

Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disease, and there are no mechanism-based therapies. AD is defined by the presence of abundant neurofibrillary lesions and neuritic plaques in cerebral cortex. Neurofibrillary lesions are made of paired helical and straight Tau filaments (PHFs and SFs), whereas Tau filaments with different morphologies characterize other neurodegenerative diseases. No high-resolution structures of Tau filaments are available. Here we present cryo-electron microscopy (cryo-EM) maps at 3.4–3.5 Å resolution and corresponding atomic models of PHFs and SFs from AD brain. Filament cores are made of two identical protofilaments comprising residues 306–378 of Tau, which adopt a combined cross-β/β-helix structure and define the seed for Tau aggregation. PHFs and SFs differ in their inter-protofilament packing, showing that they are ultrastructural polymorphs. These findings demonstrate that cryo-EM allows atomic characterization of amyloid filaments from patient-derived material, and pave the way to study a range of neurodegenerative diseases.
Neurofibrillary lesions strongly correlate with cognitive deficits, making them an important therapeutic target for AD1. Dominantly inherited mutations in MAPT cause frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17T), showing that dysfunction of Tau is sufficient to cause neurodegeneration and dementia2.
Ultrastructurally, Tau inclusions are made of PHFs and SFs3–5. By negative-stain electron microscopy, the core of PHFs and SFs consists of a double helical stack of C-shaped subunits6, whereas the amino- and carboxy-terminal regions of Tau are disordered and project away from the core to form the fuzzy coat7. In AD, Tau filaments form from full-length Tau in cells, but have lost most of their fuzzy coat in extracellular ghost tangles.
Neurofibrillary lesions appear to propagate in the course of AD through connected pathways, from subcortical areas to transentorhinal cortex, limbic system and neocortex8. Although the molecular Tau species responsible for neurodegeneration are unknown, short filaments constitute the major species of seed-competent Tau in the brains of mice transgenic for human P301S Tau9.
Six Tau isoforms ranging from 352 to 441 amino acids are expressed in adult human brain, produced by alternative mRNA splicing of transcripts from MAPT, the Tau gene10. They differ by the presence or absence of inserts of 29 or 58 amino acids in the amino-terminal half, and the inclusion or not of the 31 amino acid microtubule-binding repeat, encoded by exon 10 of MAPT, in the carboxy-terminal half. Inclusion of exon 10 results in the production of three Tau isoforms with four repeats each (4R), and its exclusion in a further three isoforms with three repeats each (3R). The four repeats (R1-R4) comprise residues 244–368, in the numbering of the 441 amino acid Tau isoform.
Neurodegenerative diseases with abundant filamentous Tau inclusions are referred to as tauopathies11. Besides AD, they include tangle-only dementia (TD), chronic traumatic encephalopathy (CTE), argyrophilic grain disease (AGD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), globular glial tauopathy (GGT) and Pick’s disease (PiD)11. Unlike AD, these other diseases lack Aβ plaques. In AD, TD and CTE, all six Tau isoforms (3R and 4R) are present in the disease filaments. In AGD, PSP, CBD and GGT, only 4R Tau isoforms are found, whereas in PiD only 3R Tau inclusions are present. The existence of human tauopathies with distinct filament morphologies12 has led to the suggestion that different molecular conformers of aggregated Tau may exist. Multiple molecular conformers may also explain why AD brain-derived filaments are more effective than in vitro assembled filaments of recombinant protein in inducing Tau pathology in mouse brain13.
Progress in understanding tauopathies is hampered by the lack of atomic structures of Tau filaments from brain. Studies of amyloids are complicated by potential differences in structure between filaments in the brain and those assembled in vitro. Low-resolution cryo-EM structures are available of in vitro assembled filaments of Aβ14, and solid-state NMR has been used to study the structures of recombinant Aβ15–17 and α-synuclein18. Even when seeding in vitro filament growth with extracts from diseased brain16,18, structures may not be amplified according to their relative abundance in the brain. Here, we present atomic models, as determined by cryo-EM, for the core of PHFs and SFs that were purified from AD brain.
