

SUMMARY
Despite progress in our comprehension of the mechanisms regulating adipose tissue development, the nature of the factors that functionally characterize adipose precursors is still elusive. Defining the early steps regulating adipocyte development is needed for the generation of tools to control adipose tissue size and function. Here, we report the discovery of V-set and transmembrane domain containing 2A (VSTM2A) as a protein expressed and secreted by committed preadipocytes. VSTM2A expression is elevated in the early phases of adipogenesis in vitro and adipose tissue development in vivo. We show that VSTM2A-producing cells associate with the vasculature and express the common surface markers of adipocyte progenitors. Overexpression of VSTM2A induces adipogenesis, whereas its depletion impairs this process. VSTM2A controls preadipocyte determination at least in part by modulating BMP signaling and PPARγ2 activation. We propose a model in which VSTM2A is produced to preserve and amplify the adipogenic capability of adipose precursors.
In Brief
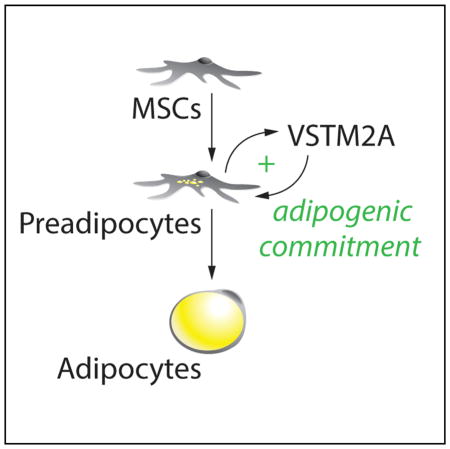
Secco et al. identify VSTM2A as a factor expressed and secreted by adipose precursors. They show that VSTM2A amplifies adipogenic commitment by promoting BMP4 signaling and PPARγ2 activation. These results indicate that VSTM2A functionally controls early events in adipocyte development.
INTRODUCTION
White adipose tissue (WAT) is the primary site for energy storage in mammals. This tissue stores triglycerides in periods of energy excess and releases fatty acids to provide energy during fasting. Beyond its role in controlling energy homeostasis, WAT serves as a central endocrine organ playing key roles in metabolism. WAT secretes a plethora of proteins termed adipokines that have profound effects on various biological processes including the regulation of food intake, glucose metabolism, insulin sensitivity, inflammation, and reproduction (Rosen and Spiegelman, 2014). Compared to WAT, brown adipose tissue (BAT) is specialized for thermogenic energy expenditure. In response to cold, BAT hydrolyses triglycerides and oxidizes fatty acids and glucose to produce heat (Cannon and Nedergaard, 2004). Owing to its significant capacity to dissipate energy and regulate metabolism, this tissue is envisioned as a potential target for the treatment of obesity and diabetes (Cypess and Kahn, 2010).
Studies in mice indicate that, during development, white and brown adipose precursors actively proliferate before turning on the expression of genes required for the development, the maturation and the maintenance of mature adipocytes (Hong et al., 2015; Hudak et al., 2014; Schulz et al., 2013; Tang et al., 2008). Reports have shown that the timing of proliferation versus hypertrophy differs between developing adipose depots (Hudak et al., 2014; Wang et al., 2013). Whereas subcutaneous WAT (sWAT) progenitors proliferate in utero and start to expand through hypertrophy at birth, these events occur postnatally in visceral epididymal WAT (eWAT) (Han et al., 2011; Hudak et al., 2014; Wang et al., 2013). In the case of BAT, adipose progenitors actively proliferate during late gestational stages (Schulz et al., 2013). The expansion of WAT observed in response to obesity also follows a tissue-specific pattern. In response to a high-fat diet, sWAT grows primarily through adipocyte hypertrophy (Jeffery et al., 2015; Wang et al., 2013). On the other hand, eWAT rapidly expands via de novo adipogenesis and hypertrophy of existing adipocytes (Jeffery et al., 2015; Wang et al., 2013).
While there has been significant advance in the understanding of the molecular mechanisms regulating the terminal differentiation of adipocytes, the identity of adipocyte precursors and the events that regulate their adipogenic conversion is only emerging (Berry et al., 2013, 2014; Cawthorn et al., 2012). Early work have shown that white adipocytes develop from a structure originally described as the “primitive organ,” a cluster of blood vessels found during the development of diverse organisms, including mice and humans (Wassermann, 1965). The close association between vasculature and adipogenesis has since been demonstrated several times (Han et al., 2011; Nishimura et al., 2007; Rupnick et al., 2002; Tang et al., 2008). Recently, cell-surface markers were used to isolate and characterize adipose precursor from WAT. This led to the identification of Lin−, CD29+, CD34+, Sca+, CD24+ as adipose progenitors capable to differentiate and reconstitute WAT in vivo (Rodeheffer et al., 2008). Adipose precursors were also shown to express high levels of preadipocyte factor-1 (Pref-1) (Hudak et al., 2014), peroxisome proliferator-activated receptor γ2 (Pparγ2) (Tang et al., 2008), and/or zinc-finger protein 423 (Zfp423) (Gupta et al., 2010, 2012) at various stage of their development.
Despite progress in our comprehension of mechanism regulating adipocyte development and expansion, the nature of the factors that functionally characterize adipose precursors is still elusive. Here, we report the discovery of V-set and transmembrane domain containing 2A (VSTM2A) as a protein highly expressed and secreted by committed preadipocytes. VSTM2A is expressed early during adipocyte development in vitro but also during adipose tissue formation in mice and humans. Anatomical analyses of developing WAT revealed that VSTM2A-producing cells closely associate with the vasculature and that VSTM2A expression is enriched in adipose precursor cells compared to mature adipocytes. We find that VSTM2A is sufficient to induce spontaneous differentiation, whereas its depletion impairs adipogenesis. Here, we show that VSTM2A controls preadipocyte determination at least in part by modulating bone morphogenetic protein (BMP) signaling and PPARγ2 activation. Our results indicate that VSTM2A is an adipogenic regulator that promotes the commitment of preadipocytes.
RESULTS
Identification of VSTM2A as a Factor Expressed by Committed Preadipocytes
As reported by Green et al. in 1974, the potential for adipogenesis greatly varies within a culture of 3T3-L1 cells (Green and Meuth, 1974), an in vitro model of adipogenesis. Following the induction of adipogenesis, some cells accumulate a large amount of lipids, whereas others are unable to do so (Figure 1A). To identify new genes that functionally characterize committed preadipocytes, we have set up a simple approach using these cells, as described previously (Gupta et al., 2010). A schematic view of the strategy is presented in Figure S1A. Briefly, we performed limiting dilutions of a 3T3-L1 culture and successfully derived new clonal sub-lines of 3T3-L1. Strikingly, when stimulated with an adipogenic cocktail, these clones showed either a low, mid, or high capacity to accumulate lipids (Figures 1B, 1C, S1B, and S1C). The adipogenic capability of each line was maintained after several passages, indicating a persistent phenotype. qRT-PCR analyses revealed that the low lines showed a lower expression of key adipogenic and lipogenic genes over the course of adipogenesis (Figure S1D). Importantly, the reduced adipogenic potential of the low lines was corrected by treating the cells with a PPARγ agonist, indicating that despite their inability to differentiate under normal conditions, these cells retained the full potential to become mature adipocytes (Figures S1E–S1G).
Figure 1. Identification of VSTM2A as a Gene Highly Expressed in Committed Preadipocytes.

(A) Examples of 3T3-L1 showing either low or high adipogenic capacity. Cells were stained with DAPI (blue) and LipidTox red (red) to stain the nuclei and the lipid droplets, respectively. Pictures of cells were taken from the same well 8 days following the induction of adipogenic differentiation.
(B) Triglyceride accumulation measured in 23 clonal lines of 3T3-L1 following the induction of differentiation. Lipid content was measured per well (n = 3/cell line) 8 days after the induction of differentiation. Data are presented as mean ± SEM. The difference between the low and high lines was confirmed in another independent experiment.
(C) Oil red O staining of low and high lines 8 days following the induction of differentiation. These pictures were produced from a single experiment.
