

SUMMARY
Although high c-Myc protein expression is observed alongside MYC amplification in some cancers, in most cases protein overexpression occurs in the absence of gene amplification, e.g. T cell lymphoma (TCL). Here, Ca2+/calmodulin-dependent protein kinase II γ (CAMKIIγ) was shown to stabilize the c-Myc protein by directly phosphorylating it at serine 62 (S62). Further, CAMKIIγ was shown to be essential for tumor maintenance. Inhibition of CAMKIIγ with a specific inhibitor destabilized c-Myc and reduced tumor burden. Importantly, high CAMKIIγ levels in patient TCL specimens correlate with increased c-Myc and pS62-c-Myc levels. Together, the CAMKIIγ: c-Myc axis critically influences the development and maintenance of TCL and represents a potential therapeutic target for TCL.
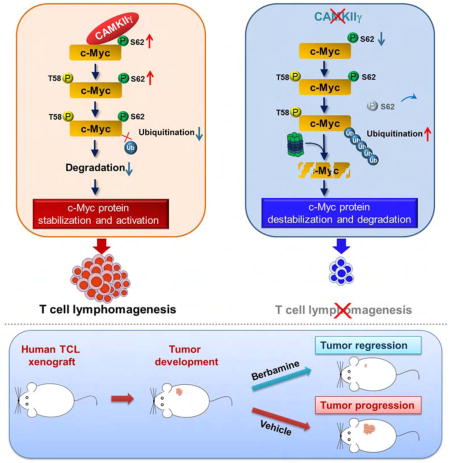
Graphical abstract
T cell lymphomas (TCL) overexpress the c-Myc protein without MYC rearrangements or amplification. Gu et al. show that CAMKIIγ stabilizes c-Myc by phosphorylating it at Ser62, that the CAMKIIγ level positively correlates with the c-Myc level in patient TCL, and that inhibition of CAMKIIγ reduces TCL burden in mice.
INTRODUCTION
The transcription factor c-Myc regulates many genes involved in essential biological processes, including cell growth, proliferation, and apoptosis (Cole, 1986; Dang, 2012; Prendergast, 1999). In malignant diseases it promotes oncogenesis by activating and repressing target genes controlling cell growth and proliferation (Nilsson and Cleveland, 2003). C-Myc is dysregulated in many human cancers, especially in a large proportion of aggressive B cell lymphoma (BCL). The significance of c-Myc dysregulation has also been recognized in T cell lymphoma (TCL): Studies in the Eμ-tTA/tetO-Myc conditional mouse model have demonstrated that the development of aggressive TCL is a consequence of c-Myc overexpression (Choi et al., 2014; Koh et al., 2015). Mechanisms underlying aberrant activity of the c-Myc oncoprotein have been defined in Burkitt and some other aggressive BCL, where molecular hallmarks include chromosomal rearrangements of MYC (Rossi et al., 2012). In contrast, TCL rarely exhibit such MYC rearrangements (Chisholm et al., 2015). To date, the mechanisms of c-Myc overexpression in TCL are still unknown.
Ca2+/calmodulin-dependent protein kinase II (CAMKII), a multi-functional serine/threonine kinase best known for its regulatory functions in learning and memory (Bui et al., 2000), can be chronically activated under pathological conditions (Hook and Means, 2001; Nowycky and Thomas, 2002; Orrenius et al., 2003). For example, in some tumors the aberrant expression of CAMKII and its tumor-promoting functions have been investigated (Colomer and Means, 2007; Meng et al., 2013). According to both our work and that of others, the major isoform of CAMKII in hematopoietic cells – namely CAMKIIγ – plays a role in leukemia (Gu et al., 2016; Si and Collins, 2008). While both CAMKIIγ action and Ca2+-related signaling pathways are important components of normal signal transduction in T lymphocytes (Lin et al., 2005), the oncogenic functions of CAMKIIγ in TCL remain uncharacterized.
With current therapy, survival of patients having TCL with high c-Myc activity is dismal. One contributor to the poor outcome is the current lack of therapeutics against c-Myc; it has been notoriously difficult to target c-Myc with small-molecule inhibitors (Toyoshima et al., 2012). Interestingly, CAMKII has been recently implicated in the survival of c-Myc-overexpressing cells (Toyoshima et al., 2012): CAMK2G was determined to be one of 102 potential genes involved in a synthetic lethal interaction with c-Myc (concomitant mutations lead to cell death). These results suggest that CAMKIIγ may be involved in c-Myc-associated malignancies, however, there is no further study regarding their interactions and functions. Our previous investigations of CAMKIIγ suggest that it is a specific and critical target through which berbamine (BBM) conveys its anti-tumor activity (Gu et al., 2012). Such findings highlight a potential therapeutic strategy whereby c-Myc-associated malignancies are targeted by inhibiting CAMKIIγ. Towards this end, we are delineating the role CAMKIIγ plays in c-Myc-associated tumors.
RESULTS
Camk2g deletion suppresses T cell lymphomagenesis in vivo
To examine the role of CAMKIIγ during T cell lymphomagenesis in vivo, we used a chemical-induced TCL-like mouse model. Through a single injection of N-Methyl-N-Nitrosourea (MNU) as described previously (Figure S1A) (Dumenco et al., 1993; Joshi and Frei, 1970; Slee and Lu, 2013), we induced a lymphomagenesis in 4 to 5-week-old Camk2g−/− mice (Backs et al., 2010) and wild-type mice. Disease onset was verified by symptoms such as hunched posture and labored breathing. At approximately 6 months after the MNU injection, wild-type mice exhibited the expected significantly enlarged thymuses, spleens and lymph nodes (Figures 1A and 1B). Histological analysis of wild-type mice showed evidence of malignant lymphoma. This included effacement of the thymic corticomedullary architecture by diffuse sheets of lymphoblasts with large euchromatic nuclei; moderate to high numbers of mitotic figures; and the presence of clusters or sheets of lymphoblasts in lymph nodes and spleens (Figures 1C and 1D). The neoplastic cells exhibited positive staining for CD3 antigen, indicating that the neoplasms originated from the T cell lineage (Figures 1C and 1D). Nevertheless, during MNU-induced TCL development, the white cell differential counts in peripheral blood did not differ significantly from those in normal (without MNU) mice (Figure S1B). In contrast, Camk2g−/− mice were resistant to MNU-induced lymphoma. By 38 weeks, all of the wild-type mice had died from the disease while 90% of the Camk2g−/− mice still survived (Figure 1E). These studies demonstrate that Camk2g deletion suppresses the development of MNU-induced TCL in mice; thus CAMKIIγ appears to be essential for in vivo T cell lymphomagenesis.
Figure 1. CAMKIIγ is essential for T cell lymphomagenesis in vivo.
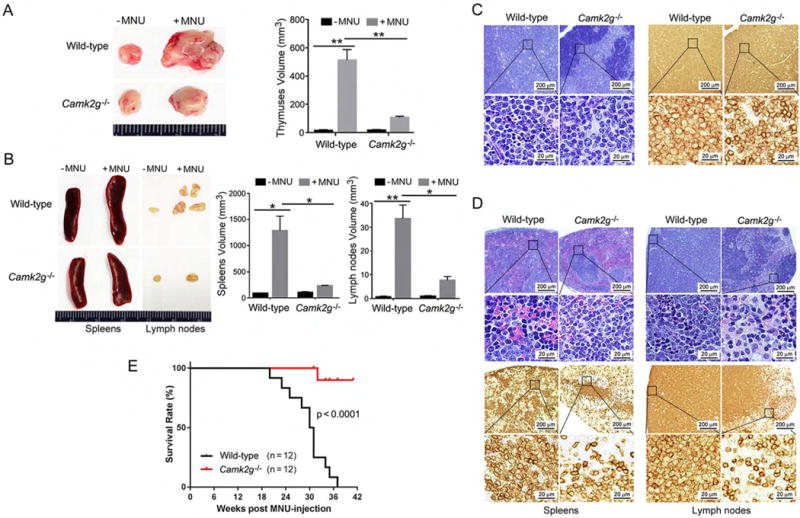
(A) Thymuses obtained from wild-type or Camk2g−/− mice 20 weeks after MNU injection or without MNU injection.
(B) Spleens and lymph nodes obtained from wild-type or Camk2g−/− mice 20 weeks after MNU injection or without MNU injection.
(C) Paraffin sections of mice thymuses in MNU-injected mice analyzed by H&E staining (left) or immunohistochemistry with murine CD3 (right).
(D) Paraffin sections of mice spleens and lymph nodes in MNU-injected mice analyzed by H&E staining (upper) or immunohistochemistry with murine CD3 (lower).
(E) Kaplan-Meier survival curves for wild-type or Camk2g−/− mice after MNU injection.
Data represent the mean ± s.d. for 8 mice. *p < 0.05, **p < 0.01. See also Figure S1.
CAMKIIγ maintains the c-Myc protein levels during T cell lymphomagenesis
We next investigated how deleting Camk2g suppressed the development of MNU-induced TCL in mice. Through RNA-sequencing, we analyzed the gene expression profiles of the thymuses of both wild-type and Camk2g−/− mice, under either malignant (with MNU) or normal (without MNU) conditions. A significant gene expression profile change was observed with 5188 differentially expressed genes (DEGs) (Figure S2A) (Huang da et al., 2009). We then performed Gene Ontology (GO) analysis to identify consensus pathways that may be altered in lymphomagenesis and affected by deletion of Camk2g. According to our results, DEGs related to lymphomagenesis in wild-type mice (compared to WT/-MNU and KO/±MNU) are enriched in GO terms associated with immune response, chemotaxis, inflammation, cell adhesion, lymphocyte activation and proliferation, and regulation of cell death and apoptosis (Table S1). We then performed Ingenuity Pathways Analysis (IPA) to predict for such changes potentially associated with upstream regulators (Table S2). Importantly, c-Myc, which is reported as a key player in B cell lymphoma and many other malignancies, ranks #2 in the list. The rank #1 position of our upstream analysis was tripartite motif-containing 24 (TRIM24). Prompted by these results, we checked the expression of TRIM24 and c-Myc in thymuses of both wild-type and Camk2g−/− mice, under either malignant or normal conditions. Notably, c-Myc displayed a highly positive correlation with CAMKIIγ expression status, compared with that of TRIM24 (Figure S2B). Consistently, we found a distinct set of known c-Myc target genes upregulated in wild-type/+MNU mouse thymuses but not in the thymuses of Camk2g−/− mice (Figure 2A). According to gene set enrichment analysis (GSEA) in wild-type/+MNU mouse thymuses, transcription targets of c-Myc were enriched compared to those in Camk2g−/−/+MNU mice (Figure S2C). Although there were no significant differences in the Myc mRNA levels (Figure S2D), a strong increase in the c-Myc protein levels was detected in wild-type but not Camk2g−/− mice (Figure 2B).