Neuropathological characteristics
We used cerebral cortex (Figure 1a) of a 74 year-old female (patient B.J.C.) with a neuropathologically confirmed diagnosis of AD. The interval between clinical diagnosis and death was 10 years. The patient’s Apolipoprotein-E genotype was ε3/ε4. Exons 16 and 17 of the Amyloid precursor protein gene, exons 3–13 of the Presenilin-1 gene, as well as exons 4, 5, 7 and 12 of the Presenilin-2 gene (including adjacent intronic sequences) were sequenced and no disease-causing mutations were found. The patient’s mother had died aged 86 with a 16 year history of AD. Thioflavin S staining showed the presence of abundant neurofibrillary tangles and neuritic plaques in cerebral cortex (Figure 1b). The sarkosyl-insoluble fraction of the patient’s neocortex contained numerous PHFs and SFs, which were composed of full-length, hyperphosphorylated Tau, as assessed by immuno-gold EM (Extended Data Figure 1). Negatively stained PHFs had a longitudinal spacing between crossovers of 650–800 Å and a width of about 300 Å at the widest part and 100–150 Å at the narrowest. SFs were about 150 Å wide with crossover distances ranging from 700–900 Å (Figure 1c). By Western blotting, pathological Tau bands of 60, 64, 68 and 72 kDa were present (Extended Data Figure 1a). These characteristics are like those described previously for PHFs and SFs from AD brains19. Sarkosyl-insoluble material seeded aggregation of full-length human Tau in cultured cells20.
Figure 1. Structure of Tau filaments from Alzheimer’s brain.
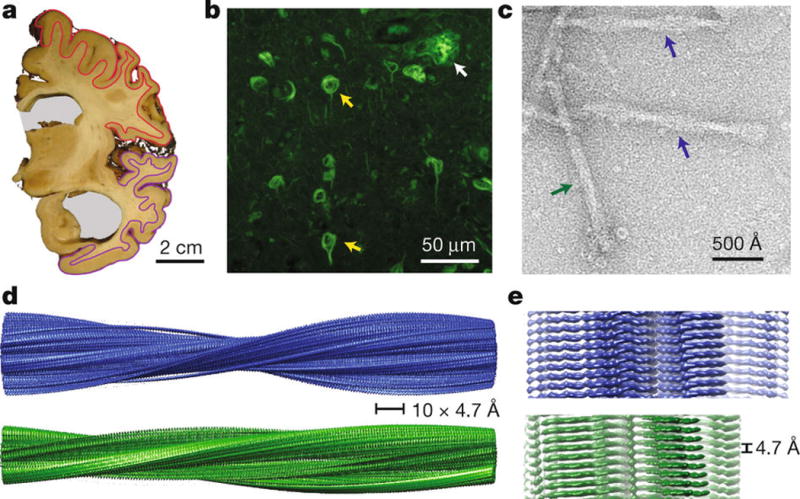
a. Coronal section at the level of the precentral gyrus of the brain used in this study shows an enlarged lateral ventricle (grey background). Grey matter from frontal cortex (red) and temporal cortex (magenta) was used for cryo-EM. b. Thioflavin S staining light microscopy showing abundant neurofibrillary tangles (yellow arrows) and a neuritic plaque (white arrow) in temporal cortex. c. Negatively stained electron micrograph of purified Tau filaments, in which PHFs (blue arrows) and SFs (green arrows) are readily distinguished. d–e. Cryo-EM reconstructions of PHFs (blue) and SFs (green).
PHFs and SFs are ultrastructural polymorphs
The sarkosyl-insoluble fraction of the patient’s cerebral cortex was purified by differential centrifugation and gel filtration (Methods) with no disruption to the structures of PHFs and SFs (Extended Data Figure 2a–c). The purified sarkosyl-insoluble fraction also seeded the aggregation of full-length human Tau in cultured cells (Extended Data Figure 2d). The two types of filaments could be readily distinguished in cryo-EM images, and corresponding 3D reconstructions were calculated separately to overall resolutions of 3.4 and 3.5 Å (Extended Data Figure 3) using helical reconstruction in RELION21. The maps showed clear side-chain densities (Figure 2) and β-strands were well separated in the direction along the helical axis (Figure 1e), allowing us to propose stereo-chemically refined atomic models for the cores of PHFs and SFs (Methods).
Figure 2. Cross-sections of the PHF and SF cryo-EM structures.
