

Abstract
BACKGROUND
A growing body of evidence indicates a positive correlation between expression of human antimicrobial peptide leucin leucin 37 (LL-37) and progression of epithelial cancers, including prostate cancer (PCa). Although the molecular mechanisms for this correlation has not yet been elucidated, the primary function of LL-37 as a chemotactic molecule for innate immune effector cells suggests its possible association in coordinating protumorigenic mechanisms, mediated by tumor-infiltrating immune cells.
METHODS
To investigate protumorigenic role(s) of cathelicidin-related antimicrobial peptide (CRAMP), a murine orthologue of LL-37, the present study compared tumor growth kinetics between mouse PCa cell lines with and without CRAMP expression (TRAMP-C1 and TRAMP-C1CRAMP-sh, respectively) in immunocompetent mice. CRAMP-mediated chemotaxis of different innate immune cell types to the tumor microenvironment (TME). The role of CRAMP in differentiation and polarization of immature myeloid progenitors (IMPs) to protumorigenic type 2 macrophages (M2) in TME was determined by adoptive transfer of IMPs into mice bearing CRAMP(+) and CRAMP(−) tumors. To differentiate protumorigenic events mediated by tumor-derived CRAMP from host immune cell-derived CRAMP, tumor challenge study was performed in CRAMP-deficient mice. To identify mechanisms CRAMP function, macrophage colony stimulating factor (M-CSF) and monocyte chemoattractant protein 1 (MCP-1) gene expression was analyzed by QRT-PCR and STAT3 signaling was determined by immunoblotting.
RESULTS
Significantly delayed tumor growth was observed in wild-type (WT) mice implanted with TRAMP-C1CRAMP-sh cells compared to mice implanted with TRAMP-C1 cells. CRAMP(+) TME induced increased number of IMP differentiation into protumorigenic M2 macrophages compared to CRAMP(−) TME, indicating tumor-derived CRAMP facilitates differentiation and polarization of IMPs toward M2. Tumor challenge study in CRAMP deficient mice showed comparable tumor growth kinetics with WT mice, suggesting tumor-derived CRAMP plays a crucial role in PCa progression. In vitro study demonstrated that overexpressed M-CSF and MCP-1 in TRAMP-C1 cells through CRAMP-mediated autocrine signaling, involving p65, regulates IMP-to-M2 differentiation/polarization through STAT3 activation.
CONCLUSION
Altogether, the present study suggests that overexpressed CRAMP in prostate tumor initially chemoattracts IMPs to TME and mediates differentiation and polarization of early myeloid progenitors into protumorigenic M2 macrophages during PCa progression. Thus, selective downregulation of CRAMP in tumor cells in situ may benefit overcoming immunosuppressive mechanisms in PCa.
Keywords: CRAMP, LL-37, Prostate cancer
INTRODUCTION
Progression of prostate cancer (PCa) is associated with protumorigenic immune modulation (1). Previously, we have shown a positive correlation between the expression of antimicrobial peptide (AMP), human leucine leucine 37 (LL-37) and its mouse orthologue cathelicidin-related AMP (CRAMP), and the grade of tumor in both human and mouse PCa (2). Originally, LL-37/CRAMP is known as an important effector molecule of innate immunity that provides a first-line host defense system by chemoattracting and activating the innate immune cells including neutrophils and macrophages to the inflammatory sites (3–5).
Recently, elevated level of LL-37/CRAMP has been documented in epithelial carcinomas of ovary, breast, and lung (6–10). The overexpression of LL-37/CRAMP correlates with increased chemotaxis of CD45+ leukocytes and mesenchymal stromal cells in human ovarian cancer and CD68+ myeloid cells in cigarette smoke-induced mouse models of lung cancer (6, 10, 11). Li et al. have shown that receptor for CRAMP, formyl peptide receptor 2 (FPR2), expressed on human macrophages polarizes them into protumorigenic type 2 macrophages (M2) by upregulating monocyte chemoattractant protein-1 (MCP-1) and macrophage colony stimulating factor (M-CSF) partially depending on NF-κB pathway in human hepatocellular carcinoma model (9). However, in contrast to this report, Wang et al. have documented that upon the stimulation with Lewis lung carcinoma cell supernatant, macrophages from FPR2 deficient mice results in M2 phenotype by phosphorylation of STAT3 and STAT6 (12). These controversial findings regarding the role of LL-37/CRAMP and its receptor FPR2 in different cancer models imply that the mechanism of action of FPR2-dependent immune modulation during tumor progression still needs to be elucidated.
Immature myeloid progenitors (IMPs), defined as Gr-1+ and CD11b+ cells, are a heterogeneous population of early myeloid lineage that can differentiate into mature granulocytes, macrophages, or dendritic cells. However, in pathological conditions such as cancer, the differentiation of IMPs is blocked and they proliferate as myeloid-derived suppressor cells (MDSCs) (13). It has been reported that monocytic MDSCs (m-MDSCs) can differentiate into M2-like tumor-associated macrophages (TAMs), regulated by activation of STAT3/6 (13–16). These TAMs promote tumor growth by enhancing collagen degradation through overexpression of mannose receptor/CD206 and matrix metalloproteinases (MMPs) in the TME (17, 18).
Based on the key role of MDSCs in immunosuppression, following their recruitment to the tumor site and a significantly-higher CRAMP/LL-37 expression by the tumor cells, present study determined the effects of CRAMP in facilitating chemotaxis and protumorigenic polarization of myeloid cells. Results of the studies using TRAMP mouse-derived PCa cells with varying levels of CRAMP expression and, syngeneic mouse lacking the CRAMP gene (Cnlp−/−), we identified that PCa-derived CRAMP facilitates IMP-to-macrophage differentiation and further polarizes them into M2 phenotype in TME indicating a crucial role for tumor-derived CRAMP in protumorigenic functions of myeloid derivatives. Here, we show that elevated CRAMP expression in mouse PCa cells not only chemoattracts IMPs to TME, but also results in overexpression of M-CSF and MCP-1 in PCa cells, which induces macrophage differentiation and M2 polarization from tumor-infiltrated IMPs by upregulating STAT3.
MATERIALS AND METHODS
Cell lines and reagents
TRAMP-C1 cell line (American Type Culture Collection) was cultured in DMEM (Life Technologies, Carlsbad, CA) supplemented with 5% horse serum (Life Technologies), 5% FBS (Sigma-Aldrich, St. Louis, MO), 5 μg/mL insulin (Sigma-Aldrich), 1% pen/strep (Life Technologies), and 10 nM dihyderotestosterone (Sigma-Aldrich). Previously established TRAMP-C1CRAMP-sh and TRAMP-C1scram-sh cells (2) were cultured with 800 μg/mL and 300 μg/mL neomycin (Life Technologies), respectively, for clonal selection.
Implantation of PCa cells
Six-to-eight-week old male C57BL/6 and athymic nude (nu/nu) mice were purchased from Harlan Laboratories. The Cnlp−/− mouse lacking the gene encoding CRAMP was developed in UAB animal facility by transferring the genotype from Cnlp−/− mice in 129/SvJ strain obtained from Dr. Richard Gallo (University of California at San Diego, CA) into C57BL/6 background. TRAMP-C1, TRAMP-C1scram-sh, or TRAMP-C1CRAMP-sh cells (5×105) were subcutaneously implanted to mice listed above. The size of external tumors was measured in two dimensions and calculated [(length × width2)/2] for spherical volume every 3–4 days.
