

Abstract
Leptin (LEP) binds to the long form of the leptin receptor (LEPRb), leading to activation of multiple signaling pathways that are potential targets for disrupting the obesity-breast cancer link. In triple-negative breast cancer (TNBC), LEP is hypothesized to predominantly mediate its tumorigenic effects via a subpopulation of LEPRb-positive tumor cells termed cancer stem cells (CSCs) that can initiate tumors and induce tumor progression. Previously, we showed that LEP promotes CSC survival in vivo. Moreover, silencing LEPRb in TNBC cells compromised the CSC state. The mechanisms by which LEPRb regulates TNBC CSC intracellular signaling are not clear. We hypothesized that activation of LEPRb signaling is sufficient to drive CSC maintenance in TNBC. Here, we show that activation of LEPRb in non-CSCs isolated using our CSC reporter system resulted in a transition to the stem cell state. In CSCs, LEP induced STAT3 phosphorylation, whereas LEP did not induce STAT3 phosphorylation in non-CSCs. Introduction of constitutively active STAT3 into LEPRb-transfected non-CSCs significantly induced NANOG, SOX2, and OCT4 expression compared with control non-CSCs. To determine the intracellular phospho-tyrosine residue of LEPRb that is necessary for the induction of the stem cell state in non-CSCs, we transfected the tyrosine residue point mutants L985, F1077, and S1138 into non-CSCs. Non-CSCs transfected with the L985 mutant exhibited increased STAT3 phosphorylation, increased SOCS3 expression, and an induction of GFP expression compared with non-CSCs expressing the F1077 and S1138 mutants. Our data demonstrate that LEPRb-induced STAT3 activation is essential for the induction and maintenance of TNBC CSCs.
Introduction
Obesity is considered a major risk factor for many cancers, including breast cancer (Calle, et al. 2003). In breast cancer patients, obesity has been shown to correlate with advanced disease at diagnosis and an overall poor prognosis (Calle and Thun 2004). Premenopausal women with both obesity and breast cancer often present with a more aggressive subtype of breast cancer termed triple-negative breast cancer (TNBC) (Carmichael 2006; Harvie, et al. 2003; Ligibel 2011). TNBCs are highly heterogeneous tumors characterized by the lack of expression of estrogen, progesterone, and human epidermal growth factor receptor-2 (HER2) receptors (Dent, et al. 2007; Foulkes, et al. 2010). In many advanced cancers, including TNBC, cancer cells are hierarchically organized with a subpopulation of multipotent, self-renewing cells at the apex termed cancer stem cells (CSCs) (Idowu, et al. 2012; Uchoa Dde, et al. 2014; Zheng, et al. 2013). CSCs possess an enhanced ability to initiate and induce tumorigenesis and express the embryonic stem cell transcription factors NANOG, SOX2, and OCT4 (Al-Hajj, et al. 2003; Collins, et al. 2005; Lathia, et al. 2015; Ohi, et al. 2011). The Yamanaka factors OCT4 and SOX2 are crucial for the maintenance of embryonic stem (ES) cells and for the reprogramming of somatic cells: Introduction of these factors into somatic cells drives the developmental signaling network essential for the induction of pluripotency in somatic cells (Takahashi, et al. 2007; Takahashi and Yamanaka 2006).
CSCs have been proposed to underlie tumor initiation, recurrence, and metastasis in TNBC (Al-Hajj et al. 2003). CSCs are both chemo- and radio-resistant and highly metastatic, driving heterogeneity and likely underlying therapeutic resistance and relapse (Visvader and Lindeman 2012). Of all the adipokines elevated in obesity that have been implicated in tumorigenesis, leptin (LEP) is unique in that it exhibits high levels in breast cancer specimens compared with normal mammary tissues (Artac and Altundag 2012; Barone, et al. 2012; Newman and Gonzalez-Perez 2014). In breast cancer cells, LEP has been implicated in promoting cell proliferation, migration, and induction of angiogenesis, but the mechanisms by which it activates signaling by the long form of the leptin receptor (LEPRb), thereby regulating these phenotypes, are still not fully understood (Frankenberry, et al. 2006; Gonzalez-Perez, et al. 2013; Saxena, et al. 2007; Snoussi, et al. 2006; Surmacz 2007; Tessitore, et al. 2004; Yin, et al. 2004; Zhou, et al. 2011). We have previously shown that LEPRb maintains cancer cells in a stem cell-like state (Zheng et al. 2013). However, the molecular details of LEPRb signaling activation, its regulation, and its downstream signaling targets and their effects on CSC maintenance have not been extensively explored in TNBC despite the strong evidence for high expression of LEP and LEPRb in TNBC (Dill, et al. 2016; Newman and Gonzalez-Perez 2014).
We previously observed that upon silencing of LEPRb signaling in CSCs, NANOG expression and function were decreased (Zheng et al. 2013). Upon re-introduction of mouse LepR into LEPRb-silenced human TNBC CSCs, NANOG expression and functional activity were rescued (Zheng et al. 2013). To delineate the mechanisms by which LEP and LEPRb regulate the stem cell state in TNBC, we utilized our previously established NANOG promoter-driven GFP reporter system (Thiagarajan, et al. 2015). We hypothesized that activation of the LEPRb signaling pathway is sufficient for the induction of stem-like properties in TNBC non-CSCs. To understand how LEPRb induces stem-like properties in TNBC non-CSCs, we examined the downstream effects of LEPRb overexpression in non-CSCs and observed a significant induction of both the expression and activity of the NANOG promoter. Upon LEP treatment, STAT3 phosphorylation was increased in LEPRb-transfected non-CSCs, indicative of LEP-induced LEPRb activation in these cells. When non-CSCs were transfected with either LEPRb or the LEPRb-L985 mutant and then treated with the STAT3 inhibitor BBI608, LEP/LEPRb-induced STAT3 expression was reduced. Together, our study demonstrates that the induction of STAT3 activation by introducing LEPRb is essential for TNBC CSC maintenance.
Materials and Methods
Materials
Plasmids encoding constitutively active STAT3 (STAT3-CA) and dominant-negative STAT3 (STAT3-DN) were purchased from Addgene (Cambridge, MA). The LEPRb tyrosine residue mutants L985, F1077, and S1138 were generous gifts from Dr. Martin G. Myers, Jr. (University of Michigan Medical School, Ann Arbor, MI). BBI608, a cancer stem cell pathway inhibitor, was a generous gift from Dr. George Stark (Lerner Research Institute, Cleveland Clinic, Cleveland, OH).
Cell culture
MDA-MB-231 and HCC70 breast cancer cells (American Type Culture Collection; Manassas, VA) were cultured in log-growth phase in modified Eagle’s medium (MEM) supplemented with 1 mM sodium pyruvate (Cellgro, Kansas City, MO) and 10% heat-inactivated fetal calf serum (FCS) at 37°C in a humidified atmosphere (5% CO2). All cells were transfected and maintained in humidified atmosphere containing 5% CO2 at 37°C.