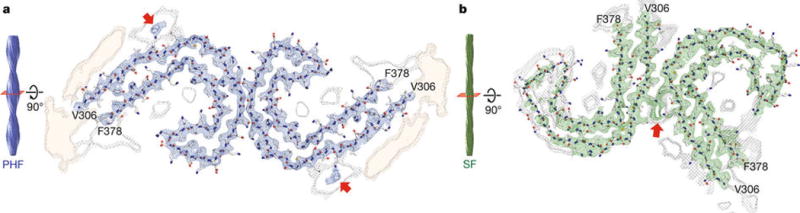
Cryo-EM density and atomic models of PHFs (a) and SFs (b). Overviews of the helical reconstructions (left) show the orientation of the cross-sectional densities (right). Sharpened, high-resolution maps are shown in blue (PHFs) and green (SFs). Red arrows point at additional densities in contact with K317 and K321. Unsharpened, 4.5 Å low-pass filtered density is shown in grey. Unsharpened density highlighted with an orange background is reminiscent of a less-ordered β-sheet and could accommodate an additional 16 amino acids, which would correspond to a mixture of residues 259–274 (R1) from 3R Tau and residues 290–305 (R2) from 4R Tau.
Consistent with previous findings6, both PHFs and SFs are composed of two protofilaments with C-shaped subunits. Successive rungs of β-strands along a protofilament are related by helical symmetry: with a rise of 4.7 Å and a twist of approximately −1° in both PHFs and SFs. In PHFs, but not SFs, the two protofilaments are related by an approximate 21 screw symmetry. A clear pattern of side chain densities and various landmark features in the two maps revealed that the core of both protofilaments in PHFs and in SFs is composed of residues V306-F378, i.e. all of R3 and R4, as well as 10 amino acids carboxy-terminal to the repeats (Extended Data Figure 4). The protofilament cores of PHFs and SFs are similar (Extended Data Figure 5), showing that they are ultrastructural polymorphs, reminiscent of fibrils formed by a peptide of transthyretin22.
Additional, weaker densities appear at both the amino- and carboxy-terminal regions of the core (orange density in Figure 2). The weaker densities suggest that these regions correspond to a mixture of peptides and/or a more dynamic or transiently occupied structure than the filament core. The weaker density at the amino-terminal region can accommodate approximately 16 residues in a β-sheet-like conformation that may represent a mixture of the C-termini of R1 and R2, which would result from the 3R and 4R alternative splicing products. This is consistent with our Western blotting results, which indicated that all six brain Tau isoforms were present in PHFs and SFs (Extended Data Figure 1a). Another region of additional density is interacting with the sidechains of K317, T319 and K321 in both types of filaments (red arrows in Figure 2). This density is strongest in SFs where it forms part of the protofilament interface, whereas in PHFs it occurs at the surface of both protofilaments.
Generality of PHF and SF structures
We used pronase treatment, mass spectrometry and antibody labeling to corroborate our model of a structured R3:R4 core with extending densities at the amino- and carboxy-termini. Pronase treatment has previously been shown to remove the fuzzy coat of PHFs and SFs, while leaving the structured core intact7. We confirmed that cryo-EM structures of the core of both PHFs and SFs do not change upon pronase-treatment (Extended Data Figure 6). Mass spectrometry of the pronase-treated filaments showed that residues I260-R406 of 3R and residues K281-R406 of 4R Tau are present in the structured core of the filaments, with R3:R4 (V306-R406) being by far the most abundant species (Extended Data Figure 7a). This finding is also in good agreement with peptides identified in earlier work23,24, and a recent report on mass spectrometry of trypsin-treated PHFs and SFs from 10 AD cases25.
In addition, to confirm that R2 is not part of the pronase-resistant core, we used Anti-4R, an antibody raised against V275-C291 of R2 (with D279)26. In agreement with a pronase-resistant R3:R4 core, Anti-4R detected both PHFs and SFs before, but not after, pronase treatment. By contrast, antibody MN423, which recognizes tau truncated at E39124, detected PHFs and SFs only after pronase treatment (Extended Data Figure 7b), as reported previously19. The model of an ordered R3:R4 core is also consistent with the observation that antibodies BR135 and TauC4, which recognize sequences inside R310 and R425, respectively, do not label either PHFs or SFs, whereas they do label PHF Tau on denaturing gels (Extended Data Figure 1a). Sequence analysis of the PHF core also showed the presence of R3:R4 of Tau24.
Thus, we propose that PHFs and SFs have a common core structure composed primarily of R3:R4, with a mixture of R1 and R2 directly preceding V306.