Adoptive transfer of Gr-1+, CD11b+ cells
TRAMP-C1 cells (5×105) were subcutaneously implanted into 6–8 week-old male C57BL/6 mice. Following the tumor formation, mice were sacrificed to sort Gr-1+, CD11b+, F4/80− cells from the spleen when average volume of tumor reached 200mm3. Sorted cells, using Gr-1-APC eFluor 780, CD11b-eFluor 450, and F4/80-PE Cy7 antibodies (eBioscience, San Diego, CA), were labeled with CFSE (5 μM) for in vivo tracking. Labeled cells (1×105 cells/mouse) were transferred into recipient male nude mice (6–8 week-old) bearing TRAMP-C1, TRAMP-C1scram-sh, and TRAMP-C1CRAMP-sh tumors by tail vein injection, when the average volume of tumors reached 800mm3. Recipient mice were sacrificed 3 days post-transfer for flow cytometry with the cells from the spleen and tumor.
Immunoprofiling by flow cytometry
Cells from the spleen or tumor from mice were stained with antibodies (eBioscience) Gr-1-APC eFluor 780, CD11b-eFluor 450, Ly6b-FITC and F4/80-PE Cy7 to detect neutrophils (Gr-1+, CD11b+, Ly6b+), macrophages (Gr-1−, CD11b+, F4/80+) and IMPs (Gr-1+, CD11b+, Ly6b−). For M1 and M2 macrophages, MHC II-PE and CD206-FITC antibodies (eBioscience), respectively, were used.
Chemotaxis assay
Splenic Gr-1+, CD11b+ IMPs (5×105) from tumor-bearing mice were plated in top Boyden chamber (5 μm, Corning Incorporated) having conditioned media from TRAMP-C1scram-sh cells with or without WRW4 (10 μg/ml, ANASPEC, Fremont, CA) and TRAMP-C1CRAMP-sh cells with or without CRAMP (4 μg/ml, ANASPEC) treatment. After 12 hours of incubation, the number of migrated cells was counted.
Culture of IMPs for differentiation in vitro
Mouse bone marrow cells were cultured with GM-CSF (20 ng/mL) for 4 days to induce Gr-1+, CD11b+ IMPs. Bone marrow-derived IMPs were cultured with conditioned media from 24 hr-cultured TRAMP-C1 and TRAMP-C1CRAMP-sh cells, and M-CSF (20 ng/mL). After 4 days, the cells were collected and flow cytometry was carried out to define M2 macrophage differentiation.
Real-time quantitative PCR
cDNAs were synthesized from total RNA from prostate cancer cells or IMPs, isolated using Trizol Reagent (Life Technologies), using iScript™ Reverse Transcription Supermix (Bio-Rad Laboratories, Hercules, CA). RT-PCR was performed with cDNAs using SYBR green (Bio-Rad Laboratories) to measure the mRNA expression. The GAPDH mRNA expression was used as internal control. The primer sequences used in RT-PCR are given in Table 1.
Table 1.
The sequences for primers used in real-time RT-PCR.
| CRAMP-forward | 5′-TCCCAAGTCTGTGAGGTTCC-3′ |
| CRAMP-reverse | 5′-ACCAATCTTCTCCCCACCTT-3′ |
| FPR2-forward | 5′-CTTTATCTGCTGGTTTCCCTTTC-3′ |
| FPR2-reverse | 5′-CTGGTGCTTGAATCACTGGTTTG-3′ |
| M-CSF-forward | 5′-CCCATATTGCGACACCGAA-3′ |
| M-CSF-reverse | 5′-AAGCAGTAACTGAGCAACGGG-3′ |
| MCP-1-forward | 5′-CTTCTGGGCCTGCTGTTCA-3′ |
| MCP-1-reverse | 5′-CCAGCCTACTCATTGGGATCA-3′ |
| GAPDH-forward | 5′-TCAACAGCAACTCCCACTCTTCCA-3′ |
| GAPDH-reverse | 5′-ACCCTGTTGCTGTAGCCGTATTCA-3′ |
Western blotting
PCa cells were lysed by sonication in PBS containing protease inhibitors (Roche), and the protein concentrations were measured using BCA protein assay (Thermo Scientific). Cell lysates (40–70 μg) were loaded on 10% or 15% SDS-PAGE gel and transferred to 0.45 μm nitrocellulose membrane (GE Healthcare). The antibodies used for immunodetection were rabbit anti-CRAMP (Innovagen, Lund, Sweden), rabbit anti-FPRL1 (NOVUS Biologicals), rabbit anti-pSTAT-3 (Cell Signaling Technology) and goat anti-actin (Santa Cruz Biotechnology). Blots were developed using HRP-conjugated secondary antibodies with enhanced chemiluminescence reagent (Millipore).
RESULTS
Downregulation of CRAMP delays PCa growth in vivo
Based on a previous study from our lab, which demonstrated that the expression of CRAMP is positively associated with progression of PCa in the TRAMP model (2), we first investigated whether knockdown of CRAMP in TRAMP-C1 cells inhibits tumorigenesis due to lack of CRAMP-mediated immune modulation. After confirming downregulated CRAMP levels in TRAMP-C1CRAMP-sh cells by protein and RNA measurements (Figure 1A & B), PCa cell lines with intact or downregulated CRAMP were subcutaneously implanted into syngeneic 6–8 week-old C57BL/6 mice. The mice bearing TRAMP-C1 and TRAMP-C1scram-sh cells developed measurable tumors from day-45 post-implantation, whereas mice implanted with TRAMP-C1CRAMP-sh cells displayed the onset of tumor only after 4 months post-implantation (Figure 1C). These data indicate that the high level of tumor-derived CRAMP promotes PCa growth, while downregulation of CRAMP in tumor cells significantly delays tumorigenesis in vivo. Intact ability in tumor engraftment of TRAMP-C1CRAMP-sh as in TRAMP-C1scram-sh control cell line was confirmed using xenograft model (Supplemental Fig. S1).
Figure 1. Silencing cathelicidin-related antimicrobial peptide (CRAMP) expression in tumor cells in situ delays prostate cancer growth.
