

Abstract
We examined the impact of two clinically approved anti-herpes drugs, acyclovir and Forscarnet (phosphonoformate) on the exonuclease activity of the herpes simplex virus-1 DNA polymerase, UL30. Acyclovir triphosphate and Foscarnet, along with the closely related phosphonoacetic acid, did not affect exonuclease activity on single-stranded DNA. Furthermore, blocking the polymerase active site due to either binding of Foscarnet or phosphonoacetic acid to the E-DNA complex or polymerization of acyclovir onto the DNA also had minimal effect on exonuclease activity. The inability of the exonuclease to excise acyclovir from the primer 3′-terminus results from the altered sugar structure directly impeding phosphodiester bond hydrolysis as opposed to inhibiting binding, unwinding of the DNA by the exonuclease or transfer of the DNA from the polymerase to the exonuclease. Removing the 3′-hydroxyl or the 2′-carbon from the nucleotide at the 3′-terminus of the primer strongly inhibited exonuclease activity, although addition of a 2′-hydroxyl did not affect exonuclease activity. The biological consequences of these results are two-fold. First, the ability of acyclovir and Foscarnet to block dNTP polymerization without impacting exonuclease activity raises the possibility that their effects on herpes replication may involve both direct inhibition of dNTP polymerization along with exonuclease mediated destruction of herpes DNA. Second, the ability of the exonuclease to rapidly remove a ribonucleotide at the primer 3´-terminus in combination with the polymerase not efficiently adding dNTPs onto this primer provides a novel mechanism by which the herpes replication machinery can prevent incorporation of ribonucleotides into newly synthesized DNA.
TOC image

Herpes viruses are complex DNA viruses that are responsible for a variety of indications including oral and genital herpes sores, chickenpox, viral encephalitis, etc. (7). Herpes simplex virus 1 (HSV1) encodes seven proteins essential for viral DNA replication: a) the heterodimeric DNA polymerase-processivity factor complex (UL30-UL42), b) the heterotrimeric helicase-primase (UL5-UL8-UL52), c), an origin-binding protein (UL9), and a single-stranded DNA binding protein (UL29/ICP8) (8–10). In addition to polymerase activity, UL30 also possesses 3′–5′ exonuclease activity that proofreads the just-incorporated nucleotide (11–12).
Acyclovir, gancicyclovir, and phosphonoformic acid are clinically useful anti-herpes drugs (13–14). Acyclovir is a remarkably powerful treatment for α-herpes virus infections due to its having minimal side effects and problems with resistance (15–18). Once converted to the triphosphate by cellular and viral kinases, acyclovir triphosphate (ACVTP) acts as a chain terminator of HSV polymerase (13–14, 19–23). The formation of acyclovir-terminated DNA followed by binding of the next required dNTP results in the formation of an extremely stable E–DNA–dNTP dead–end complex in the polymerase active site (24). Similarly, ganciclovir (as the biologically active GCVTP) primarily functions as a chain terminator during cytomegalovirus DNA replication (25). On the other hand, phosphonoformic acid (Foscarnet) is a pyrophosphate analogue that functions by directly binding to the pyrophosphate binding site in the polymerase active site (26–28).
Derse et al. observed that the exonuclease activity of herpes polymerase (UL30/UL42) did not efficiently excise acyclovir monophosphate that the polymerase activity had incorporated (20). Similarly, Hanes et al. used transient kinetic methods to directly show that the exonuclease does not efficiently hydrolyze acyclovir-terminated DNA as compared to DNA containing deoxyguanine at the primer terminus (kexo = 12 s−1 for deoxyguanine terminated DNA versus 5 × 10−3 s−1 for acyclovir terminated DNA) (29). These authors also proposed that the presence of acyclovir at the 3′-terminus of the primer interferes with movement of the DNA from the polymerase to the exonuclease active sites.
We recently showed that blocking the polymerase active site via formation of an UL30-DNA-Aphidicolin dead-end complex has little or no effect on the exonuclease activity, indicating that the polymerase and exonuclease active sites have independent DNA binding domains (30). This result also raises the possibility that other inhibitors may block polymerase activity but leave the exonuclease unbothered.
To better understand how acyclovir and Foscarnet can impact herpes replication, we employed synthetic oligonucleotides of defined sequence to examine how forming E-DNAACV-dNTP or E-DNA-PFA complexes in the polymerase active site affect exonuclease activity. In both cases, forming these complexes did not affect exonuclease activity. The poor ability of the exonuclease to hydrolyze acyclovir terminated DNA results from the modified sugar directly interfering with the hydrolysis reaction. Lastly, we observed that while the presence of a 2′-hydroxyl at the primer terminus does not affect exonuclease activity, it potently inhibits polymerase activity. The biochemical and medical significance of these results are discussed.
MATERIALS AND METHODS
Chemicals
All chemicals were of the highest grade available and were used as purchased. T4 polynucleotide kinase was from New England Biolabs, dNTPs and ddNTPs were from Invitrogen, and [γ–32P]ATP was from Perkin–Elmer. T4 DNA polymerase, T7 DNA polymerase, and Klenow Fragment were obtained from New England Biolabs. Phosphonoformic acid and phosphonoacetic acid were from Sigma. Acyclovir triphosphate and gancicyclovir triphosphate were obtained from Wayne Miller (Burroughs-Welcome Corporation, Research Triangle Park, NC).
Enzymes
His–tagged UL30 and UL30/42 were purified from SF9 insect cells infected with recombinant baculoviruses that harbor the genes encoding these proteins as described previously (31).
Oligonucleotides
Oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA). Table 1 lists the sequences of all primers and templates used in this study. All DNAs were gel purified using 20% denaturing polyacrylamide gel electrophoresis. Primers were radiolabeled at the 5′–end using [γ–32P]ATP and T4 polynucleotide kinase by standard procedures (32). DNA duplexes were formed by heating the primer–templates in a molar ratio of 1:1.4 to 95 °C followed by slow cooling to room temperature.
Table 1.
DNA substrates used. Templating bases are underlined and A*, G*, D*, and rG* indicate acyclovir, ganciclovir, dideoxy, and ribose-terminated DNAs respectively.
| DNA15ss | 5′-GCTCTGTTCCGGTTG |
| DNA15C | 5′-GCTCTGTTCCGGTTT |
| 3′-CGAGACAAGGCCAAACACAAAGTAGATTAGTAC | |
| DNA15G | 5′-GCTCTGTTCCGGTTG |
| 3′-CGAGACAAGGCCAACCACAAAGTAGATTAGTAC | |
| DNA16ACVss | 5′-GCTCTGTTCCGGTTTA* |
| DNA16ddss | 5′-GCTCTGTTCCGGTTTD* |
| DNA16riboss | 5′-GCTCTGTTCCGGTTTrG* |
| DNA16ACV | 5′-GCTCTGTTCCGGTTTA* |
| 3′-CGAGACAAGGCCAAACACAAAGTAGATTAGTAC | |
| DNA16GCV | 5′-GCTCTGTTCCGGTTTG* |
| 3′-CGAGACAAGGCCAAACACAAAGTAGATTAGTAC | |
| DNA16dd | 5′-GCTCTGTTCCGGTTTD* |
| 3′-CGAGACAAGGCCAAACACAAAGTAGATTAGTAC | |
| DNA16ribo | 5′-GCTCTGTTCCGGTTTrG* |
| 3′-CGAGACAAGGCCAAACACAAAGTAGATTAGTAC | |
| DNA30C | 5′-ATCGTATGTTTATTTGCTCTGTTCCGGTTT |
| 3′-TAGCATACAAATAAACGAGACAAGGCCAAACATAAAGTAGATTAGTAC | |
| DNA35ss | 5′-ATGTAATCGTATGTTTATTTGCTCTGTTCCGGTTT |
| DNA35C | 5′-ATGTAATCGTATGTTTATTTGCTCTGTTCCGGTTT |
| 3′-TACATTAGCATACAAATAAACGAGACAAGGCCAAACATAAAGTAGATTAGTAC |
Preparation of ACV, GCV, dideoxyguanosine terminated duplex and single stranded DNAs
ACV terminated primer-template was prepared by incubating 40 μM ACVTP with 5 μM 5′-[32P]-DNA15C in 50 mM Hepes (pH 7.6), 5% glycerol, 0.1 mg/mL BSA, 1 mM DTT and 10 mM MgCl2 at 37 °C. The reaction was initiated with 500 nM Klenow Fragment and allowed to proceed for 2 hours and then terminated by heating to 90 °C for 15 minutes. Excess ACVTP was removed from the reaction mixture by passing it through a G25 spin column. Denaturing polyacrylamide gel electrophoresis of the reaction products showed that >95% of the starting DNA15C had been converted to DNA16ACV. To prepare ACV terminated single-stranded DNA, the radiolabeled acyclovir terminated primer-template (DNA16ACV) was heated to 95 °C for 5 min. and the ACV-terminated primer strand purified using 20% denaturing acrylamide gel electrophoresis. GCV and dideoxyguanine terminated single-stranded and duplex DNAs were prepared analogously.
