

Abstract
Since the 1960’s, stimuli-responsive polymers have been utilized as functional soft materials for biological applications such as the triggered-release delivery of biologically active cargos. Over the same period, liposomes have been explored as an alternative drug delivery system with potentials to decrease the toxic side effects often associated with conventional small-molecule drugs. However, the lack of drug-release triggers and the instability of bare liposomes often limit their practical applications, causing short circulation time and low therapeutic efficacy. This perspective article highlights recent work in integrating these two materials together to achieve a targetable, triggerable nanoscale platform that fulfills all the characteristics of a near-ideal drug delivery system. Through a drop-in, post-synthesis modification strategy, a network of stimuli-responsive polymers can be integrated onto the surface of liposomes to form polymer-caged nanobins, a multifunctional nanoscale delivery platform that allows for multi-drug loading, targeted delivery, triggered drug-release, and theranostic capabilities.
Keywords: stimuli-responsive polymer gels, triggered release, drug delivery, liposomes
Graphical Abstract
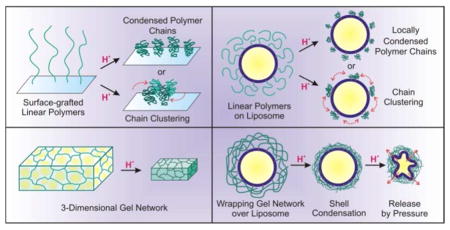
I. Introduction
Highly complex biological organisms have evolved to recognize external stimuli from the surrounding environment and efficiently respond to them either by secreting chemicals that trigger biological responses or by changing their shapes or colors.1 Such stimuli-induced responses are often based on subtle changes in the biomaterials that made up parts of those organisms, the details of which are only recently understood through the concerted efforts of many scientists. In modern materials science, the development of stimuli-responsive materials has also figured prominently, not only in the construction of model systems for understanding how biological materials undergo triggered-response behaviors but also in our desire to apply this knowledge to design new materials with ‘smart’ stimuli-responsive functionalities. Amongst the known stimuli-responsive materials, polymer-based systems are the most promising because they can be made in large scales and with a wide range of chemical functionalities, modified post-synthetically in a facile manner, and processed into materials in many different forms (surface patterns, films, solutions, or solids). Indeed, polymers can be engineered to change their molecular conformations in response to external stimuli such as pH, temperature, redox potential, light, and magnetic field, resulting in changes in density, transparency, conductivity, volume (or degree of swelling), or solvent-uptake capacity.2,3 Polymers that change their volumes, density, or other structural/mechanical properties in response to chemical signals from the external environment are classified as chemomechanical materials and have been employed as “smart” materials in numerous applications.4,5
Among the many chemomechanical polymeric materials that have been reported to date, those that form reversible hydrogels in water or protic polar solvents have been the most explored.6 While such polymers can be soluble and stable in water under one set of conditions, they can quickly undergo a phase transition to a gel in response to external stimuli such as changes in pH or temperature, which triggers conformational changes in the polymer chains.7 Generally, these chemomechanical responses are quite rapid due to the highly cooperative behaviors of adjacent polymer chains,8 leading to large volume changes under a narrow range of conditions. This property has led to the application of hydrogels as macroscopic platforms for the localized delivery of a wide range of drugs, proteins, and nucleic acids.6,9 More recently, efforts have focused on the development of nanoscale hydrogels for the systemic delivery of small-molecule drugs and biological polymers such as oligonucleotides and proteins.7,10 This is driven by the quest to improve the pharmacokinetic profiles of the active payload,11 either by enhancing the site-specific treatment of refractory diseases (i.e., diseases that resist conventional treatments), or by reducing the side effects of highly toxic drugs. However, the incorporated drug molecules can often modify the physical property of the hydrogel matrix, especially at high loadings, thus limiting the optimal structure of the final drug-loaded gel materials for pharmaceutical applications. As such, hydrogels are not compatible with many of the desirable payloads and a notable strategy for overcoming this limitation is to integrate stimuli-responsive hydrogels with a well-defined second carrier that can accommodate many of such payloads.
Liposomal vesicles, which comprise a bilayer of amphiphilic lipid molecules encapsulating an aqueous core, have been one of the most popular nanoscale delivery platforms due to their ease of synthesis and excellent biocompatibility.12,13 The membrane of liposomes can be easily modified by the incorporation of small molecules14,15 or synthetic polymers12,15,16 to enhance their physicochemical properties such as mechanical stability, chemical reactivity for surface modification, and triggered responses for cargo-release. Herein, we will discuss the integration of a chemoresponsive polymer network onto the surface of a liposomal vesicle to afford a novel biochemical stimuli-sensitive nanoscale platform for the delivery of small-molecule drugs. Readers who are interested in chemoresponsive polymeric delivery systems should consult the excellent reviews by the Lyon,7 Langer,9 and Kabanov10 groups.
II. A brief background on environmentally responsive polymer systems
Environmentally responsive linear polymers
The reversible conformational changes (i.e., the basis for macroscopic phase transitions) that stimuli-sensitive polymers exhibit in response to external conditions such as temperature, pH, and solvent composition is governed by the entropy of mixing of polymers in solvents, according to Flory-Huggins theory.17 The earliest studies of conformational changes exhibited by thermosensitive polymers were conducted in the late 1960s, when temperature-dependent aggregations were observed for aqueous solution of poly[N-isopropylacrylamide], poly(NIPAAm).18,19 Such behavior occurs via a reversible coil-to-globular transition of the poly(NIPAAm) chain across its lower critical solution temperature (LCST = 32 °C) in water.20–22 At temperatures below this LCST, hydrogen bonding exists between water molecules and the acrylamide (AAm) groups of poly(NIPAAm), enhancing the solubilization of the polymer chains in water. This hydrogen-bond network is rapidly disrupted at temperatures above the LCST, inducing an entropically favored release of water molecules out of the polymer chains (Figure 1A). At this stage, interactions among the polymer chains become more dominant, resulting in a flocculation of insoluble globular polymer aggregates. When the poly(NIPAAm) chains are grafted onto a laterally fluidic surface such as a lipid bilayer membrane, aggregated clusters can form to reduce the total surface energy (Figure 1B).23 However, because the LCST of poly(NIPAAm) is below the average human body temperature (37 °C), its structure must be further tuned to increase its LCST to a range that is compatible with most applications in human health.
Figure 1.

(A) A schematic illustration of the reversible thermosensitive transition of poly(NIPAAm) (dark green lines) between the random coil and collapsed globule conformations, governed by entropy-driven interactions with water molecules (light yellow dots). The red dashed lines in the inset indicate possible hydrogen bonding between water molecules and the substituents of the polymer chain. (B) A schematic illustration of the process in which grafted linear poly(NIPAAm) chains on a laterally fluidic surface can condense in the presence of an external stimuli and further aggregate to form clusters.
