

Abstract
A polypeptide-based hydrogel system, when prepared from a diblock polymer with a ternary copolypeptide as one block, exhibited thermo-, mechano- and enzyme-responsive properties, which enabled the encapsulation of naproxen (Npx) during the sol–gel transition and its release in the gel state. Statistical terpolymerizations of l-alanine (Ala), glycine (Gly) and l–isoleucine (Ile) NCAs at a 1:1:1 feed ratio initiated by monomethoxy monoamino-terminated poly(ethylene glycol) afforded a series of methoxy poly(ethylene glycol)-block-poly(l-alanine-co-glycine-co-l-isoleucine) (mPEG-b-P(A-G-I)) block polymers. β-Sheets were the dominant secondary structures within the polypeptide segements, which facilitated a heat-induced sol–to-gel transition, resulting from the supramolecular assembly of β-sheets into nanofibrils. Deconstruction of the three–dimensional networks by mechanical force (sonication) triggered the reverse gel–to–sol transition. Certain enzymes could accelerate the breakdown of the hydrogel, as determined by in vitro gel weight loss profiles. The hydrogels were able to encapsulate and release Npx over 6 days, demonstrating the potential application of these polypeptide hydrogels as an injectable local delivery system for small molecule drugs.
Graphical abstract
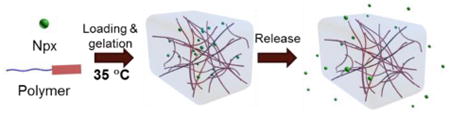
Multi-responsive polypeptide-based hydrogels exhibited thermo-, mechano-, and enzyme-responsive properties, enabling performance as a delivery system for encapsulation and release of naproxen.
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) have been widely used in the clinical treatment of acute and chronic pain and inflammation by inhibiting cyclooxygenases (COXs).1 NSAIDs are usually ingested orally in high dosages.1, 2 Despite their wide and systemic usage, in such high dosages, NSAIDs can cause undesirable side effects, such as adverse gastrointestinal symptoms and cardiovascular risks. To minimize these side effects, NSAIDs can be incorporated in a drug delivery system, for direct injection into target sites.3, 4 After injection of this drug-loaded complex, NSAIDs may be released in response to applied external stimuli or internal enzyme degradation to circumvent systemic dosing and minimize undesirable side effects.5, 6 Therefore, it is expected to be clinically beneficial to develop a stimuli-responsive biocompatible material into a localized delivery agent for the treatment of inflammation, such as osteoarthritis.
Hydrogels have attracted significant attention in the past decades for their potential applications as scaffolds for tissue engineering, matrices for controlled drug release, and absorbents for aqueous pollutant capture and removal.7-11 They are a class of materials comprising polymeric or low-molecular-weight crosslinked hydrogelators that entrap substantial amounts of water or aqueous solutions while maintaining a distinct three-dimensional structure.12, 13 The high water content and tunable properties of these materials render them suitable synthetic mimics of soft tissue microenvironments, as well as promising media for localized storage and delivery of therapeutic agents.14, 15 In addition, the controlled incorporation of supramolecularly noncovalent interactions or chemically covalent linkages into hydrogelators yields hydrogels that respond to various physical, chemical or biological stimuli.16, 17 Regulated reorganizations within the three-dimensional structures and transitions between the sol and gel states provide tunable release rates of encapsulated small molecules.18 Recently, Cao et al. reported the controlled release of naproxen (Npx), one kind of NSAID, from a γ-ray-responsive hydrogel, composed of a diselenide-containing polymer and an Npx conjugated dipeptide, by UV irradiation.6 At present, Npx-loaded hydrogels remain limited to primarily low-molecular-weight hydrogelators,3, 4 and few studies have been performed on polymeric hydrogelators for NSAID delivery.
Among biodegradable polymers, polypeptides offer significant advantages, including high biocompatibility, multiple functionalities and tunable secondary structures.19-23 These materials have been widely used in the construction of supramolecularly-assembled hydrogels.18, 24-26 Their broad applicability is attributed to the precisely-defined nano- and microstructures derived from hierarchical assembly of polypeptides, and the variety of stimuli-responsive behaviors exhibited by natural and synthetic polypeptides.27-29 Secondary structures, especially the α-helix and β-sheet conformations, can serve as the driving forces to form well-defined fibrous structures, which further assemble into noncovalently crosslinked three-dimensional networks that can encapsulate aqueous media to form hydrogels.24, 30, 31 Recently, Chen and co-workers reported a biocompatible polypeptide hydrogelator, poly(ethylene glycol)-block-poly(γ-propargyl-l-glutamate) (PEG-b-PPLG), with a temperature-triggered sol–to–gel transition driven by the formation of β-sheet structures.26 Herein, we report a thermo-, mechano-, and enzyme-responsive polypeptide-based hydrogelator, methoxy poly(ethylene glycol)-block-poly(l-alanine-co-glycine-co-l-isoleucine) (mPEG-b-P(A-G-I)), synthesized by statistical terpolymerization of α-amino acid N–carboxyanhydride (NCA) monomers of l-alanine, glycine and l-isoleucine. This termonomer mixture was selected for statistical incorporation of matrix metalloproteinase (MMP) MMP-2 and 9-active G-IAG cleavage sites.21, 32 In contrast to many polypeptide hydrogelators that undergo gel-to-sol with increased temperature, a reverse thermal gelation profile and sonication-triggered gel-to-sol transition were observed for these materials. Detailed characterization studies and analyses were performed to investigate the driving force for the heat-induced hydrogelation and to understand the correlation between nanostructural morphology and stimuli-responsive sol-gel transitions. The encapsulation and release of Npx demonstrated the potential application of these polypeptide hydrogels as a local delivery system to reduce side effects stemming from the systemic delivery of Npx.
Experimental section
Materials
Ethyl acetate, n-hexane, tetrahydrofuran (THF), diethyl ether, N,N‐dimethylformamide (DMF, anhydrous), l-alanine, l-isoleucine, bis(trichloromethyl) carbonate, glycine, trichloromethyl chloroformate, Proteinase K, Trypsin and naproxen (Npx) were purchased from Sigma-Aldrich (USA). Monomethoxy-monoamino-terminated poly(ethylene glycol) (mPEG45-NH2, Mn = 2000 g/mol) was purchased from Rapp Polymere (Germany). All chemicals were used without further purification, unless otherwise noted. Nanopure water (18.2 MΩ•cm) was acquired from a Milli-Q water filtration system, Millipore Corp. (St. Charles, MO). Tris-HCl buffered saline and phosphate buffered saline (PBS) were purchased from Sigma-Aldrich (USA) as dry powders, and dissolved in deionized water (1.0 L) to obtain the desired buffer solutions.
Instrumentation
1H and 13C NMR spectra were recorded on a Varian Inova 300 spectrometer interfaced to a UNIX computer using VnmrJ software. Chemical shifts were referenced to the solvent resonance signals. Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) spectra were recorded on an IR Prestige 21 system (Shimadzu Corp.) and analysed using IRsolution v. 1.40 software. Circular dichroism (CD) spectra were recorded on a Chirascan CD spectrometer from Applied Photophysics, Ltd. (Leatherhead, UK) equipped with a 150 watt xenon arc lamp. The polymer solutions for CD measurements were prepared at the indicated concentration in nanopure water. CD spectra were acquired between 180 and 280 nm, using a wavelength step of 1.0 nm, in a quartz cell with a path length of 1.0 mm and analyzed using Pro-Data Version 5 software.