Immunoblotting
Cells were lysed in buffer containing 20 mM Tris pH 7.4, 137 mM NaCl, 1% NP-40, 10% glycerol, 20 mM NaF, 1 mM sodium orthovanadate, and 1 mM phenylmethylsulfonyl fluoride (PMSF). Protein concentrations were measured using a BCA protein assay (Thermo, Rockford, IL). Membranes were incubated overnight at 4°C with primary antibodies. STAT3, SOCS3 and pSTAT3 antibodies were purchased from Cell Signaling (Beverly, MA), the actin antibody was purchased from Santa Cruz Biotechnology (Dallas, TX), and the GAPDH antibody was from Sigma (St. Louis, MO). Anti-rabbit IgG antibodies conjugated to horseradish peroxidase (HRP) (Amersham, Piscataway, NJ) were used as secondary antibodies and visualized using the West Pico Chemiluminescent substrate from Pierce (Rockford, IL).
RT-PCR
Total RNA was isolated using TRI reagent (Ambion, Austin, TX) and stored at −80°C until use. RNA concentration was determined using a NanoDrop 1000 Spectrophotometer (Thermo, Wilmington, DE). Reverse transcriptase (RT) reactions were prepared using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). Two micrograms of total RNA was used as a template for first-strand cDNA synthesis. Amplification of transcripts was performed using a Taq DNA polymerase kit (Qiagen, Valencia, CA) with 200 ng of total RNA. Semi-quantitative RT-PCR was quantified by digitally scanning the gels followed by analysis using ImageJ (NIH, Bethesda, MD). Real-time PCR was performed on a StepOnePlus Real-Time PCR system from Applied Biosystems with SYBR-Green master mix (Applied Biosystems). The threshold cycle (CT) values for each gene were normalized to the expression levels of GAPDH. Agarose gel electrophoresis (2%) in the presence of ethidium bromide was performed to determine the expression of LEPRb and to confirm the transfection of mLEPRb into MDA-MB-231 CSCs and non-CSCs. The primers used were as follows:
| Human LEPRb-L | 5’-GAAGATGTTCCGAACCCCAAGAATTGTTCC-3’ |
| 5’- GCACTTGGTGACTGAACTATTTATAAGCCC-3’ | |
| Mouse LepR-L | 5’-TTTTTACCAAGCACGCAGAATC-3’ |
| 5’- ACCCCGAGAATGAAAGTTGTG-3’ | |
| GAPDH | 5’-TCGACAGTCAGCCGCATCTTCTTT-3’ |
| 5’-ACCAAATCCGTTGACTCCGACCTT-3’ | |
| SOX2 | 5'-CACACTGCCCCTCTCAC-3' |
| 5'-TCCATGCTGTTTCTTACTCTCC-3' | |
| OCT4 | 5’-TCTCCCATGCATTCAAACTGAG-3’ |
| 5’-CCTTTGTGTTCCCAATTCCTTC-3’ | |
| NANOG | 5’-GAAATACCTCAGCCTCCAGC-3’ |
| 5’-GCGTCACACCATTGCTATTC-3’ | |
| β-Actin | 5’-AGAAAATCTGGCACCACACC-3’ |
| 5’-AGAGGCGTACAGGGATAGCA-3’ |
Flow cytometry analysis
MDA-MB-231 and HCC70 cells transduced with the NANOG-GFP promoter reporter were resuspended in PBS containing 2% FBS at a concentration of 1 million cells/mL and subjected to sorting for GFP on a BD FACSAria II. MDA-MB-231/HCC70 parental cells were used as a control to define negativity for GFP expression. MDA-MB-231 and HCC70 non-CSCs transfected with LEPRb and LEPRb mutants were subjected to analysis on a BD LSRFortessa (BD Biosciences, CA) to determine the levels of GFP expression. Data analysis was performed using the FlowJo software (Tree Star, Inc.).
Transfection of LEPRb and LEPRb tyrosine mutants
MDA-MB-231 and HCC70 GFP-negative cells were transfected with LEPRb or LEPRb tyrosine-residue mutant constructs (L985, F1077, and S1138, generous gifts of Dr. Martin Myers, University of Michigan Medical School, Ann Arbor, MI) using Lipofectamine transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The cells were grown in MEM supplemented with 10% FBS and selected in MEM supplemented with 10% FBS and 750 μg/ml G418 (Sigma, St. Louis, MO). Transfection of LEPRb tyrosine mutants was followed by FACS analysis for the GFP intensity of the transfected (GFP-negative) non-CSCs in comparison with the GFP-positive CSCs (Thiagarajan et al. 2015). Brightfield and fluorescent images of the transfected cells were captured using an inverted microscope.
Treatment with LEP and EPO
MDA-MB-231 and HCC70 GFP-positive and GFP-negative cells were cultured in 6-well dishes until they reached confluence and were then deprived of serum for 16 hours. Cells were then incubated in the absence or presence of LEP (0, 5, 25 ng/ml) for 30 minutes. Cells were subsequently lysed, and protein was quantified, resolved on SDS-PAGE, and blotted for pSTAT3 followed by STAT3, SOCS3, and Actin.
MDA-MB-231 and HCC70 cells transfected with the LEPRb tyrosine mutants (L985, F1077, and S1138) were treated with increasing concentrations of erythropoietin (Epo) (0, 50, and 100 ng/ml) for 30 minutes. Cells were lysed, and protein was quantified, resolved on SDS-PAGE, and blotted for pSTAT3 followed by STAT3.
Luciferase reporter assays
The Gluc-ON Promoter Reporter Clones pEZX-PG02 (HPRM11834-PG02, GeneCopoeia, Rockville, MD, USA) were used to construct NANOG, SOX2 and OCT4 luciferase reporter constructs. MDA-MB-231 and HCC70 GFP-negative and GFP-positive cells were transfected with STAT3-CA and STAT3-DN plasmids (Addgene, Cambridge, MA) followed by transfection with human NANOG, SOX2, and OCT4 promoter-luciferase reporter constructs using X-tremeGENE HP DNA Transfection reagent (Roche, Indianapolis, IN) according to the manufacturer’s protocol. After 72 hours, secreted Gaussia luciferase was assayed from the culture media using the Secrete-Pair™ Luminescence assay kit (GeneCopoeia, Rockville, MD) according to the manufacturer’s instructions. The luminescence intensity of each well was measured using a luminometer (PerkinElmer).
Limiting dilution assays
For tumorsphere formation assessments, cells were cultured in duplicate rows of serial dilutions of 1, 5, 10, 20 cells per well in one 96-well plate per condition (Corning, Tewkesbury, MA, USA) with 200 μl of serum-free DMEM/F12 supplemented with 20 ng/ml basic fibroblast growth factor (Invitrogen), 10 ng/ml epidermal growth factor (BioSource, Grand Island, NY, USA), 2% B27 (Invitrogen), and 10 μg/ml insulin (Sigma, St. Louis, MO). Tumorspheres were counted after 2 weeks under a phase contrast microscope. The frequency of sphere formation was calculated in such a way that a well with a tumorsphere was counted as a positive well and a well with no tumorsphere was counted as a negative well. The stem cell frequencies were calculated using an extreme limiting dilution algorithm (ELDA) (http://bioinf.wehi.edu.au/software/elda/) (Hu and Smyth 2009).
Statistical Analysis
Values reported in the results are mean values +/− standard deviation. One-way ANOVA was used to calculate statistical significance, and p-values are detailed in the text and figure legends.
