

Abstract
For the last 40 years the authors have collaborated on trying to understand the complexities of human cancer by formulating testable mathematical models that are based on mutation accumulation in human malignancies. We summarize the concepts encompassed by multiple mutations in human cancers in the context of source, accumulation during carcinogenesis and tumor progression, and therapeutic consequences. We conclude that the efficacious treatment of human cancer by targeted therapy will involve individualized, uniquely directed specific agents singly and in simultaneous combinations, and take into account the importance of targeting resistant subclonal mutations, particularly those subclones with alterations in DNA repair genes, DNA polymerase, and other genes required to maintain genetic stability.
Keywords: cancer, mutator hypothesis, DNA replication, DNA repair, carcinogenesis, evolutionary dynamics, duplex sequencing, mathematical models
Introduction
Until some twenty years ago it was commonly accepted that every cell in our body contained similar, if not identical, nuclear genomes. Obviously, there were exceptions– repetitive elements that expanded and contracted, line elements that duplicated, and telomeres that shrunk and elongated. But these were the exceptions. In contrast, the interrogation of multiple genomes from the same individual by massively parallel next generation DNA sequencing (NGS) provided evidence of extensive multigenic mosaicism in cancers, and even in nonmalignant tissues. The low accuracy of routine NGS precluded the detection of rare subclonal mutations and prevented us from realizing the unanticipated plasticity of our genomes.
The recent award of the Nobel Prize in Chemistry for DNA repair brought into focus the extensiveness of DNA damage that occurs in human cells. Advances in chemistry have greatly extended our knowledge of the enzymology of DNA repair and the structure of intermediates in DNA damage repair; advances in DNA sequencing are making it possible to approach exciting biological questions surrounding DNA damage and mutagenesis. Tomas Lindahl calculated that each cell in our body undergoes some 50,000 DNA damage events per day [1]. There are multiple scenarios that could occur when the DNA replicating and/or repair apparatus encounters unrepaired DNA damage. Small lesions are frequently bypassed by DNA polymerases [2]. Larger DNA adducts are more likely to stall DNA replication [3], induce the SOS-response in bacteria, and increase the expression of the Y- family DNA polymerases in eukaryotic cells [4–6]. These specialized DNA polymerases have active sites that can encompass bulky lesions [7], allowing DNA synthesis to proceed.
The accuracy of DNA repair is governed by the ability of repair complexes to recognize distortions in DNA resulting from the presence of altered nucleoside bases and sugar residues. The pioneering work in the laboratory of Phil Hanawalt, along with that of others, established pathways for nucleotide excision repair, global excision repair [8] and transcription coupled repair [9], and consequences of deficiencies in these processes. The fact that biallelic mutations that inactivate many DNA repair enzymes are lethal substantiates the importance of DNA repair mechanisms [10]. Minor changes in the structure of these proteins result in decreased fidelity of DNA repair processes.
The accuracy of DNA replication also depends on both initial conformational recognition of correct base-pairs by the polymerase active site and subsequent proofreading steps [11–13]. The work of Sam Wilson established alterations in the structure of DNA polymerase β at the template-binding site as it encounters complementary or non-complementary nucleotides [14].
Until recently, DNA repair pathways and polymerases were not considered as primary targets for cancer therapy. There were no reports of mutations in DNA polymerase genes in the extensive databases compiled by analyzing DNA from human tumors [15]. Reanalysis of the same database unexpectedly provided extensive documentation that the major replicating DNA polymerases, Pol-δ and –ε were mutated in several human cancers [16]. Moreover, human colon cancers that have mutations in the Pol-δ or Pol-ε exonuclease domain have exceptionally high mutation frequencies throughout their genomes [17–18]. Patients with brain tumors that carry inherited biallelic mismatch repair mutations and develop somatic mutations in replicative DNA polymerases have the highest mutation frequencies reported, (>250/Mb) [19]. Similarly, only a limited number of mutations were reported in the multiple DNA repair pathways. However, recent reports indicate that 10% of metastatic prostate cancers contain inherited mutations in DNA repair genes [20]. From these studies we conclude that DNA repair and DNA replication proteins are attractive targets for design of inhibitors of proliferation in human cancers.
This article presents our perspective on the association of spontaneous mutations with the initiation and progression of human cancers. It stems from collaborations that have been ongoing for more than 40 years on the pathways by which damaged DNA results in mutations in normal and malignant cells, including constructing mathematical models that probe the mechanism and the consequences of mutation for carcinogenesis, tumor evolution, and therapy. The mutator hypothesis [21] states that tumors are genetically unstable compared to normal tissue, and that this plays a critical role in carcinogenesis. The hypothesis of a mutator phenotype in human cancer is increasingly supported by the power of DNA sequencing to unveil the thousands and perhaps millions of changes in the nucleotide sequence of DNA present in the genomes of many cancer cells. [22].
In this commentary, we will focus on mutational diversity. We note that DNA mutations (single-base substitutions) are not the only clinically relevant source of phenotypic variation in human cancers. Chromosomal rearrangements, gene amplification, and stable epigenetic changes can also cause long-term phenotypic variation. Furthermore, short-term plasticity in gene expression can cause transient phenotypic variation within stable epigenetic or genetic states [23]. These latter phenomena are clearly important and can rapidly cause resistance to targeted therapy in a majority of cells within a tumor. The studies carried out in cultured cells and small animals are frequently of short duration and involve fewer cells compared to clinical cancers, and thus may preferentially score for the rapid onset of resistance that is often reversible. Resistance to therapy in humans resulting from DNA alteration is permanent, is observed late in the growth of tumors, and is likely to represent the emergence of pre-existing subclonal mutations [24].
Experimental Support for the Mutator Phenotype Hypothesis
Mechanistic studies of DNA replication, damage, and repair
Originally, the mutator hypothesis was framed around errors made by DNA polymerases during DNA replication [21]. However, with growing knowledge of DNA replication and repair, it became apparent that there are hundreds of genes involved in these processes, alterations of which could also result in enhanced mutagenesis [25]. We envisioned a cascade of mutation accumulation in cancer cells manifested by increasing heterogeneity with random mutations accumulating in DNA replication and repair proteins. It was postulated that amongst the earliest molecular events that initiated transformation of normal cells into premalignant cells was damage to critical genes required for maintaining genetic stability. The initial focus was on replicative DNA polymerases (Pol-α, -δ, and -ε) [13]; these enzymes are responsible for the accurate copying of some 6 billion nucleotides during each division cycle. Single amino acid substitutions in their catalytic sites result in increased errors in nucleotide incorporation. Mutations that reduce the accuracy of nucleotide selection or of exonucleolytic hydrolysis of mis-incorporated nucleotides without diminishing rates of polymerization could result in increased single-base substitutions throughout the genome. Some of these polymerase-induced mutations could occur in additional genes required to maintain genetic stability. A cascade of mutations would ensue, resulting in progressive accumulation of mutations in human tumors (Figure 1).