Thermogravimetric analysis (TGA) was performed under argon atmosphere using a Mettler-Toledo model TGA/SDTA851e (Mettler–Toledo, Inc., Columbus, OH), with a heating rate of 10 °C/min. Measurements were analysed using Mettler-Toledo STARe v. 7.01 software. Glass transition temperatures (Tg) were measured by differential scanning calorimetry (DSC) on a Mettler-Toredo DSC822®, with a heating rate of 10 °C/min. Measurements were analysed using Mettler-Toledo STARe v. 7.01 software. The Tg was taken as the midpoint of the inflection tangent of the second heating scan.
Wide-angle X-ray scattering (WAXS) was performed on a Bruker D8 Bragg Brentano X-ray powder diffractometer. Samples were placed in the sample holder of a two-circle goniometer, in a radiation safety enclosure. The X-ray source was a 2.2 kW Cu X-ray tube, maintained at an operating current of 40 kV and 40 mA. The standard Bragg-Brentano para-focusing mode was employed, with the X-ray source diverging from a diffraction slit (1 mm) of the tube to irradiate the sample and then converging onto a position-sensitive X-ray Detector (Lynx-Eye, Bruker-AXS). The two-circle 250 mm diameter goniometer was computer-controlled with independent step motors and optical encoders for the θ and 2θ circles with a smallest angular step size of 0.0001° 2θ. Data collection was automated using the COMMANDER program by employing a DQL file and analyzed in EVA software.
Transmission electron microscopy (TEM) was performed on a JEOL 1200 EX microscope operating at 100 kV, and micrographs were recorded at calibrated magnifications using an SLA-15C CCD camera. The final pixel size was 0.42 nm/pixel. Samples for TEM measurements were prepared as follows: 10 μL of the diluted solution (with a polymer concentration of 1 mg/mL) was deposited onto a carbon-coated copper grid, and after 2 min, the excess solution was quickly wicked away by a piece of filter paper. The samples were then negatively stained with 1 wt% phosphotungstic acid (PTA) aqueous solution. After 30 s, the excess staining solution was quickly wicked away by a piece of filter paper and the samples were left to dry under vacuum overnight.
Scanning electron microscopy (SEM) imaging was performed on a JOEL JSM 6400 SEM operated at an acceleration voltage of 15 kV. The preparation of samples for SEM involved placing 100 μL hydrogel on a carbon thin film. The gel was then subjected to immediate freezing by liquid nitrogen, followed by lyophilisation for 3 days. The surface of the gel was sputter-coated with gold for 3 min under argon before imaging.
Sonication-responsive experiments were performed with an ultrasonic homogenizer (maximum power, 150W, 20KHz, Model 150 V/T, Biologics, Inc.) equipped with a micro tip with a diameter of 3.81 mm, employing a power outlet of 45 W at a frequency of 20 KHz at room temperature.
Experimental procedures
Synthesis of l-alanine N-carboxyanhydride (Ala NCA) monomer
In a 250 mL three-necked round bottom flask equipped with a magnetic stir bar, condenser, and N2 inlet, l-alanine (5.0 g, 56 mmol, 3 equiv) and bis(trichloromethyl) carbonate (5.6 g, 19 mmol, 1 equiv) were added and suspended in 100 mL dry THF at 50 °C under N2 bubbling for 4 h. The reaction mixture was cooled to room temperature and unreacted l-alanine was removed by filtration. The filtrate was concentrated and the resulting brown oil material was further purified by recrystallization three times from THF/n–hexane 2:1 (v/v), and dried under vacuum to obtain the product as white crystals (1.4 g, yield: 63%). The product was stored in a -20 °C freezer under N2 protection. 1H NMR (DMSO-D6, 300 MHz, ppm): δ 1.33 (d, 3H, J = 7.0 Hz, COCH(CH3)NH), 4.48 (q, 1H, J = 7.0 Hz, COCH(CH3)NH), 9.00 (br, 1H, COCH(CH3)NH). 13C NMR (DMSO-D6, 75 MHz, ppm): δ 18.8, 52.8, 151.7, 172.4. FTIR (cm-1): 3500-3100, 2838, 1840, 1760, 1280, 1139, 1023, 925. HRMS: calculated [M-H]- for C4H5NO3: 114.0191, found: 114.0193.
Synthesis of glycine (Gly) NCA monomer
In a 250 mL three-necked round bottom flask equipped with a magnetic stir bar, condenser, and N2 inlet, glycine (5.0 g, 67 mmol, 3 equiv) was suspended in 100 mL THF and heated to 50 °C. Trichloromethyl chloroformate (4.4 g, 22 mmol, 1 equiv) was added into the reaction by syringe and the reaction mixture was heated at reflux for another 4 h under N2 atmosphere. The reaction mixture was filtered before cooling to room temperature. The filtrate was concentrated, recrystallized three times from THF/n-hexane 2:1 (v/v) and dried under vacuum to obtain the product as white crystals (1.5 g, yield: 67%). The product was stored in a -20 °C freezer under N2 atmosphere. 1H NMR (300 MHz, DMSO-D6, ppm): δ 4.18 (s, 2H, COCH2NH), 8.83 (br, 1H, COCH2NH). 13C NMR (75 MHz, DMSO-D6, ppm): δ 46.3, 153.0, 169.4. FTIR (cm-1): 3500-3100, 2955, 1876, 1740, 1275, 1113, 1061, 925. HRMS: calculated [M-H]- for C3H3NO3: 100.0035, found: 100.0039.
Synthesis of l-isoleucine (Ile) NCA monomer
In a 250 mL three-necked round bottom flask equipped with a magnetic stir bar, condenser, and N2 inlet, l-isoleucine (5.0 g, 42 mmol, 3 equiv) and bis(trichloromethyl) carbonate (4.6 g, 14 mmol, 1 equiv) were added and suspended in 100 mL dry THF at 50 °C under N2 bubbling for 4.5 h. The reaction mixture was cooled to room temperature and unreacted l-isoleucine was removed by filtration. The filtrate was concentrated and the resulting yellow oil was recrystallized three times from THF/n-hexane 2:1 (v/v), and dried under vacuum to obtain the product as white crystals (2.2 g, yield: quantitative). The product was stored in a -20 °C freezer under N2 protection. 1H NMR (DMSO-D6, 300 MHz, ppm): δ 0.86 (t, 3H, J = 9.0 Hz, CH3CHCH2CH3), 0.92 (d, 3H, J = 7.0 Hz, CH3CHCH2CH3), 1.28 (m, 2H, CH3CHCH2CH3), 1.79 (s, 1H, CH3CHCH2), 4.39, (s, 1H, COCHNH), 9.09 (br, 1H, COCHNH). 13C NMR (DMSO-D6, 75 MHz, ppm): δ 11.3, 14.7, 23.8, 36.5, 61.7, 152.2, 170.8. FTIR (cm-1): 3500-3100, 2970, 2881, 1848, 1750, 1215, 1091, 1070, 939. HRMS: calculated [M-H]- for C7H11NO3: 156.0661, found: 156.0665.
