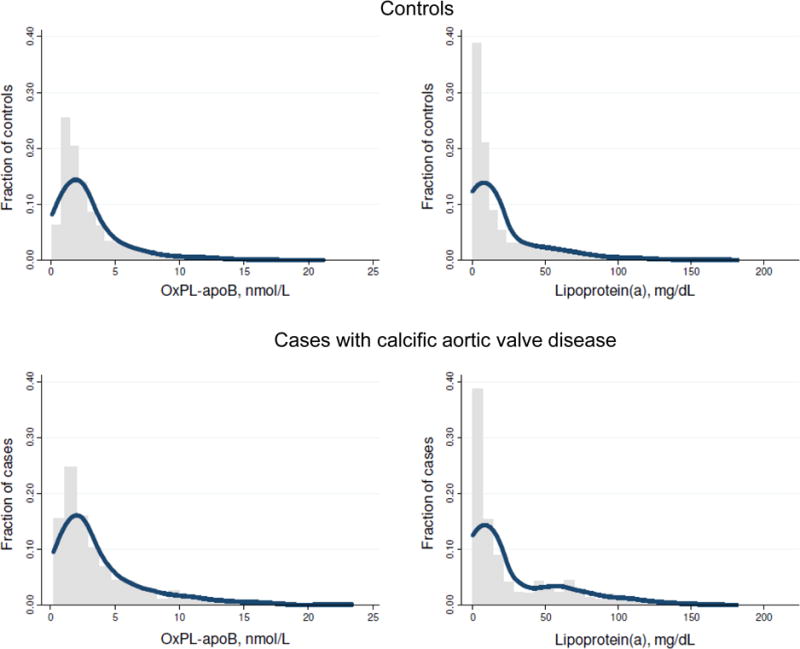
Concentration distributions of OxPL-apoB and lipoprotein(a) in controls and cases. Note, one lipoprotein(a) measurement of 327 mg/dL among cases was not included in the depicted lipoprotein(a) data (lower right panel).
Figure 2.
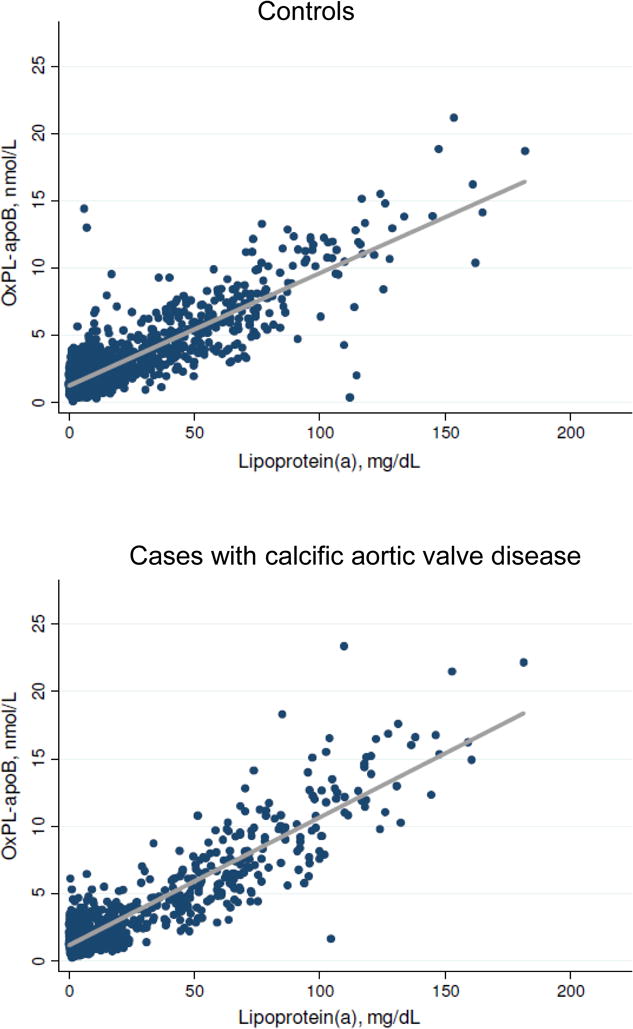
Association of OxPL-apoB with lipoprotein(a) levels in controls and cases. The best fit linear regression line is shown in grey. One lipoprotein(a) measurement of 327 mg/dL among cases was not included in the depicted data (lower panel); the OxPL-apoB value in this individual was 22.0 nmol/L.