Structure of the common protofilament
The core of PHFs and SFs is composed of eight β-sheets (β1-8) that run along the length of the protofilament, adopting a C-shaped architecture. Each C is comprised of a β-helix region, where three β-sheets are arranged in a triangular fashion, and two regions with a cross-β architecture, where pairs of β-sheets pack anti-parallel to each other (Figure 3a–b). Forming such a continuous β-strand architecture along the chain of an individual Tau molecule (80% of residues are in a β-strand conformation occupying only 13.3% of the total ϕ/ψ space) is achieved by interspersing the β-strand regions with β-breaking prolines (P312, P332, P364), β-turn glycines (G323, G355) or β-arc residues (E342, D348). The strain of having many twisted β-strand regions is further relieved by changes in the height of the chain along the helical axis, resulting in differences of up to 10 Å between the highest point in β1 and the lowest point in the tip of the β-helix (Figure 3c). Thereby, an ordered, in-register, parallel hydrogen-bonding pattern is maintained throughout the protofilament structure. Additional hydrogen bonds that stabilize the protofilament are implied by the side chains of ladders of asparagines and glutamines.
Figure 3. The common protofilament core.
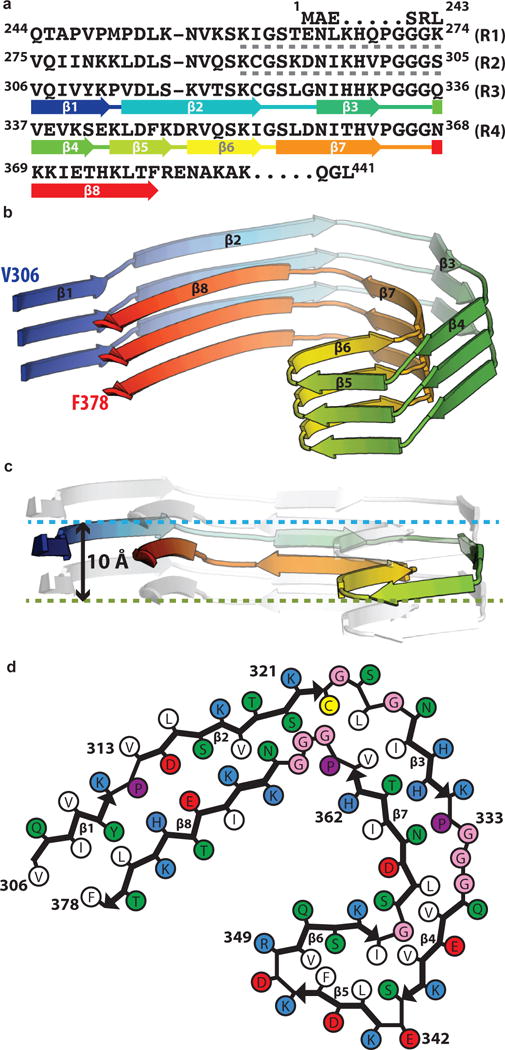
a. Sequence alignment of the four microtubule-binding repeats (R1–R4) with the observed eight β-strand regions coloured from blue to red. The sixteen residues from R1 or R2 that may form an additional, less-ordered β-sheet are indicated with grey dashed lines. b. Rendered view of the secondary structure elements in three successive rungs. c. As in b, but in a view perpendicular to the helical axis, revealing the differences in height along the helical axis within a single molecule. d. Schematic view of the protofilament core.
Starting from the termini (Figure 3d), there is a heterotypic cross-β interface formed between β1-2 and β8. The amino-terminal end of the ordered core is formed by the hexapeptide 306VQIVYK311, which forms a complementary packing interface with residues 374–378 from the opposing β8 by face-to-face packing of hydrophobic groups. Strands β2 and β8 pack against each other through a polar-zipper motif. Next, there is a right-angle turn accomplished through G323 and G326 on one side and a compact 364PGGG367 motif on the other side. It is possible that cysteine C322 forms intra-sheet disulphide bonds, but we were unable to detect a regular pattern that would allow us to visualise density for such bonds in our helical reconstruction. A hydrophobic cluster of L324, I326 and V363 stabilizes the region immediately following the turn and the cross-β interface between β3 and β7 is further cemented by hydrogen bonds between the sidechains of H328 and T361. Following β3, residues 332PGGG335 adopt an extended β-spiral conformation. Finally, the two sides meet through a β-helix structure that is defined by three β-strands in R4 (β4-6). Two-residue (E342, K343) and three-residue (347KDR349) β-arc corners punctuate the triangular β-helix geometry, which is closed with a pivotal ~70° glycine conformation (G355). Hydrophobic clustering, aliphatic stacking (V339, L344, V350, and I354) and aromatic stacking (F346) stabilize the interior of the β-helix. Alternating charged residues between E338 and R349 on the solvent exposed surface of the β-helix provide intramolecular charge compensation that offsets the repulsive effects of stacking sidechains with identical charge.