Figure 2. CAMKIIγ maintains the c-Myc protein levels during T cell lymphomagenesis.
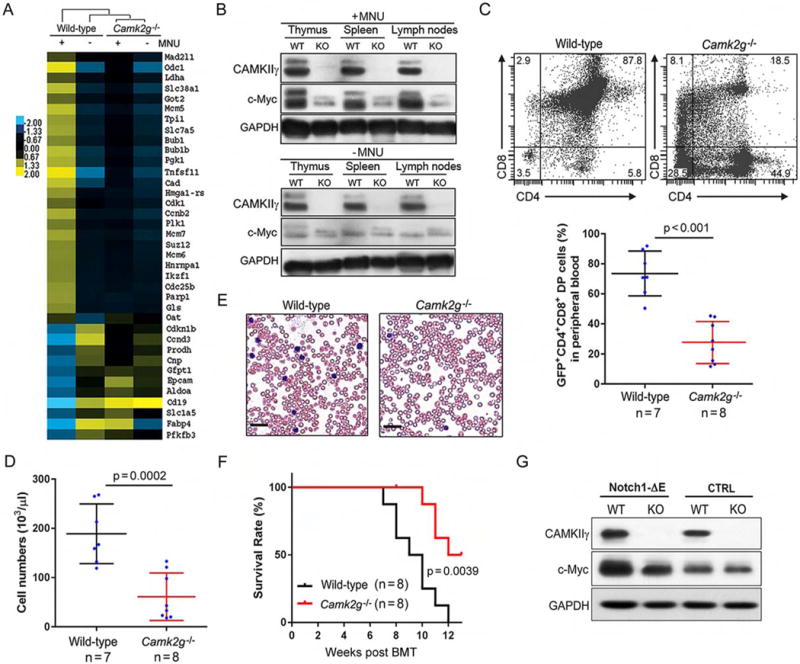
(A) Heat map showing c-Myc-regulated gene expression profiles in thymuses of wild-type and Camk2g−/− mice with (+) or without (−) MNU injection. Yellow and blue colors depict high and low gene expression levels, respectively. Levels are scaled based on expression values that have been mean-centered to zero.
(B) Representative western blots of the CAMKIIγ and c-Myc proteins in the indicated tissues of wild-type (WT) or Camk2g−/− (KO) mice at 20 weeks after MNU injection or without MNU injection. GAPDH was used as a loading control.
(C) Representative flow cytometry plots of CD4+ and CD8+ cells (upper), and the relative proportions of CD4+CD8+ (double-positive) cells (Notch1-ΔE-GFP-positive cells, lower) in peripheral blood of mice expressing wild-type or Camk2g−/− progenitors at 8 weeks after BMT.
(D) Quantification of white blood cell (WBC) counts in the peripheral blood of mice transplanted with Notch1-ΔE expressing wild-type or Camk2g−/− progenitors at 8 weeks after BMT.
(E) Representative blood smear preparations from mice transplanted with Notch-ΔE expressing wild-type or Camk2g−/− progenitors at 8 weeks after BMT. Scale bars, 50 μm.
(F) Kaplan-Meier survival curves of mice transplanted with hematopoietic progenitors from the wild-type and Camk2g−/− backgrounds expressing Notch1-ΔE.
(G) Representative western blots of CAMKIIγ, c-Myc, and GAPDH levels in BMCs of indicated mice. GAPDH was used as a loading control.
Data represent the mean ± s.d. See also Figure S2, Tables S1 and S2.
To further address the roles of CAMKIIγ in c-Myc related T cell tumorigenesis, we then performed bone marrow transplantation (BMT) experiments using hematopoietic progenitors from wild-type or Camk2g−/− mice (Figure S2E). We transplanted isogenic C57BL/6 mice with wild-type or Camk2g−/− hematopoietic progenitors infected with retroviruses driving the expression of an oncogenic constitutively active form of Notch1 (Notch1-ΔE) (Herranz et al., 2014; Schroeter et al., 1998). The tumorigenetic kinetics displayed a much faster disease onset in the wild-type background than in the Camk2g−/− background: We observed (1) significant faster increase in the frequency of CD4+CD8+ double-positive (DP) cells (Figure 2C) and tumor burden in the peripheral blood (Figures 2D and 2E); (2) more severe infiltration of lymphoblasts in the thymuses; and (3) the presence of clusters or sheets of lymphoblasts in spleens and livers (Figure S2F). Moreover, the wild-type mice succumbed to the disease much faster than Camk2g−/− mice (Figure 2F). Consistently, in Notch1-ΔE-expressing mouse bone marrow cells (BMCs) from the wild-type background, the c-Myc protein levels were much higher than those observed in the Camk2g−/− model (Figure 2G). Notably, the Myc mRNA levels observed in both of these backgrounds were not significantly different (Figure S2G). Altogether, these results suggest that high protein levels of c-Myc depend on CAMKIIγ in TCL.
CAMKIIγ: c-Myc axis enhances T cell lymphoma cell proliferation
We next examined whether CAMKIIγ: c-Myc axis is required for TCL’s cellular proliferation in vitro. A set of TCL cell lines – H9, JB6, Jurkat and SU-DHL-1 – was treated with a widely used specific CAMKII inhibitor, KN93 (Rich and Schulman, 1998; Sumi et al., 1991). For all these cell lines, KN93 suppressed their growth (Figure S3A). This suppression was not observed for KN93’s related, inactive analog KN92. Using the H9 cell line as a representative example, genetic ablation of CAMK2G (H9CAMK2G−/−), via clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated proteins (Cas) technology (Figure 3A), also dramatically suppressed cellular proliferation (Figure 3B) (Ran et al., 2013; Xue et al., 2014). Moreover, in H9CAMK2G−/− cells, flow-cytometric analysis showed a significant increase in the percentage of G2/M-phase cells and a significant decrease in the percentage of S-phase cells (Figure 3C). Deletion of Camk2G in TCL cells therefore appears to induce G2/M arrest, a feature that is consistent with inhibition of cell proliferation. Annexin V (AV) - propidium iodide (PI) double staining showed that, upon deletion of CAMK2G, the percentage of total apoptotic cells in H9CAMK2G−/− cells was 9.63%, compared to 3.5% in H9 cells (Figure 3D). Furthermore, both knocking out CAMK2G by CRISPR/Cas technology and inhibiting CAMKIIγ activity by KN93 in H9 cells decreased the protein levels of c-Myc, but not other lymphoma-associated signaling molecules (Figures S3B) nor the MYC mRNA levels (Figure S3C).
Figure 3. CAMKIIγ: c-Myc axis is essential for the proliferation of T cell lymphoma cells.
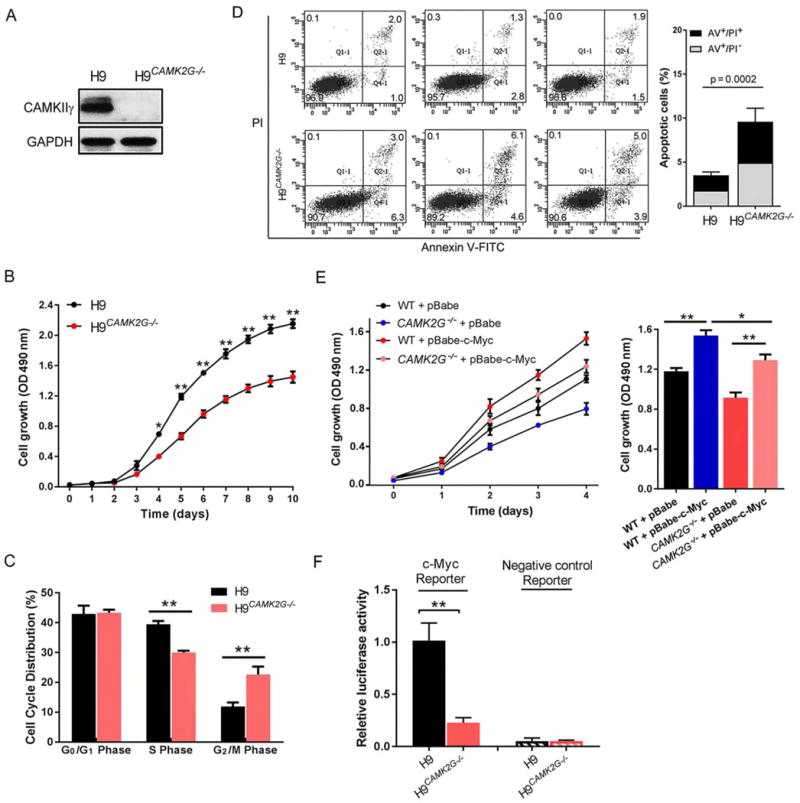
(A) Representative western blots of CAMKIIγ and GAPDH levels in H9 and H9CAMK2G−/− cell lines. GAPDH was used as a loading control.
(B) Proliferation curves of H9 and H9CAMK2G−/− cells. 1.5 × 103 H9 or H9CAMK2G−/− cells were plated in normal growth medium in 96-well plates and MTS assays were performed for a total of 10 days.
(C) Propidium iodide (PI) profiling of H9 and H9CAMK2G−/− cells shows the average percentage of cells in each phase of the cell cycle.
(D) The apoptosis rate in H9 and H9CAMK2G−/− cells was detected using flow cytometry for 3 independent experiments (left). Both early [Annexin V-FITC (AV)+/propidium iodide (PI)−] and late (AV/PI) apoptotic cells were calculated as the incidence of apoptotic cell death (right).
(E) H9 and H9CAMK2G−/− cells were infected with indicated retrovirus. 3 × 103 transfected cells were plated in normal growth medium in 96-well plates and MTS assays were performed for a total of 4 days.
(F) C-Myc transcriptional activity was analyzed by luciferase assay. C-Myc reporter: pBV-Luc wt MBS1-4 plasmids; Negative control reporter: pBV-Luc.
Data represent the mean ± s.d. for three independent experiments. *p < 0.05, **p < 0.01. See also Figure S3.
We then asked whether c-Myc overexpression can reverse the repercussions of deleting CAMK2G – i.e. the impaired cellular proliferation. As shown in Figure 3E, overexpression of c-Myc in H9 cells indeed rescued their ability to proliferate (Figure 3E). As with deleting CAMK2G, knocking down c-Myc in H9 cells also dramatically suppressed proliferation; however, in this scenario, CAMKIIγ overexpression only slightly improved the impaired ability to proliferate (Figure S3D). In addition, we investigated whether CAMKIIγ is required for c-Myc’s transcriptional activity. Knockout of the endogenous CAMK2G reduced c-Myc transcriptional activity (Figure 3F), whereas overexpression of CAMKIIγ significantly increased c-Myc transcriptional activity (Figure S3E). Considering the results altogether, CAMKIIγ positively regulates the proliferation of T cell lymphoma cells by sustaining c-Myc levels and activities.