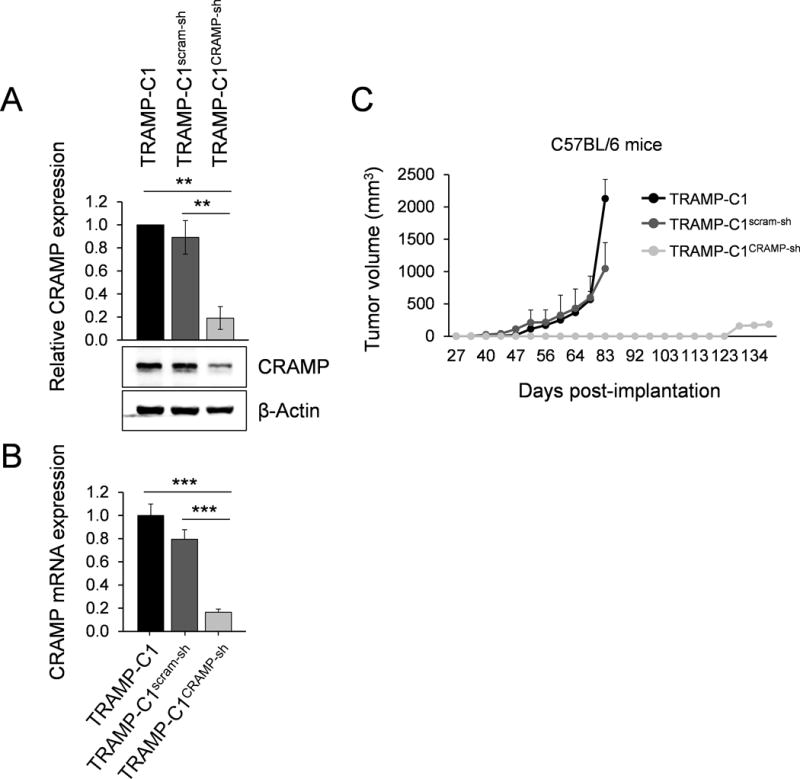
(A) Immunoblotting showing down-regulation of CRAMP following stable induction of shRNA construct targeting CRAMP gene in TRAMP-C1 cells. The levels of CRAMP were normalized to β-actin levels. Results are expressed as mean ± SD (n=3). **P < 0.01 (B) Down-regulated CRAMP was determined by real-time PCR in TRAMP-C1CRAMP-sh cells compared to TRAMP-C1 and TRAMP-C1scram-sh cells (mean ± SE) (n=3) ***P < 0.001 (C) C57BL/6 mice were subcutaneously implanted with TRAMP-C1, TRAMP-C1scram-sh or TRAMP-C1CRAMP-sh cells (5×105). Tumor volume is indicated as mm3 ± SE (n=10).
Tumor-derived CRAMP chemoattracts key innate immune effectors to TME in vivo
Next, we evaluated whether CRAMP secreted by PCa cells mediates migration of innate immune effectors to TME in C57BL/6 mice bearing CRAMP(+) tumors. Since lineage derivatives of IMPs, including neutrophils and macrophages, have been known to respond to chemotaxis by CRAMP during infection and that these cells are known to polarize towards protumorigenic populations in TME, we characterized the effect of tumor-derived CRAMP on macrophages, neutrophils, and IMPs.
Mice were challenged with TRAMP-C1, TRAMP-C1scram-sh, and TRAMP-C1CRAMP-sh PCa cells. Following the tumorigenesis in TRAMP-C1 and TRAMP-C1scram-sh groups, mice were sacrificed at two different time points and the cells from the spleen and TME were subjected to immunoprofiling. To monitor the influx of splenic innate immune effectors to TME, day-30 post-implantation, when TRAMP-C1 and TRAMP-C1scram-sh tumors were only palpable, was chosen as first time point, while day-50 post-implantation, when average volume of CRAMP(+) tumors reached 100 mm3, was selected as second time point. However, mice implanted with TRAMP-C1CRAMP-sh cells did not develop tumors even at day-50 post-implantation, hence, only splenic cells in this group were used for flow cytometry at both time points.
Flow cytometry analysis showed that TRAMP-C1 and TRAMP-C1scram-sh tumor-bearing mice resulted in a statistically significant decrease in the number of splenic neutrophil and IMP from day-30 to day-50 post-implantation (Figure 2A & B). Conversely, the number of splenic macrophages in these mice was increased from day-30 to day-50 post-implantation (Figure 2C). Since tumor-infiltrating immune effectors are known to originate from the spleen in tumor-bearing mice, we speculated that significant reduction of splenic IMPs and neutrophils may suggest that more number of these cells, than that of macrophages, have mobilized into TME in response to tumor-produced CRAMP. Surprisingly, the number of tumor-infiltrated macrophages was the highest among the immune infiltrates in TME, while the number of tumor-infiltrated neutrophils and IMPs were comparable in CRAMP(+) tumors (Figure 2D). Moreover, despite significant changes in the number of splenic innate immune effectors in tumor-bearing mice, the number of these cells in TRAMP-C1CRAMP-sh group remained the same as in WT at both time points (Figure 2A–C).
Figure 2. Chemotaxis of key innate effectors towards CRAMP-expressing TME.
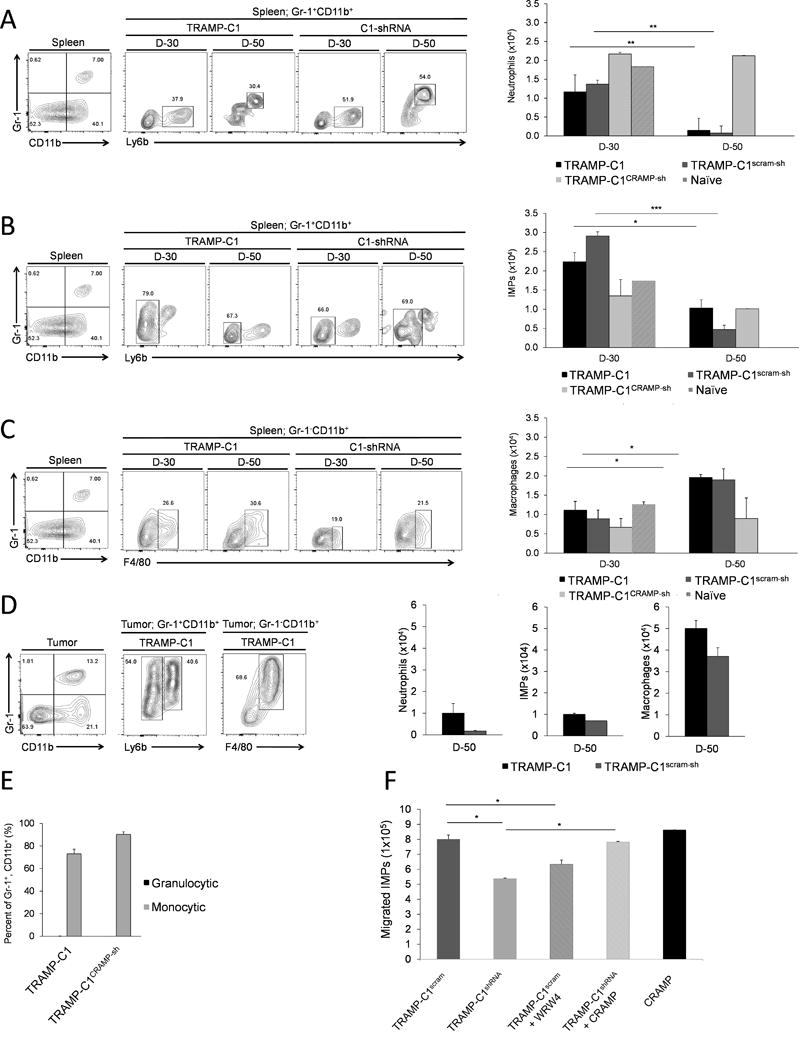
The number of (A) Neutrophils (Gr-1+, CD11b+, Ly6b+), (B) Immature myeloid progenitors (IMPs) (Gr-1+, CD11b+, Ly6b−) and (C) Macrophages (Gr-1−, CD11b+, F4/80+) in the spleen of TRAMP-C1 and TRAMP-C1scram-sh tumor-bearing mice were compared to mice bearing TRAMP-C1CRAMP-sh cells at day-30 and day-50 post-implantation. (D) At day-50 post-implantation, the absolute number of neutrophils, IMPs, and macrophages in TME was analyzed. (mean ± SE, n=3) *P < 0.01 **P < 0.005 ***P < 0.001 (E) Subtypes of Gr-1+, CD11b+ cells in TRAMP-C1 tumors were defined by expression of Ly6C or Ly6G. Data is shown as mean ± SE (n=3). (F) Splenic Gr-1+, CD11b+ cells (1×106) from TRAMP-C1 tumor-bearing mice were plated in 5 μm cell culture insert in different groups pre-plated with TRAMP-C1scram-sh cells with or without WRW4 treatment, TRAMP-C1CRAMP-sh cells with or without CRAMP treatment, and CRAMP peptide only. Chemoattracted cells at the bottom chamber were counted using hemocytometer. Data is shown as mean ± SE from triplicate experiments (*P < 0.01).