Risk of CAVD as a function of OxPL-apoB and lipoprotein(a) levels
A stepwise higher risk of CAVD for progressively higher OxPL-apoB and lipoprotein(a) levels (Figure 3) was observed. OxPL-apoB levels ranged from 0 to 23 nmol/L; a 1 nmol/L increase associated with a multivariable adjusted odds ratio for CAVD of 1.10(1.07–1.14). Lp(a) levels ranged from 0 to 327 mg/dL; a 10 mg/dL increase associated with a multivariable adjusted odds ratio for CAVD of 1.10(1.06–1.13). OxPL-apoB levels were associated with risk of CAVD with multivariable adjusted odds ratios of 1.2(95%CI:1.0–1.6) for 34th–66th percentile levels, 1.6(1.2–2.1) for 67th–90th percentile levels, 2.0(1.3–3.0) for 91st–95th percentile levels, and 3.4(2.1–5.5) for levels >95th percentile, versus levels <34th percentile (trend, p<0.001). Corresponding odds ratios for OxPL-apo(a) were 1.2(1.0–1.5), 1.2(0.9–1.6), 2.1(1.4–3.1), and 2.9(1.9–4.5) (trend, p<0.001) (Supplemental Figure III), and for lipoprotein(a) levels 1.1(0.9–1.4), 1.2(0.9–1.6), 2.0(1.3–3.0), and 3.5(2.2–5.6) (trend, p<0.001). Overall, results were similar in age and sex adjusted and in multivariable adjusted analyses. Figure 3 and Supplemental Figure III thus demonstrate the similar and stepwise associations of increasing levels of lipoprotein(a) and OxPL-apoB and OxPL-apo(a) with increasing risk of CAVD. Upon additional adjustment for lipid lowering therapy, risk estimates for lipoprotein(a), OxPL-apoB, and OxPL-apo(a) remained significant although they were somewhat attenuated (Supplemental Figures IV and V) likely due to reverse causation, as case status is highly associated with prescription of lipid lowering therapy in this cross-sectional study. Upon additional adjustment for lipoprotein(a) levels, risk estimates for OxPL-apoB measurements lost statistical significance (trend, p= 0.14), and vice versa (trend, p=0.34). In the present study, we found no association of elevated LDL cholesterol with increased risk of CAVD (Supplemental Figure VI).
Figure 3.
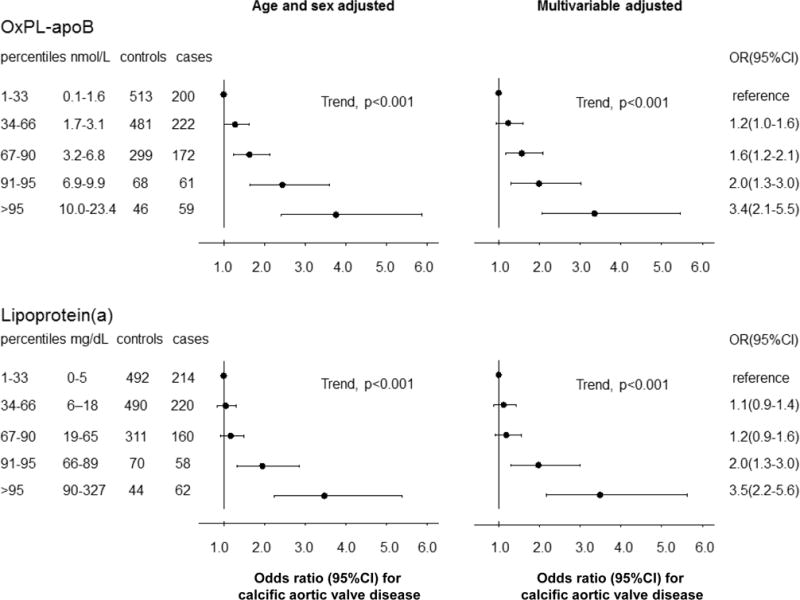
Risk of calcific aortic valve disease as a function of OxPL-apoB or lipoprotein(a) levels. Analyses were adjusted for age and sex or multivariable adjusted. Analyses excluded cases without matched controls, and vice versa (N=17). Abbreviations: OR, odds ratio; CI, confidence interval.
Association of LPA genotypes with OxPL-apoB and lipoprotein(a) levels
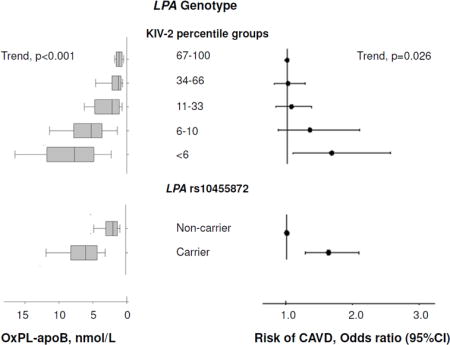
Low number of LPA KIV-2 repeats and minor allele carrier status for LPA rs10455872 were associated with high levels of OxPL-apoB (Figure 4, panel A, both p<0.001) and OxPL-apo(a) (Supplemental Figure VII, both p<0.001), and with elevated lipoprotein(a) levels (Figure 4, panel B, both p<0.001), the latter finding in accordance with previous results2. Figure 4 and Supplemental Figure VII thus demonstrate that not only lipoprotein(a) plasma levels but also measurements of OxPL-apoB and OxPL-apo(a) are at least partly determined by common variation in the LPA gene. Upon analysis of variance, the LPA KIV-2 genotype explained 24% and 35% of the variation in OxPL-apoB and OxPL-apo(a) levels, and 29% of the variation in lipoprotein(a) levels. For the LPA rs10453798 genotype, the corresponding percentages were 26% and 34%, respectively, and 30%. Combined, the two LPA genotypes explained 34%, 46%, and 39% of the variation in OxPL-apoB, OxPL-apo(a), and lipoprotein(a) levels, respectively.