To raise the LCST of poly(NIPAAm) above the body temperature, water-soluble units such as acrylic acid (AAc) or methacrylate (MAA) groups have been incorporated into the poly(acrylamide) (poly(AAm)) backbone. Both of these groups can increase the LCST of poly(AAm) by increasing the overall hydrophilicity of the polymer chain,24,25 turning it into a good candidate for a drug-delivery vector that can be controlled by external stimuli. More interestingly, the presence of the AAc groups leads to an additional sensitivity to acid that can be used as a second trigger: lowering the pH can lead to enhanced polymer-polymer interactions and lower aqueous solubility.24
Environmentally responsive polymer networks
For biomedical applications such as controlled drug-release, LCST-possessing polymers such as poly(AAm) are often cross-linked to form macroscopic gel-like networks whose structural integrity can be maintained against the dissociation of individual polymer chains during the reversible solvation/desolvation process across the LCST. The resulting cross-linked polymer gel networks can absorb a large amount of water molecules and behave like a swollen semi-solid with good fluidity and hydrophilicity. Additionally, because they are made from environmentally responsive linear polymers, the macroscopic swelling/deswelling process can be controlled using other external stimuli or a combination of stimuli.26 For example, volume shrinkage has been observed for the cross-linked network of partially hydrolyzed poly(AAm) upon acidification, where the degree of cross-linking and the number of AAc groups can both be used to tune the final swelling property of the polymer gels.27 Such acid-triggered swelling property of cross-linked gel in aqueous solution has led to their use in the development of biocompatible smart materials for the sustained delivery of drugs through the gastrointestinal tracts.9
II-1. Macroscopic cross-linked polymer gels with reversible swelling
Because cross-linked polymer gels possess the properties of both solids and fluids, they have structural integrity approaching those of solid materials while still retain a large number of solvent molecules (up to 99 wt % solvent) inside a low-density network. This high porosity allows for the rapid transport of small guest molecules in and out of the network by diffusion and facilitates reversible volume changes (Figure 2A). Based on the nature of the cross-links between polymer chains in solution, polymer gels are classified as having either non-covalent, physical cross-links or covalent, chemical cross-links.28
Figure 2.

(A) A schematic illustration of the reversible swelling of a 3-dimensional cross-linked hydrogel network. Upon a change in temperature or pH, the water molecules embedded in the swollen hydrogel on the left are expelled out of the gel and the networks collapse. (B) A schematic illustration of the non-covalent physical cross-linking between poly(ethylene glycol) and poly(methacrylate) in a solution: as the acidity of the solution increases, hydrogen bonding occurs between the oxyethylene groups of one polymer and the protonated methacrylic acids of the other polymer, leading to gel formation.
In physically cross-linked gels, non-covalent molecular interactions such as hydrophobic interactions, hydrogen bonding, and ionic interactions typically hold the gel network together and can be reversibly manipulated under specific conditions. For example, poly(ethylene glycol) and poly(methacrylic acid) can form an interpenetrating gel network in acidic solution, where the polymer chains are physically cross-linked via hydrogen bonding between the oxyethylene groups and the protonated methacrylic acids (Figure 2B).29 In the same manner, acid-induced changes has also been observed for poly[(methacrylic acid)-g-(ethylene glycol)], a poly(methacrylate) with grafted poly(ethylene glycol) side chains.30,31 This graft copolymer, with an equimolar ratio of poly(ethylene oxide)/methacrylic acid repeating units, forms a swollen gel structure at pH = 9.0 due to the enhanced solubilization of deionized methacrylate groups, which absorbs a large amount of water molecules.31 However, it contracts when acidified to a pH of 4.0 and releases a significant amount of water. The degree of cross-linking in physically cross-linked gels varies greatly depending on the acidity of the solution, its ionic characteristics, its temperature, as well as the concentrations of the two polymers. Readers interested in such hydrogel systems can consult the recent perspective article by Lee and coworkers.32
In contrast to their physically cross-linked counterparts, chemically cross-linked gels are more robust because the polymer chains are interconnected through strong covalent bonds. During the synthesis of these hydrogels, the degree of cross-linking can be precisely controlled to tune the porosity (i.e., void volume) of the networks and allow for the regulation of guest-molecule release kinetics.6 As an example, the release rates of small-molecule cargos from cross-linked poly(AAm) hydrogels, which have been investigated for therapeutic applications since the 1980s, can be finely controlled by cross-linking density and temperature.33 Generally, higher degrees of cross-linking in the polymer gels corresponds to smaller pore sizes and slower release rates of the embedded guest molecules.
In addition to the degree of cross-linking, the release behavior of a chemically cross-linked polymer gel network is heavily influenced by its LCST. Below the LCST, the swollen polymer gel network exhibited simple first-order release kinetics, where the release of guest molecules is simply governed by diffusion (Figure 3A). Above the LCST, on the other hand, the release profile is a two-step process: an initial burst of guest molecules from the rapidly shrinking gel surface, followed by a much-slower release from the inner part of gels (Figure 3B). Such a two-step release profile can be attributed to a temperature gradient that initially exists through the bulk of the gel: when a gel is exposed to a temperature above the LCST, the outer network quickly densifies, causing an initial burst, after which the whole network equilibrates and the release rate from deeper within the gel becomes much more attenuated and sustained. Until the late 1990s, these macroscopic gels have been mostly employed as devices for localized drug-delivery or tissue engineering,9 where their ability to sustain drug release over an extended duration is highly desirable. In recent years, the nanoscale forms of these gels have begun to attract much attention as a platform for the systemic delivery of biologically active agents, where they can enhance the pharmacokinetic profiles and bioavailability of these agents, produce better drug efficacy, and reduce side effects.10
Figure 3.

Temperature-dependent drug-release kinetics from chemically cross-linked thermosensitive hydrogel.34 (A) At temperatures below the LCST of the hydrogel, drug molecules are released from the swollen gel by simple diffusion process. (B) At temperatures above the LCST, rapid release of drug molecules occurs by the initial shrinkage of the hydrogel, followed by slow release of drugs from the condensed structure, showing two different pharmacokinetic profiles.
II-2. Micro- and nanoscale hydrogel particles
During the last two decades, numerous techniques–precipitation polymerization, polymerization in micro- and nanoscale reactors, or post-polymerization self-assembly processes–have been developed to produce stimuli-responsive polymer materials, both single-chain polymers and cross-linked hydrogels, that possess micro- and nanoscale particle sizes or structural domains.7,10 For hydrogels, a major advantage in converting them into micro- and nano-structured nanoparticles, henceforth referred to as micro/nanogel particles, is the ability to form colloidal solutions that are compatible with biological circulation and can thus be systemically delivered to hard-to-reach organs, tissues, and cells. A second major advantage is the faster responses of micro/nanogel particles to external stimuli compared to bulk hydrogels, allowing for rapid pharmacokinetic actions. Thirdly, core/shell-type micro/nanogel particles,35 which comprise two or more polymer materials in a single system, can exhibit distinctive response/release characteristics, arising from the combination of individual component’s inherent property. These integrated systems then allow for the fine tuning of combined properties such as multimodal/multistep responses to external stimuli. For example, a core/shell-type microgel (~60–150 nm) composed of two polymers with different LCSTs, poly(NIPAAm) as the core and poly(N-isopropylmethacrylamide) as the shell, can exhibit multiple swelling phases: fully collapsed, partially collapsed, and fully swollen phases, depending on the temperature (Figure 4A).36
Figure 4.