CAMKIIγ positively regulates the c-Myc protein stability
Increased c-Myc protein abundance may be caused by either increased c-Myc production or decreased decay. Both modes have been reported as mechanism of c-Myc overexpression in various malignancies (Lim and Elenitoba-Johnson, 2004; Thomas and Tansey, 2011). To determine whether the c-Myc protein half-life is decreased in CAMK2G−/− cells, we first examined the c-Myc protein turnover by (1) adding the translation inhibitor cycloheximide (CHX) to 293T and 293TCAMK2G−/− cells and (2) evaluating the c-Myc protein levels at different time points. Consistent with previous studies, c-Myc has a very short half-life in 293T cells, as evidenced by the rapid turnover around ~20 min in cells with CAMKIIγ. As expected, in 293TCAMK2G−/− cells, CHX treatment resulted in a much shorter, ~13 min c-Myc half-life (Figure S4A). Incorporating 35S-methionine/cysteine into c-Myc also gave similar results: ~20 min in 293T cells and ~12 min in 293TCAMK2G−/− cells (Figures S4B and S4C).
We next examined the turnover rate of the c-Myc protein in TCL cell lines – H9, JB6, Jurkat and SU-DHL-1 by CHX assay. C-Myc half-lives in H9 cells harboring various CAMKIIγ statuses shown as a representative example – knocking down CAMKIIγ significantly shortened the c-Myc half-life, whereas CAMKIIγ overexpression results in much longer half-life of the c-Myc protein (Figure 4A). The similar results obtained from all of these four cell lines – c-Myc half- lives were significantly prolonged after CAMKIIγ overexpression, ranging from 75 to 95 min, compared to that in their control cells, which ranged from 55 to 65 min; in contrast, knockdown of CAMKIIγ resulted in significant shorter c-Myc half-lives, ranging from 30 to 40 min, compared to that in their control cells, which ranged from 55 to 70 min (Figures 4B).
Figure 4. CAMKIIγ stabilizes the c-Myc protein.
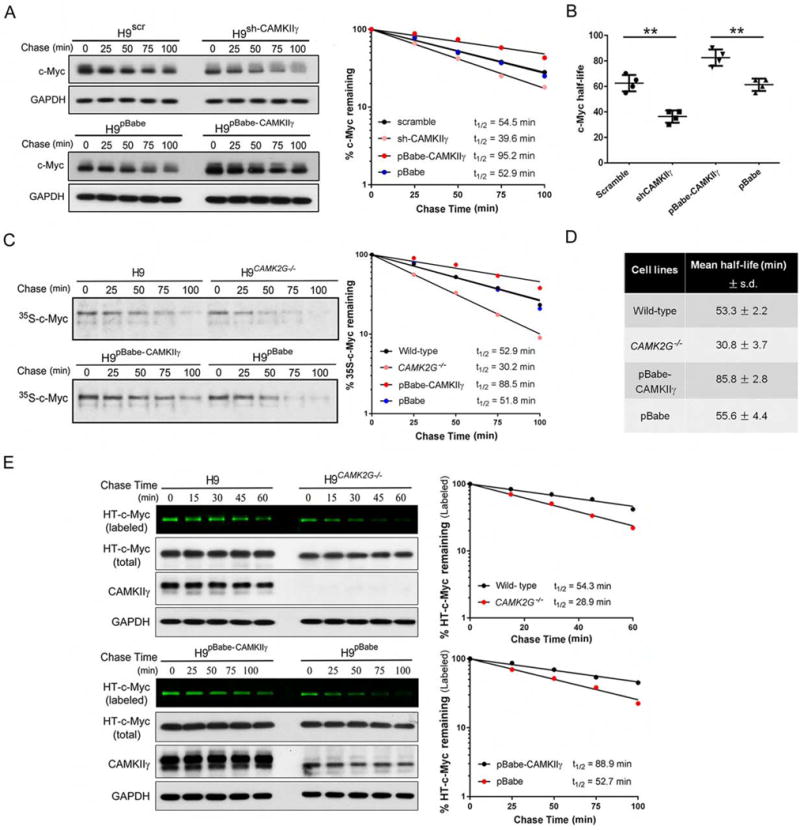
(A) H9 cells with indicated viral infection were treated with 50 μM CHX for indicated times. The c-Myc protein levels were analyzed by western blot analysis with GAPDH as loading control (left). Protein levels were measured with the densitometric intensity. C-Myc levels were quantified relative to GAPDH levels and graphed as percent c-Myc protein remaining after CHX treatment (right).
(B) Summary of c-Myc half-lives in four TCL cell lines. Data represent the mean ± s.d. for four TCL cell lines. **p < 0.01.
(C) Protein detected by autoradiography after a 35S-methionine pulse-chase from indicated cell lines at indicated time points (left). The rate of degradation of endogenous c-Myc for each cell line was represented in the graph by best-fit exponential lines (right).
(D) Summary of mean half-life ± s.d. for c-Myc calculated from three independent experiments for each cell type of H9 cells.
(E) Pulse-chase analysis of c-Myc using HT-TMR system in indicated cell lines transfected with HT-c-Myc. The HT-TMR ligand-labeled HT-c-Myc was visualized with a fluoro-image analyzer (the top sub-panel) and the total expression of HT-c-Myc was analyzed by western blot analysis with GAPDH as loading control (left). The relative amount of HT-TMR ligand-labeled HT-c-Myc was measured with the densitometric intensity of each band (right).
Half-lives of c-Myc were calculated from exponential line equations and were shown for each cell type. Pulse-chase results shown here were representative of three independent experiments. See also Figure S4.
We built upon this finding using the H9 cell line as a representative example: i.e. in H9, H9CAMK2G−/−, H9pBabe-CAMKIIγ, or H9pBabe cells. The half-life of c-Myc was measured by the pulse- chase assay via two approaches: (1) S-methionine/cysteine (Figure 4C and 4D) and (2) HaloTag (HT) and HaloTag-tetramethylrhodamine (TMR) ligand system (Figure 4E). Both approaches displayed consistent results – the half-life of c-Myc was significantly shortened in the absence of CAMK2G whereas it was significantly prolonged by CAMKIIγ overexpression, suggesting that CAMKIIγ positively regulates the half-life of the c-Myc protein (Figures 4C–4E). In contrast, the half-life of another unstable protein expressed ectopically – HT-tagged p53 (HT-p53) was not altered by the status of CAMKIIγ, suggesting the specific regulation of c-Myc stability by CAMKIIγ (Figures S4D). Thus, CAMKIIγ is able to stabilize the c-Myc protein in TCL.
CAMKIIγ stabilizes c-Myc by enhancing c-Myc phosphorylation at serine 62
The tight control of c-Myc activity is defective at multiple levels in cancers in which the protein is constitutively activated and stabilized (Wang et al., 2011a). The c-Myc protein stability can be regulated by two phosphorylation sites with opposing functions: serine 62 phosphorylation (pS62) stabilizes c-Myc whereas threonine 58 phosphorylation (pT58) promotes c-Myc degradation (Pulverer et al., 1994; Sears et al., 1999). We therefore hypothesized that the dependence of c-Myc turnover on CAMKIIγ most likely involves CAMKIIγ kinase activity. We transiently transfected H9 cells separately with various hemagglutinin (HA)-tagged CAMKIIγ mutants expression plasmids. As anticipated, a positive correlation between CAMKIIγ activity and c-Myc/pS62-c-Myc levels was observed. Compared to cells that were CAMK2G−/− or expressing K43M-mutant (kinase dead), those expressing wild-type or T287D-mutant (constitutively active) CAMKIIγ had elevated levels of c-Myc/pS62-c-Myc (Figure 5A). The ubiquitin-proteasome pathway is known to proteolytically degrade c-Myc (Lim and Elenitoba-Johnson, 2004; Thomas and Tansey, 2011). According to our results, the proteasome inhibitor MG132 abolished the differential c-Myc protein levels we observed in transfected cells (Figure 5A). We also investigated phosphorylation after CAMKIIγ inhibition by KN93. To do so, we used two antibodies specific to phosphorylated S62 and two antibodies specific to phosphorylated T58; the specificity of these antibodies was validated using S62A or T58A c-Myc mutants, respectively (Figure S5A). Using H9 cells as a representative example, KN93 decreased p-CAMKIIγ, c-Myc, and pS62-c-Myc, but not pT58-c-Myc levels in a dose- and time-dependent manner (Figures S5B and S5C). Compared to our results in the above CAMKIIγ inhibition model, we observed similar phosphorylation status at S62 and T58 in H9 cells with or without endogenous CAMKIIγ (Figure 5B).
Figure 5. CAMKIIγ stabilizes c-Myc by enhancing c-Myc phosphorylation at serine 62.
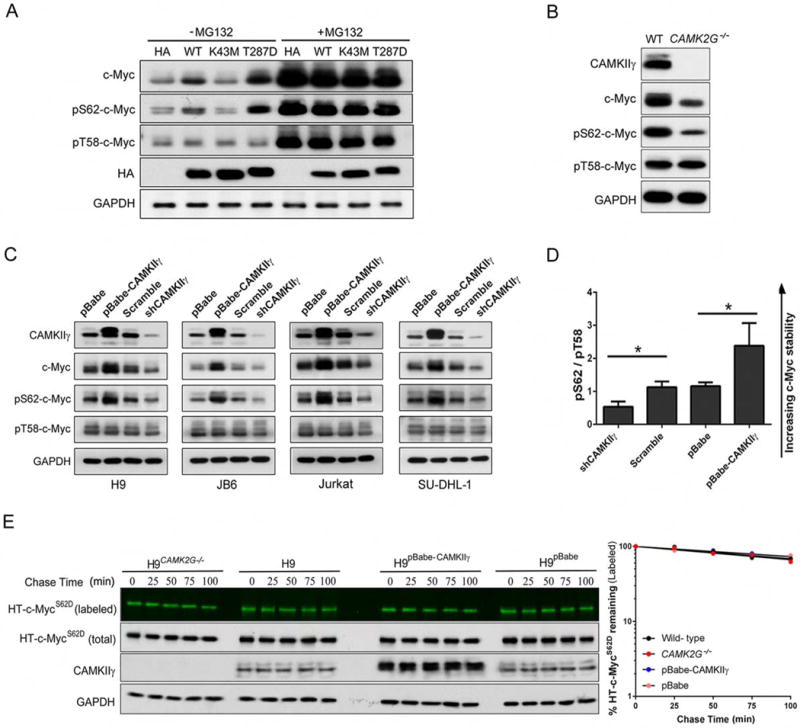
(A) The transfected H9CAMK2G−/− cell lines were treated with or without 10 μM of MG132 for 3 hr; whole cell lysates were analyzed by western blotting for indicated proteins. GAPDH and HA were used as controls.