Figure 4.
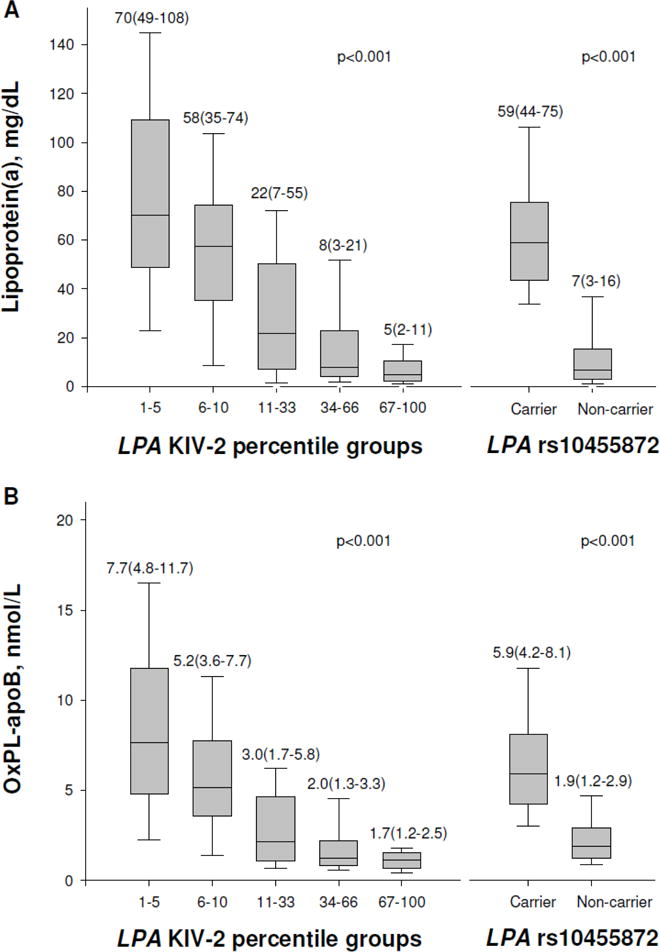
OxPL-apoB and lipoprotein(a) levels as a function of LPA genotypes. Boxes show median and interquartile range (also given in numbers) and error bars depict the 10th and 90th percentiles.
LPA genotypes and risk of CAVD
Consistent with previous results2,16, a low number of KIV-2 repeats and minor allele carrier status for rs10455872 was associated with higher risk of CAVD (Supplemental Figure VIII). On instrumental variable analysis, a doubling in genetically determined OxPL-apoB, OxPL-apo(a), and lipoprotein(a) levels were associated with odds ratios of CAVD of 1.18(1.10–1.27), 1.09(1.05–1.13), and 1.09(1.05–1.14), respectively, comparable to the corresponding observational estimates of 1.27(1.16–1.39), 1.13(1.08–1.18), and 1.11(1.06–1.17) for a doubling in OxPL-apoB, OxPL-apo(a), and lipoprotein(a) plasma levels (Figure 5, Supplemental Figure IX). Figure 5 and Supplemental Figure IX thus demonstrate that the higher levels of lipoprotein(a) and OxPL-apoB and OxPL-apo(a), explained by LPA genotype, associate with increased risk of CAVD, like elevations in plasma lipoprotein(a), OxPL-apoB, and OxPL-apo(a). These findings are consistent with a causal association of elevated lipoprotein(a), OxPL-apoB and OxPL-apo(a) levels with increased CAVD risk.
Figure 5.