Results
TNBC CSCs are enriched for LEPRb expression
To determine the crucial molecular mechanisms by which LEP-mediated LEPRb signaling activation regulates CSC maintenance, we assessed the expression of LEPRb in TNBC CSCs. To study LEPRb signaling, we utilized the TNBC CSC reporter system that we developed and validated wherein a GFP reporter reads out the activity of the NANOG promoter (Thiagarajan et al. 2015). MDA-MB-231 and HCC70 NANOG-GFP cells were sorted into GFP-positive (CSC) and GFP-negative (non-CSC) populations based on GFP expression. Non-transduced MDA-MB-231 and HCC70 parental cells were used as sorting controls. The sorted CSCs and non-CSCs were validated based on higher expression of the cell surface markers CD44 and CD49f and the stem cell transcription factors NANOG, SOX2, and OCT4 (Thiagarajan et al. 2015). RNA extracted from the sorted CSCs and non-CSCs was analyzed for the expression of LEPRb, and CSCs were enriched for LEPRb expression when compared with non-CSCs (Figure 1A and 1C).
Figure 1. LEPRb is enriched in TNBC CSCs and is sufficient to induce NANOG promoter activity in non-CSCs.
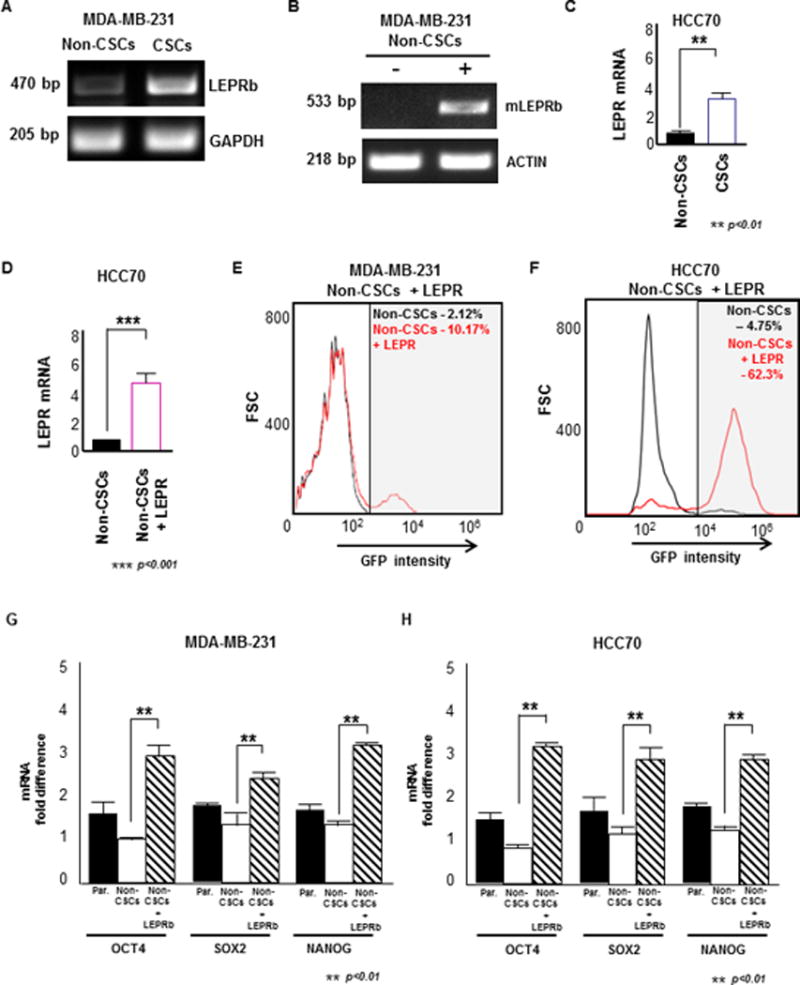
(A) RNA was extracted from sorted MDA-MB-231 CSCs (GFP+) and non-CSCs (GFP-). CSCs showed an enrichment for LEPR compared with the non-CSCs, as analyzed by RT-PCR. GAPDH was used as the loading control. (B) Transfection of mouse LEPR into MDA-MB-231 non-CSCs was confirmed by RNA extraction and an expression analysis of LEPR expression by RT-PCR. The LEPR-transfected non-CSCs showed a higher expression of mouse LEPR compared with the control non-CSCs. (C) HCC70 CSCs (GFP+) showed LEPR enrichment compared with the non-CSCs (GFP-) as determined by RT-PCR. GAPDH was used as the loading control. (D) Mouse LEPR transfection into HCC70 non-CSCs was confirmed by RT-PCR analysis of LEPR expression. LEPR expression was higher in LEPR-transfected HCC70 non-CSCs compared with control non-CSCs. (E & F) Histograms of non-CSCs (black) and mouse LEPR-transfected non-CSCs (red) show an increase in the fraction of GFP+ cells from 2.12% to 10.17% in MDA-MB-231 and 4.75% to 62.3% in HCC70. (G & H) LEPR-transfected MDA-MB-231 and HCC70 non-CSCs exhibited significantly higher expression of the stem cell transcription factors OCT4, NANOG, and SOX2 compared with control non-CSCs (** p < 0.01).
LEPRb induces NANOG promoter activity when expressed in non-CSCs
Previously, we showed that when LEPRb was silenced, the expression of the stem cell self-renewal transcription factors NANOG, SOX2, and OCT4 was inhibited (Zheng et al. 2013). LEPRb-silenced cells showed reduced proliferation, self-renewal, and tumor progression in xenotransplantation studies. We also observed that the expression of NANOG could be rescued in LEPRb-silenced human cancer cells by mouse LEPR (Zheng et al. 2013). To investigate whether LEPRb introduction was sufficient to induce a stem cell state in non-CSCs, we introduced mouse LEPR into MDA-MB-231 and HCC70 non-CSCs (Fig. 1B and 1D) and observed an induction in NANOG promoter-driven activity based on the increased intensity of GFP expression by FACS analysis. FACS analysis indicated that a very small percentage of non-CSCs expressed GFP (2.1% and 4.8% in MDA-MB-231 and HCC70 cells, respectively). However, upon introduction of mouse LEPR into non-CSCs (non-CSCs+LEPRb), the expression of GFP (CSC marker) increased to 10.2% and 62.3% of the total population of MDA-MB-231 and HCC70 cells, respectively (Fig. 1E and Fig. 1F). In addition, the expression of the stem cell transcription factors SOX2, OCT4, and NANOG increased in both MDA-MB-231 and HCC70 non-CSCs expressing LEPRb compared with control non-CSCs (Fig. 1G and 1H). Brightfield and fluorescence images of control non-CSCs and LEPRb-overexpressing non-CSCs showed a significant induction of NANOG activity as read out by GFP signal (Supplementary Fig. 1A and 1B). Based on these observations, the introduction of LEPRb is sufficient to induce the transition of TNBC non-CSCs to a stem cell state.