Synthesis of methoxy poly(ethylene glycol)-block-poly(l-alanine-co-glycine-co-l-isoleucine) (mPEG-b-P(A-G-I)) by statistical terpolymerization of NCA monomers initiated by mPEG-NH2
As a representative example, the synthesis of mPEG45-b-P(A5-G7-I5) is described. In a 10 mL flame-dried Schlenk flask equipped with a magnetic stir bar, mPEG45-NH2 (80.0 mg, 0.04 mmol, 1 equiv) was added into a solution of Ala NCA (23.0 mg, 0.2 mmol, 5 equiv), Gly NCA (20.0 mg, 0.2 mmol, 5 equiv) and Ile NCA (31.4 mg, 0.2 mmol, 5 equiv) dissolved in 6.4 mL anhydrous DMF solution. The reaction mixture was stirred at 400 rpm under continuous N2 flow (100 mL/min) at room temperature for 12 h. During the polymerization, the Schlenk flask was capped with a rubber stopper with a needle outlet connected to a syringe barrel filled with drierite. Monomer consumption was monitored by the disappearance of the NCA anhydride peak at 1788 cm-1 using ATR-FTIR. The polymerization solution was precipitated into diethyl ether under vigorous stirring, and the polymer was isolated by centrifugation and dried under vacuum at room temperature to yield a white powder (102.2 mg, yield: 80%). 1H NMR (300 MHz, TFA-D, ppm): δ 0.70-1.10 (br, 15H, CH3CHCH2CH3; br, 15H, CH3CHCH2CH3), 1.46 (br, 10H, CH3CHCH2CH3; br, 15H, CH3CH), 1.60-2.10 (br, 5H, CH3CHCH2CH3), 3.52 (s, 3H, CH3O), 3.85 (s, 180H, CH2CH2O), 4.20 (br, 14H, NHCH2CO), 4.64 (br, 10H, NHCHCO). 13C NMR (75 MHz, TFA-D, ppm): δ 12.9, 13.8, 15.8, 24.0, 36.9, 42.6, 50.1, 57.5, 69.5, 70.7, 172.5, 174.8, 175.6. FTIR (cm-1): 3286, 2868, 1699, 1625, 1515, 1451, 1349, 1249, 1099, 948, 849, 696. DSC: Tg = -17 °C, Tc = -15 °C, Tm = 30 °C, Tg = 126 °C. TGA in N2: 20-233 °C, 2% mass loss; 233-367 °C, 29% mass loss; 367-430 °C, 46% mass loss; 430-500 °C, 3% mass loss; 20% mass remaining above 500 °C.
Determination of critical gelation temperature (Tgel)
Polymer solutions were prepared by dissolving polypeptide powders in nanopure water at the desired concentrations. Homogeneous solutions were obtained by 2 min sonication with a probe sonicator at room temperature. Hydrogel formation was determined using the test tube inversion method.33 Briefly, each sample was dissolved in nanopure water at a predetermined concentration in a 2 mL vial. After equilibrium at room temperature overnight, the vial was immersed in an oil bath equilibrated at given temperatures for 30 min, and the vial was then inverted. If no flow was observed after 5 min, the sample was regarded as a gel. For measurement of the sol–gel transition temperature, the temperature was raised at a step of 0.5 °C/h. The sol-gel transition temperatures were recorded as the average of three measurements.
Hydrolytic and enzymatic degradation of polypeptide hydrogels
Degradation of the polypeptide hydrogel was performed by measuring the weight loss of the hydrogel at predetermined time points spanning 10 days after immersion of the hydrogel in degradation media.24 An aliquot of polypeptide gelator solution (4 wt% in nanopure water, 0.3 mL) was placed in a 1.5 mL vial and maintained at 37 °C overnight for gel formation. Tris-HCl buffer (0.05 M, pH = 8.0, 0.2 wt% NaN3, 0.5 mL) was added gently into the vial without disturbing the gel. The vial was then maintained at 37 °C throughout the degradation study. Tris-HCl buffer solution, together with any accumulated gel debris, was removed at desired time points and the weight of the vial was recorded to assess the change of hydrogel weight. The remaining weight percent of the hydrogels was calculated from the weight of the hydrogel at each time point relative to the weight measured before Tris-HCl buffer addition. In the enzymatic degradation study, the enzyme was first dissolved in Tris–HCl buffer solution (0.05 M, pH = 8.0, 0.2 wt% NaN3) with a concentration of 1 mg/mL. The enzyme solution was stored at -20 °C and warmed to 37 °C immediately prior to addition into the hydrogel. Proteinase K and Trypsin were added at multiple time points over the duration of the experiment, and each degradation study was repeated three times, with the remaining weight listed as the average of the three measurements. Due to limited supply and high expense of MMP-2, the degradation study was conducted with a single initial time point for addition of MMP-2 and in a single run.
Naproxen (Npx) encapsulation and release
The loading of Npx was conducted by dissolving the block polypeptides in PBS buffer solution (0.01 M, pH = 7.4, 0.3 mL) containing 1 mg/mL of Npx. Each vial was held at 37 °C overnight to facilitate hydrogel formation. PBS buffer solution (pH = 7.4, 0.5 mL) was added to each vial and the samples were incubated at 37 °C, shaking at 60 rpm during the release study. At predetermined time points, the PBS buffer was removed for analysis and replaced with fresh solutions (0.5 mL). The release experiments were repeated three times and the error bars shown are the standard deviations. The concentration of the released Npx was determined as a function of time on a Shimadzu Prominence HPLC equipped with an SPD-20 AV prominence UV-Vis detector set to 254 nm and a Waters × Bridge C8 column (4.6 × 150 mm, 5 μm, 100 Å). The system was operated at 40 °C in an eluent system composed of 55% 20 mM ammonium acetate buffer and 45% acetonitrile in isocratic mode with a flow rate of 1 mL/min and a run time of 15 min.34
Results and discussion
Stemming from our general interest in the creation of biodegradable stimuli–responsive polymeric materials with simple molecular design and synthetic feasibility, especially for future biomedical applications, we began investigation into the syntheses and physical properties of hybrid diblock polymers, having a statistical terpolypeptide as one of the block segments, with the expectation that the polypeptide would respond to multiple external stimuli, including thermal, mechanical and biological triggers. Recently, our group reported a novel approach to tune the stimuli-responsive properties of polypeptide gel systems via control of the secondary structures within the polypeptide segment, which was offered by variation in the feedstock selection during the statistical copolymerization of various N-carboxyanhydride (NCA) monomers. The last two decades have witnessed significant development of controlled ring-opening polymerization (ROP) of amino acid NCAs as a convenient and efficient synthetic approach for the preparation of well-defined polypeptide materials in large scales.35-41 In fact, NCA ROP can be conducted using normal Schlenk techniques, with the enhancement of polymerization rate and retention of controlled features achieved through a straightforward N2 flow method.40, 42 Using this technique, the methoxy poly(ethylene glycol)-block-poly(l-alanine-co-glycine-co-l-isoleucine) (mPEG-b-P(A-G-I)) was synthesized by statistical copolymerization of l-alanine (Ala), glycine (Gly) and l–isoleucine (Ile) NCAs, initiated by monomethoxy monoamino-terminated poly(ethylene glycol) (mPEG45-NH2, Mn = 2000 g/mol) as the macroinitiator under continuous N2 flow (100 mL/min), as shown in Scheme 1. A series of block polymers with polypeptide segments of various lengths was synthesized by varying the ratio of NCA monomers to the macroinitiator, while maintaining the ratio of three NCA monomers at 1 : 1 : 1 (Table 1). For each terpolymerization, the NCA monomers and macroinitiator were dissolved in anhydrous N,N–dimethylformamide (DMF) and the ROP was allowed to proceed for 12 h at room temperature. Spontaneous gelation in DMF was observed during each polymerization within 8 h, and the targeted diblock polymers were obtained as white powders by precipitation of the reaction mixture into diethyl ether, followed by drying under vacuum.
Scheme 1.

Synthetic approach for mPEG-b-P(A-G-I) diblock copolymer.
Table 1.