Simultaneous Polymerase/Exonuclease Assays
All experiments were performed under steady-state conditions at 37 °C. Assays typically contained 1 μM 5′–[32P]-primer/template, 50 mM Hepes (pH 7.6), 5% glycerol, 0.1 mg/mL BSA, 1 mM DTT, 10 mM MgCl2, 10 μM dNTPs, and varying concentrations of inhibitor (ACVTP, PFA, or PAA). Reactions were initiated by adding enzyme (typically 50 nM) and quenched at various times by adding 5 volumes of 90% formamide, 10 mM EDTA, 1X Tris/Borate/EDTA buffer, and 0.1% bromophenol blue. Samples were heated for 2 min. at 90 °C and products separated by denaturing gel electrophoresis (20% acrylamide, 8 M urea) and analyzed by phosphorimagery (Molecular Dynamics).
Exonuclease Assays
All exonuclease assays were performed under conditions of excess substrate as described above except that dNTPs were omitted from the assays. 5′–[32P]–DNA (1 μM) was incubated with reaction buffer in the presence of varying concentrations of ACVTP (0–200 μM). Reactions were initiated by adding enzyme (typically 5 nM for single stranded DNA and 50 nM for duplex DNA) and aliquots quenched at designated time intervals.
Measurement of IC50 values for various DNAs
Assays contained fixed concentration of either 5′-[32P]-DNA35ss or 5′-[32P]-DNA35C (1 μM) and varying concentrations of an unlabeled DNA containing G, ddG, ACV or GCV at the primer 3′ terminus. The reciprocal of the fraction of DNA35ss or DNA35C hydrolyzed was plotted against DNA concentration to obtain the IC50 of the unlabeled DNA to inhibit exonuclease activity on the [32P]-labeled DNA.
RESULTS
We previously showed that formation of an E-DNA-aphidicolin ternary complex where the DNA was bound in the polymerase active site had no effect on the exonuclease activity (30), raising the possibility that other inhibitors of herpes polymerase might likewise generate complexes with inhibited polymerase activity but active exonuclease. To test this possibility, we examined the effects of two clinically useful anti-herpes drugs, acyclovir and phosphonoformic acid, on exonuclease activity using synthetic oligonucleotides of defined sequence.
We initially examined the effects of ACVTP, PFA and PAA on exonuclease activity using a single-stranded DNA as substrate (DNA35ss). Figure 1 shows the time course for exonuclease activity in the presence of increasing concentrations of ACVTP. Even at saturating concentrations, ACVTP did not inhibit the exonuclease activity on DNA35ss (Figure 1 Panel B). Similar results were obtained with PFA and PAA (Figures S1 and S2 in Supplementary Material). Using long, partially double-stranded DNAs as substrate (oligo(dG).poly(dC) (33), radiolabeled activated calf thymus DNA (34), and radiolabeled E. coli DNA (35)), previous work reported that both PFA and PAA inhibited exonuclease activity. However, we found that PFA only mildly inhibited exonuclease activity on 5′-[32P]-DNA15C (Figure S3A). To ensure that the lack of inhibition was not a consequence of the short length of DNA15C and/or its sequence, we tested a somewhat longer DNA (DNA30C). Again, adding PFA did not inhibit exonuclease activity (Figure S3B).
Figure 1.

ACVTP does not affect exonuclease activity on DNA35ss. UL30 was incubated with DNA (1 μM) and aliquots were taken out at various times. (A) Phosphorimages of the products of DNA35ss degradation using UL30 at varying concentrations of ACVTP including 0, 1, 5, 20, and 200 μM at various time intervals: 0, 0.5, 1, 5, 10, and 20 min. (B) Plot of exonuclease products as a function of ACVTP concentration at various time intervals. Note: The gels shown are representative of experiments that were performed multiple times.
We next determined how these compounds impact exonuclease activity under conditions where the polymerase can simultaneously elongate a primer:template. We previously showed that under conditions of excess DNA over UL30 (or the UL30/UL42 complex), the enzyme processes some of the DNA via dNTP polymerization and some via exonuclease activity (30). Thus, these conditions allow simultaneous monitoring of both polymerase and exonuclease activity. Assays contained 1 μM 5′-[32P]-DNA15C, all 4 dNTPs and increasing concentrations of ACVTP, PFA or PAA (Figures 2 (ACVTP), 3 (PFA) and S4 (PAA, Supplementary Material)). The DNA concentration is much greater than its KD (Ca. 10–50 nM) such that all of the polymerase active sites should contain bound DNA. In the absence of any inhibitor, the polymerase elongates some of the DNA to the end of the template while the exonuclease hydrolyzes a fraction of the DNA. Adding increasing concentrations of ACVTP, PFA or PAA inhibited dNTP polymerization but had at most small effects on exonuclease activity. Polymerase inhibition without significantly impacting the exonuclease is consistent with the idea that the two active sites have independent DNA binding domains. Similar results were obtained with the UL30/UL42 complex, indicating that UL42 does not affect the independence of the polymerase and exonuclease DNA binding domains (Figure S5 in Supplementary Material).
Figure 2.
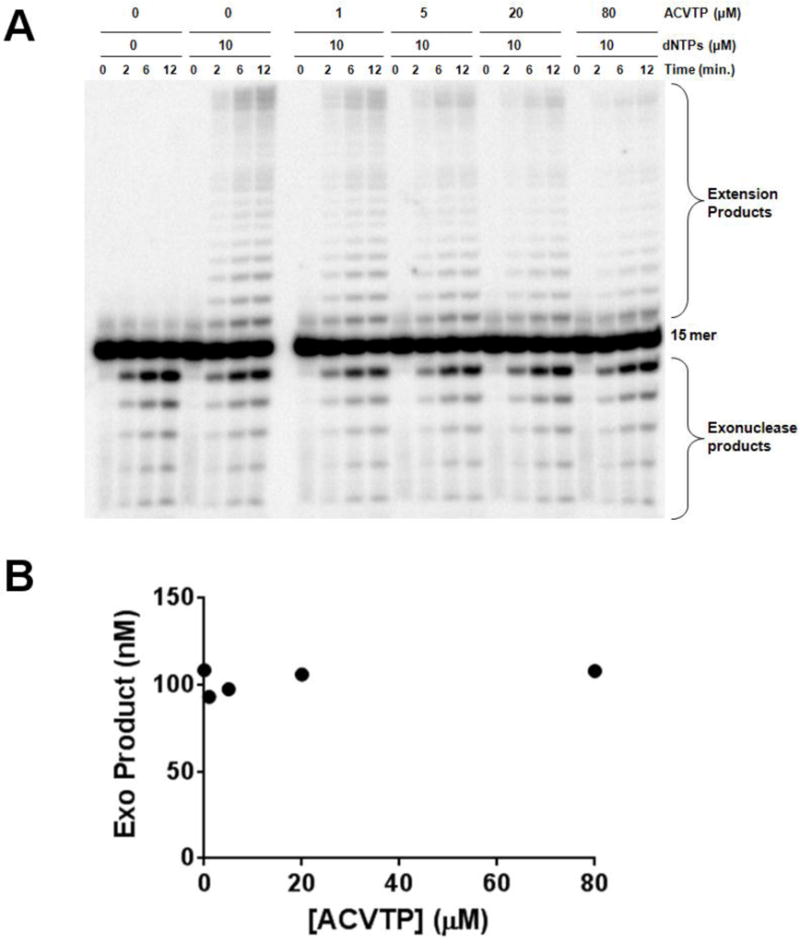
Effect of ACVTP on polymerase and exonuclease activities under processive conditions. UL30 was incubated with DNA15C (1 μM) in the presence of 0 μM or 10 μM dNTPs and varying concentrations of acyclovir triphosphate (0–80 μM). Aliquots of each reaction were analyzed at various times after initiating the reaction. (A) Phosphorimages of the products of DNA15C full extension and degradation using UL30. (B) Plot of exonuclease products as a function of ACVTP concentration at 6 min.