PHFs and SFs form different interfaces
The ultrastructural polymorphism between PHFs and SFs is due to differences in lateral contacts between the two protofilaments. In PHFs, the two protofilaments form identical structures that are related by helical symmetry, and the interface is formed by the anti-parallel stacking of residues 332PGGGQ336 (Figure 4a). The offset of half the β-strand distance along the helical axis results in a staggered packing of 333GGG335 from both protofilaments in PHFs. In particular, the glycine tripeptide adopts a characteristic polyglycine II, β-spiral structure27 that forms a hydrogen-bonding pattern both within and between the two protofilaments. The PHF interface is further stabilized by the formation of two hydrogen bonds between Q336 and the backbone carboxyl of K331 on the opposite protofilament.
Figure 4. Protofilament interface in PHFs and SFs.
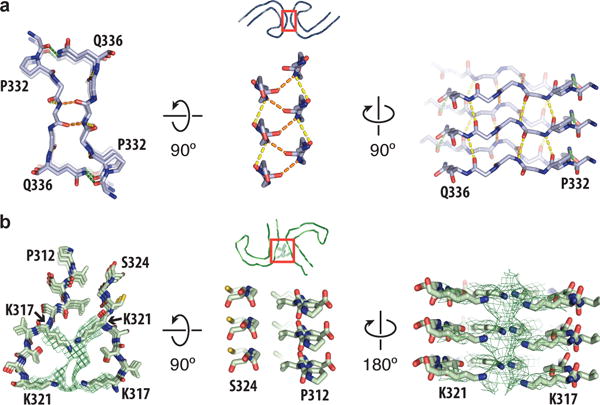
a. Packing between residues 332PGGGQ336 of the two protofilaments in PHFs. Inter-protofilament hydrogen bonds are shown in orange and green. Intra-protofilament hydrogen bonds are shown in yellow. b. Packing between residues 317–324 and residues 312–321 of the protofilaments in SFs. Additional density between the side chains of lysines 317 and 321 is shown as a green mesh.
In SFs, the two protofilaments pack asymmetrically (Figure 2b). The weaker β-sheet-like density at the amino-terminal end of the structured core is only visible in one of the protofilaments, as steric hindrance at the protofilament interface prevents the formation of a peripheral β-strand on the second protofilament. Although the β-helices of the two protofilaments appear to be in a similar chemical environment, the β-helix of the second protofilament is more disordered. The backbones of the two protofilaments are nearest each other between residues 321KCGS324 of the first and 313VELSK317 of the second protofilament, where the protofilaments are packed against each other at approximately the same position along the helical axis (Figure 4b). However, these residues do not form inter-protofilament salt-bridges or hydrogen bonds, nor does the interaction seem to be mediated by hydrophobic packing. Instead, the SF protofilament interface appears to be stabilized through the region of additional density that is interacting with the side chains of K317, T319 and K321 of both protofilaments (red arrow, Figure 2b). These residues are part of the epitope of the Alz-50 and MC-1 antibodies28 that is formed by two discontinuous portions of Tau: 7EFE9 and 313VDLSKVTSKC322. Nuclear magnetic resonance (NMR) experiments on heparin-induced filaments of recombinant 4R Tau confirm the interaction between the amino-terminus and residues 313–322 of the structured core29, which is strongly affected by ionic strength, and takes place mainly through intra-molecular interactions29,30. Therefore, although we cannot preclude the role of other poly-anionic molecules, we speculate that the additional density corresponds to 7EFE9, and that salt bridges between the negatively charged glutamates of the amino-terminus and the positively charged lysines of the core stabilize the interaction between the two protofilaments. Similar additional density is also interacting with K317 and K321 in PHFs, where it does not contribute to the protofilament interface (red arrows, Figure 2a). Mutations directly preceding the 7EFE9 motif, i.e. R5L31 and R5H32, have been observed in FTDP-17T.