Figure 1. Mutational cascade during carcinogenesis as envisioned by the mutator hypothesis.

During tumor progression there is a progressive increase in mutations resulting from unrepaired DNA damage. Most have no effect on cellular phenotypes (neutral), others enhance proliferation (drivers), and others cause increased mutagenesis (mutators). As the tumor encounters environmental restrictions such as reduced nutrition, inadequate angiogenesis, hypoxia, etc. specific mutations are selected. Circles represent mutations in genes that enhance mutagenesis, triangles indicate mutations selected that enhance proliferation under adverse conditions, and white rectangles represent passenger mutations of unknown functions. Note that many of the tumor cells contain multiple drivers and mutators. Also to be noted is that many topographically distinct mutations are maintained during tumor proliferation. Adapted from [22].
In humans, the earliest mutations characterizing the transformation of normal cells into malignant cells occur prior to diagnosis and must be extrapolated from DNA obtained from clinical samples. Even with tumors in animals we lack adequate technologies to detect and analyze the earliest changes. The 50,000 lesions produced per cell per day as a result of spontaneous and/or endogenous chemical reactions [1] are not localized to specific genes but instead are distributed stochastically. Important sources of DNA damage include the chemical instability of the DNA helix by depurination [1, 26–27], deamination of cytidine to thymidine [28], and by cellular reactive molecules (eg, oxygen and nitrogen reactive species) [29]. Damage by environmental agents frequently involves similar chemical alterations, and in order to be a significant cause of human cancer must contribute a comparable number of lesions. If unrepaired these DNA altered up bases could increase the frequency of incorporation of non-complementary nucleotides during DNA replication.
Ordered models of carcinogenesis and tumor progression
The concept that malignant cells contained tens of thousands of mutations and that these mutations were major contributors to geographical diversity within a tumor and to therapeutic resistance was not widely accepted; there was scant evidence. Moreover, chemotherapy by a single agent, 5-fluorouracil, was found to be effective against human colon cancer [30]. The most prevalent concept was one in which tumors acquired critical mutations responsible for proliferation and survival in a specific order [31], successively empowering cancer cells with the ability to overcome micro-environmental challenges such as decreased nutrition, decreased oxygen supply, etc. Sequential evolutionary bottlenecks with “clonal sweeps” involving proliferation of specific mutations were postulated, and it was further claimed that these sweeps would “purify”, or reduce, the genetic diversity within the tumor [32]. These concepts were particularly attractive, in that they suggested that therapies directed against a limited number of targets could eradicate cancers, a hope that still flourishes within the pharmaceutical and biotechnology industries.
Evidence for the mutator hypothesis from The Cancer Genome Atlas
Ironically, the most compelling evidence supporting the mutator phenotype was the extensive catalog of single-base substitutions delineated by The Cancer Genome Atlas (TCGA) [15]. It was proposed that by performing whole exome DNA sequencing one could identify a small set of “driver” mutations that were unique to different tumor types. Instead, there ensued a progressively increasing number of mutated genes in each examined cancer. The number of verified mutations in gene-coding sequences ranges from 500 in acute myeloid leukemia to more than 100,000 in single melanomas of the skin [33–37]. Moreover, the most commonly mutated sites are not present in all types of cancer nor are they present in 100% of malignant cells within a tumor. Conventional NGS only detects mutations that are present in the majority of cells within a sample, and only those present at allele frequencies of greater than 1–5% are considered to be non-artifactual [37]. Mutations present at lower frequencies would be frequently missed by routine NGS.
A few mutations may be necessary to avoid the detrimental consequences of environmental changes such as variations in climate, food supply, and threats from predators that have occurred throughout species evolution. Evidence indicates that there are approximately 40 mutations in families between each generation [38]. Somatic cell mutagenesis is more prevalent; most are neutral or detrimental, but a few are advantageous and facilitate adaptation to environmental changes such as variations in the supply of nutrients and oxygen, as well as the threat of immune surveillance for variant cells. It is estimated that only one or two mutations occur each time a normal cell divides.
Theoretical Analysis of the Mutator Hypothesis and Implications for Tumor Evolution
Formulation using focused quantitative modeling
While it is feasible to measure the mutation burden in tumors, theoretical analysis of the mechanism of mutation is hampered by the lack of knowledge of mutation rates and tumor evolutionary histories in addition to mere mutation accumulation. Key parameters such as number of cell generations are invariably unknown. Furthermore, a variety of model structures or topologies are possible and different models may fit the same data. In order to investigate the truth or falsity of the mutator hypothesis, we employed focused and parsimonious models designed specifically to address this question, utilizing a minimum of variable parameters, and a minimum number of untestable assumptions. Systematic sensitivity analyses were conducted not only over parameter values, but also over types of models (i.e. different fundamental assumptions), and only conclusions that were robust to all these sensitivity analyses were reported. We call this approach “focused quantitative modeling” (FQM) [ 39].
Negative clonal selection and lethal mutagenesis
Three theoretical objections raised legitimate doubt about the mutator hypothesis when it was initially proposed. The first is that cell clones exhibiting a mutator phenotype would become extinct due to accumulation of mutations that reduced cellular fitness, “negative clonal selection” [40]. A cell would lose fitness if a mutation occurred in certain critical “reduced fitness” (RF) loci. RF loci could either be dominant (i.e., heterozygous mutation reduces fitness) or recessive (i.e., only biallelic mutation reduces fitness). Moreover, the model considered a worst-case scenario in which cells with a single RF mutation became extinct, when in fact such cells often have compensatory mechanisms, including defects in apoptotic processes. Thus, the model would overestimate the impact of negative clonal selection in the presence of a mutator phenotype. Key parameters in the model were the baseline mutation rate, the fold increase in mutation rate due to a mutator mutation, and the number of dominant and recessive RF loci (measured in template nucleotides). Key conclusions included: recessive RF loci are not important because biallelic random RF mutations are infrequent, and “negative clonal selection” only inhibits growth by several-fold, except under extreme circumstances. These small effects might be overshadowed by more rapid acquisition of oncogenic mutations and this hypothesis was later confirmed (vide infra).
A key prediction of this work was that there was a mutation threshold beyond which additional mutations would result in a net inhibition of growth. An upper limit to mutation tolerance has been established by a series of elegant experiments measuring the DNA sequence of singularly budded yeast [41–42] with mutations that inactivate multiple repair pathways. The upper limit of single-base substitutions is similar to that exhibited by neural tumors from patients with biallelic mutations in mismatch repair and acquired mutations in replicative DNA polymerases [19]. Ongoing are experiments to increase the mutation frequency past this error limit using nucleoside analogues that form non-canonical base-pairings at high frequency, a treatment strategy termed “lethal mutagenesis” [43–45]. The success of this strategy may depend on a pre-existing mutator phenotype in the cancer cell, providing selectivity compared to normal cells.