Since m-MDSCs in tumor-bearing hosts are known as precursors of TAMs (19), these results led us to speculate the potential role of CRAMP in regulating differentiation of monocytic myeloid cells to TAMs in TME. Thus, we further analyzed the subtype of tumor-infiltrated Gr-1+, CD11b+ cells, monocytic (Ly6C+) or granulocytic (Ly6G+). Interestingly, most of Gr-1+, CD11b+ cells in TME were monocytic that could differentiate into TAMs (Figure 2E).
Whether IMPs undergo CRAMP-dependent chemotaxis like neutrophils and macrophage has not been established yet. Thus, to confirm if infiltration of IMPs into TME was mediated by CRAMP in vivo, an in vitro chemotaxis assay was performed. Results of this study showed that indeed higher number of IMPs migrated toward TRAMP-C1scram-sh cells compared to that towards TRAMP-C1CRAMP-sh cells (Figure 2F). In addition, when activity of FPR2 was blocked in TRAMP-C1scram-sh cells with an FPR2 antagonist WRWWWW (WRW4) (20), the number of migrated IMPs was decreased than that in TRAMP-C1scram-sh cells (Figure 2F). When TRAMP-C1CRAMP-sh cells were stimulated with exogenous CRAMP, there was an increase in migrated IMPs (Figure 2F). The data suggests that PCa-produced CRAMP directly plays a role in chemoattraction of IMPs suggesting the significance of CRAMP-mediated protumoigenic events.
Host immune cell-derived CRAMP does not affect protumorigenic effects mediated by CRAMP produced by PCa in situ
Since neutrophils and macrophages are known to upregulate CRAMP expression to chemoattract more number of immune cells to inflammatory site, we next sought to determine the significance of CRAMP, produced by host immune cells versus that produced by PCa cells. To this end, we used a syngeneic, knockout (KO) mouse in C57BL/6 background that lacks Cnlp gene (Cnlp−/−) encoding CRAMP which was developed from Cnlp−/− mice from 129/SvJ strain in a transplantable tumor challenge model with CRAMP-expressing TRAMP-C1 cells. TRAMP-C1 tumor growth kinetics in Cnlp−/− mice was comparable to that of WT as both groups exhibited measurable tumors from day-40 post-implantation (Figure 3A). The result suggests that host immune cell-derived CRAMP does not contribute additional protumorigenic effect, but PCa-derived CRAMP is a key factor.
Figure 3. Host immune cell-derived CRAMP does not exhibit additional effects in protumorigenic event mediated by prostate tumor-derived CRAMP.
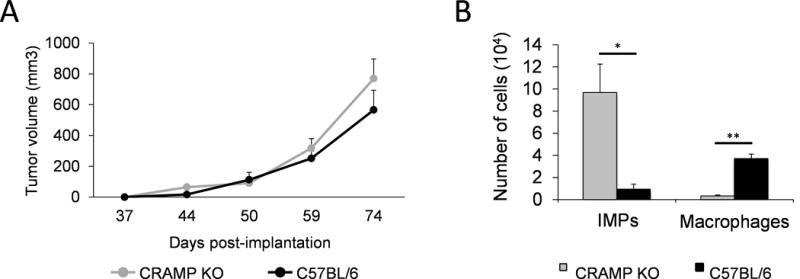
(A) Tumor volume in TRAMP-C1 tumor-bearing CRAMP deficient and wild-type (WT) mice. Data is presented as mm3 (mean ± SE, n=3). (B) The number of immature myeloid progenitors and macrophages from tumor microenvironment was compared between CRAMP deficient and WT mice. The data with absolute number are shown as mean ± SE (n=3). *P < 0.01 **P < 0.005 ***P < 0.001
Tumor-bearing Cnlp−/− mice displayed higher number of tumor-infiltrated IMPs but lower number of macrophages than WT
Chemotactic role of CRAMP for innate immune effectors is well-defined. Since we observed comparable tumor growth between Cnlp−/− and WT mice, we further characterized whether absence of CRAMP in tumor-infiltrated immune effectors modulates the levels of IMPs and macrophages in TME. Following TRAMP-C1 tumor challenge in Cnlp−/− and WT mice, the mice were sacrificed at day-60 post-implantation, when mean tumor volume reached 200 mm3 and immune infiltrates at the TME analyzed by flow cytometry. Results of this study demonstrated that TRAMP-C1 tumor-bearing Cnlp−/− mice had significantly elevated number of tumor-infiltrated IMPs compared to WT (Figure 3B). Conversely, the number of tumor-infiltrated macrophages was significantly reduced in Cnlp−/− mice compared to WT (Figure 3B). This suggests that PCa-derived CRAMP is sufficient for influx of IMPs toward TME. In accordance with the data from tumor challenge study using WT (Figure 2A–D), significantly decreased level of macrophages in TME in Cnlp−/− mice implies possible function of CRAMP in IMP-to-TAM differentiation in TME. Thus, we sought to determine whether tumor-derived CRAMP regulates IMP differentiation to TAMs.
Tumor-infiltrated IMPs differentiate into macrophages and polarize toward M2 in CRAMP-enriched TME
Considering role of M2 in angiogenesis and ECM remodeling beyond immunosuppression, IMP-to-M2 differentiation and polarization links the mechanisms needed during tumor growth. To identify whether PCa-derived CRAMP regulates IMP differentiation and polarization to M2, splenic IMPs from tumor-bearing C57BL/6 mice were labeled with carboxyfluorescein succinimidyl ester (CFSE) and adoptively transferred to nude mice bearing TRAMP-C1, TRAMP-C1scram-sh, and TRAMP-C1CRAMP-sh tumors. The recipient mice were sacrificed 3 days post-transfer to examine lineage conversion of transferred cells toward macrophage. Interestingly, although CFSE-positive IMPs were absent in both spleen and TME of recipient mice with CRAMP(+) tumors (Figure 4A), these mice had an increase in the number of macrophages in CRAMP(+) TME (Figure 4B). This pattern was observed only in CRAMP(+) tumors but not in TRAMP-C1CRAMP-sh tumors. However, TRAMP-C1CRAMP-sh tumors retained significantly high number of IMPs both in the spleen and TME (Figure 4A), but significantly low number of macrophages in TME compared to CRAMP(+) tumor-bearing mice (Figure 4B). Data implies that TRAMP-C1 and TRAMP-C1scram-sh tumor-derived CRAMP promotes differentiation of tumor-infiltrated IMPs into macrophages.