(A) Three different swelling phases of a core/shell-type microgel particle composed of poly(NIPAAm) as the core and poly(N-isopropylmethacrylamide) as the shell of the particle.36 (B) Reversible formation of nanogel particles via thermosensitive micellization.37
Another approach for constructing core/shell-type nanogel particles is based on the self-assembly of amphiphilic block-copolymers, containing both hydrophilic and hydrophobic blocks in a single polymer chain, into micelle-like structures. In this case, the block-copolymer chains behave as amphiphilic macro-surfactants that can aggregate into micelles at concentration above the critical micelle concentration (CMC).16 One example of this is poly[(ethylene glycol)-b-(NIPAAm)], which forms micelle-like large nanoparticles (100–200 nm) at temperatures above the LCST.37 As explained above, these polymer chains are highly miscible with water at temperatures below the LCST, while being aggregated out of the solution at temperatures above the LCST. Therefore, at concentrations above the CMC and temperatures above the LCST, these polymer chains can self-assemble into micelle structure with the hydrophobic poly(NIPAAm) block at the core and the hydrophilic poly(ethylene glycol) on the shell. This micellization behavior is reversible and the micelle can fall apart when the temperature decreases below the LCST (Figure 4B) or when the polymer concentration falls below the CMC. Such disruption can be prevented by cross-linking as shown by several research groups,36–38 the most comprehensive body of work is perhaps by Wooley and coworker, as summarized below.39
The Wooley group has extensively studied the synthesis and application of a block-copolymer-derived nanogel platform called shell cross-linked knedels (SCK).39 By varying the composition and block length of amphiphilic copolymer chains, various nanoscale structures with unique morphology can be achieved via self-assembly.40 For example, either poly(acrylic acid)-b-polystyrene41 or poly(acrylic acid)-b-poly(octadecylacrylate)42 can be assembled into micelle-like nanoparticles (~10–50 nm) with poly(acryclic acid) blocks on the shell. These surface acid groups can then be cross-linked with amide bonds to form stable SCKs that do not fall apart when the polymer concentration falls below the CMC. The core and shell of the SCKs derived from poly(acrylic acid)-b-poly(octadecylacrylate)42 were then further modified with a wide range of imaging and therapeutic agents. Notably, the facilitated release of guest molecules such as doxorubicin, an anticancer drug, from these SCKs could be achieved on the benchtop under physiologically relevant conditions, suggesting its promising potential as a delivery system for therapeutics. This enhanced drug-release was attributed to the change of the crystalline, doxorubicin-incorporated core of the SCK into a molten phase at body temperature, facilitating drug-release. Together with the relatively high loading capacity of the SCK (15–20 wt %), this enhanced release property constitutes one of the first examples where external stimuli such as temperature can be used to effect a potentially biologically beneficial response in a nanogel particle.
II-3. Insertion of stimuli-responsive lipids and non-cross-linked linear copolymers into the liposomal membrane
Since the mid 1960s, when artificial vesicles of amphiphilic phospholipids were first introduced,43 liposomes have been a popular vehicle for the delivery of a wide range of biological cargos12 such as small-molecule pharmaceuticals, macromolecular DNA, and small interfering RNA for various therapeutic purposes. Part of this popularity stems from the excellent biocompatibility of lipids and the large volume of aqueous core inside the liposomal vesicle (~3 μL/μmol lipids for a 100 nm particle),44 which allows for a relatively high cargo-loading capacity (~ 12.5 wt % of doxorubicin for a 100 nm particle). The recognition that therapeutic applications would benefit from a programmed release of the encapsulated cargos at specific target sites to achieve the best drug efficacy has driven research efforts in recent decades into the engineering of liposomes possessing drug-release triggers that can respond to tissue-specific stimuli.12,45 For example, the incorporation of acid-sensitive lipids such as phosphatidylethanolamine into the membrane of a liposome can lead to a pH-dependent lamellar-to-hexagonal liquid crystalline phase transition46 that releases the payload from the core in acidic environments, such as those encountered in solid cancer47,48 or late endosomes.49 Acid-degradable synthetic lipids can also be integrated into the membrane of the liposomes, leading to a facile degradation of these vesicles in mild acidic conditions and induce the release of their cargos.50 Although such ‘intrinsically’ pH-sensitive liposomes have been used successfully as prototypical drug-delivery platforms,51,52 the acid-degradable lipid components can lose their functions through lipid-exchange with biological membranes,53 resulting in cargo leakage during circulation and rendering these pH-sensitive liposomes ineffective as drug delivery vehicles.
In an alternative approach, liposomes have been post-synthetically modified with chemomechanically responsive polymers that are highly susceptible to specific external stimuli of the surrounding environments such as pH, temperature, lights, and redox potential.12,45 While such modification was primarily based on the insertion of the polymers into the liposome membrane after liposome formation, the grafted polymer chains can associate tightly with each other or with the lipid membrane through either hydrogen bonding54,55 or van der Waals interactions,55,56 upon changes in external conditions such as pH fluctuations or temperature increases above the LCST of the polymer component. This eventually destabilizes the highly ordered bilayer structure of the lipid membrane57 and leads to premature release of the payload. In one example, a pH-sensitive random copolymer of NIPAAm and MAA with hydrophobic octadecyl anchors is inserted into the membrane of a 100 nm liposome comprising a 3:2 molar ratio of egg phosphatidylcholine (EPC):cholesterol.58 In the presence of acid, or when the external temperature is increased above the LCST, these inserted MAA-containing copolymer chains condenses into clusters on the membrane surface, leading to the formation of transient pores57 that cause premature release of the encapsulated drug molecules (Figure 5A). Another problem that has been observed in these polymer-decorated liposomes is the high water solubility of the modified polymers, which facilitate their dissociation from the membrane, especially when the anchoring is through a single functional group.59,60
Figure 5.

(A) A schematic illustration of the grafting of polymer chains onto a lipid bilayer membrane: the terminal hydrophobic anchor (purple wiggles) of the polymers is the only part that is initially associated with the inner region of the bilayer through hydrophobic interactions. Upon acidification or external temperature increases, the volume of the polymer chain quickly decreases and the polymer chains either form clusters or associate tighter with the membranes, forming transient pores on the membrane. Encapsulated drug molecules (red triangles) can then be easily released through these pores into the aqueous surrounding media (water = green spheroids). (B) A schematic representation of how a multi-anchored copolymer can be grafted onto a lipid bilayer membrane through multiple hydrophobic interactions. (C) A schematic representation of the formation of a PEGylated lipid bilayer membrane that have been cross-linked via redox- or photo-induced reaction: the polymerizable lipid molecules (the polymerizable groups are shown as the small green “wiggles” in the lipid bilayers in the first panel) and the PEGylated lipids segregate, creates pore that can then allow for payload release. (D) A schematic representation of how polymer chains with single hydrophobic anchor can be cross-linked to form the shell of the polymer-caged nanobin described in section III. An external trigger can then induce the collapse of the polymer shell, releasing the encapsulated drugs.
Among the many attempts to endow liposome with stimuli-responsive properties, perhaps the most promising strategy is the insertion of stimuli-responsive copolymers possessing randomly distributed multiple hydrophobic anchor groups, which can provide greater binding affinity of polymer chains to the liposomal membrane56 and overcome the instability of the conventional lipid-only systems. In addition, the insertion of copolymers with randomly distributed multi-anchors into the membranes (Figure 5B) of drug-loaded liposomes can induce higher cargo-release rates compared to those that incorporate polymer chains with a single hydrophobic anchor at the terminal end,58 presumably due to the enhanced interpenetration of the multi-anchored polymers into membrane through increased hydrophobic interaction. Unfortunately, uncontrolled aggregation of liposome particles has been observed for these systems due to the potential of multi-anchored polymers to bridge several liposome particles during the polymer-insertion process.58,61
In an alternate strategy, polymerizable lipid amphiphiles, such as bis(sorbyl) phosphatidylcholine, have also been incorporated into sterically stabilized PEGylated liposomes and used to trigger payload release.62 Upon either photo- or redox-initiated intra-membrane cross-linking, significant leakage of payloads can be observed, attributable to a phase separation between the cross-linked lipids and the PEGylated lipids (Figure 5C).63 This hypothesis is supported by a follow-up report that when such polymerizable lipids are integrated into the membranes of simple, non-PEGylated liposomes, the subsequent cross-linking process actually leads to significant enhancements in mechanical stabilities for the liposomes and severely limit the release of encapsulated payloads.64 These observations prompted us to hypothesize that incorporating a cross-linked polymer network around a liposomal template would also lead to similarly sterically stabilized liposomes (Figure 5D). However, if such a network can then be triggered to condense/phase separate, cargo-release should be possible.