Epidemiologic evidence and models of carcinogenesis
Models of epidemiologic data have shown that cancer incidence increased as the 2nd–12th power of age, most commonly the 6th power, and this was most commonly assumed to represent the number of mutations that were required for cellular transformation [46–49]. The subsequent papers of Hanahan and Weinberg involving transfection of cells with oncogenes, associated each oncogenic mutation with a fundamental essential characteristic of the malignant phenotype [50–51], and the six oncogenic mutations associated with essential phenotypes in [50] were considered to correspond to the six mutations derived from the epidemiologic analysis. Six independent oncogenic mutations at a wild type mutation rate of 10−9 per base per cell division is clearly not possible in the absence of selection, given the total number of cells in the human body, forming the basis of an argument that a mutator mutation is “necessary” for multistage carcinogenesis [52]. Conversely, it is possible to formulate a model in which each successive oncogenic mutation confers a fitness advantage without tumor initiation. These events would fit the epidemiologic data without postulating a mutator mutation, suggesting that a mutator mutation might not be necessary to explain the cancer incidence curves, and therefore comprising a second objection to the mutator hypothesis [53].
Efficiency of carcinogenesis
Comparisons of carcinogenesis models with cancer incidence data implicitly assume that the rate of forming founder cells is equal to that of cancer incidence, an assumption which may be questionable since many incipient cancers do not survive to be clinically detectable. Accordingly, instead of trying to determine whether mutator mutations were necessary to fit epidemiologic data, we asked what pathways for malignant transformation would be most efficient. We reasoned that all possible pathways to a cancer cell may in fact be in play, with or without mutator mutations, with varying degrees of selection. However, those evolutionary pathways that create transformed cells more efficiently would be more likely to be reflected in observed clinical cancers. Efficiency was defined as the average expected number of founder cells the evolutionary pathway would develop in the amount of time (in cell generations) generally associated with carcinogenesis. Of interest for the mutator hypothesis was the relative efficiency in generating malignant cells of an evolutionary pathway with a mutator mutation (occurring at any time during carcinogenesis) compared to the efficiency of the analogous pathway without a mutator mutation [54]. A ratio of > 1 would indicate mutator pathways were favored, whereas a ratio of < 1 would indicate the reverse.
In this framework, it was clear that a third objection to the mutator hypothesis needed to be considered. If multistage carcinogenesis occurs in a given number of stages, the mutator hypothesis adds an additional step, requiring a mutator mutation. Mutations are rare events, and the number of bases in the genome potentially resulting in a mutator phenotype is unknown. Even with no selection for cells with incomplete complements of oncogenes (the “constant fitness” case), would the low probability and rate of this additional step (acquisition of the mutator mutation) outweigh the advantage of more rapid acquisition of required oncogenic mutations?
A radically different model of carcinogenesis
In investigating these questions, we constructed a model [54] that was radically different from the accepted multi-stage carcinogenesis model with a fixed sequence of acquisition of mutations [31 ]. In our model, any oncogenic mutation as well as the mutator mutation could occur at any time during carcinogenesis. All carcinogenic pathways were “in play”, substantially increasing the potential complexity and diversity of cancer, with significant implications for tumor evolution and therapy. Furthermore, a particular malignant phenotype might be obtained from any of a number of alternative oncogenic mutations. For example, there might be twelve alternative mutations any one of which might result in an invasive phenotype. A tumor cell would need to have undergone mutations in at least one of the set of oncogenic genes that specify each critical phenotype. This effectively implied that convergent evolution would occur within tumors. We subsequently made this prediction explicit [39], and it was confirmed in a study of renal cell cancer in which convergent evolution was observed within single lesions where neighboring cells harbored different mutations conferring the same phenotype [55]. Given the competition between neighboring cells to acquire a full complement of oncogenic mutations inherent in our model, it is also not unreasonable to expect that in many instances more than one founder cell would be formed at approximately the same time. Currently, tumor evolution is thought of as a single branching evolutionary tree with a single trunk that might be targeted by therapy. We now predict the eventual discovery of small evolutionary forests with multiple trees and therefore multiple trunks, a prediction which is less clear in models which seek to impose a fixed order on the oncogenic process.
Robust predictions and experimental support
We first investigated the simple “constant fitness” case where partial complements of oncogenic mutations did not result in any increase in fitness. We then created more complex models encompassing several variations of selection during carcinogenesis, including a case with negative clonal selection, to investigate the second and first objections mentioned above, respectively [56]. Below we present the conclusions and predictions robust to all these different scenarios. A separate group using a different mathematical approach independently verified these calculations [57]. Key parameters in the models included the number of oncogenic mutations needed for full transformation, the number of oncogenic loci- (in bases) mutation of which could result in the oncogenic phenotype(s), the number of loci (in bases)- mutation of which could result in a mutator mutation, the number of cell generations until tumor initiation, the wild type mutation rate per base per cell generation, and the fold increase in the mutation rate conferred by a mutator mutation. Representative results for the constant fitness case are shown in Figure 2.
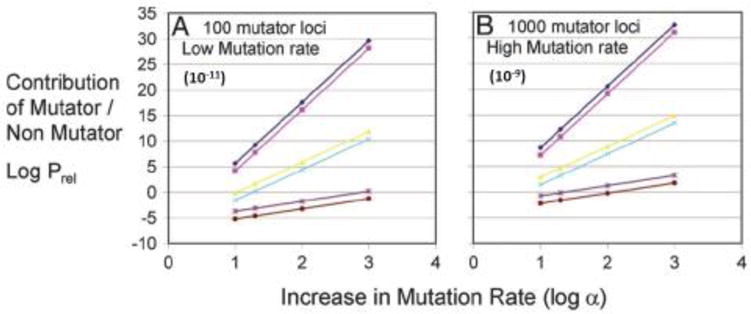
Figure 2. Efficiency of carcinogenesis with and without a mutator mutant.