Figure 4. TRAMP-C1 and TRAMP-C1scram-sh tumors induce differentiation of IMPs into M2 macrophages in the TME.
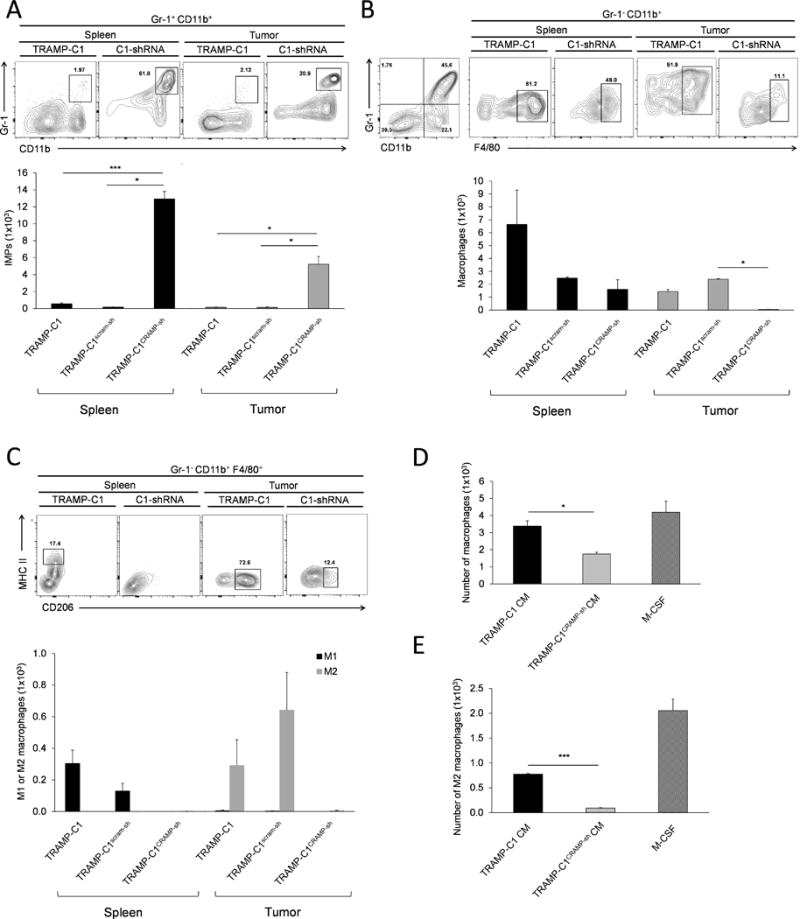
(A) The number of adoptively transferred IMPs, labeled with CFSE, in spleen and tumor was compared among the mice bearing TRAMP-C1, TRAMP-C1scram-sh and TRAMP-C1CRAMP-sh tumors. Data are shown as mean ± SE (n=3). *P < 0.01 ***P < 0.001 (B) The number of CFSE+ macrophages (Gr-1−, CD11b+, F4/80+) in the spleen and tumor was analyzed in mice bearing tumors with different level of cathelicidin-related antimicrobial peptide (CRAMP) expression. Results are presented as mean ± SE (n=3). *P < 0.01 (C) The phenotype of macrophages in the spleen and TME in mice bearing TRAMP-C1, TRAMP-C1scram-sh and TRAMP-C1CRAMP-sh tumors was determined. Data are shown as mean ± SE (n=3). (D) The number of macrophages differentiated from IMPs cultured with CM from TRAMP-C1 and TRAMP-C1CRAMP-sh cells, and M-CSF in vitro. *P < 0.05 (E) The number of M2 macrophages derived from IMPs cultured with conditioned medium from TRAMP-C1 and TRAMP-C1CRAMP-sh cells, and M-CSF in vitro. ***P < 0.001
Since macrophages can be activated either towards classical M1 subtype or alternatively towards the M2 subtype, we further evaluated phenotype of IMP-derived macrophages in TRAMP-C1, TRAMP-C1scram-sh, and TRAMP-C1CRAMP-sh tumors by flow cytometry. Results showed that 30% and 38% of macrophages in TRAMP-C1 and TRAMP-C1scram-sh tumors, respectively, were polarized toward M2, while IMPs in TRAMP-C1CRAMP-sh tumors remained as IMPs rather than being differentiated/polarized toward macrophages (Figure 4C). In vitro culture of bone marrow-derived Gr-1+, CD11b+ IMPs (21) with conditioned media (CM) of TRAMP-C1 and TRAMP-C1CRAMP-sh and with CRAMP peptide further supported our in vivo data. After 4 days of culture, less number of IMPs in TRAMP-C1CRAMP-sh CM differentiated into macrophages, compared to TRAMP-C1 CM or M-CSF (Figure 4D). In addition, the number of IMP-derived macrophages presenting M2-phenotype was higher in TRAMP-C1 CM and M-CSF groups compared to TRAMP-C1CRAMP-sh CM group (Figure 4E). Altogether, the data indicate that CRAMP secreted by PCa cells facilitates not only differentiation of tumor-infiltrated IMPs to macrophages, but also polarization of macrophages toward protumorigenic M2.
PCa-derived CRAMP induces overexpression of M-CSF and MCP-1 through NF-kB and STAT3 activation by autocrine signaling
Next, we characterized the expression of genes, specifically M-CSF and MCP-1 that are critical for determining the fate of macrophage differentiation and polarization, regulated by CRAMP-mediated autocrine signaling in PCa cells. Knockdown of CRAMP gene in TRAMP-C1CRAMP-sh cells resulted in decreased phosphorylation of NF-kB p65 and STAT3 (Figure 5A & B) that are known to regulate MCP-1 and M-CSF gene expression (9, 22). Since STAT3 is known to play a critical role in protumorigenic events including M2 skewing (23, 24), we further analyzed the level of phospho-STAT3 depending on CRAMP stimulation and FPR2 inhibition. FPR2 blockade in TRAMP-C1 cells by WRW4 treatment resulted in decreased phospho-STAT3, while addition of CRAMP in TRAMP-C1CRAMP-sh cells displayed increased phospho-STAT3. We confirmed downregulation of M-CSF and MCP-1 mRNAs in TRAMP-C1CRAMP-sh cells (Figure 5E). Also, the inhibition of FPR2 in TRAMP-C1 cells resulted in downregulation of M-CSF and MCP-1 mRNAs (Figure 5F), whereas CRAMP-induced stimulation in TRAMP-C1CRAMP-sh cells increased M-CSF and MCP-1 mRNA levels (Figure 5G). Altogether, the results indicate that CRAMP regulates M-CSF and MCP-1 expression in PCa cells through p65 and STAT3 activation.
Figure 5. Cathelicidin-related antimicrobial peptide signals through p65 and STAT3 activation and up-regulates MCP-1 and M-CSF gene expression.