(D) qRT-PCR analyses of Pparg2 and Pparg1 mRNA in subconfluent low (n = 7) and high (n = 8) cell lines. Data are presented as mean ± SEM and are representative of two independent experiments. *p < 0.05 versus low lines.
(E) Heatmap showing the differential expression profile of several genes between low (n = 5) versus high (n = 5) lines measured by microarray. These data are from a single experiment.
(F) qRT-PCR analyses of Vstm2a mRNA expression in subconfluent low (n = 7) and high (n = 8) cell lines. Data are presented as mean ± SEM and are representative of two independent experiments. *p < 0.05 versus low lines.
(G) Western blot analysis of VSTM2A protein levels in low, high, and parental 3T3-L1 cells. S6K was used as a loading control. These images are representative of at least three independent experiments.
Being interested in identifying factors characterizing preadipocytes, we used proliferating low and high lines and looked at the expression of key adipogenic regulators. Consistent with recent reports showing that Pparg2 is a key marker enriched in preadipocytes (Gupta et al., 2010; Tang et al., 2008), we observed that high lines expressed higher basal levels of Pparg2 (Figure 1D). Pparg1 was also slightly elevated in these cells. Interestingly, the elevated basal Pparg1 and 2 levels were not associated with major changes in the expression of many factors known to regulate their own expression (Figure S1H). Of note, we did not observe a significant difference in Zfp423 expression between the lines, indicating that basal Pparg2 expression can be regulated independent of, or in parallel with this factor recently shown to play key roles in regulating preadipocyte determination (Figure S1H) (Gupta et al., 2010). To better characterize the transcriptional signature defining the adipogenic potential of the low and high lines, we performed a microarray analysis of these cells still in the sub-confluent state. We observed that the expression profiles of the high lines clustered together and significantly differed from the ones of the low lines (Figure S1I; Table S1). Several genes known to participate in the adipogenic process were enriched in the high lines, including Pparg, Ap2, Cd36, and Plin4 (Figure 1E). We found that the most differentially expressed gene between the high and low lines was a gene with unknown function, V-set and transmembrane domain containing 2A (Vstm2a) (Figure 1E). qRT-PCR and western blotting analyses confirmed that VSTM2A is highly expressed in cells with a high adipogenic potential (Figures 1F and 1G). Altogether, these results identify VSTM2A as a factor expressed by committed preadipocytes in vitro.
In the studies described above, we noted a positive correlation between Vstm2a and Pparg2 mRNA expression (Figure S1J). Interestingly, forcing the adipogenic commitment of NIH 3T3 fibroblasts through the overexpression of PPARγ2 was sufficient to increase Vstm2a expression in sub-confluent cells, prior to their differentiation (Figures S1K and S1L). A similar increase in Vstm2a expression was measured in response to PPARγ2 overexpression in other preadipocyte models including 3T3-L1 and C3H10T1/2 (Figures S1M and S1N). To better define the relation between PPARγ2 and Vstm2a expression, we have overexpressed PPARγ2 in immortalized cancer cell lines (293T and U87). Because these cells cannot commit to the adipogenic lineage and differentiate into adipocytes, they represent a good model to define whether PPARγ2 directly controls Vstm2a expression or indirectly controls its expression by promoting adipogenic commitment. As expected, overexpression of PPARγ2 strongly increased the expression Ap2 a well-established direct target of PPARγ2 (Figures S1O and S1P). However, PPARγ2 completely failed in inducing the expression of Vstm2a in cancer cells (Figures S1O and S1P). These results confirm that adipogenic commitment, downstream of PPARγ2, is a strong driver of Vstm2a expression in preadipocytes.
VSTM2A Is a Conserved Protein Expressed Early during Adipocyte Development
VSTM2A is a 236 amino acid protein that carries a signal peptide in the N-terminal and an immunoglobulin v-set domain in its central section (Figure 2A). This protein is highly conserved throughout evolution with homologs being found from human to zebrafish (Figure S2A). Because available data on VSTM2A are very limited, we first looked at its expression during adipocyte differentiation in vitro. As shown in Figures 2B and 2C, the induction of adipogenesis in 3T3-L1 cells was associated with a transient increase in the expression of VSTM2A. Notably, the induction in VSTM2A levels preceded the elevation in PPARγ and Ap2 expression. These effects were also observed when adipogenesis was induced in other cellular models including mouse embryonic fibroblasts (MEFs), WAT-derived stromal vascular fraction (SVF), C3H10T1/2 mesenchymal stem cells (MSCs), and human primary MSCs (Figures S2B–S2E). A very similar profile was also observed during the differentiation of immortalized brown preadipocytes (Figure S2F). These observations indicate that VSTM2A is expressed early during fat cell formation in vitro and implicate a role for this protein in regulating the early stages of adipocyte development. These results also show that the positive relation between Pparg2 and Vstm2a reported in proliferating preadipocytes (Figures S1J and S1L– S1N) is lost in terminally differentiated cells and further support the idea that Vstm2a is not a direct target of PPARγ2.
Figure 2. VSTM2A Is Expressed Early in Adipose Cell Development In Vitro and In Vivo.

(A) Structure of human and mouse VSTM2A protein.
(B) qRT-PCR analyses of Vstm2a, Pparg2, and Ap2 mRNA expression in 3T3-L1 cells at sub-confluence (sub) or 0, 2, 4, and 8 days following the induction of adipogenic differentiation. Data are presented as mean ± SEM (n = 3/condition). The data presented are representative of at least three independent experiments. For Vstm2a mRNA, *p < 0.05 versus sub; #p < 0.05 versus day 0; $p < 0.05 versus day 2. For Pparg2 and Ap2 mRNA, only the comparison with sub-confluence is presented, *p < 0.05 versus sub.
(C) Western blot analysis of protein isolated from differentiating 3T3-L1 cells. These images are from a single western blot experiment. The data presented are representative of two independent experiments.
(D) Western blot analysis showing the tissue distribution of VSTM2A in adult mouse (n.s., non-specific band). The image is representative of two experiments performed with tissues collected from two other animals.
(E) Western blot analysis showing the tissue distribution of VSTM2A in a mouse sacrificed at postnatal day 4 (P4). The image is the representative of two experiments performed with tissues collected from two different animals.
(F) Western blot analysis of VSTM2A protein levels in mice sacrificed at P4, P7, P14, P28, and P56. The image is representative of two additional independent experiments.
(G) qRT-PCR analyses of Vstm2a, Pparg2, and Ap2 mRNA expression in eWAT isolated from mice sacrificed at P4, P7, P14, P28, and P56. Data are presented as mean ± SEM (n = 4–7/condition). These data are from a single experiment. *p < 0.05 versus P4.
(H) LipidTox red staining of eWAT isolated from mice sacrificed at P4, P7, P14, P28, and P56. These pictures were produced from a single experiment. Scale bar, 25 μM.
(I) Western blot analysis of VSTM2A protein levels in WAT isolated from human fetuses at various stages of their development and in WAT isolated from adult humans. This experiment was performed once.
See also Figure S2.
We next looked at VSTM2A expression in mouse tissues, aiming to find a distribution pattern that may reflect a role for the protein in adipose tissue biology. Our first survey of adult mice showed that VSTM2A expression was essentially restricted to the central nervous system and neurons (Figures 2D and S2G and S2H). Because we identified VSTM2A as a factor highly expressed in preadipocytes and in developing fat cells, we reasoned that its expression might be enriched in the early phases of adipose tissue formation. In mice, eWAT develops postnatally, whereas sWAT develops in utero (Han et al., 2011; Wang and Scherer, 2014). To determine whether VSTM2A is expressed in the developmental phase of adipose tissue, pups were examined 4 days postnatal (P4), and VSTM2A levels were measured in several tissues. Strikingly, we found that VSTM2A was expressed in eWAT of P4 mice (Figure 2E). When the expression of VSTM2A was analyzed in detail during eWAT development (P4–P56), we observed that its expression was critically high after birth but reduced as the tissue started to express more PPARγ and to accumulate lipids (Figures 2F–2H). An identical expression profile was observed when VSTM2A levels were measured during the developmental phase of sWAT, i.e., from E16.5 to P14 (Figure S2I), indicating that the early expression of VSTM2A is a phenomenon that is conserved between WAT depots.