CSC-specific LEPRb activation of the JAK2/STAT3 signaling pathway
We previously observed a downregulation of NANOG expression and function upon silencing of LEPRb in CSCs that was rescued upon re-introduction of LEPRb (Zheng et al. 2013). Here, we observed an induction of both the expression and activity of the NANOG promoter upon introduction of LEPRb into non-CSCs. To study the effect of LEP signaling in both CSCs and non-CSCs, we exogenously treated CSCs and non-CSCs with increasing concentrations of LEP (0, 5, and 25 ng/mL). Both groups were then immunoblotted for pSTAT3 as an indicator of activation of the JAK2/STAT3 downstream signaling pathway. Induction of pSTAT3 was detected in MDA-MB-231 non-CSCs, whereas pSTAT3 could not be detected in HCC70 non-CSCs (Fig. 2A and 2B). LEP induced LEPRb activation primarily in the CSCs, as detected by the induction of pSTAT3 downstream of LEPRb.
Figure 2. LEPRb-dependent STAT3 activation induces the stem cell state in LEPRb-transfected TNBC non-CSCs.
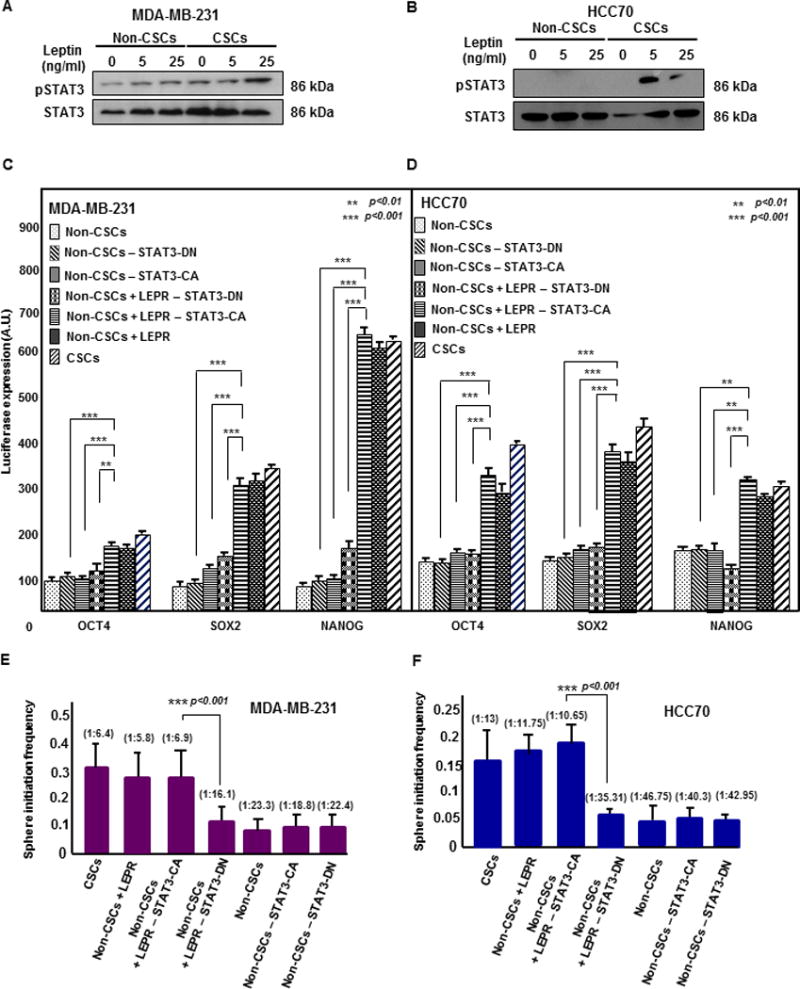
(A & B) Immunoblots of MDA-MB-231 and HCC70 CSCs (GFP+) and non-CSCs (GFP-) treated with increasing concentrations of LEP (0, 5, and 25 ng/ml) for 30 minutes were probed for pSTAT3 and total STAT3. Increased STAT3 phosphorylation was observed in LEP-treated CSCs compared with LEP-treated non-CSCs. Twenty micrograms of protein per sample was loaded into each well. (C & D) Transfection of constitutively active STAT3 (STAT3-CA) in LEPR-transfected non-CSCs (MDA-MB-231 and HCC70) increased the activity of the stem cell transcription factors NANOG, SOX2, and OCT4 as quantified by the secreted luciferase assay method. A significant increase in the transcriptional activity of all three stem cell transcription factors was observed in both LEPR-transfected non-CSC and control non-CSC groups expressing STAT3-CA compared with the corresponding dominant negative STAT3 (STAT3-DN)-expressing groups. A significant increase in transcriptional activity was also observed in STAT3-CA-expressing LEPR-transfected non-CSCs compared with STAT3-DN-expressing non-CSCs. (** p < 0.01, *** p < 0.001). (E and F) Limiting dilution analyses of STAT3-CA- and STAT3-DN-expressing LEPR-transfected non-CSCs were performed. The MDA-MB-231 and HCC70 STAT3-CA-expressing LEPR-transfected non-CSC groups showed a significant increase in stem cell frequency compared with STAT3-DN-expressing non-CSCs without LEPR overexpression (p < 0.001).
STAT3 induction of the stem cell state in LEPRb-expressing non-CSCs
We next examined whether introduction of constitutively active STAT3 (STAT3-CA) was able to activate the stem cell transcription factors NANOG, SOX2, and OCT4. To address this, we transfected both control and LEPRb-overexpressing non-CSCs with STAT3-CA and dominant negative STAT3 (STAT3-DN) as a control. We then assayed NANOG, SOX2, and OCT4 promoter activity using a luciferase reporter assay. The introduction of STAT3-CA into LEPRb-overexpressing non-CSCs significantly enhanced the transcriptional activity of NANOG, SOX2, and OCT4 compared with STAT3-DN expressed in LEPRb-overexpressing non-CSCs, STAT3-CA-expressing control non-CSCs, and STAT3-DN-expressing non-CSCs (Fig. 2C and 2D). While there was a significant difference between control and LEPRb-overexpressing non-CSCs transfected with STAT3-CA, there was no difference between control and LEPRb-overexpressing non-CSCs transfected with STAT3-DN. These data demonstrate that STAT3 induces a stem cell state by activating the transcription of NANOG, SOX2, and OCT4 in LEPRb-overexpressing TNBC non-CSCs.
LEPRb-dependent STAT3 activation is sufficient for TNBC CSC maintenance
Based on our observation that STAT3 activated NANOG, SOX2, and OCT4 in LEPRb-overexpressing non-CSCs, we investigated whether the activation of STAT3 is sufficient to maintain TNBC CSCs. To assess the effect of STAT3 in these cells, we performed limiting dilution assays comparing control and LEPRb-overexpressing non-CSCs transfected with either STAT3-CA or STAT3-DN. Coupled with the increased transcription of stem cell factors, we observed a significant increase in the stem cell frequencies of the STAT3-CA-transfected LEPRb-overexpressing non-CSCs compared with the STAT3-DN-transfected LEPRb-overexpressing non-CSCs, the STAT3-CA-transfected non-CSCs, STAT3-DN-transfected non-CSCs, and control non-CSCs in both MDA-MB-231 and HCC70 cells (Fig. 2E and 2F). These results indicate that STAT3 is sufficient to induce self-renewal and maintain the stem-cell state in TNBC CSCs.