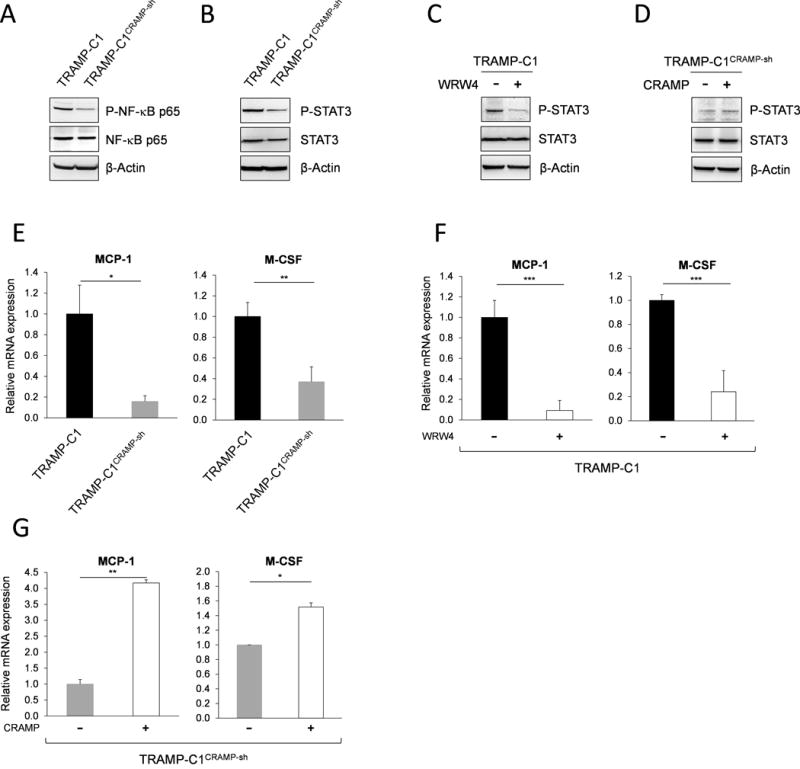
Immunoblotting representing decrease in phosphorylation of (A) NF-κB p65 and (B) STAT3 in TRAMP-C1CRAMP-sh cells compared to TRAMP-C1 cells. The expression of phospho-p65 and STAT3 was normalized to total p65 and STAT3. Immunoblotting showing the expression of phosphorylated STAT3 (C) in TRAMP-C1 cells with or without WRW4 (10 μg/mL) treatment and (D) in TRAMP-C1CRAMP-sh cells with or without CRAMP (5 μg/mL) treatment. (E) Real-time PCR presenting significant reduction of MCP-1 and M-CSF gene expression in TRAMP-C1CRAMP-sh cells compared to TRAMP-C1 cells. The mRNA levels of MCP-1 and M-CSF were normalized to GAPDH mRNA levels. Data are shown as mean ± SE of triplicates. *P < 0.05 **P < 0.01 (F) MCP-1 and M-CSF mRNA levels in TRAMP-C1 cells with or without blocking the activation of formyl peptide receptor 2 using WRW4 (10 μg/mL). Results are presented as means ± SE of triplicates. ***P < 0.001 (G) Real-time PCR showing significantly increased MCP-1 and M-CSF mRNA expression after addition of exogenous CRAMP (5 μg/mL) in TRAMP-C1CRAMP-sh cells compared to control. Data represent mean ± SE of triplicates. *P < 0.01 **P < 0.005
CRAMP upregulates FPR2 expression through autocrine signaling in PCa cells
CRAMP-mediated signal transduction is known to occur through FPR2. Our molecular analysis indicated that TRAMP-C1CRAMP-sh cells have significantly lower expression of FPR2, both in gene and protein levels (Figure 6A & B). When CRAMP was exogenously added to TRAMP-C1CRAMP-sh cells, FPR2 expression was restored to levels comparable to TRAMP-C1 cells (Figure 6C & D), indicating that FPR2 expression is regulated by autocrine mechanism.
Figure 6. Cathelicidin-related antimicrobial peptide regulates FPR2 expression in prostate cancer cells.
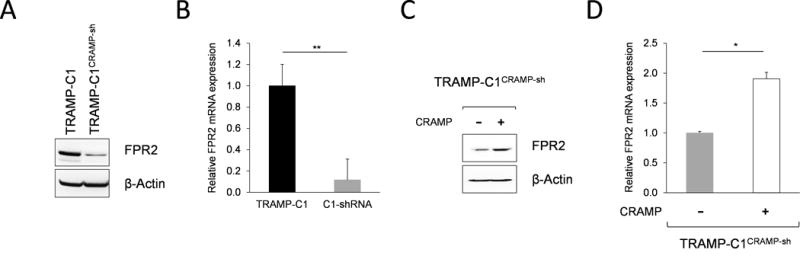
(A) Downregulation of FPR2 expression in TRAMP-C1CRAMP-sh cells was shown by western blot. The level of FPR2 was normalized to β-actin. (B) Real-time PCR showing significantly decreased FPR2 mRNA expression in TRAMP-C1CRAMP-sh cells compared to TRAMP-C1. Data is shown as mean ± SE of triplicates. **P < 0.005 (C) Increased expression of FPR2 in TRAMP-C1CRAMP-sh cells stimulated with exogenous CRAMP (5 μg/mL) was confirmed by western blot. FPR2 expression was normalized to β-actin. (D) Relative expression of FPR2 mRNA in TRAMP-C1CRAMP-sh cells with or without CRAMP (5 μg/mL) treatment was compared by quantitative RT-PCR. Data is presented as mean ± SE of triplicates. *P < 0.01
DISCUSSION
The current study underscores a critical protumorigenic role of CRAMP during PCa progression by showing that silencing CRAMP gene expression in TRAMP-C1 PCa cells significantly delays tumor growth in a syngeneic mouse model. Our results lend mechanistic support to previous correlative observations in clinical samples that higher LL-37 expression is associated or correlated with disease progression in breast, lung, and ovarian cancers (6–8). Li et al. proposed the role of CRAMP in promoting lung cancer development by highlighting the chemotactic action of immune cell-derived CRAMP that further enhances immune infiltration into TME (10). In contrast, our data from tumor challenge study with CRAMP-expressing TRAMP-C1 cells using Cnlp−/− mice exhibited comparable tumor growth between Cnlp−/− and WT mice, indicating that the levels of tumor-produced CRAMP are crucial and sufficient for modulating the chemotactic event in TME. More interestingly, analysis of tumor infiltrates showed significantly higher number of IMPs and a correspondingly lower number of macrophages in Cnlp−/− mice, as compared to WT mice. This suggests in part that CRAMP originated from tumor-infiltrating host immune cells, in addition to PCa cells, may have an additional role in modulating differentiation and polarization of myeloid cells towards M2 macrophages. The protumorigenic function of M2 is to facilitate angiogenesis and tissue remodeling for tumor metastasis. Functional characterization of CRAMP produced by innate immune effectors, thus, may be of great interest to elucidate the additional involvement of tumor-infiltrating host immune cells in promoting PCa progression.