Average degree of polymerization (DPn), number-averaged molecular weight (Mn) and composition of the mPEG-b-P(A-G-I) diblock copolymers synthesized with different monomer to initiator ratios.
| Entry | M : Ia | DPnb | Mn (kDa)b | Polymerb |
|---|---|---|---|---|
| 1 | 15 : 1 | 17 | 3.3 | mPEG45-b-Poly(A5-G7-I5) |
| 2 | 30 : 1 | 30 | 4.2 | mPEG45-b-Poly(A8-G13-I9) |
| 3 | 50 : 1 | 43 | 5.1 | mPEG45-b-Poly(A15-G19-I9) |
| 4 | 80 : 1 | 63 | 6.4 | mPEG45-b-Poly(A24-G29-I10) |
| 5 | 100 : 1 | 72 | 6.9 | mPEG45-b-Poly(A28-G34-I10) |
All polymerizations were conducted using mPEG45-NH2 as the macroinitiator at a concentration of 10 mg/mL, and a molar ratio of Ala : Gly : Ile = 1 : 1 :1. M: I indicated the ratio of the total monomer (Ala + Gly + Ile) to initiator.
Calculated by 1H NMR spectroscopy.
The molecular weight and composition of the block polymers were determined by 1H NMR spectroscopy in deuterated trifluoroacetic acid (TFA-D) to break the strong hydrogen bonding interactions within the polypeptide segments in order to solubilize the polymers (Figure 1). Comparison of the intensities of the methylene proton resonances of the mPEG chains at ca. 3.85 ppm (b in Figure 1) with those of the Gly methylene protons at ca. 4.20 ppm (e in Figure 1), the Ile side chain methine proton at ca. 1.85 ppm (g in Figure 1), and the backbone methine protons for both Ile and Ala at ca. 4.64 ppm (c+f in Figure 1) yielded the block lengths and compositions. For both Ala and Gly, the calculated numbers of repeat units from 1H NMR spectroscopy correlated linearly with the theoretical values, while for Ile, the numbers of repeat units in the obtained polymers plateaued in polymerizations conducted at Ile NCA monomer to macroinitiator ratios above 10 (Figure S1 in ESI†). These results suggested higher reactivities of Gly and Ala NCA monomers relative to the Ile NCA monomer.43-45
Figure 1.
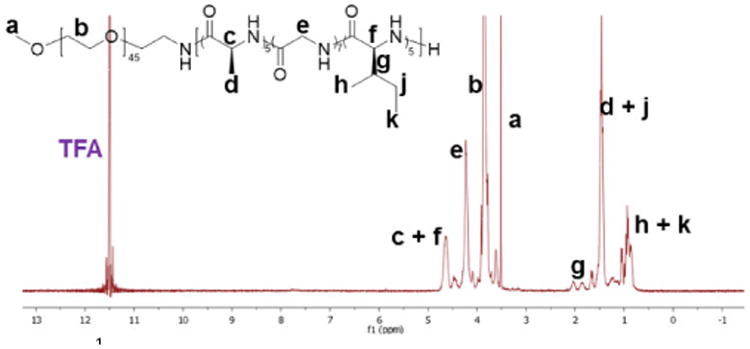
1H NMR spectrum of polymer 1 acquired in TFA-D.
The ability to form hydrogels of copolymers with different polypeptide block lengths was characterized by test tube inversion experiments.33, 46 Polymer solutions were prepared at a variety of concentrations in nanopure water at room temperature, and hydrogels formed upon heating the solutions until gelation was observed, i.e., above the critical gelation temperature (Tgel). The obtained hydrogels were stable and no gel-to-sol transitions were observed when hydrogels were left undisturbed for at least 3 days at room temperature. The effects of polymer concentration and composition on Tgel were determined by measurement of Tgel values for a series of block polymers as a function of concentration, and the results are summarized in Figure 2. For each polymer, Tgel decreased as polymer concentration increased, as seen from the sol-gel phase diagram in Figure 2. For example, the Tgel of polymer 1 decreased from 84 °C for a 2 wt% solution to 22 °C for a 6 wt% solution. For 6 wt% solutions, the Tgel value increased from 22 °C for polymer 1, with a polypeptide block length of 17 repeat units, to 32 °C for polymer 2, with a polypeptide block length of 30 repeat units, whereas the Tgel value decreased to 16 °C for polymer 5, having the longest polypeptide block (DPn = 72). Due to a Tgel value (35 °C) similar to the physiological temperature of the human body, we focused our efforts on hydrogels formed from polymer 1 solutions (4 wt%) to investigate the potential for application as a drug delivery system.
Figure 2.
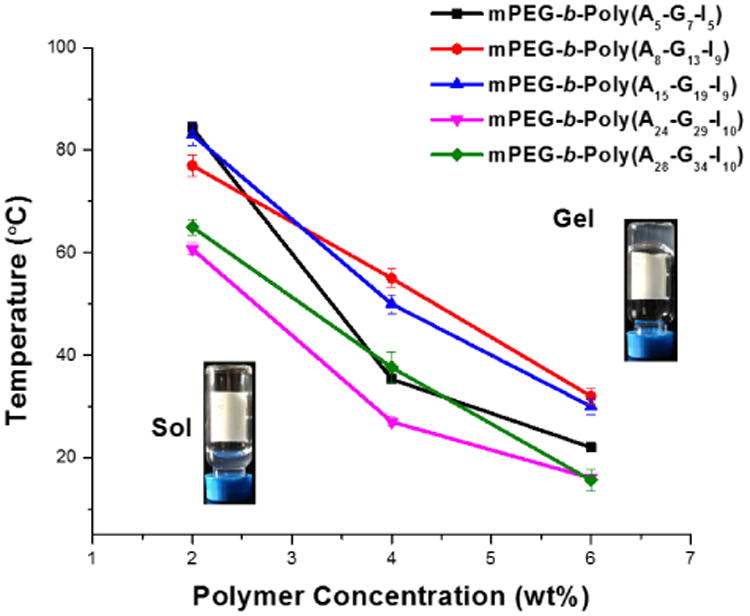
Tgel of mPEG-b-P(A-G-I) hydrogels as a function of polymer concentration.
In order to further understand the gelation mechanism, the supramolecular structure of the 4 wt% polymer 1 hydrogel in the solid state was investigated by attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) and wide-angle X-ray scattering (WAXS). The characteristic absorbance bands of secondary structures were clearly observed in the FTIR spectrum, especially in the amide I region (1600 - 1700 cm-1) (Figure 3). The absorbance bands at 1701 cm-1 and 1625 cm-1 (amide I), 1515 cm-1 (amide II) and 694 cm-1 (amide V), were attributed to β-sheet conformation.47 Further quantitative analyses of secondary structure populations were conducted by employing the secondary derivative of the absorbance as a function of wavenumber, and then using deconvolution strategies to extract the relative peak intensities in the heavily populated amide I region (Figure S2 in ESI†).48-50 The β-sheet contribution was calculated to be 63%, which was similar to the β-sheet content (66%) for PEG45-b-PGly30 in the solid state reported previously.43 In addition, the WAXS pattern of the dried hydrogels showed a d spacing of 4.6 Å with strong signals from PEG crystallization reflection (Figure S3 in ESI†), consistent with the melting and crystallizing peaks in the DSC traces of polymer 1 (Figure S4 in ESI†). A d spacing of 4.6 Å has also been observed in poly(l-alanylglycine)51 and the self‐assembled PEG-b-β-strand-peptides52, and attributed to antiparallel β-sheet conformation. The results from qualitative and quantitative FTIR analyses, together with the WAXS characterization, indicated the dominance of β-sheet structures in the solid state of the hydrogels.
Figure 3.