Discussion
AD PHFs and SFs consist of an ordered core of pairs of protofilaments comprising residues 306–378, and disordered amino- and carboxy-termini forming the fuzzy coat (Figure 5). The core forms a sufficient platform for the incorporation of both 3R and 4R Tau into the growing filament. As we were unable to detect any order in the fuzzy coat, this heteromolecular addition may occur in a random manner. This would explain why PHFs and SFs are able to seed in vitro aggregation of recombinant samples containing only 3R Tau, only 4R Tau, or a mixture of 3R and 4R Tau13. It may also explain why the intracerebral injection of PHFs and SFs from AD brain leads to neuronal uptake, seeding and propagation in wild-type mice13,33, since the Tau306-378 sequence is identical in humans and mice. The ordered core is probably necessary and sufficient for the seeded assembly of Tau in AD brain.
Figure 5. Schematic representation of full-length Tau filaments.
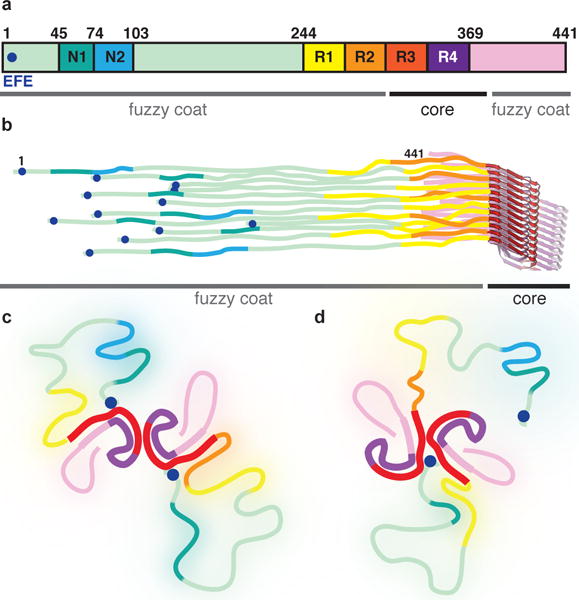
a. Primary structure of the longest Tau isoform in human brain. The two inserts near the amino-terminus are labeled N1 and N2. The microtubule-binding repeats are labeled R1-4. The 7EFE9 motif near the amino-terminus is marked with a blue circle. b. The six different isoforms pack randomly along the helical axis. c. In PHFs, the amino-terminal region of Tau monomers in both protofilaments folds back to form an additional, less ordered β-sheet against β1–β2 of the ordered core, then becomes disordered, and folds back to form an interaction between 7EFE9 and lysines 317 and 321 of the core. d. In SFs, only a single additional β-sheet can form, and the amino-terminus of one of the protofilaments forms part of the protofilament interface.
The protofilament core is composed of a combination of two structural motifs that are closely associated with amyloid and prion structures – cross-β packing34 and β-helices35. This structure is markedly different from the conformation of Tau bound to microtubules, where residues 269–284 and 300–312 adopt a hairpin conformation and the rest of the structure appears disordered by NMR36. The generic cross-β structure is widely accessible to peptides of varying lengths with unrelated amino acid sequences37. In AD Tau, in-register, anti-parallel β-sheet packing is mediated through a mixture of hydrophobic and polar interactions. The amino-terminal part of the cross-β structure is formed by the hexapeptide 306VQIVYK311, which is essential for the assembly of Tau filaments38,39. In contrast to homotypic interactions seen in microcrystals of isolated peptides40, the hexapeptide in AD Tau packs through a heterotypic, non-staggered interface with the opposing residues 373–378. The same packing interface is absent in the widely used K18 and K19 constructs41, which span three or four repeat domains of recombinantly expressed Tau, and terminate at E372. Therefore, filaments made of K18 and K19 proteins cannot represent the complete core structure of AD brain-derived PHFs and SFs, and care should be taken when extrapolating experimental results derived from the use of constructs ending before F378 to Tau aggregation in vivo42.
In contrast to generic cross-β structures, β-helices are mediated by specific interactions that are not accessible to most amino acid sequences43. β-Helices can only arise from sequences with a specific pattern of hydrophobic residues on the inside, polar residues on the outside, and a pivotal β-arch forming glycine at the end of the motif. An example of such β-helices is the prion-forming domain of HET-s from the filamentous fungus Podospora anserina44. The HET-s prions form a β-solenoid core, which is essential for infectivity, composed of two charge-complementary left-handed β-helical windings and a linker region. Despite having little sequence similarity, the β-helices in AD Tau and HET-s218-289 (PDB-entry 2RNM44) adopt a similar fold (Extended Data Figure 8), raising the possibility that this motif may serve as a scaffold for prion-like, templated misfolding45. The β-helix in Tau is closed by G355, which is located in the 353KIGS356 motif of R4 that comprises the potential phosphorylation site S356.