LEPRb intracellular residues Tyr1077 and Tyr1138 are essential for STAT3 activation
LEP binding to LEPRb initiates a cascade of signaling events that begins with the activation of the receptor-associated tyrosine kinase JAK2. Activation of JAK2 leads to the phosphorylation of three tyrosine residues on the intracellular domain of LEPRb, Tyr985, Tyr1077, and Tyr1138. To study the requirement for these residues in LEPRb-mediated stem cell maintenance, single point mutants of each of these residues, L985, F1077, and S1138, were introduced into non-CSCs. These point mutants were previously reported by Dr. Myers, Jr., and colleagues (Bjorbak, et al. 2000; Myers, et al. 2008; Myers 2004). These mutants are chimeric and contain the erythropoietin (Epo) extracellular domain, while the intracellular domain contains the LEPRb intracellular signaling domain (Fig. 4F). To study downstream signaling activation by these LEPRb residues, cells transfected with the mutant receptors were immunoblotted for pSTAT3 after treatment with different concentrations of Epo. Interestingly, pSTAT3 did not increase with Epo treatment in non-CSCs transfected with the F1077 and S1138 mutants, while only the L985 mutant showed increased pSTAT3 (Fig. 3A and 3B). These findings strongly suggest that the phosphorylation of Tyr1077 and Tyr1138 is crucial for the recruitment and activation of STAT3 in TNBC.
Figure 4. STAT3 inhibition decreases self-renewal in LEPRb-expressing non-CSCs.
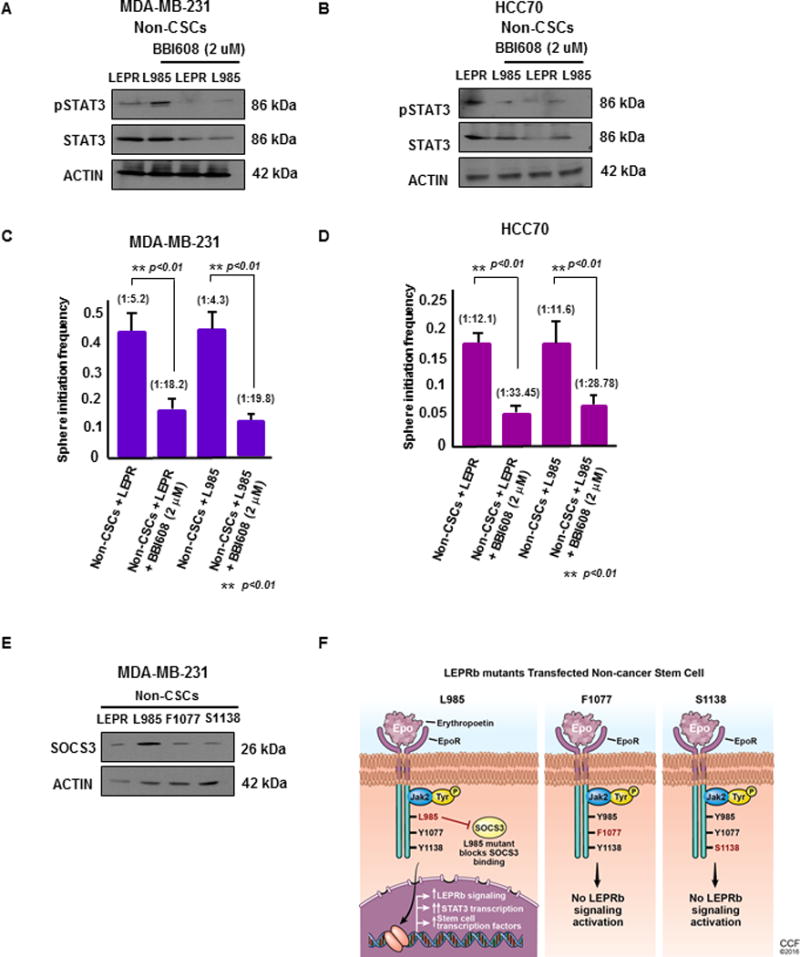
(A & B) Upon treatment with BBI608, a STAT3 pathway inhibitor, at 2 μM for 24 hours, non-CSCs (MDA-MB-231 and HCC70) transfected with LEPRb and L985 showed decreased expression of total STAT3 by immunoblotting. Actin was used as a loading control. Twenty micrograms of protein per sample was loaded into each well for the immunoblots. (C & D) Limiting dilution analyses of LEPR- and L985-transfected non-CSCs treated with BBI608 were performed. The LEPR and L985-transfected non-CSCs treated with BBI608 showed a significant decrease in stem cell frequency compared with the untreated MDA-MB-231 and HCC70 non-CSCs (p < 0.01). (E) Immunoblotting of LEPRb and LEPRb mutant (L985, F1077, S1138)-transfected non-CSCs treated with LEP (25 ng/ml) showed increased SOCS3 expression in L985-transfected non-CSCs compared with the other groups. Actin was used as a loading control. Twenty micrograms of protein per sample was loaded onto each well for the immunoblots. (F) Schematic of the transfection of the LepR L985, F1077 and S1138 mutants into non-CSCs. These tyrosine mutations were made in a chimeric protein containing the erythropoietin (Epo) receptor extracellular domain and the intracellular domain from the long form of the mouse leptin receptor containing the tyrosines. In this chimeric receptor, the activation of LepR-dependent signals occurs under the control of Epo stimulation. Upon Epo stimulation, the intracellular LepR domain maintains the same intracellular signaling program induced by native LepR. In the presence of the L985 mutation, the downstream signaling effects are still activated, due to its inability to bind to SOCS3, the inhibitor of LepR signaling. This leads to constitutively activated downstream LepR signaling, which includes activation of STAT3 and its downstream target genes. Upon introduction of F1077 and S1138, STAT5 and STAT3 transcriptional activation was blocked, leading to the inhibition of LepR signaling pathways.
Figure 3. STAT3 is necessary for LEPRb induction of the stem cell state in non-CSCs.