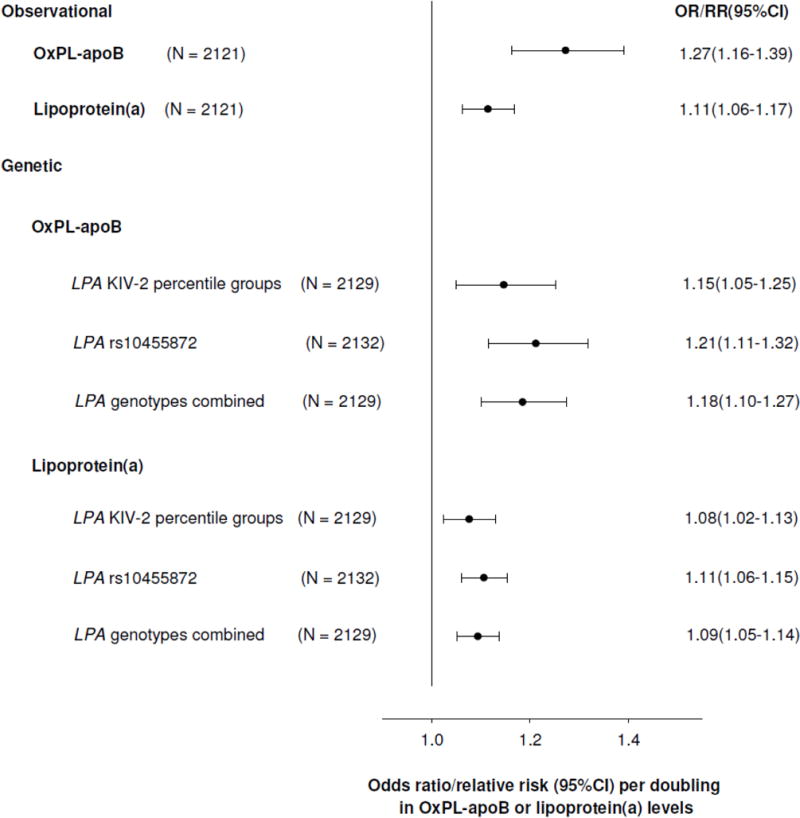
Risk of calcific aortic valve disease per doubling in OxPL-apoB or lipoprotein(a) levels in observational and genetic (instrumental variable) analyses. Abbreviations: OR, odds ratio; CI, confidence interval; RR, relative risk.
DISCUSSION
The present case-control study conducted within the CGPS demonstrates an observational and genetic association of OxPL-apoB and OxPL-apo(a) with risk of developing CAVD. Notably, this effect was independent of all other measured risk factors for CAVD, except for lipoprotein(a). Indeed, levels of OxPL-apoB, OxPL-apo(a), and lipoprotein(a) were closely correlated and could explain the effect in a similar fashion. This suggests that the risk of OxPL in mediating CAVD is mechanistically linked through its presence on lipoprotein(a). Further, LPA genotypes explained similar proportions of the total variation in plasma OxPL-apoB, OxPL-apo(a), and lipoprotein(a) levels, a novel finding, and instrumental variable analyses demonstrated higher risk of CAVD as a function of genetically higher levels. Taken together, these observations identify OxPL on lipoprotein(a) as a likely causal and genetic risk factor that may explain the association of lipoprotein(a) with CAVD.
In the present study, and despite some differences in the assays, the OxPL-apoB and OxPL-apo(a) assays generally demonstrated similar correlations with lipoprotein(a) in both cases and controls and provided similar risk prediction. Notably, both assays rely on the same monoclonal antibody to detect OxPL, and this antibody preferentially detects the PC headgroup of specific OxPL covalently bound to apolipoprotein(a) influenced by the lysine binding site of KIV-105. The close correlation of OxPL-apoB and OxPL-apo(a) with lipoprotein(a) measurements in both cases and controls reflects that lipoprotein(a) is a carrier of OxPL, while the higher risk of CAVD found in cases likely reflects the higher levels of OxPL-containing lipoprotein(a) in cases versus controls.
CAVD and atherosclerosis have many common mechanisms, although these are not entirely overlapping, as only approximately 50% of patients with CAVD have concomitant obstructive coronary artery disease11. Data from in vitro, animal, and large genetic association studies have provided ample support for proatherogenic and prostenotic effects of elevated lipoprotein(a) levels, and possibly also for a prothrombotic effect at extreme levels19–24. Furthermore, a large body of evidence suggests that a notable proportion of the atherogenicity of lipoprotein(a) may be mediated through its content of OxPL7,14,25–27. A potential mechanism defining the ability of lipoprotein(a) and/or OxPL to induce CAVD includes the ability of lipoprotein(a) in binding to exposed or denuded valve surfaces through its potent lysine binding site28. In patients with elevated lipoprotein(a) levels enriched in OxPL, lipoprotein(a) may attach tightly to exposed valve surfaces, where it may then induce chronic inflammation and calcification of valvular cells through its associated OxPL leading to the progression of CAVD. Indeed, pathological studies of vulnerable human coronary and carotid plaques8 and explanted human valves have demonstrated an increased content of lipoprotein(a)29 and OxPL8,30 and OxLDL31,32. However, elevated lipoprotein(a) levels appear not to be causally associated with increased low-grade inflammation as measured through C-reactive protein levels16, and in the present study, adjustment for hs-CRP levels did not attenuate risk estimates. Nonetheless, very recent data indicate that elevated lipoprotein(a) levels are indeed associated with increased arterial inflammation and enhanced peripheral blood mononuclear cells trafficking to the arterial wall, at least partly mediated through its OxPL content33. Furthermore, Lp(a) associated autotaxin has recently been implicated in CAVD development through conversion of lysophosphatidylcholine to lysophosphatidic acid that promotes inflammation and mineralization of the aortic valve34,35. In summary, these effects of OxPL and lipoprotein(a) may theoretically all contribute to CAVD development characterized by stages of lipid deposition, inflammation, fibrosis, and calcification eventually leading to symptomatic stenosis9,36,37.