Knowledge of the atomic coordinates of Tau filaments may be useful for the rational design of specific inhibitors of Tau aggregation, as well as tracer compounds. Luminescent conjugated oligothiophenes (LCOs) bind to HET-s218-289 through exposed lysine ladders on the surface of the β-solenoid46. Therefore, LCOs such as pentameric formyl thiophene acetic acid (pFTAA), a fluorescent dye that identifies neurofibrillary lesions in AD brain sections47, may bind to aggregated Tau in a similar way. The specific structure of the β-helix (or possibly the protofilament interface), as opposed to the more generic cross-β structure near the termini of the core, may also provide a binding site for positron emission tomography ligands that show specific binding to AD Tau filaments, but not to Tau filaments from non-AD diseases, Aβ, or α-synuclein48.
Different molecular conformers of Tau have been suggested to be related to specific neurodegenerative diseases. The structures presented here show how different protofilament packing interactions lead to ultrastructural polymorphism in Tau filaments. Protein sequencing and mass-spectrometric analyses of Tau filaments from various diseases have indicated that in PiD, CBD and PSP, R1 and R2 are more resistant to trypsin digestion than in AD25. This raises the possibility of another type of polymorphism, where Tau protofilaments may form from alternative combinations of the repeats (R1:R2 and R2:R3 from 4R Tau, and R1:R3 from 3R Tau) (Extended Data Figure 9). Whether these hypothetical conformations exist in other tauopathies remains to be seen.
Conclusion
The cryo-EM structures of AD PHFs and SFs establish a basis for understanding the differences between molecular conformers of Tau aggregates, and explain how different isoforms are incorporated into the filaments. Our results demonstrate that amyloid structures from human brain may be obtained by cryo-EM. This opens up new possibilities for studying the molecular mechanisms underlying a wide range of neurodegenerative diseases.
Methods
Extraction of Tau filaments from AD brain
Sarkosyl-insoluble material was extracted from grey matter of frontal and temporal cortex as described19. The resuspended pellet (10 μl/g tissue) was further purified by 30-fold dilution in 10 mM Tris-HCl pH 7.4, 800 mM NaCl, 5 mM EDTA, 1 mM EGTA with a final concentration of 10% (w/v) sucrose, followed by centrifugation at 20,100 × g for 30 min at 4°C. The pellet, containing large contaminants such as collagen, was discarded. The supernatant was centrifuged at 100,000 × g for 1 h at 4°C. The pellet was resuspended in 20 mM Tris-HCl pH 7.4 containing 100 mM NaCl at 10 μl/g tissue, and run over a Superose 6 Increase 3.1/300 column (GE Healthcare). PHFs and SFs eluted in the void volume, whereas ferritin, the main contaminant, and residual sucrose were retained. The filaments were concentrated using a Vivacon 500 centrifuge device (Viva Products) at 3,000 × g and stored at 4°C. Pronase treatment was carried out as described19.
Immunolabeling, histology and molecular genetics
Western blotting and immunogold EM were carried out as described19. For Western blotting, samples were resolved on 4–20% or 10% Tris-glycine gels (Novex), and the primary antibodies were diluted in PBS plus 0.1% Tween 20 and 1% BSA. BR133, BR134 and BR13510 at 1:4000; AT8 (Thermo; catalogue nr. MN1020) at 1:1000; MC-128 at 1:10; HT7 (Thermo; catalogue nr. MN1000) at 1:2500, Anti 4R-tau (Cosmo Bio; catalogue nr. CAC-TIP-4RT-P01) and TauC425 at 1:2000; and MN42324 at 1:500. For immunogold EM, primary antibodies were diluted as follows: BR133, BR134, BR135, AT8, Anti 4R, TauC4 and MN423 at 1:50; and MC-1 at 1:10. Neurohistology, immunohistochemistry, and molecular genetics were carried out as described49. For immunohistochemistry, the following antibody dilutions were used: RD3 (Millipore; catalogue nr. 05-803) at 1:3000; Anti-4R at 1:100; and AT8 and AT100 (Thermo; catalogue nr. MN1060) at 1:300. The brain sections were 8 μm thick, and were counterstained with haematoxylin.