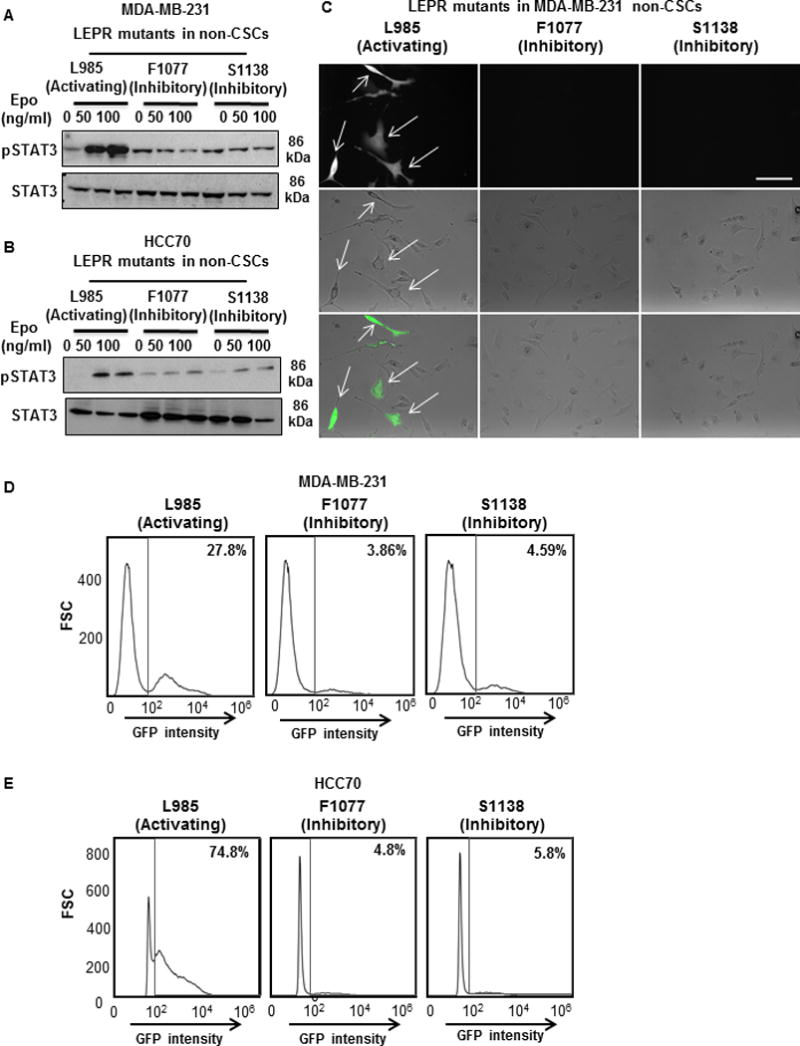
(A & B) MDA-MB-231 and HCC70 non-CSCs were transfected with the LEPRb mutants L985, F1077, and S1138 and then treated with increasing concentrations of erythropoietin (0, 50, and 100 ng/mL) for 30 minutes. Immunoblots of the erythropoietin-treated cell lysates were probed for pSTAT3 and total STAT3. The L985-transfected cells showed an induction of pSTAT3 compared with cells transfected with the other mutants. Twenty micrograms of protein per sample was loaded into each well for the immunoblots. (C) Photomicrographs of LEPR L985-transfected MDA-MB-231 non-CSCs show an induction of GFP expression, whereas cells expressing the other two mutants did not show an induction of GFP expression. (D & E) Histograms of non-CSCs transfected with LEPR mutants show an induction of GFP expression primarily in the L985-transfected non-CSCs (27.8% in MDA-MB-231; 74.8% in HCC70) compared with non-CSCs expressing F1077 (3.86% in MDA-MB-231; 4.8% in HCC70) or S1138 (4.59% in MDA-MB-231; 5.8% in HCC70).
Tyr1077 and Tyr1138 drive stem-like properties in LEPRb-expressing non-CSCs
Increased STAT3 phosphorylation was observed only in the L985 LEPRb-transfected non-CSCs compared with the F1077 and S1138 LEPRb mutants. Similarly, upon treatment with Epo, only cells transfected with L985 showed an induction of green fluorescence as an indicator of an induction of the stem cell state in MDA-MB-231 cells (Fig. 3C). These cells were analyzed by FACS to assess the induction of GFP intensity as a readout of NANOG promoter activity. Owing to their inhibition of LEPRb signaling, LEPRb-transfected non-CSCs expressing F1077 (3.9% GFP-positive in MDA-MB-231; 4.8% in HCC70) and S1138 (4.6% GFP-positive in MDA-MB-231; 5.8% in HCC70) failed to induce NANOG expression, while L985 exhibited an induction of NANOG expression (27.8% GFP-positive in MDA-MB-231; 74.8% in HCC70) (Fig. 3D and 3E). These findings support an essential role for the Tyr1077 and Tyr1138 residues in inducing and maintaining stem-like properties in LEPRb-transfected TNBC non-CSCs.
STAT3 inhibition compromises self-renewal in LEPRb-expressing TNBC non-CSCs
We studied the effect of inhibiting STAT3 on the maintenance of the CSC state in LEPRb-transfected non-CSCs. Twenty-four hours after treating wild-type LEPRb- and L985-expressing MDA-MB-231 and HCC70 non-CSCs with the cancer stem cell STAT3 pathway inhibitor BBI608 at 2 μM, we observed an inhibition of STAT3 expression (Fig. 4A and 4B). Limiting dilution assays demonstrated a significant reduction in stem cell frequency in BBI608-treated LEPRb- and L985-expressing MDA-MB-231 and HCC70 non-CSCs (Fig. 4C and 4D). These results indicate that STAT3 activation and expression are essential for the TNBC CSC transition and CSC maintenance. The expression of SOCS3, a downstream signaling target indicative of the function of LEPRb, and that of the LEPRb mutants (L985, F1077, S1138) was determined by immunoblotting after introduction into MDA-MB-231 non-CSCs (Fig. 4E). Activated pSTAT3 translocates to the nucleus, where it mediates changes in the expression of target genes, including SOCS3, which encodes a feedback inhibitor of LEPRb signaling. SOCS3 showed high expression in L985-expressing cells compared with wild-type LEPRb-, F1077-, or S1138-expressing cells (Fig. 4E). Tyr985 also serves as the binding site for SOCS3 and thus plays a prominent role in the feedback inhibition of LEPRb signaling, as shown in Fig. 4F (Bjorbak et al. 2000; Villanueva and Myers 2008).
DISCUSSION
Though the oncogenic impact of LEP and its effects through LEPRb signaling in breast cancer are well established, the mechanisms by which LEPRb regulates intracellular signaling have not been completely explored (Artac and Altundag 2012; Barone et al. 2012; Frankenberry et al. 2006). To better understand the molecular mechanisms by which LEP-induced LEPRb signaling activation regulates and maintains the CSC state in cultured TNBC cells, we used our previously validated CSC NANOG-GFP reporter system (Thiagarajan et al. 2015). As non-CSCs express LEPRb at low levels compared with CSCs, we assessed whether overexpressing LEPRb would stimulate the CSC state in these cells. LEPRb-transfected non-CSCs showed a 5-fold increase in GFP intensity as a measure of NANOG promoter activation based on our previously established model (Thiagarajan et al. 2015). Here, we demonstrate that LEP stimulates the phosphorylation of STAT3 in LEPRb-transfected non-CSCs compared with non-transfected non-CSCs. The increased expression of LEPRb and STAT3 activation recapitulated the phosphorylation status of STAT3 in CSCs, which express LEPRb at high levels. These results demonstrate that the induction of STAT3 phosphorylation in non-CSCs occurred via increased LEPRb expression and activation. To determine whether NANOG promoter activation occurred downstream of STAT3 activation, we introduced constitutively active STAT3 into LEPRb-transfected non-CSCs and observed a significant increase in self-renewal and an induction of the transcriptional activity of the NANOG, SOX2 and OCT4 stem cell transcription factors compared with LEPRb-transfected non-CSCs expressing dominant-negative STAT3. Taken together, these results demonstrate that the LEP-induced LEPRb activation of STAT3 phosphorylation is sufficient to stimulate stem-like properties in TNBC cells.