To test whether VSTM2A is also expressed in the developmental phase of WAT in humans, VSTM2A levels were measured in sWAT collected from human fetuses. Light microscopy analyses of human fetuses previously showed that WAT appears and progressively develops from the 14th to the 24th weeks of gestation (Poissonnet et al., 1983, 1984). We observed that VSTM2A was highly enriched in sWAT isolated from fetuses but was not detected in adults (Figure 2I). Interestingly, VSTM2A levels declined as the fetus developed, exactly as observed in mice. Altogether, these results show that VSTM2A is expressed early during adipocyte development in vitro and in vivo and that this process is conserved from mice to humans.
VSTM2A-Expressing Cells Closely Associate with the Vasculature in Developing WAT and Express Markers of Adipose Progenitors in Adult WAT
Studies have reported that adipose progenitors closely associate with the adipose vasculature and that these cells can proliferate and differentiate into mature adipocytes (Jiang et al., 2014; Tang et al., 2008). To determine whether VSTM2A-expressing cells show a similar distribution pattern, immunofluorescence assays were performed on eWAT isolated from P4 mice. These experiments revealed that VSTM2A-expressing cells are found exclusively in the perivascular compartment, in close association with cells expressing the endothelial cell marker CD31 (Figure 3A). As shown in Figure 3B, VSTM2A positive cells did not express alpha smooth muscle actin (αSMA), a marker of mural cells. Importantly, we did not observe co-staining between VSTM2A, CD31, or αSMA, indicating that VSTM2A-expressing cells are distinct from endothelial and mural cells. An important question arising from these results is whether VSTM2A expression also characterizes preadipocytes found in adult WAT. As shown in Figure 2D, we observed limited VSTM2A protein expression by western blotting in adult WAT. However, Vstm2a mRNA was detected in these samples using qRT-PCR, a more sensitive detection method. Because VSTM2A is transiently expressed by preadipocytes during in vitro adipogenesis and WAT development, we reasoned that its low expression in adult WAT may be a reflection of the low relative abundance of this specific cell population in this tissue. To confirm the specific expression of Vstm2a in adipose precursor cells, eWAT from adult mice was digested with collagenase and sorted to isolate both mature adipocytes and Lin−, CD29+, CD34+, Sca1+, PDGFRα+ adipose progenitors, as described (Berry and Rodeheffer, 2013; Church et al., 2014; Rodeheffer et al., 2008) (Figures 3C and S3A). This experiment revealed that Vstm2a expression was enriched in Lin−, CD29+, CD34+, Sca1+, PDGFRα+ cells compared to mature fat cells (Figure 3D), thus supporting the hypothesis that Vstm2a expression characterizes adipose precursors in vivo.
Figure 3. VSTM2A-Expressing Cells in WAT Are Adipose Precursors that Closely Associate with the Vasculature.

(A and B) Immunofluorescence assays performed on eWAT collected from P4 mice. The expression of VSTM2A was analyzed in comparison with (A) the endothelial marker CD31 or (B) the mural cell marker αSMA. eWAT was collected and analyzed from five mice at P4. Representative pictures are shown. Scale bar, 10 μM.
(C) Presentation of the strategy used for the isolation of mature adipocytes and adipose precursors.
(D) qRT-PCR analyses of Vstm2a, Pparg2, and Ap2 mRNA expression in mature adipocytes and adipose precursors isolated from adult mice. Data are presented as mean ± SEM (n = 4/condition). This experiment was performed twice. *p < 0.05 versus adipocytes.
(E–G) qRT-PCR analyses of Vstm2a expression in eWAT collected from (E) mice fed a high-fat diet for 4 weeks, (F) ob/ob mice and (G) db/db mice. Data are presented as mean ± SEM (n = 4–7/group). These experiments were performed once for high-fat fed mice and db/db and twice for ob/ob mice. *p < 0.05 versus Chow or Ctrl.
See also Figure S3.
The development of obesity is associated with de novo adipogenesis in mice (Kim et al., 2014; Wang et al., 2013). The increase in adipogenesis observed in response to a high-fat diet occurs mainly in eWAT (Jeffery et al., 2015; Wang et al., 2013). To test the impact of obesity on Vstm2a expression, mice were fed a high-fat diet for 4 weeks, a time at which a wave of adipogenesis was reported to occur (Wang et al., 2013). As expected, this diet was sufficient to increase WAT accumulation (Figure S3B). As previously shown in Figure 2D, we found that Vstm2a expression was low in eWAT of lean mice, which probably reflects its specific expression by a small population of adipose progenitors within WAT. Interestingly, Vstm2a expression was significantly induced in response to high-fat feeding (Figure 3E). To determine whether elevated Vstm2a expression could also be observed in response to obesity in other mouse models, eWAT was collected from genetically obese ob/ob and db/db mice. Supporting the findings described above, we found greater expression of Vstm2a in eWAT of these obese mouse models compared to their lean littermates (Figures 3F and 3G). Altogether, these results indicate that Vstm2a expression is induced during WAT expansion.
VSTM2A Is a Glycoprotein Secreted by Committed Preadipocytes
The presence of a signal peptide in the N-terminal section of VSTM2A suggests that the protein may be secreted. To directly test this possibility, V5-tagged VSTM2A was overexpressed in various cell lines and culture media was collected for western blot and immunoprecipitation analyses (Figure S4A). VSTM2A was rapidly detected in the culture media of overexpressing cells, thus confirming that the protein is effectively secreted (Figures 4A, S4B, and S4C). We observed that blocking protein transport with Brefeldin A or deleting the signal peptide on VSTM2A completely abrogated VSTM2A secretion (Figures 4B and S4D). As shown in Figures 4A and 4B, the secreted form of VSTM2A migrated at a higher molecular weight compared to VSTM2A found in cells. Interestingly, two conserved asparagine residues (N35 and N175) potentially subjected to N-linked glycosylation were identified in VSTM2A (Figure S4E). Supporting the idea that VSTM2A is a secreted glycoprotein, the migration pattern of VSTM2A was severely modified when cells were treated with tunicamycin, an agent that blocks N-linked glycosylation (Figure 4C). A similar profile was seen when proteins were de-glycosylated in vitro with PNGase F (Figure 4D). Additionally, asparagine to glutamine (N→Q) mutations of N35 and N175, which block glycosylation of these residues, not only modified the migration profile of VSTM2A but also impaired its secretion (Figure 4E). These results indicate that N-linked glycosylation is required for the proper maturation and the secretion of VSTM2A. In addition to the conserved asparagine found in VSTM2A, we noted the presence of unpaired cysteine residues, which suggests that VSTM2A could form dimers. Western blot analyses revealed the presence of an 80-kDa band that was sensitive to denaturation, indicating the presence of dimers in this preparation (Figure 4F).
Figure 4. VSTM2A Is a Glycoprotein Secreted by Committed Preadipocytes.

(A) Western blot analyses of cell lysates and culture media of 293T cells overexpressing V5-VSTM2A. Cells were plated the day before and culture medium was changed at time 0. Cells were lysed and culture medium was collected at the indicated time. S6K was used as a loading control. The images are representative of two independent experiments.
(B) Western blot analyses of cell lysates and culture media of 293T cells overexpressing V5-VSTM2A and treated or not with Brefeldin A (3 μg/mL). S6K was used as a loading control. The images are representative of three independent experiments.
(C) Western blot analyses of cell lysates and culture media of 293T cells overexpressing V5-VSTM2A and treated or not with tunicamycin (1 μg/mL). Cells were plated the day before and pre-treated with tunicamycin for 6 hr. S6K was used as a loading control. The images are representative of three independent experiments.
(D) Western blots analyses showing that VSTM2A is a glycoprotein. Lysates and media isolated from 293T cells overexpressing V5-VSTM2A were treated with PNGase F following manufacturer’s instructions (NEB, P0407) and analyzed by western blotting. S6K was used as a loading control. The images are representative of two independent experiments.
(E) Western blot analyses of cell lysates and culture media of 293T cells overexpressing wild-type (WT) V5-VSTM2A or V5-VSTM2A with asparagine to glutamine (N→Q) mutations of N35, N175, or N35+N175. S6K was used as a loading control. This experiment was performed once.