(B) The c-Myc, p-c-Myc (S62 or T58), CAMKIIγ and GAPDH protein levels in whole cell lysates from H9 and H9CAMK2G−/− cells. GAPDH was used as a loading control.
(C) The c-Myc, p-c-Myc (S62 or T58), CAMKIIγ and GAPDH protein levels in whole cell lysates from indicated cell lines with knockdown or overexpression of CAMKIIγ. GAPDH was used as a loading control.
(D) The p-c-Myc (S62)/p-c-Myc (T58) protein levels (pS62/pT58) in Figure 5C were quantified after normalization to GAPDH. Data represent the mean ± s.d. for indicated cell lines. *p < 0.05.
(E) Pulse-chase analysis of c-MycS62D using HT-TMR system in indicated cell lines transfected with HT-c-MycS62D. The HT-TMR ligand-labeled HT-c-MycS62D was visualized with a fluoro- image analyzer (the top sub-panels) and the total expression of HT-c-MycS62D was analyzed by western blotting with GAPDH as loading control (left). The relative amount of HT-TMR ligandlabeled HT-c-MycS62D was measured with the densitometric intensity of each band (right). Best- fit exponential lines are shown for each cell line. Pulse-chase results shown here were representative of three independent experiments.
See also Figure S5.
Since CAMKIIγ positively regulated c-Myc half-life in our four TCL cell lines (Figure 4B), we interrogated whether this phenomenon involved changes in phosphorylation at T58 or S62. Phosphor-specific antibodies were used to examine the phosphorylation status of c-Myc at T58 or S62. Compared to levels in control cells expressing CAMKIIγ, CAMKIIγ knockdown cells exhibited remarkably lower pS62-c-Myc and total c-Myc; in contrast, cells in which CAMKIIγ was overexpressed exhibited higher levels of pS62-c-Myc and total c-Myc protein levels, but not pT58-c-Myc (Figure 5C). Strikingly, in all cell lines in which CAMKIIγ was overexpressed, the pS62/pT58 c-Myc ratios were remarkably higher, which is consistent with a prolonged c-Myc half-life. Conversely, CAMKIIγ knockdown significantly decreased pS62/pT58 ratios of c-Myc, which is consistent with decreased c-Myc stability (Figure 5D). The results suggest CAMKIIγ stabilizes the c-Myc protein by enhancing S62 phosphorylation of c-Myc.
S62 phosphorylation must precede phosphorylation of T58, which in turn facilitates dephosphorylation at S62 (Pulverer et al., 1994; Sears et al., 1999). As such, S62 mutations may alter this process and, as a result, the turnover of c-Myc. To further examine the impact of CAMKIIγ-mediated S62 phosphorylation on c-Myc turnover, S62D and S62A c-Myc mutants were used. The half-life of the wild-type c-Myc was prolonged by CAMKIIγ overexpression and significantly shortened by CAMK2G deletion (Figure 4E). According to our results and consisting with previous reports, HT-c-MycS62D exhibited prolonged half-lives (>100 min) in cells. Importantly, neither knockout nor overexpression of CAMKIIγ influenced HT-c-MycS62D turnover (Figure 5E). The half-life of c-MycS62A in mammalian cells is reported to be either shorter (Sears et al., 2000) or somewhat longer (Wang et al., 2011b) than the wild-type c-Myc. In our experiments, we observed rapid turnover (~30 min) of HT-c-MycS62A and, as expected, CAMKIIγ knockout or overexpression did not influence its turnover (Figure S5D). These results thus indicate that CAMKIIγ may stabilize c-Myc by enhancing c-Myc phosphorylation at S62.
CAMKIIγ directly phosphorylates c-Myc at S62 in T cell lymphoma
To obtain biochemical evidence for a direct interaction between CAMKIIγ and c-Myc, a co-immunoprecipitation experiment was carried out using H9 cell lysates. C-Myc was co-immunoprecipitated by the CAMKIIγ antibody but not the control IgG (Figure 6A). Like c-Myc, TRIM24 was also high in our upstream analysis list (Table S2); yet no specific interaction was observed between CAMKIIγ and TRIM24 (Figure S6A). An in vitro kinase assay using recombinant CAMKIIγ and c-Myc proteins showed that CAMKIIγ directly phosphorylated c-Myc at S62. Accordingly, a CAMKII-specific inhibitor, CaMKIINtide, diminished pS62-c-Myc (Figure 6B). Moreover, the S62D mutation conferred resistance to KN93-induced downregulation of ectopically expressed c-Myc (Figure 6C). Altogether, these results indicate a role for CAMKIIγ in stabilizing the c-Myc protein by directly phosphorylating them at S62.
Figure 6. CAMKIIγ directly phosphorylates c-Myc in T cell lymphoma.
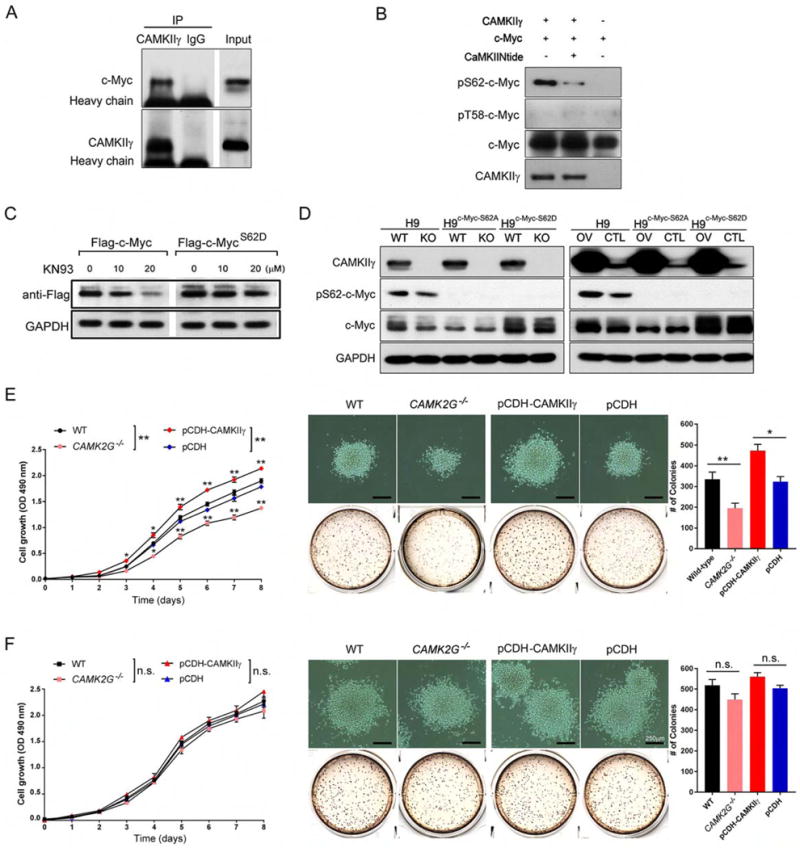
(A) H9 cell lysates were immunoprecipitated with control IgG or a CAMKIIγ antibody, and the immunoprecipitates were then analyzed by western blot analysis with a c-Myc antibody.
(B) After an in vitro CAMKIIγ kinase assay, p-c-Myc (S62) levels were analyzed using western blot analysis.
(C) H9 cells transiently transfected with the indicated vectors were treated with KN93 at the indicated concentrations for 24 hr. FLAG tagged protein expressions were analyzed by western blot analysis. GAPDH was used as a loading control.
(D) Western blot analysis of indicated proteins in the indicated stable TCL cell lines.
(E) Proliferation curve (left), representative images and quantification of the colonies (right) of the indicated H9 stable cell lines at Day 14.
(F) Proliferation curve (left), representative images and quantification of the colonies (right) of the indicated H9c-Myc-S62D stable cell lines at Day 14.
Data represent the mean ± s.d. for three independent experiments. n.s., not significant, *p < 0.05, **p < 0.01. Scale bars, 250 μm. See also Figure S6.
We next examined the relationship between proliferation of TCL and the ability of CAMKIIγ to phosphorylate c-Myc at S62. To this end, we combined in H9 cells knockout/overexpression of CAMKIIγ with S62A or S62D mutant c-Myc. Briefly, we first established two pairs of H9 stable cell lines containing different CAMKIIγ levels: H9 and H9CAMK2G−/−, H9pCDH-CAMKIIγ and H9pCDH We then applied to them CRISPR/Cas technology to establish c-MycS62D or c-MycS62A mutant cell lines (Figure 6D). H9 cells with wild-type c-Myc (c-MycWT) were very sensitive to alterations of CAMKIIγ status: H9 cells exhibited impaired proliferation and colony formation whereas CAMKIIγ overexpression significantly enhanced proliferation and colony formation (Figures 6E). Compared to H9 cells with c-MycWT, the set of c-MycS62D mutant H9 cell lines exhibited substantially enhanced proliferation as well as more and enlarged colonies (Figures 6F). In contrast, c-MycS62A significantly impaired proliferation and reduced numbers and sizes of colonies (Figures S6B and S6C). Importantly, in both c-MycS62D and c-MycS62A, cells were much less sensitive to alterations of CAMKIIγ status compared to c-MycWT: In the set of c-MycS62D mutant cell lines, neither knockout nor overexpression of CAMKIIγ influenced abilities of proliferation and colony formation significantly (Figures 6E and 6G); similarly, CAMKIIγ knockout/overexpression in c-MycS62A mutant also did not influence the abilities of proliferation and colony formation significantly (Figures S6B and S6C). Therefore, functions of CAMKIIγ: c-Myc axis in promoting TCL cell growth are dependent on S62 phosphorylation by CAMKIIγ.
Pharmacological inhibition of CAMKIIγ destabilizes the c-Myc protein, and efficiently suppresses T cell lymphomagenesis and reduces tumor burden
C-Myc is broadly implicated in human cancers yet is considered “undruggable” (Toyoshima et al., 2012). Our results suggest that CAMKIIγ inhibitors have therapeutic potential for c-Myc- overexpressing cancers. However, KN93, which is a widely used specific inhibitor for CAMKII, is not a good candidate for clinical use (Pellicena and Schulman, 2014). We therefore examined BBM, which we identified previously as a specific pharmacological inhibitor of CAMKIIγ (Gu et al., 2012). We first treated H9 cells for 24 hr with various concentrations of BBM and used the MTS assay to measure cell proliferation. BBM suppressed the growth of H9 cells (IC50 = 22.7 μM) (Figure S7A); it also decreased, in a time-dependent manner, levels of p-CAMKIIγ and CAMKIIγ protein (Figure S7B). To assess the inhibitory effects of BBM on TCL development in vivo, MNU-injected wild-type or CAMK2g−/− mice were treated with or without BBM. Based on previously observed time courses for lymphoma to develop (Joshi and Frei, 1970), we administered BBM during two 14-day treatment periods separated by 14 days (Figure S7C). Only vehicle-treated wild-type, but not CAMK2g−/− mice, developed significantly enlarged thymuses, spleens, and lymph nodes. Importantly, in these wild-type mice, BBM treatment dramatically reduced (compared to the vehicle control group) the lymphoma size and cellularity of the infiltrated thymuses, spleens, and lymph nodes (Figures 7A–7B, and Figures S7D–S7E). Moreover, BBM-treated wild-type mice displayed strong resistance to MNU-induced lymphoma regarding the disease symptoms, histological severity, and significant prolonged survival (Figure 7C). In contrast, there were no obvious differences between the vehicle control group and BBM treatment group in CAMK2g−/− mice (Figure S7F). As expected, BBM treatment significantly decreased pS62-c-Myc level and c-Myc protein levels (Figure 7D).