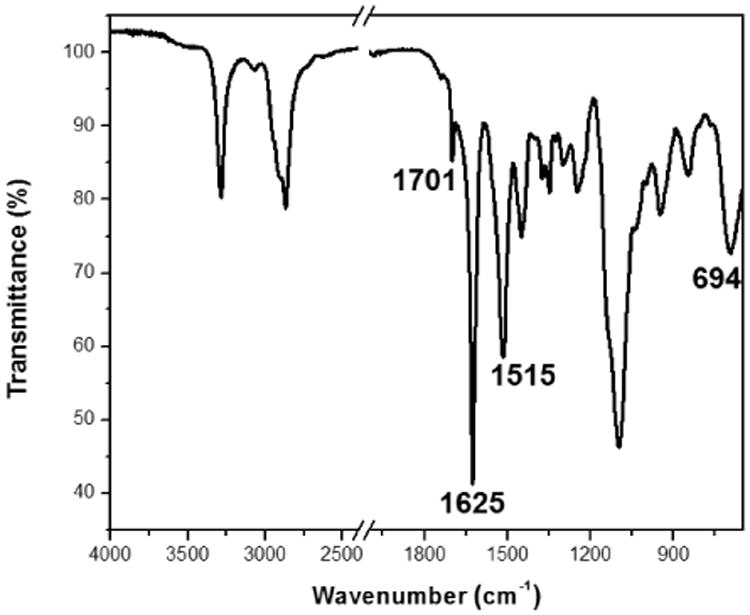
ATR-FTIR spectrum of polymer 1 in the solid state.
The strong absorption of water in the amide I region (1600 - 1700 cm-1) complicates the analysis of the secondary structure population of the hydrogels in the hydrated state. However, the O‐D bending of D2O appears at lower wavenumbers than that of H2O (Figure S5a in ESI†), enabling in situ analysis of secondary structure.53 The hydrogel sample was prepared by dissolving the polymer powder in D2O by sonication and maintaining the polymer solution at 37 °C overnight prior to analysis. With D2O as the background, the characteristic β-sheet absorbance at 1625 cm-1 (amide I) was observed in the FTIR spectrum (Figure 5b and Figure S5b in ESI†), indicating the domination of β-sheets in the secondary structure population in the hydrated gels, which was in agreement with the results from analyses conducted in the solid state.
Figure 5.

(a) TEM image of the hydrogel from polymers 1 and (b) SEM image of the lyophilized hydrogel from polymer 1.
To investigate the conformational changes of polymer 1 in nanopure water, temperature-dependent circular dichroism (CD) was employed to monitor the changes of the secondary structure as a function of temperature. Polymer solutions prepared at 0.2 wt% for CD measurements were placed in a quartz cell with a path length of 1.0 mm and spectra were acquired between 180 and 280 nm with a wavelength step of 1.0 nm. As revealed in Figure 4a, the intensity of the negative band at 220 nm increased gradually with temperature from 10 to 60 °C. The β-sheet contents at different temperatures were estimated with CDPro software with Program CONTILL and SDP48 as the reference sets, and the results are summarized in Figure 4b.54 As the temperature increased from 10 to 60 °C, the β-sheet content increased from 0% to 12%, suggesting gelation was driven by temperature-triggered β-sheet formation.
Figure 4.
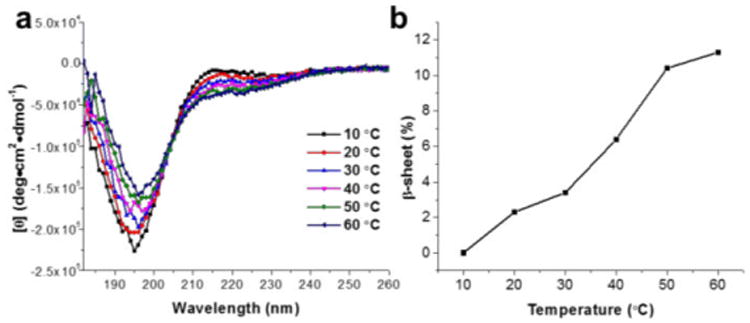
(a) CD spectra of polymer 1 in nanopure water (0.2 wt%) as a function of temperature from 10 to 60 °C. (b) Estimation of β-sheet content at each temperature from the CD spectra in the range of 190 – 240 nm with CDPro software using Program CONTILL and SDP48 as the reference sets.
Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) were utilized to study the nano- and microstructure of the hydrogels from polymer 1. The TEM sample was prepared from a 0.1 wt% polymer solution in nanopure water and stained with 1 wt% phosphotungstic acid (PTA) aqueous solution. The bright-field TEM image of the hydrogels showed fibrillary nanostructures with an average width of 13.3 nm (Figure 5a). The narrow width distribution of nanofibrils (standard deviation= 1.5 nm, Figure S6 in ESI†), obtained by counting 100 nanofibrils in the TEM image, indicated that the hydrogels had a well–defined nanofibrillar morphology. To visualize the microstructure of the hydrogels, SEM samples were prepared by lyophilising the hydrogels with the aim to maintain the gel networks. From the SEM image (Figure 5b), interconnected three-dimensional networks and highly porous structures with various sizes were observed, which could be potentially utilized to encapsulate and release cargoes, such as therapeutic drugs or cells.
Based on the results from NMR, ATR-FTIR, WAXS, CD, SEM and TEM analyses, a mechanism for the hydrogelation of mPEG-b-P(A-G-I) was proposed (Scheme 2). One-dimensional stacking of the β –sheet-forming polypeptide segments afforded well-defined fibrillary nanostructures, as observed spectrally and microscopically. Using hydrogels from polymer 1 as an example, the radius of gyration of mPEG with a molecular weight of 2000 Da can be estimated as 18 Å.52 The axial distance between adjacent α-amino acids is about 3.5 Å, thus the width of the polypeptide domain can be calculated to be 60 Å in the conformation of β-sheet secondary structures. Therefore, the diameter of the assembled nanofibrils can be estimated to be about 13.2 nm, which is in agreement with the measured width (13.3 nm) of nanofibrils in the TEM image. The hydrophilic mPEG segments are postulated to prevent the precipitation of the block polymer via increasing its aqueous solubility and maintaining the hydrogel networks by encapsulating water molecules. Heat-triggered gelation is attributed to the formation and strengthening of β-sheet secondary structures and the dehydration of PEG at elevated temperature to enhance the hydrophobic interactions. The entanglement of nanofibrils resulting from long range interactions, including peptide–peptide interaction and PEG-PEG association, constructs the three-dimensional networks of this polypeptide hydrogel system.55
Scheme 2.
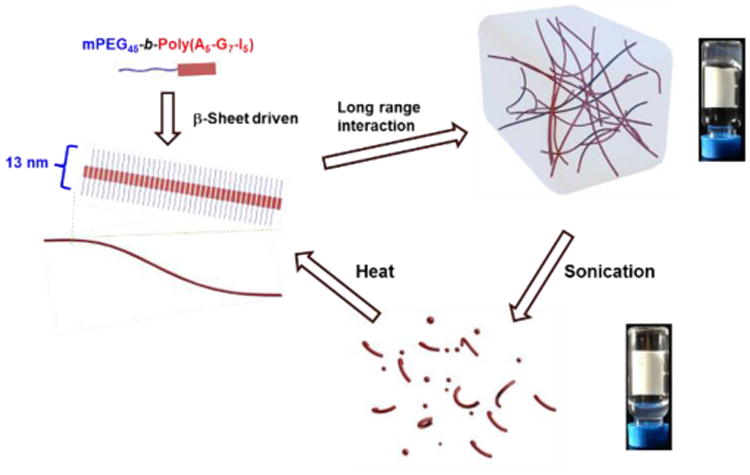
Proposed mechanism of the reversible sol-gel transitions observed in the hydrogels from polymer 1, including the heat‐induced hydrogelation resulting from supramolecularly assembled nanofibrils and sonication-triggered gel-to-sol transition.