Figure 3.
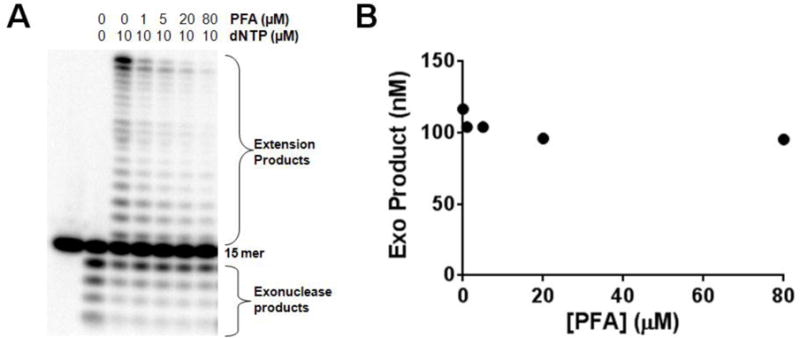
Effect of Foscarnet on polymerase and exonuclease activities under processive conditions. UL30 was incubated with DNA15C (1 μM) in the presence of 0 μM or 10 μM dNTPs, and 0–80 μM Foscarnet Aliquots were analyzed at 6 min. (A) Phosphorimages of the products of DNA15C full extension and degradation using UL30. (B) Plot of exonuclease products as a function of PFA concentration.
PFA inhibits herpes replication by forming a UL30-DNA-PFA ternary complex while acyclovir can inhibit polymerase activity by forming either an UL30-DNAACV binary or UL30-DNAACV-dNTP ternary complex. To explicitly measure the effects of these complexes on exonuclease activity, we generated the UL30-DNA-(±dNTP) complexes using a 5′-[32P] labeled primer-template while simultaneously measuring exonuclease activity on a separate 5′-[32P]-ssDNA (Figure 4). The two 5′-[32P] labeled DNAs were of different lengths, hence exonuclease activity on each DNA can be independently monitored. First, the effect of just a primer-template containing a normal nucleotide at the primer terminus was measured to control for the ability of a primer-template to directly bind in the exonuclease site. Figure 4, Panel A shows that whereas significant exonuclease activity occurred on the single-stranded DNA, virtually no degradation occurred on the primer-template (<5%), as expected in light of the exonucleases preference for single stranded DNA. Adding either PFA or PAA to assays containing a primer-template and 5′-[32P]-ssDNA had no effect on the rate at which UL30 hydrolyzed the ssDNA (Table 2 and Figures 4A, 4B and 4C), indicating that formation of either a E-DNA-PFA or E-DNA-PAA ternary complex in the polymerase active site did not affect the exonuclease. Likewise, formation of either an E-DNAACV or an E-DNAACV-dNTP complex in the polymerase active site did not inhibit exonuclease activity (Figure 4D and 4E). Thus, even under conditions where the polymerase active site is blocked due to binding and/or polymerization of various inhibitors, the exonuclease remains completely active.
Figure 4.
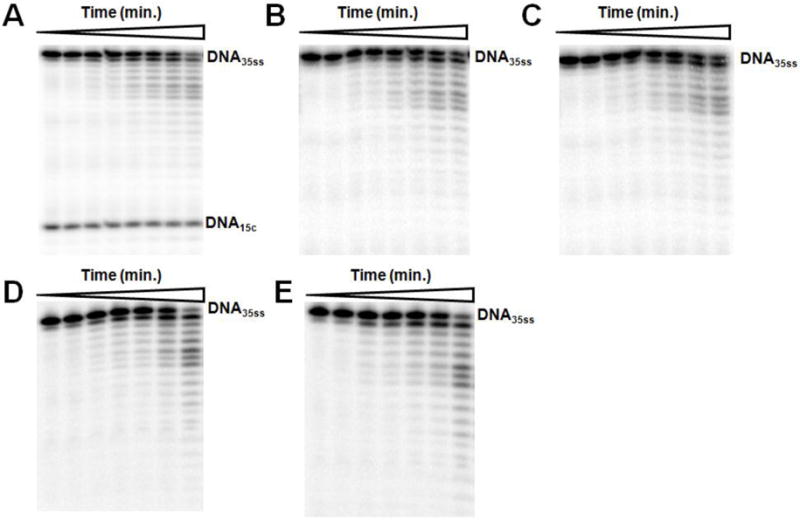
Formation of UL30-DNA15C-PFA, UL30-DNA15C-PAA, UL30-DNA16ACV, and UL30-DNA16ACV-dTTP complexes do not affect the exonuclease activity on a second DNA. Assays contained DNA35ss and the additional DNAs and compounds as noted. All DNAs were present at 1 μM. (A) DNA15C. (B) DNA15C and 50 μM PFA. (C) DNA15C and 50 μM PAA. (D) DNA16ACV. (E) DNA16ACV + 50 μM dTTP. The time points for legends A-C were: 0, 0.25, 0.75, 1, 2, 3, 5 and 7 min. The time points for legends D-E were: 0, 0.25, 0.5, 0.75, 1, 2 and 5 min. In Panels B-E, DNA15C is omitted for clarity.
Table 2.
Effects of forming E-DNA-PFA, E-DNA-PAA, E-DNAACV-dTTP, and E-DNAACV complexes on the 3′–5′ exonuclease activity of UL30 on DNA15C.
| Inhibitor | Rate (nM/min.) |
|---|---|
| No Inhibitor | 162 ± 4 |
| 50 μM PFA | 156 ± 4 |
| 50 μM PAA | 162 ± 3 |
| 1 μM DNA16ACV + 50 μM dTTP | 160 ± 15 |
| 1 μM DNA16ACV | 130 ± 20 |
Why is DNA containing ACV at the primer 3′-terminus a poor substrate for the exonuclease?
Previous work showed that double-stranded DNA containing ACV at the primer 3′-terminus is a very poor exonuclease substrate for the UL30/UL42 complex (20, 24, 29). Likewise, we observed that DNA containing ACV as the terminal nucleotide was a very poor substrate for the UL30 exonuclease (Table 3). The presence of ACV at the primer terminus (DNA16ACV) decreased the rate of the exonuclease by 23-fold compared to the identical DNA containing dG at the 3′-terminus. This much slower rate clearly indicates that the sugar of the excised nucleotide greatly impacts phosphodiester bond cleavage. We therefore endeavored to determine what features of the sugar are needed for efficient exonuclease activity and the mechanistic consequences of eliminating these features.
Table 3.
Summary of rates of exonucleolytic removal of the 3′-terminal nucleotide of DNA15G, DNA16ACV, DNA16GCV, and DNA16dd using UL30, T4 DNA polymerase, T7 DNA polymerase, and Klenow Fragment. In each case, the -fold decrease in rate is also noted.
| Rate (nM/min.) | ||||
|---|---|---|---|---|
| Enzyme | DNA15G | DNA16ACV | DNA16GCV | DNA16dd |
| UL30 | 183 ± 5 | 8 ± 1 (23-Fold) |
9 ± 1 (20-Fold) |
0.063 ± 0.002 (2900-Fold) |
| T4 | 253 ± 4 | 11 ± 1 (23-Fold) |
13 ± 1 (20-Fold) |
0.50 ± 0.02 (500-Fold) |
| T7 | 297 ± 15 | 9.0 ± 0.2 (33-Fold) |
ND 1 | ND |
| KF | 99 ± 13 | 2.0 ± 0.1 (50-Fold) |
ND | ND |
ND = Not Done.