The present study provides experimental evidence for the first time that PCa cell-produced CRAMP chemoattracts early myeloid population into TME and promotes their differentiation and polarization to M2 macrophages. Present study indicates that CRAMP derived from TRAMP-C1 PCa cells acts as a chemoattractant not only for mature myeloid cells, but also for Gr-1+ and CD11b+ cells, which are in the same myeloid lineage, known to support tumor growth by immunosuppression. Tumor challenge study using WT mice followed by immune profiling showed the infiltration of both mature myeloid cells, neutrophils and macrophages, and early myeloid IMPs, exhibiting comparable number as that of tumor-infiltrated neutrophils, into the TME. Our data demonstrate that PCa-derived CRAMP indirectly induces differentiation and polarization of tumor-infiltrated IMPs into tumor-promoting M2 population. CRAMP deficient mice challenged with CRAMP-expressing PCa cells exhibited higher number of IMPs in TME but lower number of macrophages as compared to WT. Analysis indicated that tumor-derived CRAMP regulates the expression of growth factors or cytokines, such as M-CSF and MCP-1, which are critical for differentiation of IMPs to macrophages and M2 polarization. In vitro study confirmed that differentiation of IMPs into M2 accompanies STAT3/6 signaling. CRAMP-overexpressing mouse PCa cell lines, TRAMP-C1 and TRAMP-C1scram-sh, which also overexpress FPR2, displayed elevated levels of M-CSF and MCP-1. Such protumorigenic stimulus is found to be mediated through phosphorylation of STAT3/6 eventually resulting in macrophages skewing toward M2-like phenotype through the transcription of genes that are typical of M2 polarization, such as ARG1 and CD206 (25–31).
It has been known that the migration of neutrophils and macrophages following the chemotactic gradient of CRAMP is mediated through CRAMP binding to its receptor FPR2 expressed on innate immune effectors (5). The expression of FPR2 in Gr-1+ and CD11b+ cells isolated from tumor-bearing host, in addition to neutrophils and macrophages (32–35), has been recently reported (36), indicating chemotactic response towards CRAMP-producing tumor cells. Interestingly, our study indicates that CRAMP can upregulate the expression of FPR2 in mouse PCa cell lines as a positive feedback loop, promoting autocrine signaling pathway. For STAT3 activation, FPR2 mediates transactivation of EGFR by activating membrane-bound MMPs which cleave EGF ligands that bind to EGFR (37). Given that downstream of EGFR pathway involves phosphorylation of STAT3 (38) and one of target genes of STAT3 is Cnlp (39), it is suggestive that the activation of STAT3 plays a critical role for the expression of CRAMP in PCa cells. Recent studies have shown that the overexpression of STAT3 promotes PCa metastasis, suggesting the role of STAT3 pathway during PCa progression (23, 24, 40). Considering that the expression of CRAMP is regulated by STAT3, our previous studies showing PCa progression to the advanced stage is linked to gradual increase of CRAMP expression (2) also highlights the pivotal role of STAT3 in PCa growth. However, molecular mechanisms underlying the facilitation of PCa growth by CRAMP remains to be elucidated. In this context, current study indicates that the overexpression of CRAMP in PCa cells is regulated by activation of STAT3, which also regulates MCP-1 and M-CSF. Altogether, results of the present study provide evidence of CRAMP as a protumorigenic peptide that induces immunomodulation, especially facilitating differentiation and polarization of IMPs to macrophages/M2, in a STAT3-dependent manner during PCa progression.
Chemotaxis of myeloid cells and angiogenesis during wound healing are antimicrobial-related roles of CRAMP. These CRAMP-mediated antimicrobial functions share fundamental biological features with cancer growth as our data have shown the increased chemotaxis of cells in the myeloid lineage by CRAMP during prostate cancer progression. Based on our data, it is more likely that the antimicrobial role of CRAMP eventually modulates the innate immune system facilitating tumor growth rather than two separate functions of CRAMP, depending on cancerous or non-cancerous microenvironment.
CONCLUSION
In conclusion, the present study demonstrates protumorigenic function of CRAMP via immune modulation, promoting disease progression. These data suggest that targeting LL-37, the CRAMP orthologue of humans, directly or indirectly by inhibiting STAT3 activation may have therapeutic implications against PCa progression. Therefore, further identification of CRAMP/LL37 signaling that promotes not only chemotaxis but also differentiation/polarization of innate immune effectors during the progression of human adenocarcinomas could be explored to identify a putative therapeutic target for PCa suppression. Targeted down-regulation of endogenous LL-37 in prostate tumors by approaches such as small molecular inhibitors, or targeted disruption by genetic approaches could prove to be efficacious in therapies based upon suppression of immunomodulation, angiogenesis, and metastasis induced by CRAMP. Furthermore, though this study addresses this point in a mouse model of prostate cancer, it is reasonable to postulate that the proposed use of targeted LL-37 therapy will be applicable to other cancers overexpressing this small yet potent peptide.
Acknowledgments
The assistance of Comprehensive Flow Cytometry Core at University of Alabama at Birmingham is gratefully acknowledged. We thank Dr. Richard Gallo, University of California at San Diego, for providing the Cnlp−/− mice.
Footnotes
Disclosure: There is no financial interest with any of the authors on data presented in this manuscript
AUTHOR CONTRIBUTIONS
H.C. and S.P. designed research; H.C., J.H.L., J.A.H., A.B.S., B.D.H. and C.M.L. performed research; H.C., J.H.L., and A.B.S. analyzed data; and H.C. and S.P. wrote the manuscript.