The physical properties of the block polypeptide hydrogels, in particular stimuli-responsive properties, as well as the correlation between nanostructure and physical properties were then investigated. As shown in the quantitative ATR-FTIR analyses, the content of β-sheet secondary structure in the hydrogels was calculated to be 63%, thus a mechano-triggered gel-to-sol transition was expected, based on previous studies.43, 56 The mechano–responsiveness of the hydrogels was tested by applying sonication to the hydrogel for 30 s and the gel–to-sol transition was observed by the test tube inversion experiment. This transition was reversible, with the hydrogel reforming after maintaining the sol at Tgel (ca. 37 °C) for 1 h (Figure 6a). Due to the short time in the sonication experiment and the thermal stability of the hydrogels against heating up to 90 °C, the mechanical force applied by the sonicator was proposed to break the supramolecularly-assembled structures of hydrogels formed from polymer 1, resulting in the deconstruction of the hydrogels. In order to investigate the secondary structures of the copolymer after sonication, in situ FTIR of the hydrogels in the sol state was performed. As shown in Figure 6b, the FTIR spectrum of the sol state was almost identical to the spectrum of the gel state, indicating that no significant change in the secondary structures occurred during the sonication treatment, and that the dominant β –sheet contribution was maintained in the sol state. TEM was also employed to compare the difference of the nanoscale morphologies between the gel and sol states, with the aim to further understand the sol-gel transitions upon application of external stimuli. In the TEM image of the hydrogel after sonicating for 1 min to produce a sol, the well–defined nanofibrils observed in the gel state disappeared (Figure 5a), and short nanofibrils and spherical aggregates were observed instead (Figure 6c). Conversion of the longer nanofibrils into short nanofibrils and spherical aggregates, which lacked long range connectivity, resulted in the breakdown of the hydrogel networks.55 When the sol was heated to 37 °C for over 1 h, reformation of the longer nanofibrils reconnected the hydrogel networks (Scheme 2).
Figure 6.
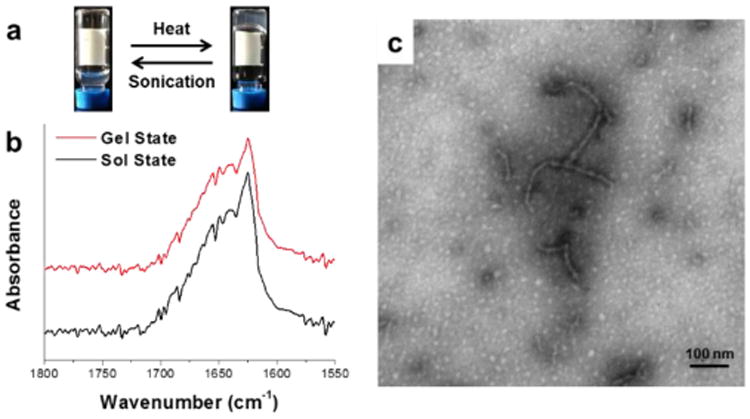
(a) Digital photographs showing reversible sol-gel transitions triggered by thermal and sonication stimuli, respectively. (b) In situ FTIR spectra in the gel state (red trace) and the sol state when sonication was applied (black trace). (c) TEM image of the hydrogel in the sol state from polymer 1 after sonication for 1 min.
Enzyme-responsive properties of synthetic polypeptide-based materials, including enzymatic degradation, are promising for biomedical applications. Therefore, in vitro gel weight loss profiles were obtained by measuring the weight loss of hydrogels formed from polymer 1 (4 wt%) at 37 °C.22, 24, 56 The enzymatic degradation medium was prepared by dissolving MMP-2, proteinase K or trypsin in Tris-HCl buffer (0.05 M, pH = 8.0, 0.2 wt% NaN3). The enzyme solution or Tris-HCl buffer without enzymes (0.5 mL) was added gently into each vial and removed along with accumulated gel debris at the selected time points. Measurements were performed in triplicate for proteinase K, trypsin and the control group (Tris-HCl buffer), but only once for MMP-2, due to limited supply and high cost. According to the weight loss profiles (Figure 7), trypsin, which cleaves peptide chains at lysine and arginine residues, had no significant effect on the hydrogel degradation, resulting in a similar degradation profile as the control group. In contrast, the hydrogels treated with proteinase K, an aggressive and broadly-active enzyme, exhibited much faster degradation, in which over 90% weight loss was observed after 72 h. As expected, MMP-2, which cleaves at G–IAG positions, resulted in intermediate degradation kinetics. It is uncertain whether the apparent plateauing of activity (after 48 h) for MMP-2 was due to a limited number of G-IAG sequences along the relatively short polypeptide chains or to a loss in enzymatic activity over time. Further experiments would be necessary to sequence the polymer and quantify the relative contributions from these effects, among others.
Figure 7.
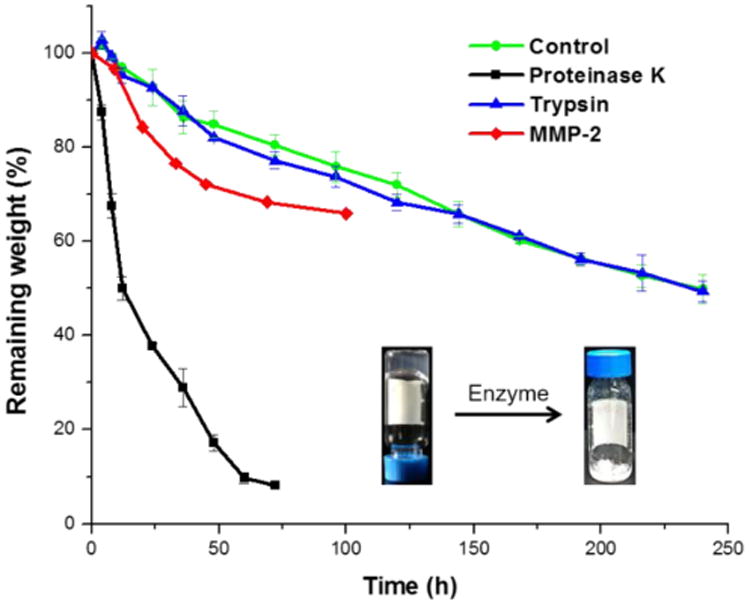
Weight loss profiles of hydrogels formed from polymer 1 in the presence and absence of enzymes. Measurements were repeated three times for proteinase K, trypsin and control group, and error bars represent the standard deviations. A single measurement was performed for MMP-2.
The utilization of this hydrogel for the entrapment and release of the small therapeutic drug, naproxen (Npx), was also investigated to explore the potential application of these materials for local drug delivery. Npx has a low solubility in nanopure water in its protonated form, but it can be dissolved in PBS buffer (0.01 M, pH = 7.4) at concentrations up to 1 mg/mL. Thus, Npx-loaded hydrogels were prepared by mixing the weight fraction of polymer 1 with the Npx solution in PBS buffer to yield a polymer concentration of 0.4 wt%, and transparent hydrogels were generated after the solution was maintained at 37 °C overnight.6 The obtained hydrogels were stable, and neither gel–to–sol transition nor precipitation of Npx was observed for at least one week at room temperature. The sonication-triggered gel–to-sol transition was also observed for the Npx-loaded hydrogels upon sonication for 10 s. The release of Npx from mPEG-b-P(A-G-I) polymer 1 hydrogels was monitored by HPLC to measure the concentration of Npx in the surrounding PBS buffer at 37 °C at predetermined time points over 6 days. As shown in Figure 8, a burst release of Npx up to 64%, was observed within 24 h, followed by slower release over the next 5 days, and reaching 80% at the end of the study. This encapsulation and release of the small molecule drug suggested the potential application of this polypeptide hydrogel system as a local drug delivery carrier to reduce the systematic drug dosage and associated undesirable side effects.