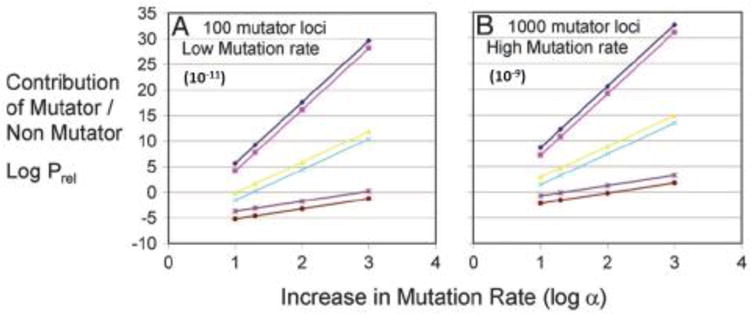
Log Prel, the common logarithm of the relative efficiency of carcinogenesis with a mutator mutation compared to that without a mutator mutation, versus log α, the common logarithm of the fold increase in mutation rate due to a mutator mutation. Parameters in (A) were: 100 base loci, mutation of which could lead to a mutator phenotype; and an initial mutation rate of 10−11 per base per cell generation. Parameters in (B) were: 1000 base loci; and an initial rate of 10−9 per base per cell generation; the number of cell generations to cancer is 170 (brown, light blue, and magenta) or 5000 (purple, yellow and dark blue); the number of required oncogenic mutations for transformation is either 2 (lower lines), 6 (middle lines), or 12 (upper lines). Log Prel > 0 indicates mutator mutations are favored, and by increasing powers of 10. Log Prel < 0 means non-mutator pathways are favored, again by increasing powers of 10 for more negative numbers. Adapted from [54].
The most robust result is that mutator pathways are favored by factors ranging from thousands to larger than billions. The extraordinary degree to which mutator pathways are favored by this analysis eliminates uncertainty in the major question despite uncertainty in parameter values. For example, we believe the assumption of 100–1000 base loci, mutation of which may result in mutator mutations, was reasonable given the number of known DNA replication and repair genes that affect mutation rates in yeast [25, 41–42]. However, if we were wrong and only one base in the entire genome could result in a mutator mutation, the major conclusion of this analysis would not change. This illustrates the advantage of FQM and calculations of rate ratios compared to attempts to parameterize models with many uncertainties in absolute rates.
Mutator pathways are invariably favored if 4 or more oncogenic mutations are required for cellular transformation. If 3 oncogenic mutations are required, the result is sensitive to the parameters of the models. If 1 or 2 oncogenic mutations are required, non-mutator pathways are favored. This makes sense intuitively; it may be counterproductive to first incur the delay of acquiring a mutator mutation in order to speed up the acquisition of only 1 or 2 oncogenic mutations. Retinoblastoma is a childhood tumor in which epidemiologic analysis has indicated that only two “hits” are required for cellular transformation [58]. Our model predicts a lower mutational burden for retinoblastoma, and this has been confirmed experimentally [59]. A recent paper attempts to determine the number of rate-limiting oncogenic mutations in solid tumors by epidemiologic comparisons of related variants of tumors with known differences in their mutation rates (colorectal cancer with and without mismatch repair deficiency, lung cancer in smokers and non-smokers), concluding colorectal and lung cancer have only three rate-limiting oncogenic mutational steps [60]. However, the analysis assumes a constant mutation rate throughout carcinogenesis, and would not be valid if the mutation rate were allowed to vary during carcinogenesis, as in our models. ‘Biological analysis of required phenotypes for transformation suggests a larger number of mutations may be required [50, 51].
The models with positive selection of cells with a partial complement of oncogenic mutations show that mutator mutations still increase carcinogenic efficiency. Conversely, selection of these cells increases the efficiency of carcinogenesis as well, and is a very reasonable proposal especially for mutations that increase proliferation rates and/or decrease death rates. Thus, we consider that both enhanced mutation and selection of partial complements of oncogenic mutations are likely to be essential features of carcinogenesis.
The model with negative clonal selection in the presence of acquisition of oncogenic mutations [56] produced additional insights, in that it was possible to estimate bounds for an optimal mutation rate in carcinogenesis, depending on the parameter values. According to the parameter values considered, the optimal mutation rate could be as low as 2 × 10−10 if 10% of genes were dominant in RF genes and only 2 oncogenic mutations were required for transformation, a somewhat extreme case. In most cases the optimum was higher than the wild type mutation rate, ranging as high as 3.6 × 10−6 per base compared to 1 × 10−9 for wild type somatic cells. If the wild type mutation rate is assumed to be optimized for species evolution, we may consider that the optimal mutation rate for tumor evolution might be up to several orders of magnitude higher than that for species evolution, perhaps due to the fact that tumor cells are not constrained by normal homeostatic interactions.
Another prediction was that mutator mutations were more “beneficial” if they occurred earlier in carcinogenesis, increasing the opportunity to accelerate oncogenic mutations. Interestingly, this currently untested prediction would not be verifiable without very deep sequencing of early and late pre-malignant lesions. The models predict that the most efficient pathway would involve an initial mutator mutation in only a small minority of the cells. The incremental mutation burden may thus be invisible in a bulk sample. During carcinogenesis, daughter cells derived from the mutator clone would progressively be enriched in the bulk sample, and the mutator mutation would become detectable in the bulk only late in carcinogenesis. This would create the illusion that the mutator mutation occurred late, and is one of many examples of the customary bulk measurements at low depth potentially being misleading. The conclusion that mutator mutations are likely to be favored is also consistent with the high mutation burden observed in the TCGA project and elsewhere.
In summary, theoretical analysis of a variety of cases using focused quantitative modeling is in accord with the mutator phenotype hypothesis and clarified three objections. Cancer evolutionary pathways containing mutator mutations are very heavily favored as long as 4 or more oncogenic mutations are required for cellular transformation, and this finding is robust across multiple variations of the model. The mutator mutations are predicted to occur early in carcinogenesis. The model featured a highly flexible order of acquisition of oncogenic mutation and mutator mutations in contrast to prevailing ideas of a fixed order of these events. The model correctly predicted the observed high burden of mutations in cancers, the high incidence of mutator mutations in various genes responsible for genome maintenance, the lack of predominance of any gene (with the exceptions of APC and p53) in mutational spectra of cancers, the presence of convergent evolution within tumors, and the lower mutational burden of retinoblastoma, which requires less than 4 oncogenic mutations.
Neutral evolution, the “Big Bang” model, and clonal sweeps
Another aspect of the theoretical analysis relates to the pattern of evolution after the tumor is initiated by a founder cell(s). Our implicit assumption [54] was that once a tumor had a full complement of oncogenic mutations, it was fit and under minimal selective pressure as it could easily outcompete normal tissue. This suggested that other loci would evolve neutrally by random genetic drift. A comprehensive study examining 349 individual intestinal glands in 15 colorectal cancer patients modeled the patterns of geographic localization of mutations by the following approaches- whole exome sequencing, targeted deep sequencing to 600X, copy number variations, and variations in methylation patterns, and geographic localization of amplification of HER2 by fluorescence in situ hybridization [61]. The results supported their “Big Bang” model—similar in spirit to ours—in that there was uniform heterogeneity everywhere, even within individual glands that might have been expected to “fix” more uniform genotypes according to the notion of clonal sweeps. Under neutral evolution, successive branches of the evolutionary tree acquire increasingly minor subclonal mutations. Increasing depth allows one to see forward in time from the clonal mutations which were present in the founder cells to increasingly rare subclonal mutations. No evidence of “clonal sweeps” was detected, although a depth of 600 represents only 600 cells in an entire tumor, perhaps not sufficient to see evidence thereof.