Figure 7. Pharmacological inhibition of CAMKIIγ destabilizes the c-Myc protein and effectively reduces tumor burden.
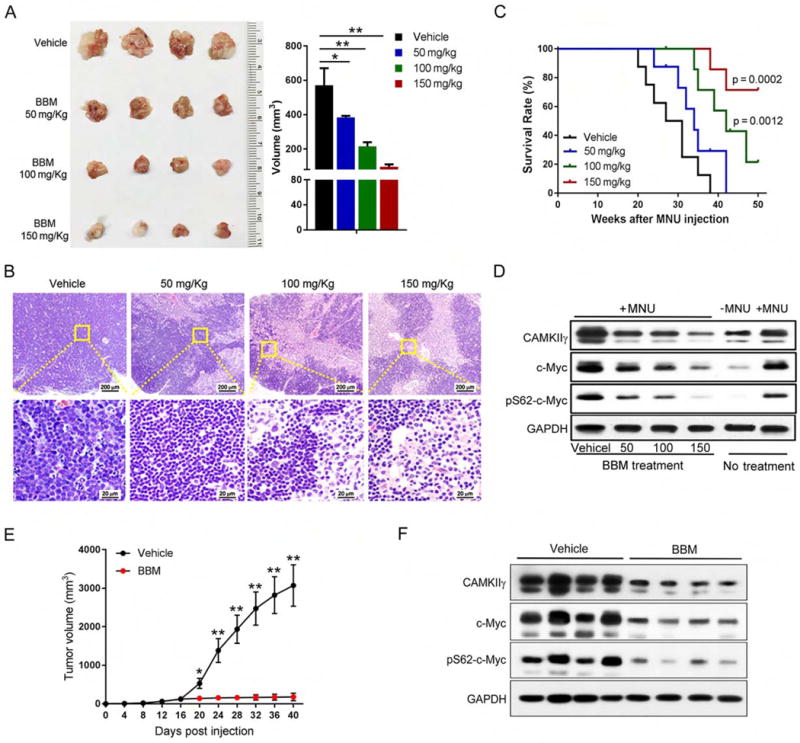
(A) Thymuses obtained from mice suffering MNU-induced lymphoma treated as indicated (n = 4).
(B) Paraffin sections of thymuses in (A) analyzed by H&E staining.
(C) Kaplan-Meier survival curves of mice suffering MNU-induced lymphoma treated as indicated (n = 8 mice per group).
(D) Indicated proteins in tissue lysates from thymuses in (A) were detected using the Western blot analysis. Tissue lysates from wild-type thymuses with or without MNU injection only were shown as expression controls.
(E) Tumor volume comparison between indicated treatment groups at indicated time points after cell inoculation (n = 6 mice per group).
(F) Indicated proteins in xenografted tumor lysates from indicated treatment groups were detected using the Western blot analysis.
Data represent the mean ± s.d. *p < 0.05, **p < 0.01. See also Figures S7 and S8.
We also examined the effect of CAMKIIγ inhibition in a pre-established tumor model. Briefly, 5 × 106 H9 cells were inoculated subcutaneously in the flank of NOD.Cg-Prkdcscid I12rgtm1Wjl/SzJ (NSG) mice. After the xenografted tumors reached a volume of ~100 mm (day 16 after cell injection), they were randomized to two groups to receive either vehicle or BBM treatment via oral gavage twice a day for 23 days (Figure S8A). A dramatic reduction of tumor volume was observed with the administration of BBM compared with that treated with vehicle (Figure 7E and Figure S8B). On day 40, all mice were euthanized and tumor tissues processed for H&E staining and immunostained for Ki-67 and TUNEL. There was no significant difference in body weight between mice in these two treatment groups, suggesting low toxicity of BBM (Figure S8C). Compared to the vehicle control group, the BBM-treatment group exhibited a significant decrease in c-Myc or pS62-c-Myc levels in the xenografted tumors (Figure 7F). In BBM-treated mice, a greater number of dead cells and decreased number of proliferating cells were observed (Figures S8D and S8E). Based on these results, targeting CAMKIIγ in TCL may be a rational therapeutic strategy.
C-Myc and CAMKIIγ overexpress in human T cell lymphoma with a positive correlation
We next investigate the potential correlation between CAMKIIγ and c-Myc/pS62-c-Myc in human TCL. We measured their protein levels in 32 TCL patients’ specimens representing three subtypes of human TCL: angioimmunoblastic T cell lymphoma (AITL, n = 10); peripheral T cell lymphoma (PTCL)-NOS, (n = 11); NK/T cell lymphoma (NK/TCL, n = 11); and benign lymph nodes specimens (n = 9). As expected, compared to the benign lymph node tissues, CAMKIIγ was highly expressed in the three TCL subtypes (Figures 8A and 8B; table S3). In accordance with a previous report (Chisholm et al., 2015), c-Myc and pS62-c-Myc expression levels were also significantly higher (compared to benign lymph nodes) in all three lymphoma subtypes (Figures 8A and 8B; tables S4 and S5). More importantly, there was a positive correlation between CAMKIIγ and c-Myc expression (R2 = 0.807, p < 0.01), as well as pS62-c-Myc (R2 = 0.826, p < 0.01; Figure 8C). The results are consistent with a role for CAMKIIγ in c-Myc overexpression in human TCL.
Figure 8. Positive correlations of protein expression among CAMKIIγ, c-Myc and pS62-c-Myc in human T cell lymphoma tissues.
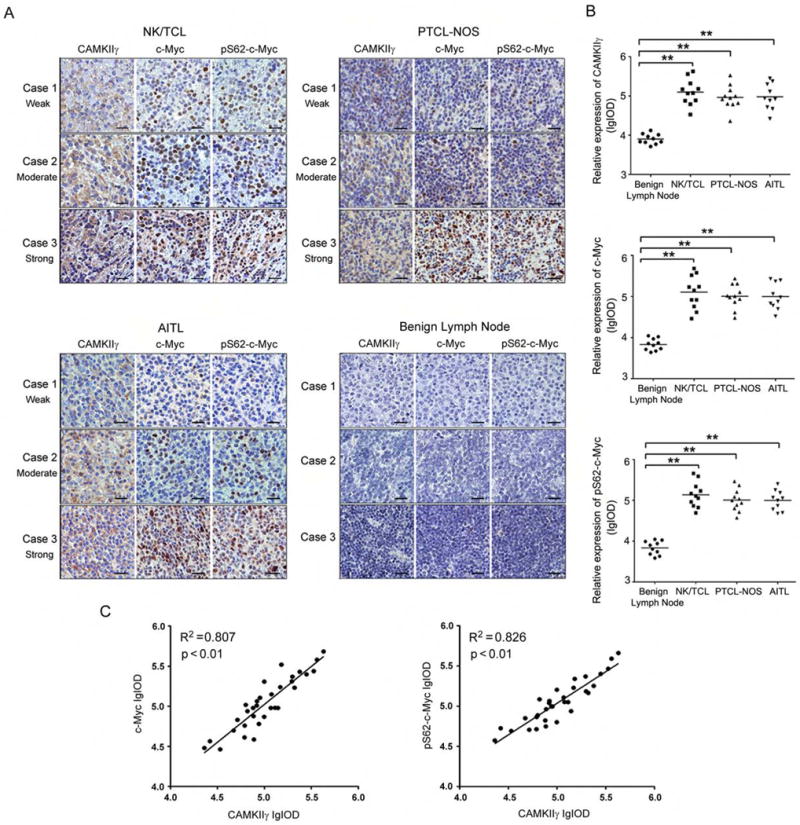
(A) Representative images of IHC staining of CAMKIIγ, c-Myc, and pS62-c-Myc in human TCL tissues and benign lymph node tissues. Three representative TCL sections from 3 individual cases were compared for weak (Case 1), moderate (Case 2), strong (Case 3) for each subtype. Three representative benign lymph node tissue sections from 3 individual cases were used as control. Scale bars, 20 μm.
(B) Protein expressions in (A) assessed as lg (Common logarithm: base 10) integrated optical density (IOD) scores. **p < 0.01. The horizontal lines shown in graphs represented median of each protein expression.
(C) The correlation between CAMKIIγ, c-Myc, or pS62-c-Myc expression in human TCL (n = 32). Correlation is shown using R2 and significance was determined using a Spearman correlation.
DISCUSSION
MYC encodes a transcription factor that is overexpressed in ~70% of human cancers and drives unrestrained cell growth and proliferation. Approximately 20% of such c-Myc cancers, such as Burkitt lymphoma and breast cancer (Nesbit et al., 1999), exhibit MYC translocations or amplifications. Such genomic alterations may help explain the high expression of the c-Myc protein in these tumors. In other cancers, however, overexpression of the c-Myc protein likely depends on alternative mechanisms. Indeed, a recent report on expression profiling of the c-Myc protein and MYC rearrangements in 1214 lymphomas indicates that the c-Myc protein is overexpressed in about 94% of TCL; importantly, none possess MYC rearrangements (Chisholm et al., 2015). Our current study provides such an alternative mechanism. That is, in lymphomas without MYC rearrangements, the CAMKIIγ c-Myc axis may contribute to c-Myc-associated tumorigenesis.