III. “Wrapping” a cross-linked 3-dimensional polymer network around a liposome: polymer-caged nanobin (PCN)
During the last two decades, several research groups have attempted to combine stimuli-sensitive polymers and liposomes into delivery systems with good biocompatibility, high drug-loading capacity, robust stability, and stimuli-sensitive cargo-release property.45,65,66 Building upon the knowledge from these studies, we integrated a thin hydrogel network over the surface of a liposome particle to develop an acid-sensitive liposome-polymer gel hybrid drug delivery platform that we named polymer-caged nanobin (PCN). In this system, cholesterol-terminated poly(acrylic acid) (Chol-PAAc) chains are immobilized into the bilayer membrane of a drug-loaded liposome (~ 100 nm) through hydrophobic cholesterol end-groups and then cross-linked with short telechelic diamine linkers to form a cage of poly[(acrylic acid)-co-(acrylamide) (poly[(AAc)-co-(AAm)]) around the liposome (Figure 6).67 When incorporated into liposome at a small amount (~10 mol% of the lipid content), this polymer additive only results in a small increase in the total mass of the vesicle and still preserves the overall biocompatible nature of liposome template. However, the final cross-linked poly[(AAc)-co-(AAm)] network greatly stabilized the liposome underneath to the extent that the PCN can be freeze-dried and rehydrated without aggregation.67 In addition, this network behaves as a nanoscale hydrogel layer that can undergo an acid-sensitive volume change that triggers drug-release by “mechanically squeezing” the drug-loaded liposome core (Figure 7A). More importantly, empty PCNs do not have an intrinsic in vitro toxicity over a broad range of therapeutically relevant concentrations,68 consistent with the observed biocompatibility of poly[(AAc)-co-(AAm)].6
Figure 6.

Fabrication scheme for polymer-caged nanobin (PCN) via a modular drop-in strategy. The alkyne-containing diamine cross-linker is shown in the second step to illustrate how a PCN can be made with both surface terminal alkyne and carboxylic acid groups available for orthogonal modifications via click chemistry (see Figure 8 and the associated discussion below). However, simple diamine cross-linkers can be used.
Figure 7.

Proposed release mechanisms of encapsulated drug molecules D from our PCN and Needham’s lipid-coated, drug-loaded microgel. (A) In PCN, protonation of the substituents in the cross-linked polymer shell induces intramolecular hydrogen bonding and densification that “squeeze” the lipid membrane, leading to an eventual release of drug molecules from the core. (B) In a drug-loaded microgel, the swollen gel particle becomes densified under acidic condition and is trapped in this form by a lipid coating that inhibits the reversed swelling. Subsequently, the lipid membrane can be ruptured by electroporation while under neutral pH, leading to a gel swelling that can release the trapped drug molecules.
Physicochemical advantages of the cross-linked polymer shell in PCN
Cross-linking the surface-embedded Chol-PAAc chains with telechelic diamine linkers overcome several challenges encountered in early polymer-modified liposome systems. First, cross-linking all of the available physically grafted polymer chains into one network prevents their dissociation from the liposomal membrane,60 a problem known to return early polymer-modified liposomes to ‘bare-vesicle’ states that readily leak cargos.69 Second, while the final cross-linked network may appear to be similar in topology to the multi-anchored copolymer-stabilized system described in section II-3 (Figures 5B and D), the post-insertion cross-linking of the end-anchored polymer chains is more advantageous from a synthetic point of view in that it completely eliminates the aforementioned inter-particle bridging problem associated with the insertion of multi-anchored polymers.61 Third, at a high enough cross-linking density, an extensive cross-linked network is created that completely extends over the whole surface of the liposome as a polymer shell, sterically prevents the undesirable aggregation of liposome particles and greatly enhances their stability to the extent that they will survive a freeze-and-thaw (lyophilization) process.67 Indeed, PCN was found to have a highly stable particle-size distribution68 and highly enhanced serum stability compared to its non-cross-linked polymer-grafted liposome (PGL) precursor.67
Cross-linking the Chol-PAAc chains with telechelic diamine linkers provides a fourth, additional advantage by converting PAAc into a gel-like network of random poly[(AAc)-co-(AAm)], which is pH- and thermo-responsive as shown in section II, that surrounds the liposome core. As such, PCN demonstrates pH- and temperature-responsive volume changes70 that are similar to those exhibited by the aforementioned mentioned poly(NIPAAm)-based core/shell-type hydrogel particles. These behaviors can then be exploited as stimuli-sensitive triggers for the release of cargo molecules from the drug-loaded liposome core. Specifically, the release rate of doxorubicin, a popular anticancer drug, was greatly enhanced for PCN compared to the rates observed for either the bare liposome or the non-cross-linked PGL precursor as the pH of the external solution decreased.68 Fast release of the encapsulated drug (t1/2 = 11.4 h) was achieved for PCN with a 50% cross-linked poly[(AAc)-co-(AAm)] shell in pH 5 solution, in stark contrast to the drug-loaded bare liposome and the non-cross-linked PGL, both of which released only a minimal amount of the drug over a two-day period.68 This pH-sensitive release is attributed to the acid-mediated densification of the cross-linked polymer cage, similar to that underlining the acid-induced volume change of hydrogels shown in section II-2. This polymer shell densification can generate a three-dimensional inward-focus pressure that leads to the irreversible rupture of the lipid bilayer membrane of the liposome core and release the encapsulated drug molecules (Figure 7A).70
The aforementioned ‘cage-densification and membrane-rupture’ mechanism of PCN is supported by the release of both NiII and AsIII from PCN loaded with ionic cargos such as [NiIIHAsIIIO3]n, an anticancer agent known to be active against acute promyelocytic leukemia.71 Under acidic condition, it is known that [NiIIHAsIIIO3]n-loaded bare liposomes preferentially release AsIII ions and not NiII:72 the former can easily pass through the lipid bilayer membrane under the form of amphiphilic arsenous acid (H3AsO3) while the less-lipophilic aqueous NiII ions is retained inside the vesicles. In contrast, [NiIIHAsIIIO3]n-loaded PCNs release both NiII and AsIII, a characteristic that has been attributed to the complete collapse of the cross-linked polymer shell under acidic condition.70 Such a scenario is supported by computational modeling studies that show a large shrinkage in the volume of the random poly[(AAc)-co-(AAm)] network upon acidification.70 Together, these results show that the drug-release profile of traditional liposome can be completely altered by the PCN polymer cage: as a facile drop-in modifier for the liposome platform, it serves as an excellent acid-sensitive trigger for controlled drug-release.
We note that Needham and coworkers have reported a lipid-coated microgel whose structure is the reverse of our PCN’s structure.73 Poly(methacrylate) microgel particles (~6.5 μm) were prepared via precipitation polymerization and an anticancer drug was incorporated into their swollen phase at neutral condition by van der Waals interaction. Upon acidification, the microgel particles became densified and were trapped in this form by lipid encapsulation (Figure 7B). Subsequently, the lipid membrane can be ruptured by electroporation while under neutral pH, leading to a rapid gel swelling that can release the trapped drug molecules. While this system also relies on a volume change of the polymer component to release the encapsulated drug, a physical perforation need to be initiated first, in contrast to the automatic response to pH and temperature changes of the polymer shell in our PCN.
High-capacity loading of a broad range of drugs
The drop-in strategy for PCN synthesis (Figure 6) allows us to leverage the large body of technology currently available for liposome synthesis and encapsulation. For example, PCN can be fabricated from liposomal template of virtually any lipid compositions without much perturbation to the drug-containing liposome core. In addition, we can take advantage of the many drug-loading protocols already developed for liposome, including ion gradient-mediated (IGM) remote drug-loading process that can maximize loading capacity in spite of the low intrinsic solubility of drug molecules.74. High drug-loading capacity in PCN particles can be achieved for a wide range of organic and inorganic drugs: 30 mol% (compared to lipids) for doxorubicin,75 45 mol% for AsO3,70 and 25 mol% for gemcitabine.76
The loading of gemcitabine (GMC), a nucleoside analog drug, into PCN highlights the additional therapeutic advantage of having enhanced cellular uptake of the drug through nanoparticle delivery. GMC-encapsulated PCN (PCNGMC) was found to have higher potency than free GMC against HeLa cells, which lack the human concentrative nucleoside transporter (hCNT), a trans-membrane transporter protein that mediates the cellular internalization of nucleoside analog drugs.76 This can be attributed to the cellular endocytosis of PCNGMC, which pass a high load of encapsulated GMC through the cellular membrane without the need for the protein transporter, resulting in highly efficient cellular internalization. Such enhanced transport is one of the reasons that nanoscale vehicles have attracted significant attention as drug delivery platforms:77 they can be easily internalized into the cytosol through endocytosis and can carry a large bolus of drug with them.