We believe it may be hard to characterize clonal sweeps in multicentric human tumors. First, clonal sweeps may be partial, resulting in incomplete expansion or diminution of a genotype. Secondly, subclonal sweeps may occur which are below the level of detection. Thirdly, if most loci are neutral, such sweeps, contrary to dogma, will not be enriching for most markers. If a subclone of n cells within a larger tumor of N cells grows to dominate the tumor, the marker that increased its fitness will indeed be purified. However, in growing from n to N cells, the subclone will undergo replication and DNA damage, and in the majority of neutral loci, regenerate a degree of diversity similar to the original. Evidence increasingly indicates that the majority of genes in the cancer genome evolve neutrally, despite the importance of the small minority that are positively or negatively selected [62–66].
Deep Sequencing and Diversity
In the scenario in which evolution of an established tumor is neutral at most loci, with little in the way of “purifying selection”, we expect the diversity seen at low sequencing depth to be only the tip of the iceberg of genetic diversity. As the depth of DNA sequencing increases, rarer and rarer subclonal mutations will be detected.
Meaningful detection of extremely rare mutations requires very high accuracy to avoid false positives. This is true whether the sequencing is done on a bulk specimen or one cell at a time. We have pioneered a technique called Duplex Sequencing [24] in which the complementarity of sense and antisense strands is exploited to achieve unprecedented accuracy on the order of less than one error per 108 nucleotides sequenced. Mutations are called only if they are present on both strands of the same molecule, in opposite positions and are complementary. This allows one to correctly score mutations that are seen only once in the sequencing results (“singlets”). Our mathematical models of neutral evolution suggest that as much as 80% of potentially detectable mutations are lost when requiring that a mutation be seen multiple times (RA Beckman, unpublished). The background mutation frequency is less than or equal to 10−8, 10,000 to 100,000 less than routine next generation DNA sequencing, and thus offers the opportunity to accurately measure mutations at specified sites in single cells. The power of Duplex Sequencing is illustrated in Figure 3.
Figure 3. Duplex Sequencing eliminates artifacts.
(A) The active site exons of the ABL1 gene were isolated by double-capture and sequenced by conventional NGS on an Illumina Hiseq 2500 (minimum Phred score=50). (B) The same DNA sample was subject to Duplex Sequencing, revealing only a single point mutation in ABL1 that confers resistance to Imatinib. Adapted from [24].
Clinical Consequences
Dynamic precision medicine
The remarkable genetic diversity of cancer provides a reservoir of possible resistance mechanisms and it is important to note that so-called “passenger” mutations, of little importance to the cancer’s initial growth, may become critical variants when the cancer is confronted with the challenge of therapy [39].
Current precision medicine matches patients to therapies based on static consensus patterns. A sample, sometimes current but often from diagnosis, is analyzed in bulk, and the patient is treated with a therapy matching the sample’s consensus molecular characteristics. It is not surprising, given the subclonal heterogeneity and evolutionary dynamics of cancer, that even the most spectacular responses to targeted therapy have often been short-lived. Moreover, if the therapy is able to stabilize tumor growth or shrink the tumor, it is maintained “as long as the patient is benefiting”. The same process is repeated when tumor growth resumes or relapse occurs.
We have developed an alternative approach, which we term dynamic precision medicine [67, 68]. This approach explicitly considers subclonal heterogeneity and evolutionary dynamics, and adapts therapy very frequently (as often as every 45 days) based on an evolutionary model. Frequent adaptation creates a jagged evolutionary landscape in which it is much harder to evolve resistance than the smooth evolutionary landscape corresponding to continuous unchanging therapy “as long as the patient is benefiting” [69]. Dynamic precision medicine also plans ahead, considering the prevention of resistance as a major priority equal to or greater than tumor size reduction. Further, it also indicates therapy based on the risk of states which are not directly observable.
The approach has been evaluated in simulations of up to 3 million virtual patients, where each virtual patient represented a different clinical presentation of relative abundance of sensitive and resistant subclones to available therapies, growth rates of the subclones, genetic/epigenetic evolution rates between phenotypic states of sensitivity or resistance, and levels of sensitivity and resistance. The range of these parameters was determined from an extensive review of preclinical literature and clinical experience, and the parameter space was meant to be a comprehensive representation across oncology. Thus, the input dataset was extremely large and was designed to maximize the generality of the result.
The original simulation [67] examined a system with two non-cross resistant therapies available, and phenotypic states corresponding to sensitive states, states resistant to either one of the therapies, and states simultaneously resistant to both therapies. Virtual patients could receive one of three alternatives: therapy 1, therapy 2, or a simultaneous combination at reduced dose (we note that simultaneous combination therapy frequently requires dose reduction), and the therapy choice was adjustable every 45 days. Each virtual patient was treated by 6 different strategies in parallel in a randomized in-silico study. A strategy was defined not as a specific therapy sequence, but rather an algorithm for determining individualized treatment sequences based on patient data, including a complete parameter set for the four phenotypic categories at diagnosis and updates on subclonal prevalence every 45 days. Strategy 0 was the current personalized medicine strategy. All the other strategies used a mathematical model of evolution every 45 days to determine the therapy choice, which either minimized the expected total tumor burden or minimized the probability of forming a doubly resistant cell.
The results (Figure 4) showed on average a doubling of survival and an increase in the cure rate from less than 1% to 17–20%. The survival curves for dynamic precision medicine, with long- term survivors, were more similar to immunotherapy survival curves. Indeed, the immune system is inherently adaptive and may present a multi-pronged attack against multiple subclones. The benefit was driven by 1 million of 3 million virtual patients who showed highly significant benefit. The other 2 million virtual patients showed equivalent results to current personalized medicine. The patients who benefited were broadly distributed across input parameter space, indicating very broad benefits across oncology. The average results were equivalent between strategies but different strategies performed better for different virtual patients. Achieving this kind of result today would require a paradigm shift and technological capabilities for frequent sampling at subclonal resolution.
Figure 4. Survival in a virtual clinical trial utilizing two non-cross resistant therapies.