In our study, we used two mouse models to establish a molecular link between CAMKIIγ and T cell lymphomagenesis: (1) the MNU-induced murine TCL model, in which a nitroso compound MNU induces thymic lymphomas in mice (Dumenco et al., 1993; Joshi and Frei, 1970; Slee and Lu, 2013); (2) the Notch1-ΔE-driven murine T cell leukemia/lymphoma model, which is well characterized and relatively fast (Herranz et al., 2014; Schroeter et al., 1998). In both models, CAMKIIγ upregulates c-Myc at the protein level, instead of enhancing gene transcription or mRNA stability (Cole and Cowling, 2008; Dai and Lu, 2008; van Riggelen et al., 2010). Previous studies have demonstrated that alteration of S62 and T58 phosphorylation levels with consequent increased c-Myc stability in tumors could explain the observed c-Myc overexpression in the absence of gene amplification (Farrell and Sears, 2014). In many cancers, the c-Myc protein exhibits high S62 phosphorylation, low T58 phosphorylation, and is aberrantly stabilized (Malempati et al., 2006; Zhang et al., 2012). This could be due to a deregulated proteolysis through the ubiquitin-proteasome pathway (Lim and Elenitoba-Johnson, 2004; Thomas and Tansey, 2011). Consistently, our study shows that CAMKIIγ stabilizes the c-Myc protein by directly phosphorylating c-Myc at S62. We conclude that CAMKIIγ is a direct upstream regulator of the c-Myc protein levels in TCL. We go on to show that the functions of CAMKIIγ: c-Myc axis are dependent on phosphorylating c-Myc at S62 by CAMKIIγ specifically, suggesting CAMKIIγ through c-Myc is responsible for maintaining malignant growth of TCL cells.
Although there has been some progress in TCL research (Crescenzo and Inghirami, 2015), TCL is known for its poorly understood mechanisms and unsatisfying treatment outcomes (Barrett et al., 2014; Lim and Levy, 2014; Piccaluga et al., 2007). Therefore, it is urgent to identify potential therapeutic targets for these diseases. C-Myc inactivation in established tumors is sufficient to induce tumor regression through processes such as proliferative arrest, cellular senescence, apoptosis, and the shutdown of angiogenesis (Giuriato et al., 2006; Wu et al., 2007). While c-Myc is considered a poor therapeutic target, our findings indicate that CAMKIIγ could be an alternative target for TCL treatment. Our previous study has shown that CAMKIIγ is a target of the natural compound BBM, which is isolated from the traditional Chinese herbal medicine Berberis amurensis (Dong et al., 2004; Xie et al., 2009). In our current studies, BBM has displayed potent effects to suppress TCL growth in mice and human TCL cells. The scaffold thus holds potential as a pharmacological agent for treating TCL. In summary, our study indicates that CAMKIIγ is a plausible molecular target for the treatment of TCL; and that BBM or its derivatives (Nam et al., 2012) may have clinical usefulness in TCL treatment. Furthermore, our results raise the intriguing possibility of targeting CAMKIIγ to treat other c-Myc-associated malignancies with the same underlying mechanism.
STAR★Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Wendong Huang (whuang@coh.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
Human TCL cell line H9 (source: male) were ordered from the American Type Culture Collection. Human TCL cell lines JB6 (source: male), Jurkat (source: male), and SU-DHL-1 (source: male) were obtained from Dr. Wing-Chung (John) Chan’s laboratory (City of Hope medical center). All cells were cultured in RPMI-1640 supplemented with 10% fetal calf serum at 37 °C in a 95% air, 5% CO2 humidified incubator.
In Vivo Mouse Studies
Female 4–5 week old Camk2g−/− mice were obtained from Dr. Eric N. Olson’s laboratory (University of Texas Southwestern Medical Center) and Dr. Johannes Backs’s laboratory (University of Heidelberg). Female 8 week old NSG mice were obtained from Dr. Jun Wu’s laboratory (City of Hope medical center). All research animals were housed in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)- accredited facility, which is located at our institution (City of Hope medical center). Animals were given ad-libitum access to water and standard rodent chow in a temperature- controlled environment. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of City of Hope Medical Center, and were conducted according to federal, state, and local regulations under the supervision of trained veterinarians.
MNU-induced Murine Lymphoma Model
N-methyl-N-nitrosourea (MNU) is a well-known carcinogen that induces TCL in mice. MNU causes neoplasia by alkylation and/or methylation-mediated mutagenesis of nucleic acids, proteins, and lipids. Two groups of C57BL/6J mice—wild-type and Camk2g−/− at 4–5 weeks of age—were given a single intraperitoneal injection of MNU (75 mg/kg) on Day 1 followed by an observation period; and the mice were observed weekly for tumor formation. MNU (Sigma) was dissolved in cold PBS immediately before treatment and injected within 1 hr.
BBM Treatment for MNU-induced Murine Lymphoma
BBM was dissolved in pure sterile water. Mice were randomly assigned to experimental groups as illustrated in Figure S7C. Administration of BBM was divided into 3 stages: (1) At day 40 after MNU injection, 50, 100, or 150 mg/kg BBM, or vehicle only, was orally administered to mice 2 times a day for 14 days; (2) administration was interrupted for 14 days; (3) at day 68 after MNU injection, 50, 100, or 150 mg/kg BBM, or vehicle only, was again administered to mice 2 times a day for 14 days.
Sizes of the thymuses, spleens, and lymph nodes were calculated using the equation Volume (mm3) = ½ ab2, where a = length and b = width of each organ.
Murine Xenograft Model
5 × 106 H9 cells were inoculated subcutaneously in the right flank of female NSG mice. After the xenografted tumors reached a volume of ~100 mm3, they were randomized to two groups to receive either vehicle or BBM treatment via oral gavage twice a day. Tumors were measured every other day with Vernier calipers and mice were euthanized and sacrificed when tumor volume exceeded 3000 mm3 in any dimension.
Murine Bone Marrow Transduction/Transplantation Model
The retroviral vector MSCV-IRES-eGFP carrying the activated form of the Notch1 (Notch1-ΔE) cDNA was used to make high-titer, helper-free, replication-defective ecotropic virus stock by transient transfection of 293T cells. Six- to eight-week-old female wild-type or Camk2g−/− C57BL/6 mice were used as donor mice for tumorigenesis experiments. Bone marrow from 5- fluorouracil (5-FU) -treated (150 mg/kg) donor mice was transduced two times with Notch1-ΔE retrovirus by cosedimentation in the presence of interleukin-3 (IL-3), IL-6 and stem cell factor. Syngeneic wild-type recipient mice were prepared by 1300 cGy γ-irradiation and a dose of 0.5 × 106 transfected cells transplanted via tail vein injection. After transplantation, recipient mice were evaluated daily for signs of morbidity, weight loss and failure to thrive.
Patient Specimens
The formalin-fixed paraffin-embedded sections of TCL patients were obtained from the Pathology departments (Second Affiliated Hospital of Zhejiang University; and Tianjin Medical University Cancer Institute and Hospital). The research proposal was approved by the Ethics Committee of the Institute of Second Affiliated Hospital of Zhejiang University and Tianjin Medical University Cancer Institute and Hospital.
METHOD DETAILS
Patient Specimens and immunohistochemistry (IHC)
IHC staining and counterstaining were performed by the Pathology core (City of Hope Medical Center). The formalin-fixed paraffin-embedded sections from patients with TCL were stained with either anti-c-Myc rabbit monoclonal antibody (clone Y69, Abcam) or anti-CAMKIIγ mouse monoclonal antibody (clone 8G10C1, Abcam). Positive expression was observed as yellow or brown staining. Image-Pro Plus 6.0 software was used for semi-quantitative analysis by randomly selecting five high-power fields from each slide and was used to calculate the sum integrated optical density (IOD) of the mean density, and the IOD of the positive staining in each field, as well as the mean value of these parameters.
Immunoprecipitation Assay
Cell extracts composed of 1 × 107 cells were prepared by solubilization in 1 ml IP lysis buffer (Thermo) in the presence of protease inhibitor cocktail (Thermo) for 10 min at 4 °C. After a brief sonication step, the lysates were cleared by centrifugation at 15,000 × g for 10 min at 4 °C; and the cell extract was immunoprecipitated with 2 μg of antibodies against CAMKIIγ or IgG control. The mixture was incubated with 60 μl of protein G plus protein A agarose for 16 hr at 4 °C by continuous inversion. Immuno-complexes were pelleted and washed 5 times with IP lysis buffer. The precipitated immuno-complexes were then boiled in lane marker sample buffer (Thermo) and subjected to western blot analysis using an anti-c-Myc antibody.
Mouse Tumor Histopathology and IHC Staining
Tumor specimens were prepared and analyzed by board-certified pathologists. Tumors were fixed in 4% PBS-buffered formalin; dehydrated and embedded in paraffin; and sectioned and processed for hematoxylin and eosin (H&E) staining. The sections were deparaffinized; heated for antigen retrieval; and incubated with the primary antibody CD3 (Abcam) or Ki-67. The secondary antibody was biotinylated with polyclonal anti-rabbit immunoglobulin (Dako) in combination with streptavidin/horseradish peroxidase (Dako). The sections were lightly counterstained using hematoxylin. Microscopy was performed by using an EVOS FL Auto Imaging System (Life technologies) for photomicrographs. Specimens of similarly prepared normal lymph nodes from C57BL/6 mice were used as controls.
CAMK2G Knockout by the CRISPR/Cas System
sgRNA sequences targeting human CAMK2G were designed using the CRISPR on-line design tool (www.genome-engineering.org/crispr). The designed sequence was cloned into the pX330 plasmid (Addgene). The 20 nt guiding sequences targeting exon1 and exon2 of human CAMK2G are shown below: 5′-TGCCAAGCTCCTCGAAGAGC-3′ (target exon1 for clone A), 5′- GTGCTTTCTCTGTGGTCCGC-3′ (target exon2 for clone B). The sgRNA-containing pX330 vector was co-transfected into 293T or H9 cells with the pMAX-GFP plasmid. After 48 hr, green fluorescent protein – positive (GFP+) cells were single-cell sorted by fluorescence-activated cell sorting (FACS) into 96-well plates. Single clones were then expanded and screened by western blot analysis. Genomic DNA was purified from clones using the QIAamp DNA mini kit and underwent sequencing to verify the deletion.
C-Myc Ser62 mutations established by the CRISPR/Cas system
sgRNAs for c-Myc Ser62 knock-in mutations were designed by Zhang laboratory CRISPR design tool ((http://crispr.mit.edu), annealed and ligated into pSpCas9n vecoter (pX461) digested by BbsI. The ssODN repair templates were designed with homologous genomic flanking sequence centered around the predicted CRISPR/Cas9 cleavage site. 500 ng of each pSpCas9n (sgRNA) plasmids with 1 μl of 10 μM ssODN were mixed for nucleofection into H9 cell lines with Cell Line Nucleofector Kit V (LONZA) followed its manual.
Two days after nucleofection, H9 cells were collected, washed by PBS and re-suspended with 1 ml of FACS medium. GFP+ cells were sorted into 96-well plates as single cell per well. The cells were incubated and expanded for 2–3 weeks, and all of the clones were further for genomic DNA extraction, PCR amplication of c-Myc sequence and Sanger sequencing. The correct S62A and S62D knock-in cell clones were selected for further experiments.