Compared to the canonical 2′-deoxyribose, the sugar in ACV lacks a 3′-hydroxyl, lacks the hydrophobic 2′-methylene and is conformationally much less constrained due to its acyclic nature. To determine which of these features contributes to the decreased exonuclease efficiency, we synthesized primer templates that contained either a 2′, 3′-dideoxynucleotide (to test the importance of a 3′-hydroxyl) or ganciclovir (to test the importance of an intact furan (Figure 5)) at the 3′-terminus. Just as the presence of acyclovir at the primer 3′-terminus greatly decreased exonuclease activity, the presence of either ganciclovir or a dideoxynucleotide at the primer 3′-terminus also greatly decreased exonuclease activity (Table 3). UL30 hydrolyzed a primer-template containing ganciclovir or 2′, 3′-dideoxyG at the primer 3′-terminus 20- and 2900-fold less efficiently, respectively, than if it contained a normal nucleotide (Table 3). UL42 does not impact the effects of the altered sugars as UL30 and the UL30/UL42 complexes gave similar results. The rates of DNA degradation obtained with DNA16ACV, DNA16GCV, and DNA16dd using UL30/UL42 were 90-, 90-, and 2200-fold lower than DNA15G, respectively. Thus, the exonuclease activity of UL30 appears exquisitely sensitive to modifications in the sugar of the nucleotide at the 3′-terminus of the DNA.
Figure 5.
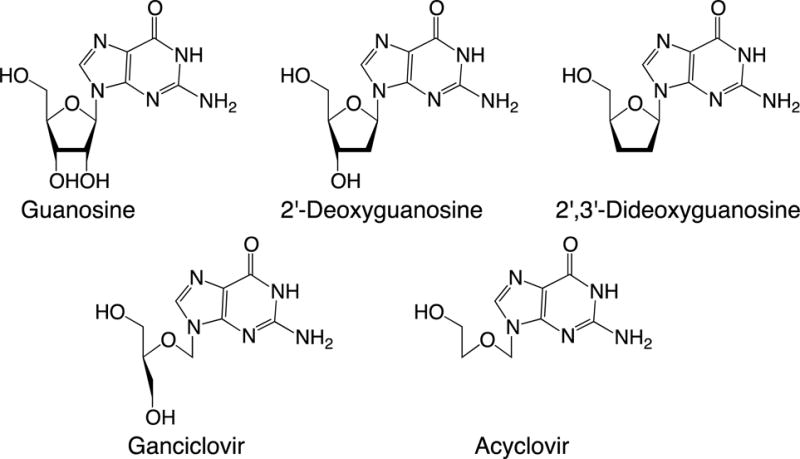
Nucleoside structures.
To determine if modifying the sugar at the primer 3′-terminus affects the exonuclease associated with other DNA polymerases, we tested these modified DNAs with several other proofreading exonucleases. Table 3 shows that just as with UL30, modifying the structure of the 3′-terminal nucleotide greatly impacts the exonuclease of T4 DNA polymerase. Similar results were obtained with T7 DNA polymerase and Klenow Fragment when ACV terminated DNA was used. These data suggest that the exonucleases associated with at least some A and B family polymerases are quite sensitive to modification of the sugar moiety.
Mechanistically, three potential explanations could account for the decreased exonuclease rates of UL30 upon removing the 3′-hydroxyl from the 3′-terminal nucleotide or converting it into an acyclic sugar: (1) it greatly weakens binding to the exonuclease; (2) since the enzyme needs to transfer the DNA from the polymerase active site to the exonuclease active site and unwind two base-pairs of the double stranded DNA for the exonuclease to hydrolyze the substrate (30), the modified sugar might inhibit unwinding and/or DNA transfer, and; (3) the altered sugar structure interferes with hydrolysis of the phosphodiester linkage. We initially tested the hypothesis that the altered sugar structure interfered with binding to the exonuclease site. Exonuclease activity was measured on a 5′-[32P]-labeled single-stranded DNA (DNA35ss), and the effects of adding increasing amounts of a primer-template containing either ddG, ACV, GCV or a normal dG at the primer 3′-terminus measured. If altering the sugar structure interfered with binding to the exonuclease site, then the DNAs with altered sugars should have inhibited exonuclease activity on the 5′-[32P]-labeled single-stranded DNA much less potently than the DNA with a normal sugar. However, all four DNAs inhibited exonuclease activity with only small differences in potency, indicating that modifying the sugar did not significantly interfere with binding of a primer-template to the exonuclease active site (Table 4). Similarly, all four DNAs also inhibited exonuclease activity of an exogenous 5′-[32P]-labeled primer-template (DNA35C) to the same extent (Table 4). To test the possibility that the modified sugars interfered with the unwinding and/or active site transfer reactions as well as explicitly determine if the modified sugar interferes with the hydrolysis reaction, we synthesized 5′-[32P]-labeled single-stranded DNAs containing dG, ACV or ddG at the 3′-terminus and measured exonuclease activity. Unlike with double-stranded DNA, neither unwinding nor transfer between polymerase and exonuclease sites are needed for hydrolysis of single-stranded DNA. Whereas the exonuclease rapidly hydrolyzed the dG terminated single-stranded DNA, it did not detectably hydrolyze either the ACV or ddG terminated DNA (Figure 6). Moreover, these single stranded DNAs containing dG, ACV or ddG at the 3′-terminus had very similar binding affinities towards the exonuclease active site as measured by their ability to inhibit exonuclease activity on a second, 5′-[32P]-ssDNA (Table 5). Thus, just as with the primer-templates, a modified nucleotide at the primer 3′-terminus does not affect the ability of single-stranded DNAs to bind to the exonuclease site. Additionally, since a modified nucleotide in single stranded DNAs blocked activity even though hydrolysis requires neither unwinding of any duplex DNA nor transfer between the polymerase and exonuclease sites, these data indicate that the modified sugar inhibits exonuclease activity by interfering with phosphodiester bond cleavage.
Table 4.
The ability of double-stranded DNAs containing dG (DNA15G), acyclovir (DNA16ACV), gancicyclovir (DNA16GCV), and dideoxyG (DNA16dd) to inhibit exonuclease activity on 1 μM 5′-[32P]-labeled single-stranded DNA (DNA35ss) or 1 μM 5′-[32P]-labeled double-stranded DNA (DNA35C).
| Inhibitor DNA | Substrate DNA | IC50 (μM) |
|---|---|---|
| DNA16GCV | DNA35C | 1.0 |
| DNA16ACV | DNA35C | 1.7 |
| DNA16dd | DNA35C | 2.5 |
| DNA15G | DNA35C | 1.3 |
| DNA16GCV | DNA35ss | 0.6 |
| DNA16ACV | DNA35ss | 1.0 |
| DNA16dd | DNA35ss | 0.3 |
| DNA15G | DNA35ss | 0.3 |
Figure 6.
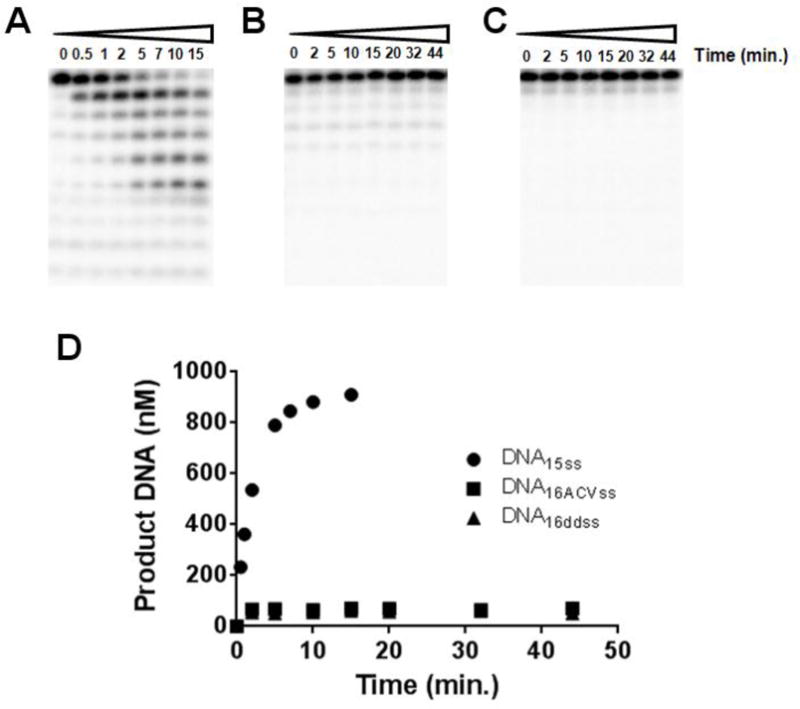
The UL30 exonuclease does not efficiently remove acyclovir or dideoxyguanosine from single-stranded DNA. UL30 was incubated with various DNAs and aliquots were taken out at the noted times. All DNAs were present at 1 μM. (A) DNA15ss. (B) DNA16ACVss. (C) DNA16ddss. (D) Amount of exonuclease products generated for each DNA.