While the liposomal core can itself encapsulate a high amount of drugs, the polymer shell of the PCN can also be used to incorporate additional therapeutic agents for use in combination therapy (i.e., therapy that used two or more drugs simultaneously78,79). Specifically, as the cross-linking step in PCN fabrication does not fully utilize all of the carboxylic acid groups on the polymer cage, the remaining carboxyl groups can be used as chemical handles to install additional functionalities such as therapeutic agents, biological targeting ligands, and imaging agents (Figure 8). For example, we have demonstrated that a large amount of a second drug, cisplatin (130 mol % of PtII compared to the lipid components of PCN, up to 6 times the maximum amount of doxorubicin that can be encapsulated inside the liposomal core), can be chemically attached on the polymer network of the PCN shell through the left-over carboxylic acids (i.e., those that do not react with the diamine cross-linkers, Figure 8), making possible the co-delivery of two different drugs in a single particle for combination therapy.80 In vitro study shows that such a concurrent delivery of both doxorubicin and cisplatin is highly synergistic against MDA-MB-231 breast cancer cells, a model for difficult-to-treat triple-negative cancer that is partially resistant to cisplatin. Such synergistic therapeutic potency was not observed in either the analogous combinations of free drugs or separately PCN-packaged drugs (Figure 9) and can be attributed to the ability of the multi-drug-loaded platform to maintain the necessary molar ratio of the drug combination in one particle that was internalized by the cell through endocytosis.80
Figure 8.

(A) Modular, orthogonal surface-modification of a “clickable” PCN nanoplatform. The inset (B) shows an idealized chemical structure of the modified polymer shell.
Figure 9.

A combination index (C.I.) plot showing the enhanced synergy observed when a dual drug-loaded PCN is used against MDA-MB-231 human breast cancer cells. Combination index were plotted on a logarithmic scale against drug effect levels. (Drug effect level is the reverse of inhibitory concentration. A drug effect level of 70% is equivalent to an IC30 value, the concentration where only 30% of the cell is viable). A combination index of 1 represents just an additive effect (i.e., the drugs are neither reinforcing or working against each other) and combination index values that are lower than 1 mean enhanced synergy in drug combination.80 The dual-drug PCN is clearly more synergistic than either the analogous combinations of single-drug-loaded PCNs or the free drugs.
Modular surface modification of PCN to incorporate a broad range of functionalities
As mentioned above, the poly[(AAc)-co-(AAm)] network offers a multitude of possibilities for post-synthesis modification. This can turn PCN into a platform endowed with multiple functionalities for a wide range of applications (Figure 8), especially when coupled with a tissue-specific targeting agent. Indeed, it has been demonstrated that the therapeutic potency of both small-molecule81 and nano-encapsulated82 chemotherapeutic drugs can be highly enhanced after modification with biological site-specific targeting ligands,83 either monoclonal antibodies or small molecules. This enhanced potency is particularly important in the treatment of refractory and metastatic cancers where both selective targeting and reduction of unfavorable toxic side effects to normal tissue are highly desirable.11 To demonstrate the versatility of the PCN platform, we incorporated terminal alkyne groups into the poly[(AAc)-co-(AAm)] shell of PCNs as part of the cross-linkers,75 which then allows for CuI-catalyzed ‘click’ ligation84 with azide-modified folic acid, a well-known targeting agent for folate receptor (FR)-overexpressing cancer cells85 (Figure 8B). The resulting folate-modified, doxorubicin-loaded PCNs clearly exhibit enhanced efficacy against an FR-overexpressing cancer cell line, with up to 50-fold higher potency compared to that of the untargeted platform.
In addition to targeting and additional drug incorporation, the aforementioned ‘surface-clickable’ capability of the poly[(AAc)-co-(AAm)] shell can be used to engender PCN platform with a wide range of biomedical agents, including imaging/tracking moieties.86 For example, a GdIII-based magnetic resonance imaging (MRI) contrasting agent has been attached to the surface of a GMC-loaded PCN.76 Such a ‘theranostic’ (a combination of therapeutic and diagnostic) platform exhibited significantly enhanced magnetic resonance (MR) relaxivity per particle, presumably due to endocytotically enhanced uptake,77 suggesting that delivery of the GdIII-based MRI contrasting agents via the PCN platform can greatly reduce the amount of diagnostic agents currently used in conventional imaging sessions. The enhanced co-delivery of both therapeutic drugs and imaging agents through our modular PCN platform, especially when guided by a targeting group, should offer a means for remedying the inherent low sensitivity of MRI contrast agents and the large concentration mismatches between therapeutics and diagnostics in current theranostic platforms.87
Recently, we have fabricated a PCN comprising a doxorubicin-loaded liposomal core and a shell that is functionalized with ~8000 molecules of a GdIII MRI contrast agents and ~10–15 copies of Herceptin (Trastuzumab), a monoclonal antibody that specifically binds to human epidermal growth factor receptor 2 (Her2), known to be overexpressed in ~25% of human breast cancers.88 In vitro testing of this PCN against cell lines that overexpress Her2 receptors reveals a 14-fold increase in doxorubicin-based cytotoxicity versus a non-targeted analogue and an 120-fold improvement in cellular GdIII-uptake in comparison with clinically approved DOTA-GdIII, leading to significant T1 MRI contrast enhancement.89 These results clearly demonstrate that the multifunctional PCN platform can indeed fulfills the criteria for an ideal theranostic platform that efficiently and selectively delivers both chemotherapeutic and MRI contrast agents to Her2-overexpressing cancer cells when guided by a cancer-targeting ligand (Herceptin).
IV. Summary and outlook
Over the last two decades, stimuli-responsive copolymers have become an important class of materials in the development of “smart” soft structures.5 As shown in this Perspective article, these materials have been utilized as acid-sensitive cargo-release triggers in nanoparticle delivery systems that may enhance drug release, and thus potency, at disease sites. As an indication of the critical importance of site-specific release, the recent failures of a cisplatin-encapsulated liposomal drug formulation in clinical treatments of non-small-cell lung cancer90 and squamous cancers of the head and neck91 have been attributed to the insufficient release of active drug molecules out of liposomal vesicle, resulting in their low bioavailability.92 In such instances, the incorporation of a triggered release-capable polymer shell, such as that used in our PCNs, into the drug-loaded liposome formulations may improve their clinical outcomes with an enhancement of circulation stability and the capability to trigger-release drug at the disease tissue, while still maintaining the platform biodegradability. In addition, our PCN strategy combines the anchoring ability of the cholesterol end groups of the Chol-PAAc modifiers and the PEG-mimic cross-linkers to attenuate the surface charge contributions by the exposed carboxylic acids groups (Figure 10) and allow the PCNs to escape recognition by immune cells such as reticuloendothelial system (RES) in the spleen and liver.11 At the same time, the unreacted carboxylic acids are still accessible to small molecules such as a second drug or an imaging agent, allowing for the incorporation of multiple functionalities.93
Figure 10.

The surface charge of a carboxylated grafted polymer shell on a liposome can be attenuated by the PEG-modified cross-linker, which can screen the negative charges of the exposed carboxylic acids.