(F) Western blots analyses showing that VSTM2A dimerizes following its secretion. Media was isolated from 293T cells overexpressing V5-VSTM2A was kept on ice or denaturated by heat and/or β-mercaptoethanol. This experiment was performed three times.
(G) Western blot analyses of cell culture media of NIH 3T3, a low adipogenic 3T3-L1 line (4H4) a high adipogenic 3T3-L1 line (4D2) and parental 3T3-L1 cells. Cells were plated until confluence and then the culture medium was changed (time 0), collected over 3 days, and analyzed for endogenous VSTM2A. This experiment was performed three times with the same outcome.
(H) Western blot analysis of cell lysate and culture media collected from eWAT explants. Samples of eWAT were collected from P4 mice and incubated in DMEM 10% for the indicated time.
See also Figure S4.
To determine whether VSTM2A secretion is specific to committed preadipocytes, we next measured the secretion of the endogenous protein in non-adipogenic (NIH 3T3 and low line) versus adipogenic cells (high line and 3T3-L1). Our experiments showed that VSTM2A is secreted only by committed preadipocytes (Figure 4G). In line with these results, we observed that eWAT explants isolated from P4 mice also secreted high levels of VSTM2A (Figure 4H). Overall, these results indicate that VSTM2A is a glycoprotein produced and secreted by preadipocytes in vitro and in vivo.
Overexpression of VSTM2A Induces the Spontaneous Differentiation of Preadipocytes, Whereas Its Depletion Severely Impairs This Process
In order to evaluate whether VSTM2A impacts on adipogenesis, we have overexpressed the protein in preadipocytes (3T3-L1) and immortalized MSCs (C3H10T1/2). We initially observed that VSTM2A overexpression did not affect adipogenesis when induced with the classical differentiation cocktail (data not shown). Due to the fact that the differentiation cocktail was specifically designed to maximize and accelerate the adipogenic conversion in vitro, a complete differentiation being obtained 6–8 days following the induction, we thought that its use might have cover possible pro-adipogenic functions of VSTM2A. To test this possibility, cells were grown to confluence and spontaneous adipogenesis, i.e., differentiation without stimulation, was analyzed over 20 days. Strikingly, overexpression of VSTM2A increased the spontaneous differentiation and lipid accumulation in both 3T3-L1 and C3H10T1/2 cells (Figures 5A and 5B). The elevation in adipogenesis was associated with a significant increase in the expression of several adipogenic markers in both cell lines (Figure 5C). Using primers to specifically measure endogenous Vstm2a mRNA, we found that overexpression of VSTM2A was sufficient to increase endogenous Vstm2a levels in 3T3-L1 (Figure 5D). Interestingly, we also found that expression of Pparg2, but not Pparg1, highly correlated with the expression of endogenous Vstm2a (Figure 5E), supporting the idea that VSTM2A specifically affects Pparg2 expression.
Figure 5. VSTM2A Induces the Spontaneous Conversion of Preadipocytes and MSCs into Adipocytes.

(A) Western blot analyses of cell lysates of 3T3-L1 and C3H10T1/2 cells overexpressing VSTM2A.
(B) Pictures of 3T3-L1 and C3H10T1/2 cells overexpressing VSTM2A. Cells overexpressing VSTM2A were plated and spontaneous differentiation was analyzed over 20 days. Representative pictures are shown. This experiment was performed twice for 3T3-L1 and four times for C3H10T1/2, in each case with similar outcomes. Scale bar, 100 μM.
(C) qRT-PCR analyses of adipogenic genes measured in 3T3-L1 and C3H10T1/2 cells overexpressing VSTM2A and treated as described in (B). Data are presented as mean ± SEM (n = 4/condition). This experiment was performed twice. *p < 0.05 versus control.
(D) qRT-PCR analyses of endogenous Vstm2a mRNA expression measured in 3T3-L1 cells overexpressing VSTM2A and treated as described in (B). Data are presented as mean ± SEM (n = 4/condition). This experiment was performed once. *p < 0.05 versus control.
(E) Correlation between endogenous Vstm2a mRNA and Pparg2 or Pparg1 in 3T3-L1 following spontaneous differentiation.
(F) Schematic representation of the co-culture experiments performed in C3H10T1/2 cells.
(G) Co-culture experiments performed in C3H10T1/2 cells. The producer cells are expressing either MSCV or VSTM2A, whereas the recipients are the cells that are exposed to the media. Spontaneous differentiation was assessed 24 days post-confluence. The number of differentiated cells was counted across wells and results are presented as fold increase versus control. This experiment was reproduced three times with the same outcome. Data are presented as mean ± SEM (n = 18 well/condition). *p < 0.05 versus control.
(H) Spontaneous differentiation was assessed 24 days post-confluence in C3H10T1/2 cells overexpressing native VSTM2A or Δ-VSTM2A. The number of differentiated cells was counted across wells and results are presented as fold increase versus control. This experiment was reproduced three times with the same outcome. Data are presented as mean ± SEM (n = 24 well/condition). *p < 0.05 versus control.
See also Figure S5.
To determine whether secreted or intracellular VSTM2A was responsible for the induction of adipogenesis, we have performed co-culture experiments using a trans-well system (Figure 5F). Although VSTM2A-producing cells showed a significant increase in adipogenesis, control cells exposed to the conditioned media did not show any pro-adipogenic phenotype (Figure 5G). Supporting these results, overexpression of Δ-VSTM2A, which lacks a signal peptide and cannot be secreted (Figures S5A and S5B), was sufficient to induce adipogenesis in cells expressing this isoform (Figure 5H). Consistent with these findings, we observed that adding escalating doses of purified VSTM2A to the culture media did not induce Pparg2 expression and did not promote spontaneous adipogenesis (Figures S5C and S5D). Moreover, adding anti-VSTM2A antibodies to the culture media to neutralize the effect of secreted VSTM2A did not reduce adipogenesis in VSTM2A-producing cells (data not shown). All these results confirm that intracellular, but not extracellular VSTM2A, promotes adipogenic commitment in vitro.
We next sought to determine the impact of VSTM2A depletion on adipogenesis. We first identified two short hairpin RNA (shRNA) that efficiently knockdown VSTM2A (Figure S6A). Using these hairpins, we observed that VSTM2A depletion severely impaired adipogenesis, both in 3T3-L1 and C3H10T1/2 cells (Figures 6A and 6B). As expected, this effect was associated with an important reduction in the expression of adipogenic and lipogenic genes post-differentiation (Figures 6C and 6D). The impact of VSTM2A depletion on adipogenesis was not associated with a consistent defect in mitotic clonal expansion, a key step required for the optimal adipogenic conversion of 3T3-L1 preadipocytes (Figure S6B). Notably, we observed that VSTM2A depletion also reduced the development of brown adipocytes in vitro (Figures S6C and S6D). A significant reduction in the expression of uncoupling protein 1 (Ucp1), a key marker of differentiated brown adipocytes, was measured in VSTM2Adepleted cells (Figure S6E). These results indicate that VSTM2A plays role in the development of white and brown fat cells in vitro.
Figure 6. VSTM2A Knockdown Impairs Adipogenesis.

(A and B) Oil red O staining and triglyceride accumulation following the differentiation of (A) 3T3-L1 or (B) C3H10T1/2 cells infected with control or Vstm2a shRNA. Pictures were taken 6 days after the induction of adipogenesis and are representative of more than ten independent experiments for 3T3-L1 and two independent experiments for C3H10T1/2. Scale bar, 40 μM. Lipid content was measured per well (n = 4/group) 6 days after the induction of differentiation. Data are presented as mean ± SEM. *p < 0.05 versus sh_Ctrl.
(C and D) qRT-PCR analyses of adipogenic genes measured 6 days after the differentiation of (C) 3T3-L1 or (D) C3H10T1/2 cells infected with control or Vstm2a shRNA. Data are presented as mean ± SEM (n = 4/condition). The results presented are representative of three independent experiments in the case of 3T3-L1 and one experiment in the case of C3H10T1/2. * p < 0.05 versus sh_Ctrl.
See also Figure S6.