Table 5.
Effects of varying the 3′-terminal nucleotide of single stranded DNA on their ability to inhibit exonuclease activity on a second, 5′-[32P]-labeled single-stranded DNA.
| Inhibitor DNA | Substrate DNA | IC50 (nM) |
|---|---|---|
| DNA35ss | DNA15ss | 55 |
| DNA16ACVss | DNA35ss | 35 |
| DNA16ddss | DNA35ss | 70 |
Substitution of the 2′-H with an OH group inhibits polymerase but not exonuclease activity
In order to determine the effect of replacing a 2′-H with an OH group, we prepared DNA containing a single ribonucleotide at the 3′-terminus and measured both exonuclease and polymerase activity. Figures 7A and 7B show the time courses for degradation of DNA16riboss (a single-stranded DNA with a NMP at the 3′-terminus) and DNA16ribo (duplex DNA with a NMP at the primer 3′-terminus), respectively. The polymerase degraded DNA16riboss and DNA15ss at similar rates, (kcat = 49 ± 3 min−1 and 67 ± 2 min−1 respectively) while it degraded DNA16ribo slightly faster than DNA15G (kcat = 10 ± 2 min−1 and 4 ± 1 min−1, respectively). Thus, unlike the other modifications, the presence or absence of a 2′-hydroxyl does not significantly impact the exonuclease.
Figure 7.
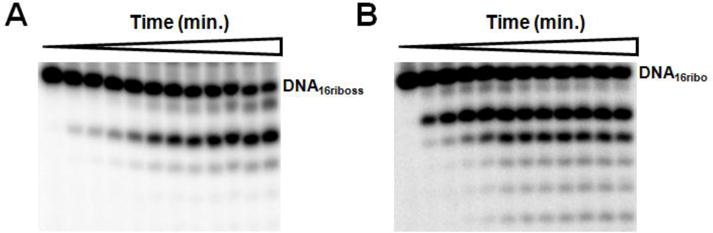
Time course for degradation of DNA16riboss and DNA16ribo. UL30 was incubated with DNA (1 μM) and aliquots were taken out at various times. (A) Phosphorimages of the products of DNA16riboss degradation using UL30 (1 nM) after 0, 0.25, 0.5, 1, 2, 5, 7, 10, 15, 20, 25, and 30 min. (B) Phosphorimages of the products of DNA16ribo degradation using UL30 (25 nM) after 0, 0.25, 0.5, 1, 2, 5, 7, 10, 15, 20, 25, and 30 min.
In contrast to the minimal effect on exonuclease activity, the presence of even a single ribonucleotide at the primer 3′-terminus strongly inhibited dNTP polymerization. Adding a single NMP to the 3′-end of a DNA primer (DNA16ribo) decreased kcat/Km for polymerization of the next correct dNTP by 3200-fold as compared to DNA containing a dNMP at the 3′-terminus (Figure 8). Similarly, we previously observed that herpes polymerase does not efficiently elongate RNA primers (36). To further test these results, we incubated UL30 (exonuclease proficient) with excess primer-template that contained a ribonucleotide at the primer 3′-terminus and the next correct dNTP. This resulted in the vast majority of the DNA being processed in the exonuclease site and only small amounts of elongated product (<5%, Figure 9).
Figure 8.
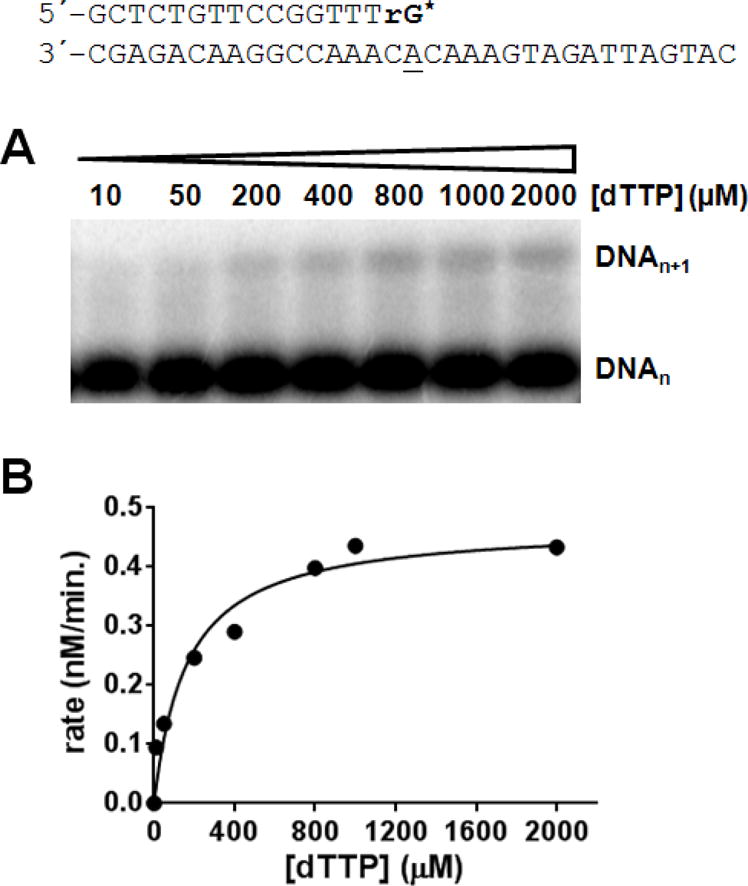
HSV pol does not efficiently polymerize dNTPs onto DNA containing a ribonucleotide at the 3′-terminus. UL30/UL42 (exo-) (50 nM) was incubated with DNAribo (1 μM) in the presence of 100 μM TTP and aliquots were taken out after 15 min. (A) Phosphorimages of the products. (B) Plot of products as a function of TTP concentration. Data were fit to the Michaelis–Menten equation and gave Vmax = 4.7 ± 0.4 (nM/min.) and KmdTTP = 163 ± 53 μM.
Figure 9.
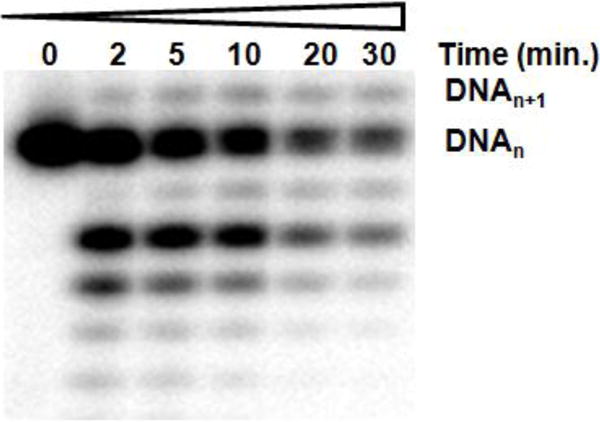
UL30 preferentially degrades DNA containing a ribonucleotide at the primer 3′-terminus rather than adding the next correct dNTP. UL30 (exo+) (70 nM) was incubated with DNAribo (1 μM) in the presence of 100 μM dTTP and aliquots were taken at various time intervals. Phosphorimages of the products of DNA extension and degradation using 100 μM dTTP which is the next correct incoming dNTP.