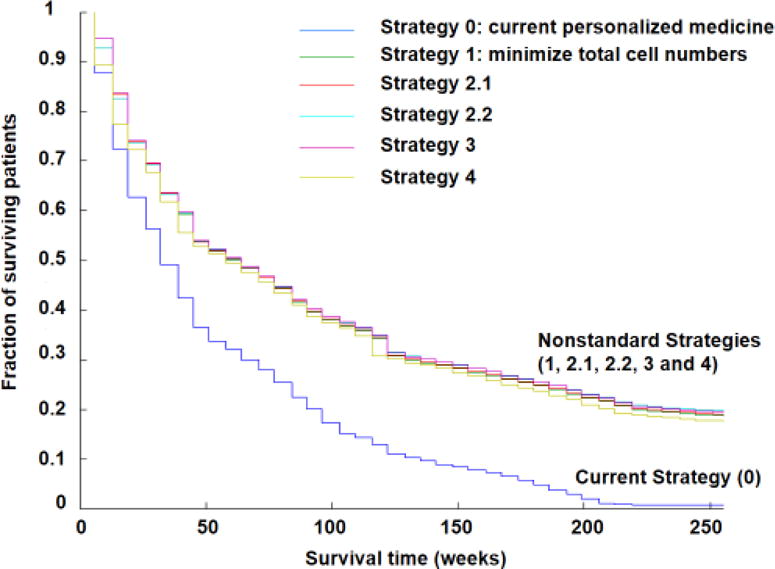
Approximately 3 million virtual patients were treated with each of these strategies. The x axis shows time (weeks) and the y axis shows surviving patient fraction. Strategy 0 (dark blue) is the current personalized medicine strategy: treatment with the best drug for the observed predominant cell type and switching to the alternative drug on tumor progression or relapse. Strategy 1 (green) minimizes total cell numbers at the next timepoint. Strategy 2.1 (red) minimizes the chance of developing doubly resistant cells at the next timepoint unless the patient has detectable disease (109 cells); in that case, total cell number is minimized. Strategy 2.2 (light blue) minimizes the chance of developing doubly resistant cells at the next timepoint unless the patient has a large disease burden (1011 cells); in that case, total cell number is minimized. Strategy 3 minimizes the total cell population unless the predicted number of doubly resistant cells at the next timepoint is ≥ 1; then the likelihood of formation of doubly resistant cells is minimized. Strategy 4 (olive) predicts the time to mortality (1013 cells) and the time to doubly resistant cells, and prioritizes the most imminent threat. Adapted from [67].
Our simulation was unique among theoretical studies in that we allowed different subclones to have different mutation rates, reasoning that they would have different random mutations in DNA replication or repair enzymes. Indeed, we found that hypermutator subclones were particularly dangerous because of their ability to more rapidly evolve independent mutations providing resistance to multiple non-cross resistant therapies.
Complex therapy sequences interleaving combinations and high dose monotherapy;
Recent authors have stressed the importance of combination therapy to address intra-tumoral heterogeneity [70]. This idea has been extensively tested in oncology, having initial success in pediatric leukemias in the 1960s [71] and lymphomas in the 1970s [72]. HIV infection provides an example of successful combination therapy for a disease that evolves quickly but is less genetically complex than cancer, a disease of eukaryotic cells [73]. However, additional progress in other cancers using this idea has been mixed, largely because it is not possible to give simultaneous combinations of adequate complexity at meaningful doses. Due to robustness and redundancy within single genetic states, 3 or 4 agents may be required just to exterminate a single genetic state. With multiple genetic states with different phenotypic properties, effective simultaneous combinations may involve 10 or more agents given at effective doses, which is not feasible. Our simulations, however, indicate that highly complex regimens may be administered as 45-day pulses of single agents at full dose interleaved with combination pulses at lower dose. The sequences are highly individualized and too complex to be derived by intuitive reasoning [67, 68, 74]. We conclude that therapeutic regimens must be of sufficient complexity to counteract intra-tumoral heterogeneity, and simultaneous combinations may be an important component of such regimens.
Diversity as a therapeutic endpoint
Other pioneers have preceded us in proposing evolutionary strategies for therapy of cancer. Gatenby [75] has proposed an adaptive approach to cancer therapy based largely on the notion of pitting cancer subclones against each other. The approach aims to promote diversity and in particular, relies on sensitive cells to hold the growth of resistant cells in check by competition for a limited ecologic niche, thus advocating therapeutic doses that do not fully eradicate sensitive cells. In stark contrast, we believe that competition between subclones is far less important than cooperation between them [76], and we would seek to limit diversity rather than encouraging it. Competition is restricted to a minority of larger lesions visible radiologically, whereas patient survival is limited by countless small metastases growing exponentially without competition. Observations that resistance became apparent after eliminating the sensitive cells are, in our view, based on the fact that pre-therapy bulk measurements fail to detect resistant subclones that are already present and growing.
Summary and Conclusion
In summary, we must conclude that cancer biology is very complex, due to the extraordinary diversity between and within tumors, and to the continued evolution of tumors in response to therapy. Acknowledging and embracing this complexity, we have presented experiments and mathematical models of carcinogenesis that focus on deficits in DNA repair and alterations in DNA synthetic processes as an important contributor to the innumerable mutations that occur in most human cancers. Our mathematical models allow mutations to occur in any order during carcinogenesis, resulting in greater potential complexity and diversity than previous models seeking to impose a fixed order of mutation acquisition. Importantly, our mathematical model of therapy includes variable mutation rates of subclones existing within a single cancer, leading to much richer dynamics, especially in response to therapy. Finally, we include the large number of genes that can potentially affect mutagenesis. Other sophisticated mathematical models have also focused on targeting mutation-selection networks [77–80]. These models have generally assumed a constant mutation rate for all subclones, which we believe to be unrealistic based on the large number of genes that affect mutation rates. Further, our simulation shows that hypermutator clones are priority targets for therapy, even when present as rare variants (1:100,000). Drugs targeting mutant forms of DNA repair and replication proteins may permit specific targeting of hypermutator clones. We believe that approaches focused on the diversity and dynamics within cancers will have a major impact on patient outcomes.
Acknowledgments
Supported by the National Institute of Health under award numbers NCI P01-CA77852,NCI R01-CA160674 and NCI R01-CA102029 (to L.A.L.). We thank J. Shen, A. Kamath-Loeb and M. Blair for comments and editing.
Abbreviations
- APC
adenomatous polyposis coli
- DNA
deoxyribonucleic acid
- FQM
focused quantitative modeling
- HER2
human epidermal growth factor receptor 2
- HIV
human immunodeficiency virus
- NGS
next generation sequencing
- Pol
polymerase
- RF
reduced fitness
- TCGA
The Cancer Genome Atlas
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest disclosure
RAB is a stockholder in Johnson & Johnson, and consults for Astra Zeneca and EMD-Serono. He is also the founder and Chief Scientific Officer of Onco-Mind, LLC, founded to promote novel precision therapy strategies for cancer incorporating evolutionary dynamics.