VSTM2A Controls Adipogenic Commitment by Amplifying BMP Signaling and Pparg2 Expression
In agreement with the above findings, we observed that VSTM2A knockdown reduced basal expression of Pparg2 in proliferating preadipocytes prior to differentiation (Figure 7A). The opposite phenotype was found in cells overexpressing either VSTM2A or Δ-VSTM2A (Figure 7B). Together, these results confirm a positive role for VSTM2A in the control of basal Pparg2 expression and preadipocyte determination. Interestingly, treating cells with the PPARγ agonist rosiglitazone significantly improved adipogenesis in VSTM2A-depleted cells (Figures 7C–7E and S7A– S7C), indicating that VSTM2A affects adipose cell development at least in part by modulating PPARγ activation. Because intracellular VSTM2A does not localize to the nucleus (Figure S7D), we reasoned that VSTM2A unlikely regulates Pparg2 expression directly. Bone morphogenetic protein (BMP) signaling pathway has been described as key signaling node regulating cell fate decision and adipogenesis, in part through the phosphorylation of SMADs and the promotion of PPARγ activation (Bowers et al., 2006; Gupta et al., 2010; Jin et al., 2006; Schulz et al., 2013; Tang et al., 2004; Tseng et al., 2008). To evaluate whether VSTM2A controls adipogenesis by affecting BMP signaling, we measured the phosphorylation of SMAD1/5/8 in response to VSTM2A depletion or overexpression. Strikingly, we observed an important reduction in basal SMAD1/5/8 phosphorylation in 3T3-L1 cells depleted from VSTM2A (Figure 7F). Moreover, VSTM2A depletion impaired the normal response to recombinant BMPs (Figure 7G). The same effect was observed when VSTM2A was depleted in brown preadipocytes (Figures S7E and S7F). Confirming the relation between VSTM2A and BMP signaling, cells overexpressing VSTM2A showed elevated SMAD1/5/8 phosphorylation (Figure 7H). Importantly, such elevation in SMAD1/5/8 activation was not observed when VSTM2A was overexpressed in non-adipogenic NIH 3T3 (Figures S7G and S7H), a cell type in which VSTM2A failed to promote Pparg2 expression and to induce adipogenesis (Figures S7I and S7J). The same was observed when VSTM2A was expressed in non-adipogenic cancer cell lines (Figures S7K and S7L). Together, these results indicate that the effect of VSTM2A on BMP signaling and Pparg2 expression is context specific and occurs only in cells with a certain degree of adipogenic commitment.
Figure 7. VSTM2A Controls Adipogenic Commitment by Amplifying BMP Signaling and Pparg2 Expression.

(A) qRT-PCR analyses of Pparg1 and Pparg2 expression measured in sub-confluent 3T3-L1 cells infected with control or Vstm2a shRNA. Data are presented as mean ± SEM (n = 4/condition). This experiment was performed once. *p < 0.05 versus sh_Ctrl.
(B) qRT-PCR analysis of Pparg2 expression measured in sub-confluent C3H10T1/2 cells overexpressing native VSTM2A or Δ-VSTM2A. Data are presented as mean ± SEM (n = 4/condition). The results presented are representative of five independent experiments. *p < 0.05 versus Ctrl.
(C) Oil red O staining following the differentiation of 3T3-L1 cells infected with control or Vstm2a shRNA and treated or not with the PPARγ agonist rosiglitazone (5 μM). Pictures were taken 8 days after the induction of adipogenesis and are representative of three independent experiments. Scale bar, 40 μM.
(D) Triglyceride accumulation assessed in the experiment described in (C). Data are presented as mean ± SEM (n = 3/condition). This experiment was performed three times. *p < 0.05 versus shCtrl; #p < 0.05 versus no rosiglitazone.
(E) qRT-PCR analyses of adipogenic genes assessed in the experiment described in (C). Data are presented as mean ± SEM (n = 4/condition). *p < 0.05 versus shCtrl; #p < 0.05 versus no rosiglitazone.
(F) Western blot analyses of 3T3-L1 cells infected with control or Vstm2a shRNA. Cells were lysed in basal conditions. This experiment was performed four times with similar outcome.
(G) Western blot analyses of 3T3-L1 cells infected with control or Vstm2a shRNA treated or not with escalating doses of BMP2 or BMP4. Cells were serum-deprived for 3 hr and treated with BMP2 or BMP4 for 1 hr. This experiment was performed twice.
(H) Western blot analyses of 3T3-L1 and C3H10T1/2 cells overexpressing or not VSTM2A. Cells were lysed in basal conditions. These experiments were performed at least with five times with similar outcome.
(I) qRT-PCR analysis of Pparg2 expression measured in proliferating 3T3-L1 cells treated or not with BMP4 for 38 hr. Data are presented as mean ± SEM (n = 4/condition). This experiment was performed once. *p < 0.05 versus shCtrl; #p < 0.05 versus no BMP4.
(J) Oil red O staining following the differentiation of 3T3-L1 cells infected with control or Vstm2a shRNA and treated or not with BMP4 (50 ng/mL) from subconfluence to day 0. Pictures were taken 6 days after the induction of adipogenesis and are representative of two independent experiments. Scale bar, 40 μM.
(K) Triglyceride accumulation assessed in the experiment described in (J). Data are presented as mean ± SEM (n = 3/condition). This experiment was performed once. *p < 0.05 versus shCtrl; #p < 0.05 versus no BMP4.
(L) RT-qPCR analyses assessed in the experiment described in (J). Data are presented as mean ± SEM (n = 3–4/condition). *p < 0.05 versus shCtrl; #p < 0.05 versus no BMP4.
(M) Western blot analyses of protein lysates isolated form C3H10T1/2 treated or not with LDN-193189 (0.1 μM) for 24 hr.
(N) Oil red O staining following the spontaneous differentiation C3H10T1/2 cells (20 days) overexpressing MSCV or VSTM2A and treated or not with LDN-193189 (0.1 μM) for the duration of the study. This experiment was performed twice with similar outcome. Scale bar, 60 μM.
(O) The number of differentiated cells was counted across wells for each conditions (n = 12 wells/condition). Data are presented as mean ± SEM. *p < 0.05 versus control.
(P) RT-qPCR analysis of Pparg2 expression measured in the experiment described in (N). Data are presented as mean ± SEM (n = 4–5/condition). *p < 0.05 versus Ctrl; #p < 0.05 versus no LDN.
(Q) Schematic overview of the model proposed.
See also Figure S7.
To test whether VSTM2A amplifies the adipogenic commitment of preadipocytes by controlling BMP signaling, we tried to rescue the adipogenic defect of VSTM2A knockdown cells with BMP4. Similarly to previous findings (Gupta et al., 2010), we found that BMP4 treatment increased basal Pparg2 expression (Figure 7I). Furthermore, treating cells with BMP4 was sufficient to correct Pparg2 expression in VSTM2A-depleted cells (Figure 7I). We next induced differentiation and found that BMP4 treatment improved adipogenesis in VSTM2A knockdown cells (Figures 7J and 7K). This effect was associated with a significant increase in the expression of adipogenic and lipogenic genes post-differentiation (Figure 7L), confirming the importance of BMP signaling in the pro-adipogenic effects of VSTM2A. We next tried to determine whether blocking BMP signaling with LDN-193189, a cell permeable small molecule inhibitor of BMP type I receptors, was sufficient to reduce adipogenesis in cells overexpressing VSTM2A. We found that LDN-193189 reduced SMAD1/5/8 phosphorylation, Pparg2 expression and spontaneous adipogenesis in VSTM2A-overexpressing cells (Figures 7M–7P). Altogether, these results indicate that functional BMP signaling is necessary to transduce the pro-adipogenic effects of VSTM2A. We propose a model in which VSTM2A is produced by preadipocyte to maintain and amplify the adipogenic capability of these cells (Figure 7Q).