Although the association of lipoprotein(a) as a risk factor for CAVD was previously suggested29,38, it was not until the recent meta-analysis of data from genome-wide association studies by Thanassoulis et al1 that a potential genetic and causal association was identified that appeared to be clinically relevant. The LPA rs10455872 intron SNP was the only SNP that reached genome-wide significance for risk of aortic valve calcification and stenosis1. We and others have since demonstrated a clear association of elevated lipoprotein(a) levels with increased risk of CAVD in prospective general population studies and provided further genetic data in support of a causal association of lifelong high lipoprotein(a) levels with increased risk of CAVD2,3. The present study shows for the first time in an epidemiological cohort without prior CAVD the potential role of OxPL in the development of CAVD, and findings are pathophysiologically consistent with recent data showing that patients with pre-existing CAVD and elevated OxPL-apoB or OxPL-apo(a) and lipoprotein(a) levels have much faster progression of CAVD and more frequent need for aortic valve replacement15.
Another important observation in this study was that there was no association of LDL cholesterol and CAVD after multivariable adjustment including the use of lipid lowering therapy. This is consistent with 4 randomized statin trials in patients with pre-existing CAVD where significant LDL cholesterol lowering did not affect echocardiographically-determined progression of CAVD39–42, suggesting LDL is unlikely to have a major causal association with either the development of CAVD, as shown in this study, or the progression of pre-existing CAVD as shown in these trials. Finally, in the ASTRONOMER trial rosuvastatin actually increased lipoprotein(a) and OxPL-apoB levels, whereas they did not change in patients on placebo. If indeed these are causal mediators, their increase may have negated any effect of LDL cholesterol lowering.
We applied a Mendelian randomization study design where association of genotypes, affecting a putative causal risk factor, with risk of disease may be taken as evidence of causality17. The argument for causality is based on the fact that associations of genotypes with disease are generally unconfounded, since genotypes are distributed independent of environmental and lifestyle factors in homogenous populations, and may not result from reverse causality, as genotypes are invariant and not affected by disease status. Limitations of Mendelian randomization studies43 include genetic confounding and false positive findings if the examined genotypes are in linkage disequilibrium with other genetic variation affecting disease. In the present study, the stepwise association of KIV-2 repeat genotype with risk of CAVD makes genetic confounding seem highly unlikely. Furthermore, pleiotropic effects of genetic variants can make it difficult to be certain which intermediate parameter is causing the effect of the genotype on outcome, illustrated in the present study by both lipoprotein(a) per se and OxPL-apoB or OxPL-apo(a) being “causally” associated with CAVD. To the best of our knowledge there are no known genetic variants that affect only OxPL, thus, clinical trials of e.g. lipoprotein(a) lowering vs. inactivation of OxPL are needed to provide final proof for OxPL being the key biological variable in inducing CAVD.
Another limitation of this study is the inclusion of exclusively white individuals of Danish descent, which may limit the generalizability of our results. However, the inclusion of genetically homogenous individuals in the present study, represents a strength in Mendelian randomization studies by minimizing risk of population stratification and false genetic associations43.
In conclusion, the current study identifies OxPL on lipoprotein(a) as a genetic and likely causal risk factor for CAVD. This finding suggests novel therapeutic approaches to halt the development or progression of CAVD, where no medical therapy exists today36,37. Novel therapies may be specifically directed at lowering lipoprotein levels and their associated OxPL, as recently shown with an antisense oligonucleotide that lowers both lipoprotein(a) and OxPL-apoB and OxPL-apo(a) by ~80%44 or by directly inactivating OxPL with specific antibodies5,11.
Supplementary Material
HIGHLIGHTS.
The present study
shows for the first time in an epidemiological cohort without prior CAVD the potential role of OxPL (on apolipoprotein B (OxPL-apoB) or on apolipoprotein(a) (OxPL-apo(a)) in the development of CAVD
demonstrates a genetic association of OxPL with future risk of developing CAVD
identify OxPL as a likely causal risk factor for CAVD that may explain the association of lipoprotein(a) with CAVD
Acknowledgments
The authors thank laboratory technicians Anja Jochumsen, Trine Søberg Nielsen, Hanne Damm, Anders Marcuslund-Reuss, and Mia Hansen (Department of Clinical Biochemistry, Herlev Hospital) and Xiaohong Yang (UCSD) for excellent technical assistance.
SOURCES OF FUNDING
The Danish Council for Independent Research – Medical Sciences, Ib Michael Kristiansen (IMK) Almene Fund, and Johan and Lise Boserup’s Fund, all Copenhagen, Denmark. Drs. Tsimikas and Witztum are supported by the National Institutes of Health (National Heart, Lung and Blood Institute grants R01-HL119828, R01-HL093767, P01-HL088093 and P01-HL055798).