Figure 8.
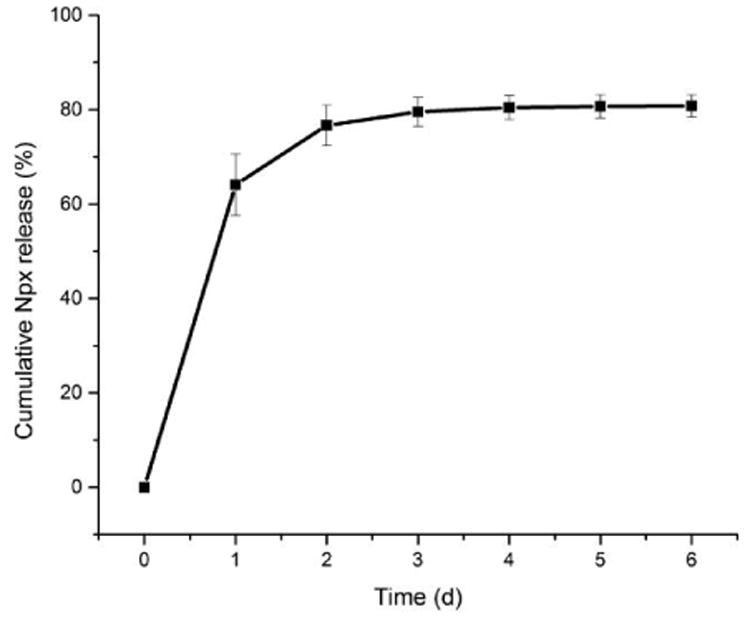
Cumulative release of Npx from a hydrogel formed from polymer 1 (0.4 wt% in 0.01 M PBS buffer, pH = 7.4) containing 1 mg/mL Npx at 37 °C over 6 days. Measurements were repeated three time and the error bars represented standard derivations.
Conclusions
In summary, a polypeptide hydrogel system prepared from ternary sequences of l-alanine, glycine and l-isoleucine within a polypeptide segment of a PEG-peptide diblock structure exhibited thermo-, mechano- and enzyme-responsive properties, which enabled the encapsulation and release of Npx. The diblock polymer, mPEG–b-P(A-G-I), was facilely synthesized from the statistical ROP of Ala, Gly and Ile NCA monomers initiated by mPEG-NH2. Reversible and repeatable sol-gel transitions were observed in response to externally-applied stimuli, with heat-induced sol-to-gel transitions and sonication-triggered gel-to-sol transitions. The Tgel was largely dependent on the polymer concentration, and the block length of the polypeptide segments, which could be tuned easily into the targeted temperature range, ca. 37 °C, to enable biomedical application of these materials. Detailed characterization studies revealed that the hydrogelation was driven by the supramolecular assembly of β-sheets into well–defined fiber–like nanostructures, which further entangled into a three–dimensional network through long range interactions. The mechanical force produced by sonication deconstructed the supramolecularly-assembled structures of the hydrogels, resulting in a gel-to-sol transition. In addition, enzymes that are either broadly aggressive or selective for G-IAG motifs were found to accelerate the breakdown of the polypeptide segments, resulting in the reversion of the hydrogels into solutions. Npx was loaded into the hydrogels at room temperature in the sol state and encapsulated within the hydrogel after heating to the gelation temperature. The release of Npx from the hydrogels diffused into the surrounding environment, demonstrating the potential application of this polypeptide-based hydrogel as a local drug delivery system, desired to reduce the systemic dosage of small molecule drugs with associated adverse side effects.
Supplementary Material
Acknowledgments
This work is financially supported from the National Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology (HHSN268201000046C), the National Science Foundation (DMR-1105304, DMR-1309724, and DMR-1507429), and the Welch Foundation through the W. T. Doherty-Welch Chair in Chemistry (Grant no. A-0001). The Microscopy & Imaging Center (MIC) at Texas A&M University is also gratefully acknowledged. The authors thank Professor Tadhg P. Begley at Texas A&M University for access to the circular dichroism spectrometer.
Footnotes
References
- 1.Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, Whelton A, Makuch R, Eisen G, Agarwal NM, Stenson WF, Burr AM, Zhao WW, Kent JD, Lefkowith JB, Verburg KM, Geis GS. J Am Med Assoc. 2000;284:1247–1255. doi: 10.1001/jama.284.10.1247. [DOI] [PubMed] [Google Scholar]
- 2.Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R, Morton D, Lanas A, Konstam MA, Baron JA, Investigators AT. N Engl J Med. 2005;352:1092–1102. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 3.Li JY, Kuang Y, Gao Y, Du XW, Shi JF, Xu B. J Am Chem Soc. 2013;135:542–545. doi: 10.1021/ja310019x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majumder J, Das MR, Deb J, Jana SS, Dastidar P. Langmuir. 2013;29:10254–10263. doi: 10.1021/la401929v. [DOI] [PubMed] [Google Scholar]
- 5.Bhuniya S, Seo YJ, Kim BH. Tetrahedron Lett. 2006;47:7153–7156. [Google Scholar]
- 6.Cao W, Zhang XL, Miao XM, Yang ZM, Xu HP. Angew Chem Int Ed. 2013;52:6233–6237. doi: 10.1002/anie.201300662. [DOI] [PubMed] [Google Scholar]
- 7.Estroff LA, Hamilton AD. Chem Rev. 2004;104:1201–1217. doi: 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]
- 8.Kiyonaka S, Sugiyasu K, Shinkai S, Hamachi I. J Am Chem Soc. 2002;124:10954–10955. doi: 10.1021/ja027277e. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Z, Yi Q, Xia T, Yin W, Kadi AA, Li J, Zhang Y. Org Biomol Chem. 2017;15:2191–2198. doi: 10.1039/c6ob02667b. [DOI] [PubMed] [Google Scholar]
- 10.Du X, Zhou J, Shi J, Xu B. Chem Rev. 2015;115:13165–13307. doi: 10.1021/acs.chemrev.5b00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park MH, Yu Y, Moon HJ, Ko DY, Kim HS, Lee H, Ryu KH, Jeong B. Adv Healthcare Mater. 2014;3:1782–1791. doi: 10.1002/adhm.201400140. [DOI] [PubMed] [Google Scholar]
- 12.Kopecek J, Yang J. Angew Chem Int Ed. 2012;51:7396–7417. doi: 10.1002/anie.201201040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lange SC, Unsleber J, Drucker P, Galla HJ, Waller MP, Ravoo BJ. Org Biomol Chem. 2015;13:561–569. doi: 10.1039/c4ob02069c. [DOI] [PubMed] [Google Scholar]
- 14.Kharkar PM, Kiick KL, Kloxin AM. Chem Soc Rev. 2013;42:7335–7372. doi: 10.1039/c3cs60040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shinde UP, Moon HJ, Ko DY, Jung BK, Jeong B. Biomacromolecules. 2015;16:1461–1469. doi: 10.1021/acs.biomac.5b00325. [DOI] [PubMed] [Google Scholar]
- 16.Park MH, Joo MK, Choi BG, Jeong B. Acc Chem Res. 2012;45:424–433. doi: 10.1021/ar200162j. [DOI] [PubMed] [Google Scholar]