Given the crucial role for LEPRb signaling activation of STAT3 in TNBC CSCs, we examined signal transduction by the intracellular domains of LEPRb that are critical for the maintenance of CSCs (Zheng et al. 2013). For this purpose, we utilized a chimeric protein consisting of the Epo receptor extracellular domain with the intracellular domain of LEPRb (Gong, et al. 2007). Binding of LEP to LEPRb activates JAK2, which in turn phosphorylates the three tyrosine residues Tyr985, Tyr1077, and Tyr1138 on the intracellular tail of LEPRb (Myers 2004). Each of these phosphorylation sites is located in a unique amino acid motif, and each of these residues can therefore recruit a distinct set of downstream signaling proteins upon phosphorylation. Understanding the contributions of these individual tyrosine residues and their downstream signaling effects was crucial for discerning the pathways that regulate CSC maintenance. We introduced single point mutants of the three tyrosine residues, L985, F1077 and S1138, of LEPRb into non-CSCs. Of the three mutants, only L985 showed an activation of pSTAT3 compared with the other mutants, which did not induce STAT3 phosphorylation. Upon downregulation of STAT3 expression by the addition of BBI608 (a CSC STAT3 pathway inhibitor) in non-CSCs expressing LEPRb and L985, self-renewal was compromised compared with the untreated control groups. Notably, STAT3 phosphorylation induces the expression of SOCS3, which is known to inhibit LEPRb signaling and attenuate STAT3 phosphorylation (Bjorbak et al. 2000). In the L985-transfected cells, we observed an increase in SOCS3 expression. As phosphorylation of Tyr985 is necessary for binding SOCS3, this leads us to conclude that eliminating the negative feedback inhibition of LEPRb signaling leads to a constitutive activation of LEPRb-JAK2/STAT3 signaling (Bjorbak et al. 2000; Villanueva and Myers 2008). We conclude that the inability of SOCS3 to bind to the mutant L985 residue resulted in positive feedback and further activation of LEPRb signaling, which could then be blocked by a STAT3 pathway inhibitor. This strongly validates the idea that STAT3 activation is crucial for the activation of CSC maintenance by LEP signaling.
Significant correlations between obesity and the incidence of various cancers have been reported (Arnold, et al. 2016; O'Brien S, et al. 1999; Renehan, et al. 2008; Tessitore, et al. 2000). Even mild inflammation, as observed in obesity, is characterized by a high level of adipose tissue secretion of cytokines with disparate effects that are potentially relevant to the development of cancer (Vona-Davis and Rose 2007). Among these cytokines, LEP functions are commonly reinforced through crosstalk with multiple oncogenes, cytokines, and growth factors. Many LEP-induced signals are essential not only for normal biological function but also for oncogenesis (Artac and Altundag 2012; Barone et al. 2012). In this study, we have shown that the LEPRb/STAT3 signaling axis plays a critical role in the regulation of stem cell transcription factor activity and the maintenance of a CSC-like state. Our studies lack in vivo studies that could provide further evidence of whether transfection of LEPRb in non-CSCs promotes tumor-initiation and progression by inducing the transition to a stem cell state. In summary, our present data outline for the first time that constitutive activation of the LEPR/STAT3 signaling axis stimulates the downstream activation of the stem cell transcription factors NANOG, SOX2, and OCT4, as shown in Figure 5. We propose a model whereby constitutive activation of STAT3 as observed in chronic obesity conditions may serve as a signaling hub with potential implications in the transition and maintenance of TNBC CSCs by virtue of an activated LEPRb signaling complex. Indeed, these studies support the idea that targeting the STAT3 signaling pathway may be a viable therapeutic approach to target TNBC CSCs. Identification of LEP-LEPRb-specific individual pathways will be crucial for our comprehensive understanding of the regulatory processes underlying TNBC CSC maintenance.
Figure 5. Graphical summary of LEPRb-mediated activation of STAT3-dependent stem cell pathways in TNBC non-CSCs.

Panel 1 shows the decreased expression of the leptin receptor in non-cancer stem cells. Panel 2 shows the activation of the leptin receptor signaling pathway upon introduction of LepR into non-cancer stem cells. Ligand binding to the pre-dimerized extracellular domain of LepRb initiates the phosphorylation and activation of the constitutively associated JAK2 tyrosine kinase. Activated JAK2 leads to the phosphorylation of the three tyrosine residues, Y985, Y1077 and Y1138, on the cytoplasmic region of LEPR. Y985 plays a complex role in LepRb signaling, as it not only serves as a docking site for the inhibitory SOCS3 (suppressor of cytokine signaling 3) protein that is induced by STAT3 activation but also binds SHP-2 (src homology domain protein tyrosine phosphatase 2), an upstream activator of the ERK cascade. Y1138 recruits STAT3 for phosphorylation by JAK2, which leads to STAT3 nuclear translocation. Nuclear localization of STAT3 increases the transcription of its target genes, including the stem cell transcription factors NANOG, SOX2, and OCT4.
Supplementary Material
(A&B) Photomicrographs of the LEPR-transfected non-CSCs showed an increase in the number of cells expressing GFP compared with the control non-transfected non-CSCs. Scale bar – 100 µM.
Acknowledgments
Funding
This work was funded by National Institutes of Health grant CA191263 to O.R. and J.D.L. The work in the Reizes laboratory is also supported by the Cleveland Clinic Foundation, Clinical and Translational Science Collaborative of Cleveland (UL1TR000439), Case Comprehensive Cancer Center (P30 CA043703), Cleveland Clinic VeloSano Bike Race, and The Laura J. Fogarty Endowed Chair for Uterine Cancer Research. The Lathia laboratory also receives funding from the National Institutes of Health grants NS089641, NS083629, and CA157948; a Distinguished Scientist Award from the Sontag Foundation; a Research Scholar Award from the American Cancer Society; Blast GBM; the Cleveland Clinic VeloSano Bike Race; and the Case Comprehensive Cancer Center.
We thank the members of the Reizes and Lathia laboratories for insightful discussion and constructive comments on the manuscript. We thank Cathy Shemo, Patrick Barrett, Joseph Gerow, and Sage O’Bryant for flow cytometry assistance. We thank Dr. Martin Myers (University of Michigan Medical School, Ann Arbor, MI) for providing us with the leptin receptor tyrosine residue mutants as a generous gift.