Colony Formation Assay
The colony formation assay was carried out using MethoCult™ H4230 methylcellulose medium (80 ml, StemCell Technologies), according to the manufacturer’s instructions. Briefly, thaw the H4230 methylcellulose overnight at 4 °C and add directly in the H4230 bottle with 20 ml Iscove’s Modified Dulbecco’s Medium (IMDM; StemCell Technologies, Cat #36150). Shake vigorously and leave overnight to settle, and then make aliquots. Cells were added to the mixed H4230 methylcellulose at 1000 cells/ml and dispensed to 6-well plates at 1 ml per well in triplicates. Colonies were counted after 14 days of culture at 37 °C.
Cycloheximide (CHX) Chase Assay
Cells were treated with 50 μM cycloheximide (CHX) and harvested at indicated time points. Protein was extracted from the cells and subjected to western blot analysis. Protein levels were measured with the densitometric intensity.
HaloTag (HT) Pulse-Chase Assay
The protein of interesting (POI) was cloned into HaloTag (HT) vector to establish HT-POI vector. Cells (1 × 106 cells in 6-well plates) were transfected with 0.5 μg of HT-POI, and after cultured for 40 hr, cells were incubated with 5 μM HaloTag-tetramethylrhodamine (TMR) ligand (Promega) for 15 min to allow the pulse labeling of HT-POI, and washed twice with PBS. After the incubation for the indicated time, the cells were washed twice with PBS and suspended in lysis buffer. The whole cell extract was subjected to SDS-PAGE, and the TMR-labeled HT-POI was visualized with a fluoro-image analyzer FLA-3000G (FUJI FILM, Tokyo, Japan).
35S-methionine Pulse-Chase Assay
Cells were washed by Methionine/Cysteine-free cell culture medium (Sigma, R7513) twice, and then pulse-labeled with Pulse medium containing 35S-Methionine/Cysteine (35S-Met, 0.1 mCi/ml, Perkin Elmer) for 30 min at 37 °C. The labeling was stopped by replacing the Pulse medium with regular cell culture medium. Cells were collected at the indicated time points. Whole cell extracts were incubated with protein A/G agarose beads (Thermo) pre-coupled with c-Myc antibody for 4 hr at 4 °C to purify the c-Myc protein. Precipitates were separated by SDS-PAGE and proteins were detected by autoradiography.
Apoptosis Assay
The terminal deoxynucleotidyltransferase–mediated dUTP nick end labeling (TUNEL) staining was carried out on xenograft sections using the In Situ Cell Death Detection Fluorescein Kit (ROCHE), according to the manufacturer’s instructions. Briefly, slides containing paraffin embedded tissues were de-waxed in xylene and graded ethanol series, then digested with 15 μg/ml Proetinase K solution for 20 min. After wash in PBS, the slides were further incubated with TUNEL reaction mixture at 37 °C for 30 min. After washing in PBS for 3 times, slides was mounted with mounting medium contain DAPI (Invitrogen) and imaged with Zeiss LSM510 confocal microscope.
Luciferase Reporter Assay
PBV-Luc wt MBS1–4 plasmids (Addgene: Plasmid #16564) or negative control reporter pBV- Luc plasmids (Addgene: Plasmid #16539) were co-transfected in H9 cells together with β-gal plasmid. After washing in PBS, the cells were lysed in Passive Lysis Buffer, followed by measurement of reporter activity according to the manufacturer’s instructions (Promega). Relative Luc values (Luc/Gal) were used to normalize for transfection efficiency in all experiments.
Cell Cycle Distribution Assay
Cells were washed by PBS and fixed with 70% ethanol for 24 hr at −20 °C. Then cells were centrifuge for 5 min at 200 × g, washed by PBS, and incubated with propidium iodide (PI)/Tirton X-100 staining solution with RNase A (Sigma) for 2–4 hr at room temperature. The samples were sent for FACS analysis (FACScan, BD). Acquisition and analyses were performed using the CellQuest (BD) software.
In vitro Kinase assay
For the in vitro CAMKIIγ kinase assay, human recombinant CAMKIIγ protein was pre-activated in a reaction containing calcium, calmodulin, and ATP with or without CaMKIINtide for 10 min at 30 °C. Human recombinant c-Myc protein was added for 5 min and quenched with lane marker sample buffer. PS62-c-Myc was examined using western blot analysis.
In vivo ubiquitination assay
H9CAMK2G−/− cells were co-transfected with HA-c-Myc, Flag-CAMKIIγ and GFP-tagged ubiquitin. Cells were pre-treated with MG132 (Calbiochem) for 6 hr at a concentration of 10 μM or without treatment. Cell extracts were prepared using RIPA buffer. We used monoclonal antiFlag agarose beads to purify the transfected c-Myc protein. Precipitates were analyzed by western blotting with anti-GFP to detect ubiquitinylated c-Myc protein.
Survival/Proliferation assay
The MTS assay, which measures cell survival, was conducted with the CellTiter 96 Aqueous Cell Proliferation Kit (Promega). The IC50 was defined as the drug concentration that induced a 50% viability decrease.
Western Blot Analysis
Cell specimens were washed twice with PBS buffer; and total cellular protein was extracted using Radio-Immunoprecipitation Assay buffer (RIPA). Cell extracts were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 10% polyacrylamide gels); and then transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad) and blocked with 5% nonfat milk (Bio-Rad) in TBS–Tween 20 (TBST). The membranes were then reacted with primary antibodies overnight at 4 °C. After 3 washes with TBST, membranes were probed with a horseradish peroxidase-conjugated secondary antibody for 1 hr at room temperature, and reacted with SuperSignal West Pico Chemiluminescent Substrate (Thermo).
Real-time PCR
Total RNA (1 mg), collected by the RNeasy kit (QIAGEN), was used in a reverse transcriptase reaction with the SuperScript® III Reverse Transcriptase kit (Life Technologies). The SYBR Green Real-Time PCR Master Mixes kit (Life Technologies) was used for the thermocycling reaction in an ABI-7500 RealTime PCR machine (Applied Biosystems). The qPCR analysis was carried out in triplicate with the following primer sets:
C-Myc (Forward: 5′-CCCTATTTCATCTGCGACGAG-3′; Reverse: 5′- GAGAAGGACGTAGCGACCG-3′),
m36b4 (Forward: 5′-GCCCTGCACTCTCGCTTTCT-3′; Reverse: 5′- CAACTGGGCACCGAGGCAACAGTTG-3′).
All gene expression studies were repeated in independent experiments.
RNA-seq Library Preparation and Sequencing
RNA quality was determined with an Agilent Bioanalyzer (RNA integrity number [RIN] > 7.5 for all samples). RNA-sequencing was performed according to the manufacturer’s protocol using the Illumina TruSeq RNA Sample Preparation Kit V2 (San Diego, CA). Briefly, 500 ng of each total RNA sample was used for poly (A) mRNA selection and fragmentation, followed by first and second strand synthesis, end repair, adenylation of the 3′ ends, and adapter ligation. The ligated material was amplified by PCR and then purified. The final library was 200–500 bp with a peak at approximately 280 bp. Libraries were loaded on an Illumina HiSeq 2500 for parallel sequencing.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq Data Analysis
The 51 bp-long single-ended sequence reads were mapped to the human genome (hg19) using TopHat; and the frequency of RefSeq genes detected was counted with customized R scripts. The raw counts were then normalized using the trimmed mean of M values (TMM) method implemented in the Bioconductor package edgeR. Low expression genes with rpkm values less than 5 in all samples were excluded from the differential expression analysis. Differentially expressed genes were identified by exact tests using edgeR, with an absolute fold-change ≥ 1.5 as the cut off value. GO category enrichment analysis was performed with DAVID (Huang da et al., 2009), gene set enrichment analysis was performed in GSEA (Broad Institute) and upstream regulator analysis was generated through the use of QIAGEN’s Ingenuity Pathway Analysis (IPA, QIAGEN Redwood City).
Statistical Analysis
Student’s t-test (two-sided) was applied, and changes were considered statistically significant for p < 0.05. For intercomparison of more than 2 groups, a one-way ANOVA followed by a post-hoc test was applied. To assess the association between c-Myc and CAMKIIγ, Pearson correlation analysis was used. In the figures, changes are noted using *p < 0.05 and **p < 0.01. The data were normally distributed and variation within and between groups was not estimated. The sample size was not pre-selected and no inclusion/exclusion criteria were used. Survival in mouse experiments was represented with Kaplan-Meier curves, and significance was estimated with the lop-rank test (Prism GraphPad). The data shown in the bar graphs are the mean and s.d. of at least three biological replicates. Statistical analysis was conducted using the Microsoft Excel or GraphPad Prism software packages.
DATA AND SOFTWARE AVAILABILITY
Data Resources
RNA-seq data files have been deposited in the NCBI Gene Expression Omnibus under accession number GEO: GSE99364.
Supplementary Material
SIGNIFICANCE.
Rearrangements of MYC are rarely observed in certain cancers, including TCL in which the c-Myc protein is overexpressed. Our cumulative findings provide an explanation for this phenomenon, CAMKIIγ appears to be a kinase essential for maintaining the c-Myc protein level. Thus the CAMKIIγ: c-Myc axis appears to fundamentally support the development of TCL. The mechanism for TCL development is not fully understood and TCL in general has poor treatment outcomes. Because c-Myc is an “undruggable” target, it is essential to identify important c-Myc regulators that can be targeted. Here we identify CAMKIIγ as such a regulator and show that a CAMKIIγ-specific inhibitor potently eliminates TCL burden with minimal toxicity. Therefore, these studies define a potential therapeutic approach for treating TCL.
HIGHLIGHTS.
CAMKIIγ: c-Myc axis is essential for T cell lymphomagenesis
CAMKIIγ stabilizes the c-Myc protein by direct phosphorylation it at S62 in TCL
Targeting CAMKIIγ destabilizes the c-Myc protein and reduces tumor burden
C-Myc and CAMKIIγ overexpress in human TCL with a positive correlation
Acknowledgments
We thank Dr. Eric N. Olson from University of Texas Southwestern Medical Center and Dr. Johannes Backs from University of Heidelberg for providing the CAMK2g−/− mice. We thank Dr. Adolfo A. Ferrando from Columbia University for providing the MSCV-IRES-eGFP-Notch1-ΔE plasmid. We also thank Dr. Ian Talisman for his help in manuscript editing and proofreading. This work was supported in part by Toni Stephenson Lymphoma Center Pilot Grant, Tim Nesvig Lymphoma Pilot award, and NCI 2R01CA139158 from the National Cancer Institute. R.X. is supported by National Natural Science Foundation of China (81470306 and 81670138), and Science Technology Department of Zhejiang Province (2016C33096). L.L. is supported by the STOP CANCER Foundation RESEARCH CAREER DEVELOPMENT Award, the The Margaret E. Early Medical Research Trust Award, and the National Cancer Institute grants (R00 CA184411).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
W.H. and R.X. designed and supervised most of the experiments and wrote the manuscript. Y.G. and J.Z. contributed greatly to experimental design and performed most of the experiments and data analysis. X.M. performed knockout cells experiments. B.K. performed FCM experiments. H.W., J.L., Y.P., and Y.X. provided primary tumor samples. L.D. performed animal experiments. L.Y., C.G. and X.W. performed RNA-seq data analysis. J.W. performed murine xenograft experiments. K.W., X.G., and G.L. quantified tumor immunohistochemistry. S.F., L.L., and WC. provided conceptual advice and technical assistance for some experiments.