Dr. Kamstrup has received lecture honoraries or consultancy fees from Fresenius Medical Care and Sanofi-Aventis/Regeneron. Dr. Nordestgaard has received consultancy fees or lecture honoraries from Sanofi-Aventis/Regeneron, Fresenius, Ionis Pharmaceuticals, Merck, AstraZeneca, Omthera, Aegerion, B. Braun, Dezima, Kaneka, Kowa, and Amgen. Drs. Tsimikas and Witztum are a co-inventors and receive royalties from patents owned by UCSD on oxidation-specific antibodies and of biomarkers related to oxidized lipoproteins. Dr. Tsimikas currently has a dual appointment with UCSD and as an employee of Ionis Pharmaceuticals. Dr. Witztum is a consultant to Ionis, Cymabay, Intercept, and Prometheus Pharmaceuticals.
NON STANDARD ABBREVIATIONS
- CAVD
calcific aortic valve disease
- PC
phosphocholine
- OxPL
oxidized phospholipids
- OxPL-apoB
OxPL measured on apoB
- OxPL-apo(a)
OxPL measured on apolipoprotein(a)
- apo(a)
apolipoprotein(a)
- KIV-2
kringle IV type 2 repeat polymorphism
- SNP
single nucleotide polymorphism
- CGPS
Copenhagen General Population Study
Footnotes
DISCLOSURES
No other authors have reported relationships relevant to the contents of this paper to disclose.
Subject codes: Valvular Heart Disease, Risk Factors, Genetic - Association Studies
References
- 1.Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, Kerr KF, Pechlivanis S, Budoff MJ, Harris TB, Malhotra R, O’Brien KD, Kamstrup PR, Nordestgaard BG, Tybjaerg-Hansen A, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. doi: 10.1056/NEJMoa1109034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol. 2014;63:470–477. doi: 10.1016/j.jacc.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 3.Arsenault BJ, Boekholdt SM, Dube MP, Rheaume E, Wareham NJ, Khaw KT, Sandhu MS, Tardif JC. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective Mendelian randomization study and replication in a case-control cohort. Circ Cardiovasc Genet. 2014;7:304–310. doi: 10.1161/CIRCGENETICS.113.000400. [DOI] [PubMed] [Google Scholar]
- 4.Bergmark C, Dewan A, Orsoni A, Merki E, Miller ER, Shin MJ, Binder CJ, Horkko S, Krauss RM, Chapman MJ, Witztum JL, Tsimikas S. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. 2008;49:2230–2239. doi: 10.1194/jlr.M800174-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Leibundgut G, Scipione C, Yin H, Schneider M, Boffa MB, Green S, Yang X, Dennis E, Witztum JL, Koschinsky ML, Tsimikas S. Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a) J Lipid Res. 2013;54:2815–2830. doi: 10.1194/jlr.M040733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leibundgut G, Witztum JL, Tsimikas S. Oxidation-specific epitopes and immunological responses: Translational biotheranostic implications for atherosclerosis. Curr Opin Pharmacol. 2013;13:168–179. doi: 10.1016/j.coph.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsimikas S, Duff GW, Berger PB, Rogus J, Huttner K, Clopton P, Brilakis E, Kornman KS, Witztum JL. Pro-inflammatory interleukin-1 genotypes potentiate the risk of coronary artery disease and cardiovascular events mediated by oxidized phospholipids and lipoprotein(a) J Am Coll Cardiol. 2014;63:1724–1734. doi: 10.1016/j.jacc.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Dijk RA, Kolodgie F, Ravandi A, Leibundgut G, Hu PP, Prasad A, Mahmud E, Dennis E, Curtiss LK, Witztum JL, Wasserman BA, Otsuka F, Virmani R, Tsimikas S. Differential expression of oxidation-specific epitopes and apolipoprotein(a) in progressing and ruptured human coronary and carotid atherosclerotic lesions. J Lipid Res. 2012;53:2773–2790. doi: 10.1194/jlr.P030890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O’Brien KD, Schoen FJ, Towler DA, Yoganathan AP, Otto CM. Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation. 2011;124:1783–1791. doi: 10.1161/CIRCULATIONAHA.110.006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yutzey KE, Demer LL, Body SC, Huggins GS, Towler DA, Giachelli CM, Hofmann-Bowman MA, Mortlock DP, Rogers MB, Sadeghi MM, Aikawa E. Calcific aortic valve disease: a consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arterioscler Thromb Vasc Biol. 2014;34:2387–2393. doi: 10.1161/ATVBAHA.114.302523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hung MY, Witztum JL, Tsimikas S. New therapeutic targets for calcific aortic valve stenosis: the lipoprotein(a)-lipoprotein-associated phospholipase A2-oxidized phospholipid axis. J Am Coll Cardiol. 2014;63:478–480. doi: 10.1016/j.jacc.2013.08.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parhami F, Morrow AD, Balucan J, Leitinger N, Watson AD, Tintut Y, Berliner JA, Demer LL. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation. A possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler Thromb Vasc Biol. 1997;17:680–687. doi: 10.1161/01.atv.17.4.680. [DOI] [PubMed] [Google Scholar]