Footnotes
Conflict of interest
None to declare
Author contributions: Conception and design (PST, QZ, JDL, OR), Financial support (JDL, OR), Administrative support (JDL, OR), Provision of study material (MGM, JDL, OR), Collection and/or assembly of data (PST, QZ, MB), Data analysis and interpretation (PST, QZ, JDL, OR), Manuscript writing (PST, EEMH, JDL, OR), Final approval of manuscript (all authors)
References
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold M, Leitzmann M, Freisling H, Bray F, Romieu I, Renehan A, Soerjomataram I. Obesity and cancer: An update of the global impact. Cancer Epidemiol. 2016;41:8–15. doi: 10.1016/j.canep.2016.01.003. [DOI] [PubMed] [Google Scholar]
- Artac M, Altundag K. Leptin and breast cancer: an overview. Med Oncol. 2012;29:1510–1514. doi: 10.1007/s12032-011-0056-0. [DOI] [PubMed] [Google Scholar]
- Barone I, Catalano S, Gelsomino L, Marsico S, Giordano C, Panza S, Bonofiglio D, Bossi G, Covington KR, Fuqua SA, et al. Leptin mediates tumor-stromal interactions that promote the invasive growth of breast cancer cells. Cancer Res. 2012;72:1416–1427. doi: 10.1158/0008-5472.CAN-11-2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, Myers MG., Jr SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem. 2000;275:40649–40657. doi: 10.1074/jbc.M007577200. [DOI] [PubMed] [Google Scholar]
- Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- Calle EE, Thun MJ. Obesity and cancer. Oncogene. 2004;23:6365–6378. doi: 10.1038/sj.onc.1207751. [DOI] [PubMed] [Google Scholar]
- Carmichael AR. Obesity and prognosis of breast cancer. Obes Rev. 2006;7:333–340. doi: 10.1111/j.1467-789X.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- Dill CD, Harbuzariu A, Harmon TL, Lipsey CC, Loye A, Gonzalez-Perez RR. Abstract C52: Targeting the obesity/leptin link to TNBC: A breast cancer disparity problem. Cancer Epidemiology Biomarkers & Prevention. 2016;25:C52–C52. [Google Scholar]
- Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- Frankenberry KA, Skinner H, Somasundar P, McFadden DW, Vona-Davis LC. Leptin receptor expression and cell signaling in breast cancer. Int J Oncol. 2006;28:985–993. [PubMed] [Google Scholar]
- Gong Y, Ishida-Takahashi R, Villanueva EC, Fingar DC, Munzberg H, Myers MG., Jr The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J Biol Chem. 2007;282:31019–31027. doi: 10.1074/jbc.M702838200. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez RR, Lanier V, Newman G. Leptin's Pro-Angiogenic Signature in Breast Cancer. Cancers (Basel) 2013;5:1140–1162. doi: 10.3390/cancers5031140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvie M, Hooper L, Howell AH. Central obesity and breast cancer risk: a systematic review. Obes Rev. 2003;4:157–173. doi: 10.1046/j.1467-789x.2003.00108.x. [DOI] [PubMed] [Google Scholar]
- Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Idowu MO, Kmieciak M, Dumur C, Burton RS, Grimes MM, Powers CN, Manjili MH. CD44(+)/CD24(-/low) cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Hum Pathol. 2012;43:364–373. doi: 10.1016/j.humpath.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligibel J. Obesity and breast cancer. Oncology (Williston Park) 2011;25:994–1000. [PubMed] [Google Scholar]
- Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- Myers MG., Jr Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog Horm Res. 2004;59:287–304. doi: 10.1210/rp.59.1.287. [DOI] [PubMed] [Google Scholar]
- Newman G, Gonzalez-Perez RR. Leptin-cytokine crosstalk in breast cancer. Mol Cell Endocrinol. 2014;382:570–582. doi: 10.1016/j.mce.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien SN, Welter BH, Price TM. Presence of leptin in breast cell lines and breast tumors. Biochem Biophys Res Commun. 1999;259:695–698. doi: 10.1006/bbrc.1999.0843. [DOI] [PubMed] [Google Scholar]
- Ohi Y, Umekita Y, Yoshioka T, Souda M, Rai Y, Sagara Y, Tanimoto A. Aldehyde dehydrogenase 1 expression predicts poor prognosis in triple-negative breast cancer. Histopathology. 2011;59:776–780. doi: 10.1111/j.1365-2559.2011.03884.x. [DOI] [PubMed] [Google Scholar]
- Renehan AG, Roberts DL, Dive C. Obesity and cancer: pathophysiological and biological mechanisms. Arch Physiol Biochem. 2008;114:71–83. doi: 10.1080/13813450801954303. [DOI] [PubMed] [Google Scholar]
- Saxena NK, Vertino PM, Anania FA, Sharma D. leptin-induced growth stimulation of breast cancer cells involves recruitment of histone acetyltransferases and mediator complex to CYCLIN D1 promoter via activation of Stat3. J Biol Chem. 2007;282:13316–13325. doi: 10.1074/jbc.M609798200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoussi K, Strosberg AD, Bouaouina N, Ben Ahmed S, Helal AN, Chouchane L. Leptin and leptin receptor polymorphisms are associated with increased risk and poor prognosis of breast carcinoma. BMC Cancer. 2006;6:38. doi: 10.1186/1471-2407-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmacz E. Obesity hormone leptin: a new target in breast cancer? Breast Cancer Res. 2007;9:301. doi: 10.1186/bcr1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tessitore L, Vizio B, Jenkins O, De Stefano I, Ritossa C, Argiles JM, Benedetto C, Mussa A. Leptin expression in colorectal and breast cancer patients. Int J Mol Med. 2000;5:421–426. doi: 10.3892/ijmm.5.4.421. [DOI] [PubMed] [Google Scholar]
- Tessitore L, Vizio B, Pesola D, Cecchini F, Mussa A, Argiles JM, Benedetto C. Adipocyte expression and circulating levels of leptin increase in both gynaecological and breast cancer patients. Int J Oncol. 2004;24:1529–1535. [PubMed] [Google Scholar]
- Thiagarajan PS, Hitomi M, Hale JS, Alvarado AG, Otvos B, Sinyuk M, Stoltz K, Wiechert A, Mulkearns-Hubert E, Jarrar AM, et al. Development of a Fluorescent Reporter System to Delineate Cancer Stem Cells in Triple-Negative Breast Cancer. Stem Cells. 2015;33:2114–2125. doi: 10.1002/stem.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchoa Dde M, Graudenz MS, Callegari-Jacques SM, Hartmann CR, Ferreira BP, Fitarelli-Kiehl M, Edelweiss MI. Expression of cancer stem cell markers in basal and penta-negative breast carcinomas--a study of a series of triple-negative tumors. Pathol Res Pract. 2014;210:432–439. doi: 10.1016/j.prp.2014.03.005. [DOI] [PubMed] [Google Scholar]
- Villanueva EC, Myers MG., Jr Leptin receptor signaling and the regulation of mammalian physiology. Int J Obes (Lond) 2008;32(Suppl 7):S8–12. doi: 10.1038/ijo.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10:717–728. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- Vona-Davis L, Rose DP. Adipokines as endocrine, paracrine, and autocrine factors in breast cancer risk and progression. Endocr Relat Cancer. 2007;14:189–206. doi: 10.1677/ERC-06-0068. [DOI] [PubMed] [Google Scholar]
- Yin N, Wang D, Zhang H, Yi X, Sun X, Shi B, Wu H, Wu G, Wang X, Shang Y. Molecular mechanisms involved in the growth stimulation of breast cancer cells by leptin. Cancer Res. 2004;64:5870–5875. doi: 10.1158/0008-5472.CAN-04-0655. [DOI] [PubMed] [Google Scholar]
- Zheng Q, Banaszak L, Fracci S, Basali D, Dunlap SM, Hursting SD, Rich JN, Hjlemeland AB, Vasanji A, Berger NA, et al. Leptin receptor maintains cancer stem-like properties in triple negative breast cancer cells. Endocr Relat Cancer. 2013;20:797–808. doi: 10.1530/ERC-13-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Guo S, Gonzalez-Perez RR. Leptin pro-angiogenic signature in breast cancer is linked to IL-1 signalling. Br J Cancer. 2011;104:128–137. doi: 10.1038/sj.bjc.6606013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A&B) Photomicrographs of the LEPR-transfected non-CSCs showed an increase in the number of cells expressing GFP compared with the control non-transfected non-CSCs. Scale bar – 100 µM.