DISCUSSION
Two clinically useful inhibitors of herpes polymerase, ACVTP and PFA, result in E-DNA complexes where the DNA is tightly bound in the polymerase active site. In the case of ACVTP, the DNA resulting from incorporation (E-DNAACV) binds with moderate affinity. However, the next correct dNTP binds very tightly (KD = 76 nM) to generate a very stable E-DNAACV-dNTP ternary complex (24). Importantly, the presence of this bound DNA in the polymerase active site had no effect on 3′–5′ exonuclease activity. In the case of acyclovir, this occurred for both the E-DNAACV binary complex generated immediately after ACVTP polymerization as well as the E-DNAACV-dNTP ternary complex generated by ACVTP polymerization followed by binding of the next correct dNTP. Mechanistically, these data provide further support for UL30 containing at least two independent and non-overlapping DNA binding domains, one for the exonuclease and one for the polymerase.
The exonuclease likely remains active throughout the polymerase catalytic cycle (i.e., when the polymerase is in the open and closed complexes). We previously showed that the exonuclease retains activity upon formation of either an E-DNA binary complex or an E-DNA-aphidicolin ternary complex (30). Based on the structures of other B-family polymerases with just DNA or DNA and aphidicolin bound, the polymerase active site is likely in the open complex in both cases (37). In contrast, formation of the herpes polymerase-DNAACV-dNTP complex likely generates the closed complex. While the actual structure of this complex is not known, two data suggest it will resemble the closed complex. (1) The next correct dNTP binds very tightly to the E-DNAACV complex (24). (2) Other B-family polymerases form the closed complex upon generating the E-DNA-dNTP ternary complexes where the DNA cannot be elongated (e.g., it contains a 2′, 3′-dideoxynucleotide at the 3′ terminus) (38–44). Likewise, binding of PFA to the E-DNA binary complex to generate the E-DNA-PFA ternary complex probably converts UL30 into the closed conformational state, albeit the complex present after phosphodiester bond formation but prior to release of pyrophosphate. While the structure of this ternary complex has not been solved for UL30, the structure of a chimeric version of the B-family polymerase RB69-DNA-PFA ternary complex closely resembles the closed E-DNA-dNTP ternary complex (45). Importantly, these observations suggest that the exonuclease remains active regardless of whether or not the polymerase is actively polymerizing dNTPs.2
Depending upon assay conditions, we found that PFA and PAA either did not inhibit exonuclease activity or gave very mild inhibition. This, however, contrasts with previous studies that reported significant exonuclease inhibition by PFA and/or PAA (33–35). The likely cause of these differences is the DNAs used. Whereas previous work used long DNAs whose structure is not well defined (oligo(dG).poly(dC) (33), activated calf thymus DNA (34) and E. coli DNA (35)), we used two shorter DNAs of defined sequence (DNA15C and DNA30C). The latter DNA was designed to be able to completely fill the DNA binding domain of the UL30/UL42 complex as defined by the nuclease protection studies of Challberg (46). This begets the question, however, of which DNA more closely resembles the DNA found in vivo? On one hand the length of calf thymus DNA or poly(dC) is certainly more similar to that found in vivo. However, the structure/sequence of these DNAs will be very different than is found in vivo. For example, the activated calf thymus DNA will contain loops, nicks, variable lengths of single- and double-stranded DNA, etc., depending upon how it was handled. The DNAs we used will closely resemble a typical primer-template in terms of structure, but they are certainly much shorter than in vivo DNA.
The observation that both acyclovir and Foscarnet mediated polymerase inhibition minimally impact exonuclease activity may have significant implications for how these compounds impact herpes replication. The potent polymerase inhibition exhibited by both compounds is certainly critical for their biological effects as evidenced by the mechanisms by which herpes can become resistant to these drugs (13–14). With both drugs, mutations that affect ACVTP polymerization and PFA binding can give high level resistance3. In the case of drug sensitive virus, however, if these inhibitors simply stopped the polymerase from synthesizing new DNA, herpes could potentially continue replicating its DNA once the drug concentration dropped below the value needed for effective polymerase inhibition. Inhibiting the polymerase but leaving the exonuclease active could reduce this possibility since upon encountering a 3′ end of a DNA molecule, the exonuclease could degrade this already synthesized DNA. This destruction of preexisting DNA would be expected to negatively impact the potential for herpes to resume DNA replication upon diminution of the intracellular drug concentration.
Similar to previous work, we found that the exonuclease very poorly excises acyclovir from the 3′-terminus of DNA (20, 29). This was true for single-stranded DNA, a double-stranded primer-template and if the polymerase active site was occupied by a separate piece of DNA. Additionally, the presence of acyclovir in the DNA did not significantly impact binding of the DNA to the exonuclease active site. Together, these data indicate that the acyclovir inhibits exonuclease activity by directly blocking the hydrolytic reaction. This contrasts with the conclusions of Hanes et al. who inferred that acyclovir terminated DNA is a poor substrate for the exonuclease because of inefficient transfer of the DNA from the polymerase to the exonuclease based on transient kinetic methods (29). Three observations from our studies indicate that this conclusion is incorrect. First, the presence of acyclovir at the 3′-terminus of both single-stranded DNA and a primer-template does not affect binding of the DNA to the exonuclease active site. Second, the exonuclease inefficiently hydrolyzes acyclovir terminated single-stranded DNA, a DNA that never has to be transferred between the two active sites. Third, blocking the polymerase active site with another DNA still results in inefficient excision of acyclovir even though the blocked polymerase site eliminates the possibility of DNA transfer between the polymerase and exonuclease. Thus, inefficient hydrolysis of acyclovir results from the altered sugar structure directly impacting the exonuclease, not from an effect on transfer.
The herpes exonuclease exhibits surprising specificity with respect to the sugar but not the base. As would be expected for a proofreading exonuclease, the enzyme readily excises any of the four natural dNMPs. In contrast, removing the 3′-hydroxyl and/or the 2′-methylene greatly inhibits exonuclease activity. While interactions of the exonuclease active site with the sugar of the 3′-terminal nucleotide are not important for binding of the nucleic acid, these interactions likely either directly stabilize the transition state for phosphodiester bond cleavage and/or properly align the phosphodiester bond for attack by H2O.
Unlike the inhibitory effects of removing the 3′-hydroxyl or the 2′-methylene from the terminal nucleotide, the presence of a 2′-hydroxyl had no effect on exonuclease activity such that the enzyme removed NMPs and 2′-dNMPs at similar rates. Conversely, the presence of a single NMP at the 3′-end of the primer reduced the efficiency of addition of the next correct dNTP by 3200-fold as compared to a primer containing a dNMP at the 3′-terminus. Similarly, previous studies showed that herpes polymerase elongates RNA primers much less efficiently than DNA primers (700- to 26,000-fold). The comparable effects indicate that the 2′-hydroxyl at the primer 3′-terminus causes much, if not all, of the slow elongation of RNA primers (36).
The greatly reduced rate of dNTP polymerization onto a ribonucleotide in combination with an active exonuclease also suggests a simple and novel “proofreading” mechanism by which the herpes replication machinery may minimize the stable incorporation of NMPs into DNA. Upon incorporation of a ribonucleotide, the greatly reduced rate of further dNTP polymerization will provide time for the exonuclease to remove the NMP prior to the polymerase eventually incorporating the next correct dNTP. This ribonucleotide proofreading is analogous to how UL30 can proofread incorrectly polymerized dNTPs, although it should be noted that the efficiency of NTP proofreading will likely be less efficient than that for incorrect dNTP proofreading. Whereas UL30 discriminated against ribonucleotide elongation by 3,200-fold, UL30 discriminates against mismatch elongation by 104- to 105-fold (47).
As far as we are aware, this ability of UL30 to efficiently remove incorporated ribonucleotides via proofreading distinguishes it from all other replicative DNA polymerases. DNA pol α, δ and ε replicate eukaryotic nuclear DNA and none of them efficiently proofread incorporated ribonucleotides. Pol α efficiently elongates primers containing one (or more) ribonucleotides at the primer 3′-terminus and lacks a proofreading exonuclease (48). The 3′–5′ exonuclease of pol δ does not proofread ribonucleotides incorporated by pol δ’s polymerase activity while the 3′–5′ exonuclease of pol ε only weakly proofreads ribonucleotides incorporated by pol ε’s polymerase activity (49–50). This inability of pol α, δ and ε to efficiently remove just incorporated ribonucleotides from newly synthesized DNAs results in eukaryotic cells employing RNase H2 to perform this essential function (49). Likewise, bacterial pol III and Φ29 polymerase incorporate NTPs and then continue dNTP polymerization (51–52). In the case of herpes, UL30 can use its proofreading function to minimize stable ribonucleotide incorporation into DNA and potentially eliminate the need for RNaseH2 or other post-replicative mechanisms for excising ribonucleotides.