References
- 1.Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3618. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- 2.Singer B, Grunberger D. Molecular Biology of Mutagens and Carcinogens. Plenum Press; 1983. [Google Scholar]
- 3.Croy RG, Essigmann JM, Reinhold VN, Wogan GN. Identification of the principal aflatoxin B1-DNA adduct formed in vivo in rat liver. Proceedings of the National Academy of Sciences of the United States of America. 1978;75:1745–1749. doi: 10.1073/pnas.75.4.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sweasy JB, Witkin EM. Novel SOS phenotypes caused by second-site mutations in the recA430 gene of Escherichia coli. Biochimie. 1991;73:437–448. doi: 10.1016/0300-9084(91)90111-d. [DOI] [PubMed] [Google Scholar]
- 5.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, et al. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 6.Pillaire MJ, Betous R, Hoffmann JS. Role of DNA polymerase kappa in the maintenance of genomic stability. Molecular & cellular oncology. 2014;1:e29902. doi: 10.4161/mco.29902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang W. An overview of Y-Family DNA polymerases and a case study of human DNA polymerase eta. Biochemistry. 2014;53:2793–2803. doi: 10.1021/bi500019s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pettijohn DE, Hanawalt PC. Evidence for repair-replication of ultraviolet damaged DNA in bacteria. J Mol Biol. 1964;9:395–410. doi: 10.1016/s0022-2836(64)80216-3. [DOI] [PubMed] [Google Scholar]
- 9.Hanawalt PC, Spivak G. Transcription coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 10.Friedberg EC. DNA damage and repair. Nature. 2003;421:436–440. doi: 10.1038/nature01408. [DOI] [PubMed] [Google Scholar]
- 11.Beckman RA, Loeb LA. Multi-stage proofreading in DNA replication. Quarterly reviews of biophysics. 1993;26:225–331. doi: 10.1017/s0033583500002869. [DOI] [PubMed] [Google Scholar]
- 12.St Charles JA, Liberti SE, Williams JS, Lujan SA, Kunkel TA. Quantifying the contributions of base selectivity, proofreading and mismatch repair to nuclear DNA replication in Saccharomyces cerevisiae. DNA repair. 2015;31:41–51. doi: 10.1016/j.dnarep.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burgers PM, Kunkel TA. Eukaryotic DNA Replication Fork. Annual review of biochemistry. 2017 doi: 10.1146/annurev-biochem-061516-044709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Batra VK, Beard WA, Pedersen LC, Wilson SH. Structures of DNA Polymerase Mispaired DNA Termini Transitioning to Pre-catalytic Complexes Support an Induced-Fit Fidelity Mechanism. Structure (London, England: 1993) 2016;24:1863–1875. doi: 10.1016/j.str.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The Cancer Genome Atlas Research Network. https://cancergenome.nih.gov/
- 16.Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature genetics. 2013;45:136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flohr T, Dai JC, Buttner J, Popanda O, Hagmuller E, Thielmann HW. Detection of mutations in the DNA polymerase delta gene of human sporadic colorectal cancers and colon cancer cell lines. International journal of cancer. 1999;80:919–929. doi: 10.1002/(sici)1097-0215(19990315)80:6<919::aid-ijc19>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 18.Rayner E, van Gool IC, Palles C, Kearsey SE, Bosse T, Tomlinson I, et al. A panoply of errors: polymerase proofreading domain mutations in cancer. Nature reviews. Cancer. 2016;16:71–81. doi: 10.1038/nrc.2015.12. [DOI] [PubMed] [Google Scholar]
- 19.Shlien A, Campbell BB, de Borja R, Alexandrov LB, Merico D, Wedge D, et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nature genetics. 2015;47:257–262. doi: 10.1038/ng.3202. [DOI] [PubMed] [Google Scholar]
- 20.Pritchard CC, Offit K, Nelson PS. DNA-Repair Gene Mutations in Metastatic Prostate Cancer. The New England journal of medicine. 2016;375:1804–1805. doi: 10.1056/NEJMc1611137. [DOI] [PubMed] [Google Scholar]
- 21.Loeb LA, Springgate CF, Battula N. Errors in DNA replication as a basis of malignant changes. Cancer research. 1974;34:2311–2321. [PubMed] [Google Scholar]
- 22.Loeb LA. Human Cancers Express a Mutator Phenotype: Hypothesis, Origin, and Consequences. Cancer research. 2016;76:2057–2059. doi: 10.1158/0008-5472.can-16-0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. doi: 10.1038/nature10868. [DOI] [PubMed] [Google Scholar]
- 24.Schmitt MW, Fox EJ, Prindle MJ, Reid-Bayliss KS, True LD, Radich JP, et al. Sequencing small genomic targets with high efficiency and extreme accuracy. Nature methods. 2015;12:423–425. doi: 10.1038/nmeth.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolodner RD. Mismatch repair: mechanisms and relationship to cancer susceptibility. Trends in biochemical sciences. 1995;20:397–401. doi: 10.1016/s0968-0004(00)89087-8. [DOI] [PubMed] [Google Scholar]
- 26.Sagher D, Strauss B. Insertion of nucleotides opposite apurinic/apyrimidinic sites in deoxyribonucleic acid during in vitro synthesis: uniqueness of adenine nucleotides. Biochemistry. 1983;22:4518–4526. doi: 10.1021/bi00288a026. [DOI] [PubMed] [Google Scholar]
- 27.Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G—T and A—C substitutions. The Journal of biological chemistry. 1992;267:166–172. [PubMed] [Google Scholar]
- 28.Lewis CA, Jr, Crayle J, Zhou S, Swanstrom R, Wolfenden R. Cytosine deamination and the precipitous decline of spontaneous mutation during Earth’s history. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:8194–8199. doi: 10.1073/pnas.1607580113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishimura S. Involvement of mammalian OGG1(MMH) in excision of the 8-hydroxyguanine residue in DNA. Free radical biology & medicine. 2002;32:813–821. doi: 10.1016/s0891-5849(02)00778-5. [DOI] [PubMed] [Google Scholar]
- 30.Moran RG, Heidelberger C. Determinants of 5-fluorouracil sensitivity in human tumors. Bulletin du cancer. 1979;66:79–83. [PubMed] [Google Scholar]
- 31.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 32.Nowell PC. The clonal evolution of tumor cell populations. Science (New York, NY) 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 33.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science (New York, NY) 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 36.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (New York, NY) 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science (New York, NY) 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beckman RA. Efficiency of carcinogenesis: is the mutator phenotype inevitable? Seminars in cancer biology. 2010;20:340–352. doi: 10.1016/j.semcancer.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 40.Beckman RA, Loeb LA. Negative clonal selection in tumor evolution. Genetics. 2005;171:2123–2131. doi: 10.1534/genetics.105.040840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herr AJ, Kennedy SR, Knowels GM, Schultz EM, Preston BD. DNA replication error-induced extinction of diploid yeast. Genetics. 2014;196:677–691. doi: 10.1534/genetics.113.160960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kennedy SR, Schultz EM, Chappell TM, Kohrn B, Knowels GM, Herr AJ. Volatility of Mutator Phenotypes at Single Cell Resolution. PLoS genetics. 