The drop-in incorporation of the stimuli-responsive poly[(AAc)-co-(AAm)] network in our PCN strategy greatly enhances the properties of liposomes as a delivery system for small-molecule anticancer drugs: ease of modification, biocompatibility, acid-sensitive release trigger, and accessible chemical handles on the surface allows for the orthogonal and modular incorporation of a wide range of agents such as additional drugs, targeting ligands, and imaging agents. Our experimental results from benchtop to in vivo evaluations thus far have uniformly highlighted the tremendous potential offered by PCNs for improving cancer chemotherapy. Indeed, recent perspective94 and news articles95 have assessed the PCN platform to have all the characteristics of a near-ideal platform for drug delivery. However, it still needs more optimization before its full potential can be reached: thus far, we have not yet optimized the molecular weight of the Chol-PAAc polymer chains and the degree of cross-linking, both of which will affect particle stability and the sensitivity of the acid-sensitive trigger. We note that the polymer scaffolds need to be degraded into small oligomers (< ~60 kDa) after the delivery, which can then be easily excreted out of the body via renal filtration.96
The size of the nanoparticles also plays a critical role in the success of nanoscale drug delivery platforms that target cancer therapy.97,98 If the particle size is too small (<10 nm), it will be rapidly excreted out of the body through renal filtration. In contrast, large particles (>100 nm) do not penetrate deep inside the tumor tissue, resulting in limited therapeutic efficacy despite the advantage offered by the “enhanced permeation and retention (EPR)” effect.11 Among the particles that are in the 10–100 nm range, the larger particles (>70–100 nm) are easily scavenged by the RES (e.g., Kupffer cells in liver) while the smaller particles (10–40 nm) are not easily internalized by the target cells through endocytosis (due to the insufficient engagement of receptors in charge of molecular recognition on the cell surface). As the optimal size of nanoparticle drug-delivery vehicles has been suggested to be ~40–50 nm,97 the size of the currently available smallest PCNs (~100 nm) still needs to be tuned downward.
Beyond the small-molecule chemotherapeutics that have been demonstrated in the PCN platform thus far, it should be possible to include large macromolecules such as proteins, peptides, and small interfering RNA (siRNA) as therapeutic payloads. Proteins and peptides have long been used to treat diseases and siRNA has emerged as a new type of therapeutics;99,100 however, they all suffer from non-specificity, short circulation time, ease of degradation, and fast clearance.100 Such problems can be overcome if these payloads can be incorporated into our PCN platform. PCNs can also potentially sidestep the known low uptake of siRNA, which are intrinsically negatively charged and cannot be easily uptaken into cells through the lipid membrane by encapsulating them and allow for facile uptake via endocytosis and release into the target cells through endosomal escape.101 For these applications to be realized, the PCN technology needs to be expanded to allow for efficient loadings of proteins and oligonucleotides.
To achieve an ideal targeted delivery and enhanced potency for PCNs, we must also optimize the molecular recognition between the target cells and the surface-attached ligands on the surface of the PCNs. Such specific recognition can mediate the fusion of liposome vesicles to the cellular membrane through close apposition,102 which can lead to a more effective “injection” of drug into the cell and thus, higher potency. Additionally, the overall affinity of targeted PCNs for disease cells should be enhanced by having an optimal number of polyvalent interactions between the nanoscale scaffolds and the surface receptors,8 a parameter that needs further investigation.
We note, however, that while the PCNs have many desirable features, these are not necessarily required for every drug-delivery applications. Simple PEGylated liposomal drug, such as Doxil (Janssen Products, LP),103 or the non-PEGylated analog Myocet (Teva Pharmaceutical Industries, Ltd.), are currently in clinically use for several diseases that do not require sophisticated pharmacological profiles or real-time monitoring.104,105 In this sense, applications that can take advantage of all the features of PCN is likely to be in personalized medicine, including gene or hormone therapy; vaccination, where timely targeted release is critical; or theranostic, where the post-treatment adjustment of prescribed regimen is best accomplished through real-time feedback.106
Finally, for the PCN platform to reach its full potential, detailed pharmacokinetic studies still need to be completed to provide information on how the morphologies and physicochemical characteristics of these nanocarriers can be translated to the desired in vivo circulation and pharmacokinetic profiles for a particular application. While we have successfully developed a modular, multifunctional PCN system as a proof-of-concept platform with a lot of promising indications, extensive biological evaluation and platform engineering must be carried out to elucidate (and improve) detailed transport and release properties, loading capability, targeting efficiencies, and in vivo behaviors. We are confident that a successful demonstration of how stimuli-responsive polymers can improve the in vivo therapeutic efficacy of nanoscale smart delivery platforms will stimulate further developments of personalized medicine and therapy.
Acknowledgments
This work was financially supported by the NIH (NCI Center of Cancer Nanotechnology Excellence Grant C54CA151880, Cancer Nanotechnology Platform Partnerships Grant U01CA151461, and Core Grant P30CA060553 to the Robert H. Lurie Comprehensive Cancer Center of Northwestern University). S.M.L. thanks the 2012 Research Fund of The Catholic University of Korea for additional support. We thank Drs. Richard W. Ahn and S. Archana Krovi for instructive comments during the preparation of this manuscript and all of the collaborators who have helped us with the investigation of the PCN platform over the last eight years.
Biographies

Sang-Min Lee completed his B.S. in chemistry at Yonsei University in Korea and his M.S. degree, also in chemistry, at Korea Advanced Institute of Science and Technology (KAIST). In 2010, he received his doctoral degree in chemistry at Northwestern University under the guidance of SonBinh Nguyen and Thomas O’Halloran. In 2012, after an NRC postdoctoral stint under the joint direction of Robert Cook at the National Institute of Standards and Technology and Martin W. Brechbiel at the National Cancer Institute, National Institute of Health, he took up a tenure track position at the Catholic University of Korea as an assistant professor of chemistry.

During his undergraduate years, SonBinh T. Nguyen carried out chemistry research with Gregory Geoffroy at Penn State and with Henry Bryndza and Steve Ittel at DuPont Central Research. He then received a doctoral degree in chemistry under the joint guidance of Robert Grubbs and Nathan Lewis at Caltech, where he held NSF and NDSEG predoctoral fellowships. After an NSF postdoctoral fellowship with Barry Sharpless at the Scripps Research Institute, SonBinh began his independent career at Northwestern, where he is now a Professor of Chemistry. He held the Directorship of the Integrated Science Program, the Dow Chemical Company Research Professorship, and the McCormick Professorship of Teaching Excellence. He is also a senior member of the technical staff at Argonne National Laboratory.