- 17.Appel EA, del Barrio J, Loh XJ, Scherman OA. Chem Soc Rev. 2012;41:6195–6214. doi: 10.1039/c2cs35264h. [DOI] [PubMed] [Google Scholar]
- 18.Chen Y, Pang XH, Dong CM. Adv Funct Mater. 2010;20:579–586. [Google Scholar]
- 19.Deming TJ. Nature Mater. 2010;9:535–536. doi: 10.1038/nmat2789. [DOI] [PubMed] [Google Scholar]
- 20.Lalatsa A, Schatzlein AG, Mazza M, Thi BHL, Uchegbu IF. J Controlled Release. 2012;161:523–536. doi: 10.1016/j.jconrel.2012.04.046. [DOI] [PubMed] [Google Scholar]
- 21.Lin YA, Ou YC, Cheetham AG, Cui H. Biomacromolecules. 2014;15:1419–1427. doi: 10.1021/bm500020j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moon HJ, Choi BG, Park MH, Joo MK, Jeong B. Biomacromolecules. 2011;12:1234–1242. doi: 10.1021/bm101518c. [DOI] [PubMed] [Google Scholar]
- 23.Kang EY, Yeon B, Moon HJ, Jeong B. Macromolecules. 2012;45:2007–2013. [Google Scholar]
- 24.Huang J, Hastings CL, Duffy GP, Kelly HM, Raeburn J, Adams DJ, Heise A. Biomacromolecules. 2013;14:200–206. doi: 10.1021/bm301629f. [DOI] [PubMed] [Google Scholar]
- 25.Nowak AP, Breedveld V, Pakstis L, Ozbas B, Pine DJ, Pochan D, Deming TJ. Nature. 2002;417:424–428. doi: 10.1038/417424a. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Y, He C, Xiao C, Ding J, Cui H, Zhuang X, Chen X. Biomacromolecules. 2013;14:468–475. doi: 10.1021/bm3017059. [DOI] [PubMed] [Google Scholar]
- 27.Cao J, Hu P, Lu L, Chan BA, Luo BH, Zhang D. Polym Chem. 2015;6:1226–1229. [Google Scholar]
- 28.Deming TJ. Chem Rev. 2016;116:786–808. doi: 10.1021/acs.chemrev.5b00292. [DOI] [PubMed] [Google Scholar]
- 29.Merkel L, Budisa N. Org Biomol Chem. 2012;10:7241–7261. doi: 10.1039/c2ob06922a. [DOI] [PubMed] [Google Scholar]
- 30.Hong JH, Lee HJ, Jeong B. ACS Appl Mater Interfaces. 2017 doi: 10.1021/acsami.7b02488. [DOI] [PubMed] [Google Scholar]
- 31.Bhattacharyya J, Weitzhandler I, Ho SB, McDaniel JR, Li X, Tang L, Liu J, Dewhirst M, Chilkoti A. Adv Funct Mater. 2017;27:1605421–n/a. doi: 10.1002/adfm.201605421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turk BE, Huang LL, Piro ET, Cantley LC. Nat Biotech. 2001;19:661–667. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 33.Yu L, Zhang H, Ding JD. Angew Chem Int Ed. 2006;45:2232–2235. doi: 10.1002/anie.200503575. [DOI] [PubMed] [Google Scholar]
- 34.Zou J, Zhang F, Zhang S, Pollack SF, Elsabahy M, Fan J, Wooley KL. Adv Healthcare Mater. 2014;3:441–448. doi: 10.1002/adhm.201300235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aliferis T, Iatrou H, Hadjichristidis N. Biomacromolecules. 2004;5:1653–1656. doi: 10.1021/bm0497217. [DOI] [PubMed] [Google Scholar]
- 36.Deming TJ. Nature. 1997;390:386–389. doi: 10.1038/37084. [DOI] [PubMed] [Google Scholar]
- 37.Dimitrov I, Schlaad H. Chem Commun. 2003:2944–2945. doi: 10.1039/b308990h. [DOI] [PubMed] [Google Scholar]
- 38.Conejos-Sanchez I, Duro-Castano A, Birke A, Barz M, Vicent MJ. Polym Chem. 2013;4:3182–3186. [Google Scholar]
- 39.Lu H, Cheng J. J Am Chem Soc. 2007;129:14114–14115. doi: 10.1021/ja074961q. [DOI] [PubMed] [Google Scholar]
- 40.Zou J, Fan J, He X, Zhang S, Wang H, Wooley KL. Macromolecules. 2013;46:4223–4226. doi: 10.1021/ma4007939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao W, Gnanou Y, Hadjichristidis N. Chem Commun. 2015;51:3663–3666. doi: 10.1039/c4cc09055a. [DOI] [PubMed] [Google Scholar]
- 42.Fan J, Li R, He X, Seetho K, Zhang F, Zou J, Wooley KL. Polym Chem. 2014;5:3977–3981. doi: 10.1039/C4PY00628C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fan J, Zou J, He X, Zhang F, Zhang S, Raymond JE, Wooley KL. Chem Sci. 2014;5:141–150. doi: 10.1039/C3SC52504J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wamsley A, Jasti B, Phiasivongsa P, Li XL. J Polym Sci, Part A: Polym Chem. 2004;42:317–325. [Google Scholar]
- 45.Hayashi S, Ohkawa K, Yamamoto H. Macromol Biosci. 2006;6:228–240. doi: 10.1002/mabi.200500211. [DOI] [PubMed] [Google Scholar]
- 46.Choi YY, Jang JH, Park MH, Choi BG, Chi B, Jeong B. J Mater Chem. 2010;20:3416–3421. [Google Scholar]
- 47.Aggeli A, Bell M, Boden N, Keen JN, Knowles PF, McLeish TCB, Pitkeathly M, Radford SE. Nature. 1997;386:259–262. doi: 10.1038/386259a0. [DOI] [PubMed] [Google Scholar]
- 48.Kotharangannagari VK, Sanchez-Ferrer A, Ruokolainen J, Mezzenga R. Macromolecules. 2012;45:1982–1990. [Google Scholar]
- 49.Kauppinen JK, Moffatt DJ, Mantsch HH, Cameron DG. Anal Chem. 1981;53:1454–1457. [Google Scholar]
- 50.Kauppinen JK, Moffatt DJ, Mantsch HH, Cameron DG. Appl Spectrosc. 1981;35:271–276. [Google Scholar]
- 51.Panitch A, Matsuki K, Cantor EJ, Cooper SJ, Atkins EDT, Fournier MJ, Mason TL, Tirrell DA. Macromolecules. 1997;30:42–49. [Google Scholar]
- 52.Rosler A, Klok HA, Hamley IW, Castelletto V, Mykhaylyk OO. Biomacromolecules. 2003;4:859–863. doi: 10.1021/bm034058s. [DOI] [PubMed] [Google Scholar]
- 53.Kim EH, Joo MK, Bahk KH, Park MH, Chi B, Lee YM, Jeong B. Biomacromolecules. 2009;10:2476–2481. doi: 10.1021/bm9004436. [DOI] [PubMed] [Google Scholar]
- 54.Greenfield NJ. Nat Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He X, Fan J, Wooley KL. Chem Asian J. 2016;11:437–447. doi: 10.1002/asia.201500957. [DOI] [PubMed] [Google Scholar]
- 56.He X, Fan J, Zhang F, Li R, Pollack KA, Raymond JE, Zou J, Wooley KL. J Mater Chem B. 2014;2:8123–8130. doi: 10.1039/C4TB00909F. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.