References
- 1.Sfanos KS, De Marzo AM. Prostate cancer and inflammation: the evidence. Histopathology. 2012;60:199–215. doi: 10.1111/j.1365-2559.2011.04033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hensel JA, Chanda D, Kumar S, Sawant A, Grizzle WE, Siegal GP, Ponnazhagan S. LL-37 as a therapeutic target for late stage prostate cancer. Prostate. 2011;71:659–70. doi: 10.1002/pros.21282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murakami M, Lopez-Garcia B, Braff M, Dorschner RA, Gallo RL. Postsecretory processing generates multiple cathelicidins for enhanced topical antimicrobial defense. J Immunol. 2004;172:3070–7. doi: 10.4049/jimmunol.172.5.3070. [DOI] [PubMed] [Google Scholar]
- 4.Mocsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med. 2013;210:1283–99. doi: 10.1084/jem.20122220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurosaka K, Chen Q, Yarovinsky F, Oppenheim JJ, Yang D. Mouse cathelin-related antimicrobial peptide chemoattracts leukocytes using formyl peptide receptor-like 1/mouse formyl peptide receptor-like 2 as the receptor and acts as an immune adjuvant. J Immunol. 2005;174:6257–65. doi: 10.4049/jimmunol.174.10.6257. [DOI] [PubMed] [Google Scholar]
- 6.Coffelt SB, Waterman RS, Florez L, Honer zu Bentrup K, Zwezdaryk KJ, Tomchuck SL, et al. Ovarian cancers overexpress the antimicrobial protein hCAP-18 and its derivative LL-37 increases ovarian cancer cell proliferation and invasion. Int J Cancer. 2008;122:1030–9. doi: 10.1002/ijc.23186. [DOI] [PubMed] [Google Scholar]
- 7.von Haussen J, Koczulla R, Shaykhiev R, Herr C, Pinkenburg O, Reimer D, Wiewrodt R, Biesterfeld S, Aigner A, Czubayko F, Bals R. The host defence peptide LL-37/hCAP-18 is a growth factor for lung cancer cells. Lung Cancer. 2008;59:12–23. doi: 10.1016/j.lungcan.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 8.Heilborn JD, Nilsson MF, Jimenez CI, Sandstedt B, Borregaard N, Tham E, Sørensen OE, Weber G, Ståhle M. Antimicrobial protein hCAP18/LL-37 is highly expressed in breast cancer and is a putative growth factor for epithelial cells. Int J Cancer. 2005;114:713–9. doi: 10.1002/ijc.20795. [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Cai L, Wang H, Wu P, Gu W, Chen Y, Hao H, Tang K, Yi P, Liu M, Miao S, Ye D. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene. 2011;30:3887–99. doi: 10.1038/onc.2011.112. [DOI] [PubMed] [Google Scholar]
- 10.Li D, Beisswenger C, Herr C, Schmid RM, Gallo RL, Han G, Zakharkina T, Bals R. Expression of the antimicrobial peptide cathelicidin in myeloid cells is required for lung tumor growth. Oncogene. 2014;33:2709–16. doi: 10.1038/onc.2013.248. [DOI] [PubMed] [Google Scholar]
- 11.Coffelt SB, Marini FC, Watson K, Zwezdaryk KJ, Dembinski JL, LaMarca HL, Tomchuck SL, Honer zu Bentrup K, Danka ES, Henkle SL, Scandurro AB. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc Natl Acad Sci U S A. 2009;106:3806–11. doi: 10.1073/pnas.0900244106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Chen K, Wang C, Gong W, Yoshimura T, Liu M, Wang JM. Cell surface receptor FPR2 promotes antitumor host defense by limiting M2 polarization of macrophages. Cancer Res. 2013;73:550–60. doi: 10.1158/0008-5472.CAN-12-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer. 2013;13:739–52. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wynn TA. Myeloid-cell differentiation redefined in cancer. Nat Immunol. 2013;14:197–9. doi: 10.1038/ni.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207:2439–53. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madsen DH, Leonard D, Masedunskas A, Moyer A, Jurgensen HJ, Peters DE, Amornphimoltham P, Selvaraj A, Yamada SS, Brenner DA, Burgdorf S, Engelholm LH, Behrendt N, Holmbeck K, Weigert R, Bugge TH. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol. 2013;202:951–66. doi: 10.1083/jcb.201301081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–73. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 19.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181:5791–802. doi: 10.4049/jimmunol.181.8.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bae YS, Lee HY, Jo EJ, Kim JI, Kang HK, Ye RD, Kwak JY, Ryu SH. Identification of peptides that antagonize formyl peptide receptor-like 1-mediated signaling. J Immunol. 2004;173:607–14. doi: 10.4049/jimmunol.173.1.607. [DOI] [PubMed] [Google Scholar]
- 21.Yu H, Liu Y, McFarland BC, Deshane JS, Hurst DR, Ponnazhagan S, Benveniste EN, Qin H. SOCS3 Deficiency in Myeloid Cells Promotes Tumor Development: Involvement of STAT3 Activation and Myeloid-Derived Suppressor Cells. Cancer Immunol Res. 2015;3:727–40. doi: 10.1158/2326-6066.CIR-15-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang XC, Jobin C, Allen JB, Roberts WL, Jaffe GJ. Suppression of NF-kappaB-dependent proinflammatory gene expression in human RPE cells by a proteasome inhibitor. Invest Ophthalmol Vis Sci. 1999;40:477–86. [PubMed] [Google Scholar]
- 23.Abdulghani J, Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, Lisanti MP, Zellweger T, Alanen K, Mirtti T, Visakorpi T, Bubendorf L, Nevalainen MT. Stat3 promotes metastatic progression of prostate cancer. Am J Pathol. 2008;172:1717–28. doi: 10.2353/ajpath.2008.071054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14:736–46. doi: 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- 25.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–95. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117:1155–66. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Junttila IS, Mizukami K, Dickensheets H, Meier-Schellersheim M, Yamane H, Donnelly RP, Paul WE. Tuning sensitivity to IL-4 and IL-13: differential expression of IL-4Ralpha, IL-13Ralpha1, and gammac regulates relative cytokine sensitivity. J Exp Med. 2008;205:2595–608. doi: 10.1084/jem.20080452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 29.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–96. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 30.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 31.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–61. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 32.Agerberth B, Charo J, Werr J, Olsson B, Idali F, Lindbom L, Kiessling R, Jörnvall H, Wigzell H, Gudmundsson GH. The human antimicrobial and chemotactic peptides LL-37 and alpha-defensins are expressed by specific lymphocyte and monocyte populations. Blood. 2000;96:3086–93. [PubMed] [Google Scholar]
- 33.Devosse T, Guillabert A, D’Haene N, Berton A, De Nadai P, Noel S, Brait M, Franssen JD, Sozzani S, Salmon I, Parmentier M. Formyl peptide receptor-like 2 is expressed and functional in plasmacytoid dendritic cells, tissue-specific macrophage subpopulations, and eosinophils. J Immunol. 2009;182:4974–84. doi: 10.4049/jimmunol.0803128. [DOI] [PubMed] [Google Scholar]
- 34.Liu M, Chen K, Yoshimura T, Liu Y, Gong W, Wang A, Gao JL, Murphy PM, Wang JM. Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci Rep. 2012;2:786. doi: 10.1038/srep00786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang D, Chen Q, Le Y, Wang JM, Oppenheim JJ. Differential regulation of formyl peptide receptor-like 1 expression during the differentiation of monocytes to dendritic cells and macrophages. J Immunol. 2001;166:4092–8. doi: 10.4049/jimmunol.166.6.4092. [DOI] [PubMed] [Google Scholar]
- 36.Lee JM, Kim EK, Seo H, Jeon I, Chae MJ, Park YJ, Song B, Kim YS, Kim YJ, Ko HJ, Kang CY. Serum amyloid A3 exacerbates cancer by enhancing the suppressive capacity of myeloid-derived suppressor cells via TLR2-dependent STAT3 activation. Eur J Immunol. 2014;44:1672–84. doi: 10.1002/eji.201343867. [DOI] [PubMed] [Google Scholar]
- 37.George AJ, Hannan RD, Thomas WG. Unravelling the molecular complexity of GPCR-mediated EGFR transactivation using functional genomics approaches. FEBS J. 2013;280:5258–68. doi: 10.1111/febs.12509. [DOI] [PubMed] [Google Scholar]
- 38.Sen M, Joyce S, Panahandeh M, Li C, Thomas SM, Maxwell J, Wang L, Gooding WE, Johnson DE, Grandis JR. Targeting Stat3 abrogates EGFR inhibitor resistance in cancer. Clin Cancer Res. 2012;18:4986–96. doi: 10.1158/1078-0432.CCR-12-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gudmundsson GH, Agerberth B, Odeberg J, Bergman T, Olsson B, Salcedo R. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur J Biochem. 1996;238:325–32. doi: 10.1111/j.1432-1033.1996.0325z.x. [DOI] [PubMed] [Google Scholar]
- 40.Kroon P, Berry PA, Stower MJ, Rodrigues G, Mann VM, Simms M, Bhasin D, Chettiar S, Li C, Li PK, Maitland NJ, Collins AT. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 2013;73:5288–98. doi: 10.1158/0008-5472.CAN-13-0874. [DOI] [PubMed] [Google Scholar]