References
- 1.Girard PP, Cavalcanti-Adam EA, Kemkemer R, Spatz JP. Soft Matter. 2007;3:307. doi: 10.1039/b614008d. [DOI] [PubMed] [Google Scholar]
- 2.Hirst AR, Escuder B, Miravet JF, Smith DK. Angew Chem, Int Ed. 2008;47:8002. doi: 10.1002/anie.200800022. [DOI] [PubMed] [Google Scholar]
- 3.Esser-Kahn AP, Odom SA, Sottos NR, White SR, Moore JS. Macromolecules. 2011;44:5539. [Google Scholar]
- 4.Schneider HJ, Strongin RM. Acc Chem Res. 2009;42:1489. doi: 10.1021/ar800274u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spruell JM, Hawker CJ. Chem Sci. 2011;2:18. [Google Scholar]
- 6.Hoffman AS. Adv Drug Delivery Rev. 2002;54:3. doi: 10.1016/s0169-409x(01)00239-3. [DOI] [PubMed] [Google Scholar]
- 7.Nayak S, Lyon LA. Angew Chem, Int Ed. 2005;44:7686. doi: 10.1002/anie.200501321. [DOI] [PubMed] [Google Scholar]
- 8.Mammen M, Choi SK, Whitesides GM. Angew Chem, Int Ed. 1998;37:2754. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 9.Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Chem Rev. 1999;99:3181. doi: 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- 10.Kabanov AV, Vinogradov SV. Angew Chem, Int Ed. 2009;48:5418. doi: 10.1002/anie.200900441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis ME, Chen Z, Shin DM. Nat Rev Drug Discovery. 2008;7:771. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 12.Torchilin VP. Nat Rev Drug Discovery. 2005;4:145. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 13.Soussan E, Cassel S, Blanzat M, Rico-Lattes I. Angew Chem, Int Ed. 2009;48:274. doi: 10.1002/anie.200802453. [DOI] [PubMed] [Google Scholar]
- 14.Ma M, Gong Y, Bong D. J Am Chem Soc. 2009;131:16919. doi: 10.1021/ja9072657. [DOI] [PubMed] [Google Scholar]
- 15.Ma M, Chatterjee S, Zhang M, Bong D. Chem Commun. 2011;47:2853. doi: 10.1039/c0cc05137c. [DOI] [PubMed] [Google Scholar]
- 16.Ringsdorf H, Schlarb B, Venzmer J. Angew Chem, Int Ed. 1988;27:113. [Google Scholar]
- 17.Flory PJ. Principles of Polymer Chemistry. Cornell University Press; Ithaca, NY: 1953. [Google Scholar]
- 18.Heskins M, Guillet JE. J Macromol Sci, Chem. 1968;2:1441. [Google Scholar]
- 19.Yu Ye E. Polym Sci USSR. 1969;11:409. [Google Scholar]
- 20.Nishio I, Sun ST, Swislow G, Tanaka T. Nature. 1979;281:208. [Google Scholar]
- 21.Tanaka T. Phys Rev Lett. 1978;40:820. [Google Scholar]
- 22.Jeong B, Kim SW, Bae YH. Adv Drug Delivery Rev. 2002;54:37. doi: 10.1016/s0169-409x(01)00242-3. [DOI] [PubMed] [Google Scholar]
- 23.Ringsdorf H, Venzmer J, Winnik FM. Angew Chem, Int Ed. 1991;30:315. [Google Scholar]
- 24.Chen G, Hoffman AS. Nature. 1995;373:49. doi: 10.1038/373049a0. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka T, Fillmore D, Sun ST, Nishio I, Swislow G, Shah A. Phys Rev Lett. 1980;45:1636. [Google Scholar]
- 26.Yoshida R, Uchida K, Kaneko Y, Sakai K, Kikuchi A, Sakurai Y, Okano T. Nature. 1995;374:240. [Google Scholar]
- 27.Dong L-c, Hoffman AS. J Controlled Release. 1991;15:141. [Google Scholar]
- 28.Hennink WE, van Nostrum CF. Adv Drug Delivery Rev. 2002;54:13. doi: 10.1016/s0169-409x(01)00240-x. [DOI] [PubMed] [Google Scholar]
- 29.Oyama HT, Tang WT, Frank CW. Macromolecules. 1987;20:1839. [Google Scholar]
- 30.Mathur AM, Hammonds KF, Klier J, Scranton AB. J Controlled Release. 1998;54:177. doi: 10.1016/s0168-3659(97)00186-7. [DOI] [PubMed] [Google Scholar]
- 31.Klier J, Scranton AB, Peppas NA. Macromolecules. 1990;23:4944. [Google Scholar]
- 32.Huynh CT, Nguyen MK, Lee DS. Macromolecules. 2011;44:6629. [Google Scholar]
- 33.Hoffman AS. J Controlled Release. 1987;6:297. [Google Scholar]
- 34.Hoffman AS, Afrassiabi A, Dong LC. J Controlled Release. 1986;4:213. [Google Scholar]
- 35.Jones CD, Lyon LA. Macromolecules. 2000;33:8301. [Google Scholar]
- 36.Berndt I, Pedersen JS, Richtering W. J Am Chem Soc. 2005;127:9372. doi: 10.1021/ja051825h. [DOI] [PubMed] [Google Scholar]
- 37.Topp MDC, Dijkstra PJ, Talsma H, Feijen J. Macromolecules. 1997;30:8518. [Google Scholar]
- 38.Nayak S, Gan D, Serpe MJ, Lyon LA. Small. 2005;1:416. doi: 10.1002/smll.200400089. [DOI] [PubMed] [Google Scholar]
- 39.Nyström AM, Wooley KL. Acc Chem Res. 2011;44:969. doi: 10.1021/ar200097k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Z, Cui H, Hales K, Li Z, Qi K, Pochan DJ, Wooley KL. J Am Chem Soc. 2005;127:8592. doi: 10.1021/ja050290p. [DOI] [PubMed] [Google Scholar]
- 41.O’Reilly RK, Joralemon MJ, Wooley KL, Hawker CJ. Chem Mater. 2005;17:5976. doi: 10.1021/ja053919x. [DOI] [PubMed] [Google Scholar]
- 42.Nyström AM, Xu Z, Xu J, Taylor S, Nittis T, Stewart SA, Leonard J, Wooley KL. Chem Commun. 2008;3579 doi: 10.1039/b805428b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Papahadjopoulos D, Miller N. Biochim Biophys Acta. 1967;135:624. doi: 10.1016/0005-2736(67)90094-6. [DOI] [PubMed] [Google Scholar]
- 44.Enoch HG, Strittmatter P. Proc Natl Acad Sci USA. 1979;76:145. doi: 10.1073/pnas.76.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gerasimov OV, Boomer JA, Qualls MM, Thompson DH. Adv Drug Delivery Rev. 1999;38:317. doi: 10.1016/s0169-409x(99)00035-6. [DOI] [PubMed] [Google Scholar]
- 46.Allen TM, Hong K, Papahadjopoulos D. Biochemistry. 1990;29:2976. doi: 10.1021/bi00464a013. [DOI] [PubMed] [Google Scholar]
- 47.Tannock IF, Rotin D. Cancer Res. 1989;49:4373. [PubMed] [Google Scholar]
- 48.Vaupel P, Kallinowski F, Okunieff P. Cancer Res. 1989;49:6449. [PubMed] [Google Scholar]
- 49.Casey JR, Grinstein S, Orlowski J. Nat Rev Mol Cell Biol. 2010;11:50. doi: 10.1038/nrm2820. [DOI] [PubMed] [Google Scholar]
- 50.Rui Y, Wang S, Low PS, Thompson DH. J Am Chem Soc. 1998;120:11213. [Google Scholar]
- 51.Adlakha-Hutcheon G, Bally MB, Shew CR, Madden TD. Nat Biotechnol. 1999;17:775. doi: 10.1038/11710. [DOI] [PubMed] [Google Scholar]
- 52.Slepushkin VA, Simoes S, Dazin P, Newman MS, Guo LS, de Lima MCP. J Biol Chem. 1997;272:2382. doi: 10.1074/jbc.272.4.2382. [DOI] [PubMed] [Google Scholar]
- 53.Silvius JR, Leventis R. Biochemistry. 1993;32:13318. doi: 10.1021/bi00211a045. [DOI] [PubMed] [Google Scholar]
- 54.Maeda M, Kumano A, Tirrell DA. J Am Chem Soc. 1988;110:7455. [Google Scholar]
- 55.Winnik FM. Langmuir. 1999;15:4222. [Google Scholar]
- 56.Bhattacharya S, Moss RA, Ringsdorf H, Simon J. J Am Chem Soc. 1993;115:3812. [Google Scholar]
- 57.Binder WH. Angew Chem, Int Ed. 2008;47:3092. doi: 10.1002/anie.200800269. [DOI] [PubMed] [Google Scholar]