2015;11:e1005151. doi: 10.1371/journal.pgen.1005151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eigen M. Error catastrophe and antiviral strategy. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13374–13376. doi: 10.1073/pnas.212514799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loeb LA, Essigmann JM, Kazazi F, Zhang J, Rose KD, Mullins JI. Lethal mutagenesis of HIV with mutagenic nucleoside analogs. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:1492–1497. doi: 10.1073/pnas.96.4.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fox EJ, Loeb LA. Lethal mutagenesis: targeting the mutator phenotype in cancer. Seminars in cancer biology. 2010;20:353–359. doi: 10.1016/j.semcancer.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nordling CO. A new theory on cancer-inducing mechanism. British journal of cancer. 1953;7:68–72. doi: 10.1038/bjc.1953.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. British journal of cancer. 1954;8:1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cook PJ, Doll R, Fellingham SA. A mathematical model for the age distribution of cancer in man. International journal of cancer. 1969;4:93–112. doi: 10.1002/ijc.2910040113. [DOI] [PubMed] [Google Scholar]
- 49.Beckman RA, Loeb LA. Genetic instability in cancer: theory and experiment. Seminars in cancer biology. 2005:15423–435. doi: 10.1016/j.semcancer.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 50.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 51.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 52.Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer research. 1991;51:3075–3079. [PubMed] [Google Scholar]
- 53.Luebeck EG, Moolgavkar SH. Multistage carcinogenesis and the incidence of colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:15095–15100. doi: 10.1073/pnas.222118199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beckman RA, Loeb LA. Efficiency of carcinogenesis with and without a mutator mutation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:14140–14145. doi: 10.1073/pnas.0606271103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beckman RA. Mutator mutations enhance tumorigenic efficiency across fitness landscapes. PloS one. 2009;4:e5860. doi: 10.1371/journal.pone.0005860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Datta RS, Gutteridge A, Swanton C, Maley CC, Graham TA. Modelling the evolution of genetic instability during tumour progression. Evolutionary applications. 2013;6:20–33. doi: 10.1111/eva.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J, Benavente CA, McEvoy J, Flores-Otero J, Ding L, Chen X, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481:329–334. doi: 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tomasetti C, Marchionni L, Nowak MA, Parmigiani G, Vogelstein B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:118–123. doi: 10.1073/pnas.1421839112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sottoriva A, Kang H, Ma Z, Graham TA, Salomon MP, Zhao J, et al. A Big Bang model of human colorectal tumor growth. Nature genetics. 2015;47:209–216. doi: 10.1038/ng.3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Williams MJ, Werner B, Barnes CP, Graham TA, Sottoriva A. Identification of neutral tumor evolution across cancer types. Nature genetics. 2016;48:238–244. doi: 10.1038/ng.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bozic I, Nowak MA. Timing and heterogeneity of mutations associated with drug resistance in metastatic cancers. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:15964–15968. doi: 10.1073/pnas.1412075111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ling S, Hu Z, Yang Z, Yang F, Li Y, Lin P, et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E6496–6505. doi: 10.1073/pnas.1519556112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo HH, Choe J, Loeb LA. Protein tolerance to random amino acid change. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9205–9210. doi: 10.1073/pnas.0403255101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gatenby RA, Cunningham JJ, Brown JS. Evolutionary triage governs fitness in driver and passenger mutations and suggests targeting never mutations. Nature communications. 2014;5:5499. doi: 10.1038/ncomms6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beckman RA, Schemmann GS, Yeang CH. Impact of Genetic Dynamics and Single-Cell Heterogeneity on Development of Nonstandard Personalized Medicine Strategies for Cancer. Proc Natl Acad Sci U S A. 2012;109:14586–14591. doi: 10.1073/pnas.1203559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beckman RA, Yeang C-H. Nonstandard personalized medicine strategies for cancer may lead to improved patient outcomes. Personalized Medicine. 2014;11:705–719. doi: 10.2217/pme.14.57. [DOI] [PubMed] [Google Scholar]
- 69.Kauffman SA. The origins of order. Oxford University Press; New York: 1993. pp. 85–103. [Google Scholar]
- 70.Bozic I, Reiter JG, Allen B, Antal T, Chatterjee K, Shah P, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife. 2013;2:e00747. doi: 10.7554/eLife.00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Freireich EJ. The management of acute leukemia. Canadian Medical Association journal. 1967;96:1605–1610. [PMC free article] [PubMed] [Google Scholar]
- 72.DeVita VT., Jr Human models of human diseases; breast cancer and the lymphomas. International journal of radiation oncology, biology, physics. 1979;5:1855–1867. doi: 10.1016/0360-3016(79)90571-6. [DOI] [PubMed] [Google Scholar]
- 73.Autran B, Carcelain G, Li TS, Blanc C, Mathez D, Tubiana R, et al. Positive effects of combined antiretroviral therapy on CD4+ T cell homeostasis and function in advanced HIV disease. Science (New York, NY) 1997;277:112–116. doi: 10.1126/science.277.5322.112. [DOI] [PubMed] [Google Scholar]
- 74.Yeang CH, Beckman RA. Long range personalized cancer treatment strategies incorporating evolutionary dynamics. Biology direct. 2016;11:56. doi: 10.1186/s13062-016-0153-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gatenby RA, Silva AS, Gillies RJ, Frieden BR. Adaptive therapy. Cancer Res. 2009;69:4894–4903. doi: 10.1158/0008-5472.can-08-3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K. Noncell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 2014;514:54–58. doi: 10.1038/nature13556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Komarova NL, Wodarz D. Combination therapies against chronic myeloid leukemia: short-term versus long-term strategies. Cancer research. 2009;69:4904–4910. doi: 10.1158/0008-5472.can-08-1959. [DOI] [PubMed] [Google Scholar]
- 78.Haeno H, Gonen M, Davis MB, Herman JM, Iacobuzio-Donahue CA, Michor F. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell. 2012;148:362–375. doi: 10.1016/j.cell.2011.11.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Science translational medicine. 2011;3:90ra59. doi: 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Leder K, Pitter K, Laplant Q, Hambardzumyan D, Ross BD, Chan TA, et al. Mathematical modeling of PDGF-driven glioblastoma reveals optimized radiation dosing schedules. Cell. 2014;156:603–616. doi: 10.1016/j.cell.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]