The apparently complete independence of the polymerase and 3′–5′ exonuclease active sites of UL30 has two important implications. First, it suggests a novel mechanism by which current herpes treatments may fatally disrupt herpes replication, the destruction of already synthesized herpes DNA. Second, it raises the questions of where on UL30 the DNA binding domain for the exonuclease resides and how the exonuclease and polymerase activities are coordinated at the replication fork. Studies to answer these questions are in progress.
Supplementary Material
Footnotes
Funding Information: This work was supported by NIH grant AI59764 to RDK.
Abbreviations Used
ACV, acyclovir; ACVTP, acyclovir triphosphate; BSA, bovine serum albumin; ddNTP, 2′, 3′–di-deoxyribonucleoside–5′–triphosphate; DNAACV, DNA containing acyclovir at the primer 3′-terminus; DNAGCV, DNA containing ganciclovir at the primer 3′-terminus; DTT, dithiothreitol; EDTA, ethylenediamine tetraacetic acid; GCV, ganciclovir; GCVTP, gancicyclovir triphosphate; Hepes, 4–(2–hydroxyethyl)–1–piperazine–ethanesulfonic acid; HSV, herpes simplex virus; PAA, phosphonoacetic acid; PFA, phosphonoformic acid (Foscarnet); P/T, primer/template.
While unlikely, an alternative mechanism that we cannot absolutely rule out is the presence of two populations of polymerase, one with an active polymerase and one with an active exonuclease. As far as we are aware this has never been observed with a polymerase.
The primary mechanism by which HSV-1 becomes resistant to acyclovir is via mutations in the viral thymidine kinase gene such that HSV infected cells no longer accumulate the biologically active ACVTP when treated with acyclovir (1–4). Secondarily, resistance is also mediated by mutations in the UL30 gene such that the polymerase does not readily incorporate ACVTP (5–6).
Supporting Information
Contains additional data showing; (1) Effects of foscarnet on exonuclease activity using single-stranded DNA as substrate; (2) Effects of PAA on exonuclease activity using single-stranded DNA as substrate; (3) Effects of foscarnet on exonuclease activity using double-stranded DNA as substrate; (4) Effects of PAA on simultaneous polymerase and exonuclease activity, and; (5) Effects of foscarnet on simultaneous polymerase and exonuclease activity.
References
- 1.Furman PA, Coen DM, St Clair MH, Schaffer PA. Acyclovir-resistant mutants of herpes simplex virus type 1 express altered DNA polymerase or reduced acyclovir phosphorylating activities. J Virol. 1981;40:936–941. doi: 10.1128/jvi.40.3.936-941.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crumpacker CS, Schnipper LE, Chartrand P, Knopf KW. Genetic mechanisms of resistance to acyclovir in herpes simplex virus. Am J Med. 1982;73:361–368. doi: 10.1016/0002-9343(82)90123-1. [DOI] [PubMed] [Google Scholar]
- 3.Crumpacker CS, Chartrand P, Subak-Sharpe JH, Wilkie NM. Resistance of herpes simplex virus to acycloguanosine–genetic and physical analysis. Virology. 1980;105:171–184. doi: 10.1016/0042-6822(80)90165-8. [DOI] [PubMed] [Google Scholar]
- 4.Coen DM, Schaffer PA, Furman PA, Keller PM, St Clair MH. Biochemical and genetic analysis of acyclovir-resistant mutants of herpes simplex virus type 1. Am J Med. 1982;73:351–360. doi: 10.1016/0002-9343(82)90122-x. [DOI] [PubMed] [Google Scholar]
- 5.Huang L, Ishii KK, Zuccola H, Gehring AM, Hwang CB, Hogle J, Coen DM. The enzymological basis for resistance of herpesvirus DNA polymerase mutants to acyclovir: relationship to the structure of alpha-like DNA polymerases. Proc Natl Acad Sci U S A. 1999;96:447–452. doi: 10.1073/pnas.96.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coen DM, Furman PA, Gelep PT, Schaffer PA. Mutations in the herpes simplex virus DNA polymerase gene can confer resistance to 9-beta-D-arabinofuranosyladenine. J Virol. 1982;41:909–918. doi: 10.1128/jvi.41.3.909-918.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Severson JL, Tyring SK. Viral disease update. Curr Probl Dermatol. 1999;11:43–70. [Google Scholar]
- 8.Boehmer PE, Lehman IR. Herpes simplex virus DNA replication. Annu Rev Biochem. 1997;66:347–384. doi: 10.1146/annurev.biochem.66.1.347. [DOI] [PubMed] [Google Scholar]
- 9.Chattopadhyay S, Chen Y, Weller SK. The two helicases of herpes simplex virus type 1 (HSV-1) Front Biosci. 2006;11:2213–2223. doi: 10.2741/1964. [DOI] [PubMed] [Google Scholar]
- 10.Lehman IR, Boehmer PE. Replication of herpes simplex virus DNA. J Biol Chem. 1999;274:28059–28062. doi: 10.1074/jbc.274.40.28059. [DOI] [PubMed] [Google Scholar]
- 11.Kuhn FJ, Knopf CW. Herpes simplex virus type 1 DNA polymerase. Mutational analysis of the 3′–5′-exonuclease domain. J Biol Chem. 1996;271:29245–29254. doi: 10.1074/jbc.271.46.29245. [DOI] [PubMed] [Google Scholar]
- 12.Hwang YT, Liu BY, Coen DM, Hwang CB. Effects of mutations in the Exo III motif of the herpes simplex virus DNA polymerase gene on enzyme activities, viral replication, and replication fidelity. J Virol. 1997;71:7791–7798. doi: 10.1128/jvi.71.10.7791-7798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bacon TH, Levin MJ, Leary JJ, Sarisky RT, Sutton D. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin Microbiol Rev. 2003;16:114–128. doi: 10.1128/CMR.16.1.114-128.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clercq E. Antiviral drugs: current state of the art. J Clin Virol. 2001;22:73–89. doi: 10.1016/s1386-6532(01)00167-6. [DOI] [PubMed] [Google Scholar]
- 15.McGuirt PV, Furman PA. Acyclovir inhibition of viral DNA chain elongation in herpes simplex virus-infected cells. Am J Med. 1982;73:67–71. doi: 10.1016/0002-9343(82)90066-3. [DOI] [PubMed] [Google Scholar]
- 16.St Clair MH, Furman PA, Lubbers CM, Elion GB. Inhibition of cellular alpha and virally induced deoxyribonucleic acid polymerases by the triphosphate of acyclovir. Antimicrob Agents Chemother. 1980;18:741–745. doi: 10.1128/aac.18.5.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elion GB, Furman PA, Fyfe JA, de Miranda P, Beauchamp L, Schaeffer HJ. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc Natl Acad Sci U S A. 1977;74:5716–5720. doi: 10.1073/pnas.74.12.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weller SK, Kuchta RD. The DNA helicase-primase complex as a target for herpes viral infection. Expert Opin Ther Targets. 2013;17:1119–1132. doi: 10.1517/14728222.2013.827663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furman PA, St Clair MH, Spector T. Acyclovir triphosphate is a suicide inactivator of the herpes simplex virus DNA polymerase. J Biol Chem. 1984;259:9575–9579. [PubMed] [Google Scholar]
- 20.Derse D, Cheng YC, Furman PA, St Clair MH, Elion GB. Inhibition of purified human and herpes simplex virus-induced DNA polymerases by 9-(2-hydroxyethoxymethyl)guanine triphosphate. Effects on primer-template function. J Biol Chem. 1981;256:11447–11451. [PubMed] [Google Scholar]
- 21.Fyfe JA, Keller PM, Furman PA, Miller RL, Elion GB. Thymidine kinase from herpes simplex virus phosphorylates the new antiviral compound, 9-(2-hydroxyethoxymethyl)guanine. J Biol Chem. 1978;253:8721–8727. [PubMed] [Google Scholar]