- 58.Leroux JC, Roux E, Le Garrec D, Hong K, Drummond DC. J Controlled Release. 2001;72:71. doi: 10.1016/s0168-3659(01)00263-2. [DOI] [PubMed] [Google Scholar]
- 59.Allen TM, Cleland LG. Biochim Biophys Acta. 1980;597:418. doi: 10.1016/0005-2736(80)90118-2. [DOI] [PubMed] [Google Scholar]
- 60.Silvius JR, Zuckermann MJ. Biochemistry. 1993;32:3153. doi: 10.1021/bi00063a030. [DOI] [PubMed] [Google Scholar]
- 61.Hara M, Miyake M, Iijima S, Yang Q, Arai T, Yuan H, Miyake J. Supramol Sci. 1998;5:777. [Google Scholar]
- 62.O’Brien DF, Armitage B, Benedicto A, Bennett DE, Lamparski HG, Lee YS, Srisiri W, Sisson TM. Acc Chem Res. 1998;31:861. [Google Scholar]
- 63.Bondurant B, O’Brien DF. J Am Chem Soc. 1998;120:13541. [Google Scholar]
- 64.Liu S, O’Brien DF. J Am Chem Soc. 2002;124:6037. doi: 10.1021/ja0123507. [DOI] [PubMed] [Google Scholar]
- 65.Thomas JL, Tirrell DA. Acc Chem Res. 1992;25:336. [Google Scholar]
- 66.Yessine MA, Leroux JC. Adv Drug Delivery Rev. 2004;56:999. doi: 10.1016/j.addr.2003.10.039. [DOI] [PubMed] [Google Scholar]
- 67.Lee SM, Chen H, Dettmer CM, O’Halloran TV, Nguyen ST. J Am Chem Soc. 2007;129:15096. doi: 10.1021/ja070748i. [DOI] [PubMed] [Google Scholar]
- 68.Lee SM, Ahn RW, Chen F, Fought AJ, O’Halloran TV, Cryns VL, Nguyen ST. ACS Nano. 2010;4:4971. doi: 10.1021/nn100560p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Holland JW, Hui C, Cullis PR, Madden TD. Biochemistry. 1996;35:2618. doi: 10.1021/bi952000v. [DOI] [PubMed] [Google Scholar]
- 70.Lee SM, Lee OS, O’Halloran TV, Schatz GC, Nguyen ST. ACS Nano. 2011;5:3961. doi: 10.1021/nn200478m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu J, Chen Z, Lallemand-Breitenbach V, de The H. Nat Rev Cancer. 2002;2:705. doi: 10.1038/nrc887. [DOI] [PubMed] [Google Scholar]
- 72.Chen H, MacDonald RC, Li S, Krett NL, Rosen ST, O’Halloran TV. J Am Chem Soc. 2006;128:13348. doi: 10.1021/ja064864h. [DOI] [PubMed] [Google Scholar]
- 73.Kiser PF, Wilson G, Needham D. Nature. 1998;394:459. doi: 10.1038/28822. [DOI] [PubMed] [Google Scholar]
- 74.Barenholz Y, Amselem S, Goren D, Cohen R, Gelvan D, Samuni A, Golden EB, Gabizon A. Med Res Rev. 1993;13:449. doi: 10.1002/med.2610130404. [DOI] [PubMed] [Google Scholar]
- 75.Lee SM, Chen H, O’Halloran TV, Nguyen ST. J Am Chem Soc. 2009;131:9311. doi: 10.1021/ja9017336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee SM, Song Y, Hong BJ, MacRenaris KW, Mastarone DJ, O’Halloran TV, Meade TJ, Nguyen ST. Angew Chem, Int Ed. 2010;49:9960. doi: 10.1002/anie.201004867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petros RA, DeSimone JM. Nat Rev Drug Discovery. 2010;9:615. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 78.Scripture CD, Figg WD. Nat Rev Cancer. 2006;6:546. doi: 10.1038/nrc1887. [DOI] [PubMed] [Google Scholar]
- 79.Keith CT, Borisy AA, Stockwell BR. Nat Rev Drug Discovery. 2005;4:71. doi: 10.1038/nrd1609. [DOI] [PubMed] [Google Scholar]
- 80.Lee SM, O’Halloran TV, Nguyen ST. J Am Chem Soc. 2010;132:17130. doi: 10.1021/ja107333g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, Pegram M, Oh DY, Dieras V, Guardino E, Fang L, Lu MW, Olsen S, Blackwell K. N Engl J Med. 2012;367:1783. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 83.Allen TM. Nat Rev Cancer. 2002;2:750. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- 84.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem, Int Ed. 2002;41:2596. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 85.Low PS, Henne WA, Doorneweerd DD. Acc Chem Res. 2008;41:120. doi: 10.1021/ar7000815. [DOI] [PubMed] [Google Scholar]
- 86.Krovi SA, Smith D, Nguyen ST. Chem Commun. 2010;46:5277. doi: 10.1039/c0cc00232a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McCarthy JR. Nanomedicine. 2009;4:693. doi: 10.2217/nnm.09.58. [DOI] [PubMed] [Google Scholar]
- 88.Strohecker AM, Yehiely F, Chen F, Cryns VL. J Biol Chem. 2008;283:18269. doi: 10.1074/jbc.M802156200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hong BJ, Swindell EP, MacRenaris KW, Hankins PL, Chipre AJ, Mastarone DJ, Ahn RW, Meade TJ, O’Halloran TV, Nguyen ST. Part Part Syst Charact. 2013;30 doi: 10.1002/ppsc201300158. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim ES, Lu C, Khuri FR, Tonda M, Glisson BS, Liu D, Jung M, Hong WK, Herbst RS. Lung Cancer. 2001;34:427. doi: 10.1016/s0169-5002(01)00278-1. [DOI] [PubMed] [Google Scholar]
- 91.Harrington KJ, Lewanski CR, Northcote AD, Whittaker J, Wellbank H, Vile RG, Peters AM, Stewart JSW. Ann Oncol. 2001;12:493. doi: 10.1023/a:1011199028318. [DOI] [PubMed] [Google Scholar]
- 92.Bandak S, Goren D, Horowitz A, Tzemach D, Gabizon A. Anti-Cancer Drugs. 1999;10:911. doi: 10.1097/00001813-199911000-00007. [DOI] [PubMed] [Google Scholar]
- 93.Hawker CJ, Wooley KL. Science. 2005;309:1200. doi: 10.1126/science.1109778. [DOI] [PubMed] [Google Scholar]
- 94.Adair JH, Parette MP, Altınoglu EI, Kester M. ACS Nano. 2010;4:4967. doi: 10.1021/nn102324e. [DOI] [PubMed] [Google Scholar]
- 95.Views N. Nanomedicine. 2011;6:307. [Google Scholar]
- 96.Fox ME, Szoka FC, Fréchet JMJ. Acc Chem Res. 2009;42:1141. doi: 10.1021/ar900035f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Davis ME. MRS Bulletin. 2012;37:828. [Google Scholar]
- 98.Zamboni WC, Torchilin V, Patri AK, Hrkach J, Stern S, Lee R, Nel A, Panaro NJ, Grodzinski P. Clin Cancer Res. 2012;18:3229. doi: 10.1158/1078-0432.CCR-11-2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peer D, Lieberman J. Gene Ther. 2011;18:1127. doi: 10.1038/gt.2011.56. [DOI] [PubMed] [Google Scholar]
- 100.Pecot CV, Calin GA, Coleman RL, Lopez-Berestein G, Sood AK. Nat Rev Cancer. 2011;11:59. doi: 10.1038/nrc2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hong BJ, Chipre AJ, Nguyen ST. J Am Chem Soc. 2013;135 doi: 10.1021/ja404491r. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gong Y, Luo Y, Bong D. J Am Chem Soc. 2006;128:14430. doi: 10.1021/ja0644576. [DOI] [PubMed] [Google Scholar]
- 103.Barenholz Y. J Controlled Release. 2012;160:117. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 104.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Pharmacol Rev. 1999;51:691. [PubMed] [Google Scholar]
- 105.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Pharmacol Rev. 2004;56:185. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 106.Sumer B, Gao J. Nanomedicine. 2008;3:137. doi: 10.2217/17435889.3.2.137. [DOI] [PubMed] [Google Scholar]