DISCUSSION
Despite significant advances in our comprehension of the mechanisms regulating the terminal differentiation of adipocytes, the identity of adipose precursor cells and the mechanisms regulating their recruitment are still not well characterized. Here, we report the discovery of VSTM2A as a secreted protein expressed by committed preadipocytes. Our results show that VSTM2A is produced in the early phases of adipogenesis in vitro and adipose tissue development in vivo. Micro-anatomical analyses of developing WAT revealed that VSTM2A-expressing cells associate with the vasculature, a common characteristic of adipose progenitors. In adult mice, VSTM2A expression is enriched in adipose precursor cells and its expression is increased in WAT of obese mice. Despite being actively secreted, we found that intracellular VSTM2A is sufficient to induce the spontaneous adipogenic differentiation of mesenchymal stem cells and preadipocytes. Conversely, depleting VSTM2A in preadipocytes severely impairs adipogenesis. We show that VSTM2A controls adipogenesis at least in part by modulating BMP signaling and PPARγ2 activation. Our results indicate that VSTM2A is an adipogenic regulator that promotes the commitment of preadipocytes.
We have identified Vstm2a as the most differentially expressed gene between high and low 3T3-L1 cell lines. We showed that prior to differentiation, preadipocytes with a high adipogenic potential expressed high basal levels of Vstm2a. In these studies, a positive association was observed between Vstm2a and Pparg2, a nuclear receptor that controls both the commitment and the terminal differentiation of preadipocytes (Berry et al., 2013). Supporting a close relation between these players, we found that overexpression of PPARγ2 in several cell types (NIH 3T3, 3T3-L1, C3H10T1/2) was sufficient to induce their adipogenic commitment and to promote a robust increase in Vstm2a expression. Although these observations suggest that PPARγ2 lies upstream of Vstm2a, several evidence indicate that this interaction is not as straightforward. Indeed, we found that overexpression of VSTM2A was sufficient to increase basal Pparg2 expression before the induction of adipogenesis. Conversely, VSTM2A knockdown reduced Pparg2 transcription in subconfluent preadipocytes. These results indicate that PPARγ2 and VSTM2A reciprocally regulate their expression in a feedforward fashion. In details, we found that PPARγ2 indirectly promotes Vstm2a expression by amplifying adipogenic commitment in preadipocytes. On the other hand, VSTM2A promotes Pparg2 expression by activating BMP signaling. Importantly, this feedforward loop did not take place in non-adipogenic cell lines, indicating that adipogenic commitment is a prerequisite to support the relation between PPARγ2 and VSTM2A. Our findings clearly show that preserving the integrity of the feed forward loop between PPARγ2 and VSTM2A is crucial to maintain and amplify the adipogenic potential of preadipocytes.
It is important to point out that the positive association between Vstm2a and Pparg2 expression is observed only in the early phases of adipogenesis. Indeed, we found that the massive induction in PPARγ2 that occurs in the late phases of adipogenesis was associated with a reduction, rather than an increase, in the expression of Vstm2a. These results further support that Vstm2a is not a direct target of PPARγ2 and that VSTM2A does not participate in the late activation of PPARγ2 in terminally differentiated cells. It was recently observed that the Pparg promoter contains five highly conserved non-coding elements that behave as enhancers to control Pparg expression at the earliest stages of adipocyte determination (Chou et al., 2013). Interestingly, these elements do not participate in the activation of Pparg in mature adipocytes (Chou et al., 2013). Whether VSTM2A regulates basal Pparg2 expression in preadipocytes by promoting the transactivation of these specific sequences through a SMAD1/5/8-dependent process is an interesting possibility that warrants further investigation.
VSTM2A is a glycoprotein secreted by committed preadipocytes in vitro and in vivo. Despite numerous attempts, we found no role for secreted VSTM2A in the regulation of adipogenesis in vitro. Of note, we showed that overexpression of Δ-VSTM2A, which lacks a signal peptide and cannot be secreted, was efficient in promoting adipogenesis. This observation univocally revokes the role of secreted VSTM2A in regulating adipogenic commitment in vitro. Although this finding is somewhat surprising, the identification of an intracellular function for a protein secreted by preadipocytes is not unique. For example, Wnt1-inducible signaling pathway protein 2 (WISP2) was recently identified as an adipokine controlling adipogenesis through intra and extracellular mechanisms (Grünberg et al., 2014; Hammarstedt et al., 2013). Here, we show that VSTM2A regulates adipogenic commitment at least by modulating BMP signaling through an unknown mechanism. At the moment, the functions of secreted VSTM2A remain to be determined. Although secreted VSTM2A did not regulate adipogenesis in vitro, we cannot exclude the possibility that this factor may contribute to adipose tissue formation, maintenance, or expansion in vivo by regulating other, non-cell-autonomous processes. For example, secreted VSTM2A could modulate angiogenesis or neurogenesis, which are processes that must be coordinated with adipocyte development to provide adequate blood supply and facilitate innervation during the formation of adipose tissues.
VSTM2A is highly expressed in the brain. Vstm2a was recently identified by others as one enigmatic gene highly produced in mouse brain (Pandey et al., 2014). Similarly to VSTM2A, the early regulator of preadipocyte determination ZFP423 is also expressed at high levels in the brain (Vishvanath et al., 2016; Gupta et al., 2010). Loss of ZFP423 was shown to impair adipogenesis but also to cause severe defects in brain formation (Alcaraz et al., 2006; Cheng et al., 2007; Gupta et al., 2010; Warming et al., 2006). Interestingly, genome-wide analysis of gene expression during adipogenesis of mouse embryonic stem cells revealed a clear link between early adipogenesis and neural development (Billon et al., 2010). These observations indicate that some regulatory pathways are likely playing dual roles in the development of adipocytes and neurons. At the moment, the roles of VSTM2A in neurons and brain development remain uncharacterized.
In conclusion, we report the identification of VSTM2A as a factor modulating adipogenic commitment. We provide evidence that VSTM2A expression is required to maintain and amplify the adipogenic capacity of adipose precursor cells. These findings extend the current understanding of the mechanisms regulating preadipocyte development, which could eventually contribute to the generation of new tools to control adipose tissue size and function and improve metabolic health.
EXPERIMENTAL PROCEDURES
Animal Care
All experimental protocols were approved by the Animal Ethics Committee of Université Laval (CPAUL) and in accordance with the guidelines of the Canadian Council on Animal Care. All mice were on a C57BL/6J background and were purchased from the Jackson Laboratory. The animals were fed a high-fat diet (60% kCal from fat, Research Diets, D12492) for 4 weeks. All mice were sacrificed, and tissues were collected at the same time of day. Obese mice (ob/ob and db/db) and lean controls (12 weeks) were purchased from the Jackson Laboratory and maintained on a chow diet for the duration of the studies.
Human Samples
The experimental protocols performed with human adipose tissue samples were approved by the Research Ethics Committee of the Institut universitaire de cardiologie et de pneumologie de Québec – Université Laval (IUCPQ-UL). Biological tissues were obtained from a Biobank approved by the Research Ethics Committees of CHU Sainte-Justine and CSSS Jeanne-Mance. Subcutaneous adipose cells were collected from fetal tissues obtained in a context of voluntary termination of pregnancy (gestation period 16–23 weeks) and in all cases the participants have signed an informed consent. Adult subcutaneous adipose tissue samples were collected from non-smoker men aged between 48 and 63 years old with a median body mass index of 25.4. Patients treated with glitazones were excluded.
Tissue Distribution of VSTM2A
The tissue distribution of VSTM2A was performed on mouse tissues isolated from either 12-week-old mice or pups sacrificed 4 days postnatal (P4). To study VSTM2A expression during development, eWAT and sWAT were collected from mice at embryonic day 16.5 (E16.5) or P1, P4, P7, P14, P28, and P56. Subcutaneous WAT was collected from the inguinal region, whereas eWAT was collected directly under the testis. Tissues were either frozen for western blotting or RT-qPCR analyses or put into PBS and stained 15 min at 37°C with LipidTOX red neutral lipid stain (1:250) (Life Technologies H34476). Pictures were taken using the Evos fluorescence microscopy system (Life Technologies).