- 13.Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol. 2014;34:715–723. doi: 10.1161/ATVBAHA.113.302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taleb A, Witztum JL, Tsimikas S. Oxidized phospholipids on apoB-100-containing lipoproteins: a biomarker predicting cardiovascular disease and cardiovascular events. Biomark Med. 2011;5:673–694. doi: 10.2217/bmm.11.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capoulade R, Chan KL, Yeang C, Mathieu P, Bosse Y, Dumesnil JG, Tam JW, Teo KK, Mahmut A, Yang X, Witztum JL, Arsenault BJ, Despres JP, Pibarot P, Tsimikas S. Oxidized Phospholipids, Lipoprotein(a), and Progression of Calcific Aortic Valve Stenosis. J Am Coll Cardiol. 2015;66:1236–1246. doi: 10.1016/j.jacc.2015.07.020. [DOI] [PubMed] [Google Scholar]
- 16.Langsted A, Varbo A, Kamstrup PR, Nordestgaard BG. Elevated Lipoprotein(a) Does Not Cause Low-Grade Inflammation Despite Causal Association With Aortic Valve Stenosis and Myocardial Infarction: A Study of 100,578 Individuals from the General Population. J Clin Endocrinol Metab. 2015;100:2690–2699. doi: 10.1210/jc.2015-1096. [DOI] [PubMed] [Google Scholar]
- 17.Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 18.Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298(3):299–308. doi: 10.1001/jama.298.3.299. [DOI] [PubMed] [Google Scholar]
- 19.Boffa MB, Marcovina SM, Koschinsky ML. Lipoprotein(a) as a risk factor for atherosclerosis and thrombosis: mechanistic insights from animal models. Clin Biochem. 2004;37:333–343. doi: 10.1016/j.clinbiochem.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 20.Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, Parish S, Barlera S, Franzosi MG, Rust S, Bennett D, Silveira A, Malarstig A, Green FR, Lathrop M, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–2528. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 21.Deb A, Caplice NM. Lipoprotein(a): new insights into mechanisms of atherogenesis and thrombosis. Clin Cardiol. 2004;27:258–264. doi: 10.1002/clc.4960270503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 23.Kamstrup PR. Lipoprotein(a) and ischemic heart disease–a causal association? A review. Atherosclerosis. 2010;211:15–23. doi: 10.1016/j.atherosclerosis.2009.12.036. [DOI] [PubMed] [Google Scholar]
- 24.Kamstrup PR, Tybjaerg-Hansen A, Nordestgaard BG. Genetic evidence that lipoprotein(a) associates with atherosclerotic stenosis rather than venous thrombosis. Arterioscler Thromb Vasc Biol. 2012;32:1732–1741. doi: 10.1161/ATVBAHA.112.248765. [DOI] [PubMed] [Google Scholar]
- 25.Tsimikas S, Brilakis ES, Miller ER, McConnell JP, Lennon RJ, Kornman KS, Witztum JL, Berger PB. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med. 2005 Jul;353:46–57. doi: 10.1056/NEJMoa043175. [DOI] [PubMed] [Google Scholar]
- 26.Tsimikas S, Mallat Z, Talmud PJ, Kastelein JJ, Wareham NJ, Sandhu MS, Miller ER, Benessiano J, Tedgui A, Witztum JL, Khaw KT, Boekholdt SM. Oxidation-specific biomarkers, lipoprotein(a), and risk of fatal and nonfatal coronary events. J Am Coll Cardiol. 2010;56:946–955. doi: 10.1016/j.jacc.2010.04.048. [DOI] [PubMed] [Google Scholar]
- 27.Tsimikas S, Willeit P, Willeit J, Santer P, Mayr M, Xu Q, Mayr A, Witztum JL, Kiechl S. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J Am Coll Cardiol. 2012;60:2218–2229. doi: 10.1016/j.jacc.2012.08.979. [DOI] [PubMed] [Google Scholar]
- 28.Boonmark NW, Lawn RM. The lysine-binding function of Lp(a) Clin Genet. 1997;52:355–360. doi: 10.1111/j.1399-0004.1997.tb04353.x. [DOI] [PubMed] [Google Scholar]
- 29.O’Brien KD, Reichenbach DD, Marcovina SM, Kuusisto J, Alpers CE, Otto CM. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler Thromb Vasc Biol. 1996;16:523–532. doi: 10.1161/01.atv.16.4.523. [DOI] [PubMed] [Google Scholar]