- 22.Miller WH, Miller RL. Phosphorylation of acyclovir (acycloguanosine) monophosphate by GMP kinase. J Biol Chem. 1980;255:7204–7207. [PubMed] [Google Scholar]
- 23.Miller WH, Miller RL. Phosphorylation of acyclovir diphosphate by cellular enzymes. Biochem Pharmacol. 1982;31:3879–3884. doi: 10.1016/0006-2952(82)90305-7. [DOI] [PubMed] [Google Scholar]
- 24.Reardon JE, Spector T. Herpes simplex virus type 1 DNA polymerase. Mechanism of inhibition by acyclovir triphosphate. J Biol Chem. 1989;264:7405–7411. [PubMed] [Google Scholar]
- 25.Chen H, Beardsley GP, Coen DM. Mechanism of ganciclovir-induced chain termination revealed by resistant viral polymerase mutants with reduced exonuclease activity. Proc Natl Acad Sci U S A. 2014;111:17462–17467. doi: 10.1073/pnas.1405981111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crumpacker CS. Mechanism of action of foscarnet against viral polymerases. Am J Med. 1992;92:3S–7S. doi: 10.1016/0002-9343(92)90329-a. [DOI] [PubMed] [Google Scholar]
- 27.Leinbach SS, Reno JM, Lee LF, Isbell AF, Boezi JA. Mechanism of phosphonoacetate inhibition of herpesvirus-induced DNA polymerase. Biochemistry. 1976;15:426–430. doi: 10.1021/bi00647a029. [DOI] [PubMed] [Google Scholar]
- 28.Overby LR, Robishaw EE, Schleicher JB, Rueter A, Shipkowitz NL, Mao JC. Inhibition of herpes simplex virus replication by phosphonoacetic acid. Antimicrob Agents Chemother. 1974;6:360–365. doi: 10.1128/aac.6.3.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanes JW, Zhu Y, Parris DS, Johnson KA. Enzymatic therapeutic index of acyclovir. Viral versus human polymerase gamma specificity. J Biol Chem. 2007;282:25159–25167. doi: 10.1074/jbc.M703972200. [DOI] [PubMed] [Google Scholar]
- 30.Vashishtha AK, Kuchta RD. Polymerase and exonuclease activities in herpes simplex virus type 1 DNA polymerase are not highly coordinated. Biochemistry. 2015;54:240–249. doi: 10.1021/bi500840v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramirez-Aguilar KA, Low-Nam NA, Kuchta RD. Key role of template sequence for primer synthesis by the herpes simplex virus 1 helicase-primase. Biochemistry. 2002;41:14569–14579. doi: 10.1021/bi026680v. [DOI] [PubMed] [Google Scholar]
- 32.Kuchta RD, Mizrahi V, Benkovic PA, Johnson KA, Benkovic SJ. Kinetic mechanism of DNA polymerase I (Klenow) Biochemistry. 1987;26:8410–8417. doi: 10.1021/bi00399a057. [DOI] [PubMed] [Google Scholar]
- 33.Knopf KW. Properties of herpes simplex virus DNA polymerase and characterization of its associated exonuclease activity. Eur J Biochem. 1979;98:231–244. doi: 10.1111/j.1432-1033.1979.tb13181.x. [DOI] [PubMed] [Google Scholar]
- 34.Derse D, Cheng YC. Herpes simplex virus type I DNA polymerase. Kinetic properties of the associated 3′–5′ exonuclease activity and its role in araAMP incorporation. J Biol Chem. 1981;256:8525–8530. [PubMed] [Google Scholar]
- 35.Ostrander M, Cheng YC. Properties of herpes simplex virus type 1 and type 2 DNA polymerase. Biochim Biophys Acta. 1980;609:232–245. doi: 10.1016/0005-2787(80)90234-8. [DOI] [PubMed] [Google Scholar]
- 36.Cavanaugh NA, Kuchta RD. Initiation of new DNA strands by the herpes simplex virus-1 primase-helicase complex and either herpes DNA polymerase or human DNA polymerase alpha. J Biol Chem. 2009;284:1523–1532. doi: 10.1074/jbc.M805476200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baranovskiy AG, Babayeva ND, Suwa Y, Gu J, Pavlov YI, Tahirov TH. Structural basis for inhibition of DNA replication by aphidicolin. Nucleic Acids Res. 2014;42:14013–14021. doi: 10.1093/nar/gku1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hopfner KP, Eichinger A, Engh RA, Laue F, Ankenbauer W, Huber R, Angerer B. Crystal structure of a thermostable type B DNA polymerase from Thermococcus gorgonarius. Proc Natl Acad Sci U S A. 1999;96:3600–3605. doi: 10.1073/pnas.96.7.3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H, Cao W, Zakharova E, Konigsberg W, De La Cruz EM. Fluorescence of 2-aminopurine reveals rapid conformational changes in the RB69 DNA polymerase-primer/template complexes upon binding and incorporation of matched deoxynucleoside triphosphates. Nucleic Acids Res. 2007;35:6052–6062. doi: 10.1093/nar/gkm587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J, Sattar AK, Wang CC, Karam JD, Konigsberg WH, Steitz TA. Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- 41.Wang M, Xia S, Blaha G, Steitz TA, Konigsberg WH, Wang J. Insights into base selectivity from the 1.8 A resolution structure of an RB69 DNA polymerase ternary complex. Biochemistry. 2011;50:581–590. doi: 10.1021/bi101192f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berman AJ, Kamtekar S, Goodman JL, Lazaro JM, de Vega M, Blanco L, Salas M, Steitz TA. Structures of phi29 DNA polymerase complexed with substrate: the mechanism of translocation in B-family polymerases. EMBO J. 2007;26:3494–3505. doi: 10.1038/sj.emboj.7601780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 44.Xia S, Vashishtha A, Bulkley D, Eom SH, Wang J, Konigsberg WH. Contribution of partial charge interactions and base stacking to the efficiency of primer extension at and beyond abasic sites in DNA. Biochemistry. 2012;51:4922–4931. doi: 10.1021/bi300296q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zahn KE, Tchesnokov EP, Gotte M, Doublie S. Phosphonoformic acid inhibits viral replication by trapping the closed form of the DNA polymerase. J Biol Chem. 2011;286:25246–25255. doi: 10.1074/jbc.M111.248864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gottlieb J, Challberg MD. Interaction of herpes simplex virus type 1 DNA polymerase and the UL42 accessory protein with a model primer template. J Virol. 1994;68:4937–4945. doi: 10.1128/jvi.68.8.4937-4945.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baker RO, Hall JD. Impaired mismatch extension by a herpes simplex DNA polymerase mutant with an editing nuclease defect. J Biol Chem. 1998;273:24075–24082. doi: 10.1074/jbc.273.37.24075. [DOI] [PubMed] [Google Scholar]
- 48.Lehman IR, Kaguni LS. DNA polymerase alpha. J Biol Chem. 1989;264:4265–4268. [PubMed] [Google Scholar]
- 49.Clausen AR, Zhang S, Burgers PM, Lee MY, Kunkel TA. Ribonucleotide incorporation, proofreading and bypass by human DNA polymerase delta. DNA Repair (Amst) 2013;12:121–127. doi: 10.1016/j.dnarep.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williams JS, Clausen AR, Nick McElhinny SA, Watts BE, Johansson E, Kunkel TA. Proofreading of ribonucleotides inserted into DNA by yeast DNA polymerase varepsilon. DNA Repair (Amst) 2012;11:649–656. doi: 10.1016/j.dnarep.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dahl JM, Wang H, Lazaro JM, Salas M, Lieberman KR. Kinetic mechanisms governing stable ribonucleotide incorporation in individual DNA polymerase complexes. Biochemistry. 2014;53:8061–8076. doi: 10.1021/bi501216a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yao NY, Schroeder JW, Yurieva O, Simmons LA, O’Donnell ME. Cost of rNTP/dNTP pool imbalance at the replication fork. Proc Natl Acad Sci U S A. 2013;110:12942–12947. doi: 10.1073/pnas.1309506110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.