Immunofluorescence
Epididymal WAT samples were collected from P4 mice and slowly frozen in OCT before being cut at the Histology Core Laboratory of Université Laval. Slides were fixed with cold acetone, blocked, and stained overnight at 4°C in 5% normal donkey serum diluted in PBS. The following primary antibodies were used for immunofluorescence: VSTM2A (Acris, AP54523PU-N, dilution 1:200), CD31 (BD Pharmingen, 553370, 1:200), and αSMA (Abcam, AB8211, 1:200). Following the incubation with the primary antibodies, slides were washed and incubated with the corresponding secondary antibodies (donkey anti-rabbit 568, Life Technologies, A10042 1:100; goat anti-rat 488, Life Technologies A11006, 1:100) for 45 min in the dark at room temperature. After PBS washing, slides were stained with DAPI (Life Technologies, D3571, 1:10,000) for 10 min and then mounted using Fluoromount G mounting media (Southern Biotech) and imaged by confocal microscopy (Microscope LSM800, Zeiss Axio Observer Z1). For Immunofluorescence assays performed on cells, cells were plate the day before on poly-l-ornithine coated slides. The cells were next fixed with PFA 4%, permeabilized during 1 min with Triton X-100 (0.05%) and blocked 30 min with normal donkey serum. Slides were incubated for 1 hr with V5 antibody (13202S, Cell Signaling Technology, dilution 1:400) and for 45 min with secondary antibody (donkey anti-rabbit 568, Life Technologies, A10042 1:100). Slides were stained with DAPI mounted using Fluoromount G mounting media, as described above.
Cell Culture
All immortalized cell lines used (3T3-L1, C3H10T1/2, NIH 3T3) were obtained from ATCC and cultured following the instructions of this provider. Human primary MSCs were also purchased from ATCC (PCS500011). These cells were maintained in MSC basal medium (PCS500030) complemented with 3% MSC growth kit low serum (PCS500040) and were used at P4. MEFs were derived from embryos (E13.5) and the SVF was isolated from WAT by collagenase digestion, as described previously (Laplante et al., 2006). MEFs and SVF were used at passage 1 or 2. Immortalized brown preadipocytes were kindly provided by Dr. Shingo Kajimura. The cells were isolated as described (Galmozzi et al., 2014). For the isolation of 3T3-L1 clones, serial dilutions were performed to allow the isolation of a single cell per well (96-well plate format). Each well was visually inspected by microscopy to confirm the presence of a single colony per well. Wells with more than one colony were excluded. The clones were amplified and always maintained at sub-confluence. The following procedure was used for the induction of adipogenesis in 3T3-L1 cells. Subconfluent cells were maintained in DMEM with 10% fetal bovine serum (FBS). Two days after reaching confluence, cells were exposed to an adipogenic cocktail containing insulin (830 nM), dexamethasone (1 μM), and 3-isobutyl-1-methylxanthine (IBMX) (500 μM). This corresponds to day 0 (D0). Two days later (D2), the culture medium was changed for DMEM 10% FBS containing insulin (830 nM). The same procedure was followed to induce adipogenesis in human primary MSCs, SVF, NIH 3T3, and C3H10T1/2 cells, except that the adipogenic cocktail was supplemented with the PPARγ agonist rosiglitazone (5 μM). Immortalized brown preadipocytes (90% confluent) were induced to differentiate using an adipogenic cocktail containing insulin (5 μg/mL), triiodothyronine (T3) (1 nM), dexamethasone (2 μg/mL), and IBMX (500 μM). After 3 days, the media was changed and cells were maintained in DMEM 10% FBS supplemented with T3 (1 nM). To measure spontaneous differentiation, 3T3-L1 or C3H10T1/2 cells were plated to confluence and maintained in DMEM 10% FBS until day 20. Media was changed every 10 days. LDN-193189 (Sigma, SML-0559) was used to test the impact of BMP signaling on spontaneous adipogenesis.
Microarray Analyses
Whole-genome gene expression was performed using the Affymetrix GeneChip Mouse Gene 1.0 ST Array. The RNA was labeled and hybridized using a standard metrix protocol. The quality of arrays was judged using standard quality control parameters and all arrays passed the quality control filters. Expression values were extracted using the Robust Multichip Average (RMA) method (Irizarry et al., 2003) implemented in the oligo package in R (Carvalho and Irizarry, 2010). The Significance Analysis of Microarrays (SAM) method was used to identify probe sets differentially expressed between groups. The false discovery rate (FDR) and the fold change threshold were set at 5% and 1.5, respectively. All analyses were carried out with the R statistical software v.3.2.3 and Bioconductor packages.
Statistical Analyses
Data are expressed as mean ± SEM. Comparison between two experimental conditions were analyzed by Student’s unpaired t test. Two-way ANOVA was used to compare more than two experimental conditions. All statistical tests were performed using GraphPad Prism (v.6.0c), and p < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
VSTM2A is highly expressed and secreted by committed preadipocytes
VSTM2A is expressed early during adipocyte development in vitro and in vivo
VSTM2A overexpression promotes adipogenesis while its knockdown impairs this process
VSTM2A amplifies BMP signaling and Pparg2 expression in preadipocytes
Acknowledgments
The authors are grateful to Marc Veillette, Renée Dicaire, Michèle Orain, Julie Plamondon, Christine Racine, and Kerstin Bellmann for helpful technical assistance. The authors wish to thank Matthew Rodeheffer for advice on the FACS procedure. The authors also thank Yan Laplante for preparing the graphical abstract. This work was supported by grants from the Canadian Institutes of Health Research (CIHR) (MOP123387), the Natural Sciences and Engineering Research Council of Canada (NSERC) (418158-2012), Les Fonds de recherche du Québec – Santé (FRQS) (24726), Le Réseau de recherche en santé cardiométabolique, diabète et obésité (CMDO), Le Réseau de bio-imagerie du Québec (RBIQ), Diabète Québec, and La Fondation de l’Institut universitaire de cardiologie et de pneumologie de Québec – Université Laval (IUCPQ-UL) to M.L.
Footnotes
ACCESSION NUMBERS
The accession number for the microarray data reported in this paper is NCBI GEO: GSE90580. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE90580).
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.12.015.
AUTHOR CONTRIBUTIONS
Conceptualization, B.S., M.L., and K.B.; Methodology, B.S. and M.L.; Formal Analysis; B.S., M.L., and Y.B.; Investigation, B.S., M.L., E.C., M.-A.B., A.C., A.B., and Y.G.; Writing – Original Draft, B.S. and M.L.; Writing – Review & Editing, B.S. and M.L.; Funding Acquisition, M.L.; Supervision, M.L., Y.B., D.M., A.S., and D.A.G.; Resources, K.M.T., P.L.L., A.-M.L., S.G., D.M., A.S., D.A.G., J.V.G., N.P., and E.H.