- 30.Ravandi A, Leibundgut G, Hung MY, Patel M, Hutchins PM, Murphy RC, Prasad A, Mahmud E, Miller YI, Dennis EA, Witztum JL, Tsimikas S. Release and capture of bioactive oxidized phospholipids and oxidized cholesteryl esters during percutaneous coronary and peripheral arterial interventions in humans. J Am Coll Cardiol. 2014;63:1961–1971. doi: 10.1016/j.jacc.2014.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cote C, Pibarot P, Despres JP, Mohty D, Cartier A, Arsenault BJ, Couture C, Mathieu P. Association between circulating oxidised low-density lipoprotein and fibrocalcific remodelling of the aortic valve in aortic stenosis. Heart. 2008;94:1175–1180. doi: 10.1136/hrt.2007.125740. [DOI] [PubMed] [Google Scholar]
- 32.Mohty D, Pibarot P, Despres JP, Cote C, Arsenault B, Cartier A, Cosnay P, Couture C, Mathieu P. Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis. Arterioscler Thromb Vasc Biol. 2008;28:187–193. doi: 10.1161/ATVBAHA.107.154989. [DOI] [PubMed] [Google Scholar]
- 33.van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, Ravandi A, Nederveen AJ, Verberne HJ, Scipione C, Nieuwdorp M, Joosten LA, Netea MG, Koschinsky ML, Witztum JL, et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation. 2016;134:611–624. doi: 10.1161/CIRCULATIONAHA.116.020838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouchareb R, Mahmut A, Nsaibia MJ, Boulanger MC, Dahou A, Lepine JL, Laflamme MH, Hadji F, Couture C, Trahan S, Page S, Bosse Y, Pibarot P, Scipione CA, Romagnuolo R, et al. Autotaxin Derived From Lipoprotein(a) and Valve Interstitial Cells Promotes Inflammation and Mineralization of the Aortic Valve. Circulation. 2015;132:677–690. doi: 10.1161/CIRCULATIONAHA.115.016757. [DOI] [PubMed] [Google Scholar]
- 35.Nsaibia MJ, Mahmut A, Boulanger MC, Arsenault BJ, Bouchareb R, Simard S, Witztum JL, Clavel MA, Pibarot P, Bosse Y, Tsimikas S, Mathieu P. Autotaxin interacts with lipoprotein(a) and oxidized phospholipids in predicting the risk of calcific aortic valve stenosis in patients with coronary artery disease. J Intern Med. 2016;280:509–517. doi: 10.1111/joim.12519. [DOI] [PubMed] [Google Scholar]
- 36.Dweck MR, Boon NA, Newby DE. Calcific aortic stenosis: a disease of the valve and the myocardium. J Am Coll Cardiol. 2012;60:1854–1863. doi: 10.1016/j.jacc.2012.02.093. [DOI] [PubMed] [Google Scholar]
- 37.Vahanian A, Alfieri O, Andreotti F, Antunes MJ, Baron-Esquivias G, Baumgartner H, Borger MA, Carrel TP, De BM, Evangelista A, Falk V, Iung B, Lancellotti P, Pierard L, Price S, et al. Guidelines on the management of valvular heart disease (version 2012) Eur Heart J. 2012;33:2451–2496. doi: 10.1093/eurheartj/ehs109. [DOI] [PubMed] [Google Scholar]
- 38.Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, Kitzman DW, Otto CM. Clinical factors associated with calcific aortic valve disease. Cardiovascular Health Study. J Am Coll Cardiol. 1997;29:630–634. doi: 10.1016/s0735-1097(96)00563-3. [DOI] [PubMed] [Google Scholar]
- 39.Chan KL, Teo K, Dumesnil JG, Ni A, Tam J. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 2010;121:306–314. doi: 10.1161/CIRCULATIONAHA.109.900027. [DOI] [PubMed] [Google Scholar]
- 40.Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–2397. doi: 10.1056/NEJMoa043876. [DOI] [PubMed] [Google Scholar]
- 41.Dichtl W, Alber HF, Feuchtner GM, Hintringer F, Reinthaler M, Bartel T, Sussenbacher A, Grander W, Ulmer H, Pachinger O, Muller S. Prognosis and risk factors in patients with asymptomatic aortic stenosis and their modulation by atorvastatin (20 mg) Am J Cardiol. 2008;102:743–748. doi: 10.1016/j.amjcard.2008.04.060. [DOI] [PubMed] [Google Scholar]
- 42.Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Barwolf C, Holme I, Kesaniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 43.Nitsch D, Molokhia M, Smeeth L, DeStavola BL, Whittaker JC, Leon DA. Limits to causal inference based on Mendelian randomization: a comparison with randomized controlled trials. Am J Epidemiol. 2006;163:397–403. doi: 10.1093/aje/kwj062. [DOI] [PubMed] [Google Scholar]
- 44.Tsimikas S, Viney NJ, Hughes SG, Singleton W, Graham MJ, Baker BF, Burkey JL, Yang Q, Marcovina SM, Geary RS, Crooke RM, Witztum JL. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet. 2015;386:1472–1483. doi: 10.1016/S0140-6736(15)61252-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.