The authors declare no conflicts of interest.
References
- Backs J, Stein P, Backs T, Duncan FE, Grueter CE, McAnally J, Qi X, Schultz RM, Olson EN. The gamma isoform of CaM kinase II controls mouse egg activation by regulating cell cycle resumption. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:81–86. doi: 10.1073/pnas.0912658106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annual review of medicine. 2014;65:333–347. doi: 10.1146/annurev-med-060512-150254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui JD, Calbo S, Hayden-Martinez K, Kane LP, Gardner P, Hedrick SM. A role for CaMKII in T cell memory. Cell. 2000;100:457–467. doi: 10.1016/s0092-8674(00)80681-9. [DOI] [PubMed] [Google Scholar]
- Chisholm KM, Bangs CD, Bacchi CE, Molina-Kirsch H, Cherry A, Natkunam Y. Expression profiles of MYC protein and MYC gene rearrangement in lymphomas. The American journal of surgical pathology. 2015;39:294–303. doi: 10.1097/PAS.0000000000000365. [DOI] [PubMed] [Google Scholar]
- Choi PS, Li Y, Felsher DW. Addiction to multiple oncogenes can be exploited to prevent the emergence of therapeutic resistance. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E3316–3324. doi: 10.1073/pnas.1406123111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MD. The myc oncogene: its role in transformation and differentiation. Annual review of genetics. 1986;20:361–384. doi: 10.1146/annurev.ge.20.120186.002045. [DOI] [PubMed] [Google Scholar]
- Cole MD, Cowling VH. Transcription-independent functions of MYC: regulation of translation and DNA replication. Nature reviews Molecular cell biology. 2008;9:810–815. doi: 10.1038/nrm2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colomer J, Means AR. Physiological roles of the Ca2+/CaM-dependent protein kinase cascade in health and disease. Sub-cellular biochemistry. 2007;45:169–214. doi: 10.1007/978-1-4020-6191-2_7. [DOI] [PubMed] [Google Scholar]
- Crescenzo R, Inghirami G. Anaplastic lymphoma kinase inhibitors. Current opinion in pharmacology. 2015;23:39–44. doi: 10.1016/j.coph.2015.05.010. [DOI] [PubMed] [Google Scholar]
- Dai MS, Lu H. Crosstalk between c-Myc and ribosome in ribosomal biogenesis and cancer. Journal of cellular biochemistry. 2008;105:670–677. doi: 10.1002/jcb.21895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong QH, Zheng S, Xu RZ, Lu Q, He L. Study on effect of berbamine on multidrug resistance leukemia K562/Adr cells. Zhongguo Zhong xi yi jie he za zhi Zhongguo Zhongxiyi jiehe zazhi = Chinese journal of integrated traditional and Western medicine/Zhongguo Zhong xi yi jie he xue hui, Zhongguo Zhong yi yan jiu yuan zhu ban. 2004;24:820–822. [PubMed] [Google Scholar]
- Dumenco LL, Allay E, Norton K, Gerson SL. The prevention of thymic lymphomas in transgenic mice by human 06-alkylguanine-DNA alkyltransferase. Science. 1993;259:219–222. doi: 10.1126/science.8421782. [DOI] [PubMed] [Google Scholar]
- Farrell AS, Sears RC. MYC degradation. Cold Spring Harbor perspectives in medicine. 2014;4 doi: 10.1101/cshperspect.a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Chen T, Meng Z, Gan Y, Xu X, Lou G, Li H, Gan X, Zhou H, Tang J, et al. CaMKII gamma, a critical regulator of CML stem/progenitor cells, is a target of the natural product berbamine. Blood. 2012;120:4829–4839. doi: 10.1182/blood-2012-06-434894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz D, Ambesi-lmpiombato A, Palomero T, Schnell SA, Belver L, Wendorff AA, Xu L, Castillo-Martin M, Llobet-Navas D, Cordon-Cardo C, et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nature medicine. 2014;20:1130–1137. doi: 10.1038/nm.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook SS, Means AR. Ca(2+)/CaM-dependent kinases: from activation to function. Annual review of pharmacology and toxicology. 2001;41:471–505. doi: 10.1146/annurev.pharmtox.41.1.471. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Joshi VV, Frei JV. Gross and microscopic changes in the lymphoreticular system during genesis of malignant lymphoma induced by a single injection of methylnitrosourea in adult mice. Journal of the National Cancer Institute. 1970;44:379–394. [PubMed] [Google Scholar]
- Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, Al-Haddawi M, Tan SY, Osato M, Sabo A, et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature. 2015;523:96–100. doi: 10.1038/nature14351. [DOI] [PubMed] [Google Scholar]
- Lim MS, Elenitoba-Johnson KS. Ubiquitin ligases in malignant lymphoma. Leukemia & lymphoma. 2004;45:1329–1339. doi: 10.1080/10428190410001663635. [DOI] [PubMed] [Google Scholar]
- Lim SH, Levy R. Translational Medicine in Action: Anti-CD20 Therapy in Lymphoma. J Immunol. 2014;193:1519–1524. doi: 10.4049/jimmunol.1490027. [DOI] [PubMed] [Google Scholar]
- Lin MY, Zal T, Ch’en IL, Gascoigne NR, Hedrick SM. A pivotal role for the multifunctional calcium/calmodulin-dependent protein kinase II in T cells: from activation to unresponsiveness. J Immunol. 2005;174:5583–5592. doi: 10.4049/jimmunol.174.9.5583. [DOI] [PubMed] [Google Scholar]
- Malempati S, Tibbitts D, Cunningham M, Akkari Y, Olson S, Fan G, Sears RC. Aberrant stabilization of c-Myc protein in some lymphoblastic leukemias. Leukemia. 2006;20:1572–1581. doi: 10.1038/sj.leu.2404317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Li T, Ma X, Wang X, Van Ness C, Gan Y, Zhou H, Tang J, Lou G, Wang Y, et al. Berbamine inhibits the growth of liver cancer cells and cancer-initiating cells by targeting Ca(2)(+)/calmodulin-dependent protein kinase II. Molecular cancer therapeutics. 2013;12:2067–2077. doi: 10.1158/1535-7163.MCT-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam S, Xie J, Perkins A, Ma Y, Yang F, Wu J, Wang Y, Xu RZ, Huang W, Horne DA, Jove R. Novel synthetic derivatives of the natural product berbamine inhibit Jak2/Stat3 signaling and induce apoptosis of human melanoma cells. Molecular oncology. 2012;6:484–493. doi: 10.1016/j.molonc.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- Nowycky MC, Thomas AP. Intracellular calcium signaling. Journal of cell science. 2002;115:3715–3716. doi: 10.1242/jcs.00078. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nature reviews Molecular cell biology. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Frontiers in pharmacology. 2014;5:21. [Google Scholar]
- Piccaluga PP, Agostinelli C, Califano A, Rossi M, Basso K, Zupo S, Went P, Klein U, Zinzani PL, Baccarani M, et al. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. The Journal of clinical investigation. 2007;117:823–834. doi: 10.1172/JCI26833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast GC. Mechanisms of apoptosis by c-Myc. Oncogene. 1999;18:2967–2987. doi: 10.1038/sj.onc.1202727. [DOI] [PubMed] [Google Scholar]
- Pulverer BJ, Fisher C, Vousden K, Littlewood T, Evan G, Woodgett JR. Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene. 1994;9:59–70. [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich RC, Schulman H. Substrate-directed function of calmodulin in autophosphorylation of Ca2+/calmodulin-dependent protein kinase II. The Journal of biological chemistry. 1998;273:28424–28429. doi: 10.1074/jbc.273.43.28424. [DOI] [PubMed] [Google Scholar]
- Rossi M, Laginestra MA, Gazzola A, Sapienza MR, Pileri SA, Piccaluga PP. Molecular profiling of aggressive lymphomas. Advances in hematology. 2012;2012:464680. doi: 10.1155/2012/464680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Molecular cell. 1999;3:169–179. doi: 10.1016/s1097-2765(00)80308-1. [DOI] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes & development. 2000;14:2501–2514. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si J, Collins SJ. Activated Ca2+/calmodulin-dependent protein kinase llgamma is a critical regulator of myeloid leukemia cell proliferation. Cancer research. 2008;68:3733–3742. doi: 10.1158/0008-5472.CAN-07-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slee EA, Lu X. Requirement for phosphorylation of P53 at Ser312 in suppression of chemical carcinogenesis. Scientific reports. 2013;3:3105. doi: 10.1038/srep03105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochemical and biophysical research communications. 1991;181:968–975. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- Thomas LR, Tansey WP. Proteolytic control of the oncoprotein transcription factor Myc. Advances in cancer research. 2011;110:77–106. doi: 10.1016/B978-0-12-386469-7.00004-9. [DOI] [PubMed] [Google Scholar]
- Toyoshima M, Howie HL, Imakura M, Walsh RM, Annis JE, Chang AN, Frazier J, Chau BN, Loboda A, Linsley PS, et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:9545–9550. doi: 10.1073/pnas.1121119109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nature reviews Cancer. 2010;10:301–309. doi: 10.1038/nrc2819. [DOI] [PubMed] [Google Scholar]
- Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011a;10:57–67. doi: 10.4161/cc.10.1.14449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Cunningham M, Zhang X, Tokarz S, Laraway B, Troxell M, Sears RC. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer research. 2011b;71:925–936. doi: 10.1158/0008-5472.CAN-10-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Ma T, Gu Y, Zhang X, Qiu X, Zhang L, Xu R, Yu Y. Berbamine derivatives: a novel class of compounds for anti-leukemia activity. European journal of medicinal chemistry. 2009;44:3293–3298. doi: 10.1016/j.ejmech.2009.02.018. [DOI] [PubMed] [Google Scholar]
- Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, Cai W, Yang G, Bronson R, Crowley DG, et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature. 2014;514:380–384. doi: 10.1038/nature13589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Farrell AS, Daniel CJ, Arnold H, Scanlan C, Laraway BJ, Janghorban M, Lum L, Chen D, Troxell M, Sears R. Mechanistic insight into Myc stabilization in breast cancer involving aberrant Axin1 expression. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2790–2795. doi: 10.1073/pnas.1100764108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.