References
- Alcaraz WA, Gold DA, Raponi E, Gent PM, Concepcion D, Hamilton BA. Zfp423 controls proliferation and differentiation of neural precursors in cerebellar vermis formation. Proc Natl Acad Sci USA. 2006;103:19424–19429. doi: 10.1073/pnas.0609184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry R, Rodeheffer MS. Characterization of the adipocyte cellular lineage in vivo. Nat Cell Biol. 2013;15:302–308. doi: 10.1038/ncb2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DC, Stenesen D, Zeve D, Graff JM. The developmental origins of adipose tissue. Development. 2013;140:3939–3949. doi: 10.1242/dev.080549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry R, Jeffery E, Rodeheffer MS. Weighing in on adipocyte precursors. Cell Metab. 2014;19:8–20. doi: 10.1016/j.cmet.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billon N, Kolde R, Reimand J, Monteiro MC, Kull M, Peterson H, Tretyakov K, Adler P, Wdziekonski B, Vilo J, Dani C. Comprehensive transcriptome analysis of mouse embryonic stem cell adipogenesis unravels new processes of adipocyte development. Genome Biol. 2010;11:R80. doi: 10.1186/gb-2010-11-8-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers RR, Kim JW, Otto TC, Lane MD. Stable stem cell commitment to the adipocyte lineage by inhibition of DNA methylation: Role of the BMP-4 gene. Proc Natl Acad Sci USA. 2006;103:13022–13027. doi: 10.1073/pnas.0605789103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Brown adipose tissue: Function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26:2363–2367. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthorn WP, Scheller EL, MacDougald OA. Adipose tissue stem cells meet preadipocyte commitment: Going back to the future. J Lipid Res. 2012;53:227–246. doi: 10.1194/jlr.R021089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LE, Zhang J, Reed RR. The transcription factor Zfp423/OAZ is required for cerebellar development and CNS midline patterning. Dev Biol. 2007;307:43–52. doi: 10.1016/j.ydbio.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou WL, Galmozzi A, Partida D, Kwan K, Yeung H, Su AI, Saez E. Identification of regulatory elements that control PPARγ expression in adipocyte progenitors. PLoS ONE. 2013;8:e72511. doi: 10.1371/journal.pone.0072511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church CD, Berry R, Rodeheffer MS. Isolation and study of adipocyte precursors. Methods Enzymol. 2014;537:31–46. doi: 10.1016/B978-0-12-411619-1.00003-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, Kahn CR. Brown fat as a therapy for obesity and diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;17:143–149. doi: 10.1097/MED.0b013e328337a81f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galmozzi A, Sonne SB, Altshuler-Keylin S, Hasegawa Y, Shinoda K, Luijten IH, Chang JW, Sharp LZ, Cravatt BF, Saez E, Kajimura S. ThermoMouse: An in vivo model to identify modulators of UCP1 expression in brown adipose tissue. Cell Rep. 2014;9:1584–1593. doi: 10.1016/j.celrep.2014.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green H, Meuth M. An established pre-adipose cell line and its differentiation in culture. Cell. 1974;3:127–133. doi: 10.1016/0092-8674(74)90116-0. [DOI] [PubMed] [Google Scholar]
- Grünberg JR, Hammarstedt A, Hedjazifar S, Smith U. The Novel Secreted Adipokine WNT1-inducible Signaling Pathway Protein 2 (WISP2) Is a Mesenchymal Cell Activator of Canonical WNT. J Biol Chem. 2014;289:6899–6907. doi: 10.1074/jbc.M113.511964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Arany Z, Seale P, Mepani RJ, Ye L, Conroe HM, Roby YA, Kulaga H, Reed RR, Spiegelman BM. Transcriptional control of preadipocyte determination by Zfp423. Nature. 2010;464:619–623. doi: 10.1038/nature08816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Mepani RJ, Kleiner S, Lo JC, Khandekar MJ, Cohen P, Frontini A, Bhowmick DC, Ye L, Cinti S, Spiegelman BM. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab. 2012;15:230–239. doi: 10.1016/j.cmet.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarstedt A, Hedjazifar S, Jenndahl L, Gogg S, Grünberg J, Gustafson B, Klimcakova E, Stich V, Langin D, Laakso M, Smith U. WISP2 regulates preadipocyte commitment and PPARγ activation by BMP4. Proc Natl Acad Sci USA. 2013;110:2563–2568. doi: 10.1073/pnas.1211255110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee JE, Jin J, Lim JS, Oh N, Kim K, Chang SI, Shibuya M, Kim H, Koh GY. The spatiotemporal development of adipose tissue. Development. 2011;138:5027–5037. doi: 10.1242/dev.067686. [DOI] [PubMed] [Google Scholar]
- Hong KY, Bae H, Park I, Park DY, Kim KH, Kubota Y, Cho ES, Kim H, Adams RH, Yoo OJ, Koh GY. Perilipin+ embryonic preadipocytes actively proliferate along growing vasculatures for adipose expansion. Development. 2015;142:2623–2632. doi: 10.1242/dev.125336. [DOI] [PubMed] [Google Scholar]
- Hudak CS, Gulyaeva O, Wang Y, Park SM, Lee L, Kang C, Sul HS. Pref-1 marks very early mesenchymal precursors required for adipose tissue development and expansion. Cell Rep. 2014;8:678–687. doi: 10.1016/j.celrep.2014.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Jeffery E, Church CD, Holtrup B, Colman L, Rodeheffer MS. Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat Cell Biol. 2015;17:376–385. doi: 10.1038/ncb3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Berry DC, Tang W, Graff JM. Independent stem cell lineages regulate adipose organogenesis and adipose homeostasis. Cell Rep. 2014;9:1007–1022. doi: 10.1016/j.celrep.2014.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Takagi T, Kanesashi SN, Kurahashi T, Nomura T, Harada J, Ishii S. Schnurri-2 controls BMP-dependent adipogenesis via interaction with Smad proteins. Dev Cell. 2006;10:461–471. doi: 10.1016/j.devcel.2006.02.016. [DOI] [PubMed] [Google Scholar]
- Kim SM, Lun M, Wang M, Senyo SE, Guillermier C, Patwari P, Steinhauser ML. Loss of white adipose hyperplastic potential is associated with enhanced susceptibility to insulin resistance. Cell Metab. 2014;20:1049–1058. doi: 10.1016/j.cmet.2014.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Festuccia WT, Soucy G, Gélinas Y, Lalonde J, Berger JP, Deshaies Y. Mechanisms of the depot specificity of peroxisome proliferator-activated receptor gamma action on adipose tissue metabolism. Diabetes. 2006;55:2771–2778. doi: 10.2337/db06-0551. [DOI] [PubMed] [Google Scholar]
- Nishimura S, Manabe I, Nagasaki M, Hosoya Y, Yamashita H, Fujita H, Ohsugi M, Tobe K, Kadowaki T, Nagai R, Sugiura S. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes. 2007;56:1517–1526. doi: 10.2337/db06-1749. [DOI] [PubMed] [Google Scholar]
- Pandey AK, Lu L, Wang X, Homayouni R, Williams RW. Functionally enigmatic genes: A case study of the brain ignorome. PLoS ONE. 2014;9:e88889. doi: 10.1371/journal.pone.0088889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poissonnet CM, Burdi AR, Bookstein FL. Growth and development of human adipose tissue during early gestation. Early Hum Dev. 1983;8:1–11. doi: 10.1016/0378-3782(83)90028-2. [DOI] [PubMed] [Google Scholar]
- Poissonnet CM, Burdi AR, Garn SM. The chronology of adipose tissue appearance and distribution in the human fetus. Early Hum Dev. 1984;10:1–11. doi: 10.1016/0378-3782(84)90106-3. [DOI] [PubMed] [Google Scholar]
- Rodeheffer MS, Birsoy K, Friedman JM. Identification of white adipocyte progenitor cells in vivo. Cell. 2008;135:240–249. doi: 10.1016/j.cell.2008.09.036. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156:20–44. doi: 10.1016/j.cell.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnick MA, Panigrahy D, Zhang CY, Dallabrida SM, Lowell BB, Langer R, Folkman MJ. Adipose tissue mass can be regulated through the vasculature. Proc Natl Acad Sci USA. 2002;99:10730–10735. doi: 10.1073/pnas.162349799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz TJ, Huang P, Huang TL, Xue R, McDougall LE, Townsend KL, Cypess AM, Mishina Y, Gussoni E, Tseng YH. Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature. 2013;495:379–383. doi: 10.1038/nature11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QQ, Otto TC, Lane MD. Commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci USA. 2004;101:9607–9611. doi: 10.1073/pnas.0403100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Zeve D, Suh JM, Bosnakovski D, Kyba M, Hammer RE, Tallquist MD, Graff JM. White fat progenitor cells reside in the adipose vasculature. Science. 2008;322:583–586. doi: 10.1126/science.1156232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng YH, Kokkotou E, Schulz TJ, Huang TL, Winnay JN, Taniguchi CM, Tran TT, Suzuki R, Espinoza DO, Yamamoto Y, et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454:1000–1004. doi: 10.1038/nature07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishvanath L, MacPherson KA, Hepler C, Wang QA, Shao M, Spurgin SB, Wang MY, Kusminski CM, Morley TS, Gupta RK. Pdgfrβ+ mural preadipoctyes contribute to adipocyte hyperplasia induced by high-fat-diet feeding and prolonged cold exposure in adult mice. 2016;23:350–359. doi: 10.1016/j.cmet.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QA, Scherer PE. The AdipoChaser mouse: A model tracking adipogenesis in vivo. Adipocyte. 2014;3:146–150. doi: 10.4161/adip.27656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med. 2013;19:1338–1344. doi: 10.1038/nm.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S, Rachel RA, Jenkins NA, Copeland NG. Zfp423 is required for normal cerebellar development. Mol Cell Biol. 2006;26:6913–6922. doi: 10.1128/MCB.02255-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassermann F. The Development of Adipose Tissue. In: Renold AE, Cahill GF, editors. Handbook of Physiology: Adipose Tissue. American Physiological Society; 1965. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.