Seeded aggregation
Seeded aggregation in cells was carried out as described50. HEK 293T cells (ECACC) expressing full-length wild-type 0N4R tau were incubated for 3 h with the Tau filament preparation used for cryo-EM diluted 1:1000 in OptiMEM, followed by a change to complete medium (DMEM plus Glutamax and 10% foetal calf serum) and growth for 72 h. Cells were lysed in 10 mM Tris-HCl pH 7.4, 800 mM NaCl, 5 mM EDTA, 1 mM EGTA with a final concentration of 10% (w/v) sucrose by freeze-thaw, followed by sarkosyl extraction and Western blotting. Band densities were calculated using ImageJ (NIH).
Mass spectrometry
Purified Tau filaments were disaggregated using 4 M guanidinium hydrochloride and digested with trypsin. The proteolytic peptides were injected for analysis by liquid chromatography tandem mass spectrometry (LC-MS/MS) and identified by matching molecular mass to a peptide-fragment database. Pronase-treated Tau filaments were processed in a similar way, but with an additional ultracentrifugation step to separate pronase-cleaved “fuzzy” coat regions from the core of the filament. In this case, the pellet containing the pronase-resistant core of the PHFs and SFs was disaggregated, trypsin digested and analysed by LC-MS/MS. Relative abundances of peptide fragments were quantified by measuring the areas under the readout curves from ion-mass chromatography.
Cryo-Electron Microscopy
For cryo-EM, aliquots of 3 μl of purified Tau filaments at a concentration of 1 mg/ml were applied to glow-discharged holey carbon grids (Quantifoil Au R1.2/1.3, 300 mesh), blotted with filter paper to remove excess sample, and plunge-frozen in liquid ethane using an FEI Vitrobot Mark IV. Cryo-EM images were acquired on a Gatan K2-Summit detector in super-resolution counting mode on an FEI Titan Krios at 300 kV. A GIF-Quantum energy filter (Gatan) was used with a slit width of 20 eV to remove inelastically scattered electrons. For full-length filaments (FL), fifty movie frames were recorded each with an exposure time of 200 ms using a dose rate of 1.2 electrons per Å2 per frame for a total accumulated dose of 60 electrons per Å2 at a pixel size of 1.04 Å on the specimen. For pronase-treated filaments (PT), twenty movie frames were recorded, each with an exposure time of 800 ms using a dose rate of 2.75 electrons per Å2 per frame, resulting in a total accumulated dose of 55 electrons per Å2 at a pixel size of 1.15 Å on the specimen. The final data sets are composed of 1560 (FL) and 523 (PT) micrographs with defocus values in both data sets ranging from −1.0 to −3.0 μm.
Helical Reconstruction
Similar procedures were applied to the FL and PT data sets. All super-resolution frames were corrected for gain reference, binned by a factor of 2, motion-corrected and dose-weighted using MOTIONCOR251. Aligned, non-dose-weighted micrographs were then used to estimate the contrast transfer function (CTF) using Gctf52. All subsequent image-processing steps were performed using helical reconstruction methods in RELION 2.021.
Filaments were picked manually, and segments were extracted using a box size of 280 Å and an inter-box distance of ~10% (30 Å) of the box length. Reference-free 2D classification to identify homogeneous subsets was initially performed using a regularisation value of T=2. Selected class averages were repositioned onto the original micrographs according to the classification of individual segments to aid manual separation of the filaments into PHFs and SFs, which were then processed as separate data sets. An additional round of reference-free 2D classification was performed on the PHF and SF data sets yielding distinct classes for different views of the filaments.
Initial models for 3D classification were created from previously derived PHF and SF cross-sections6. Similar results could also be obtained from alternative initial models that were reconstructed from 2D class averages of segments that were extracted in a box that was large enough to almost comprise an entire pitch length. We first used 3D classification of the PHF and SF datasets with a regularisation parameter of T=4. These calculations yielded sub-nanometer (in the x-y plane only) reconstructions, in which individual β-sheets were clearly separated within the C-shaped subunits of both PHFs and SFs, but no structure was discernable along the (helical) z-axis. These results reflect the difficulty in reconstructing amyloid filaments, which have notoriously few layer lines visible in their power spectra between very low resolutions (~50Å) and the distance between β-strands (4.7Å).
We experimented with higher values of T in order to force the use of higher spatial frequencies in the regularised likelihood optimisation. 2D classifications with T-values as high as 20 yielded 2D class averages with clearly discernable β-strand separation, but possibly due to overfitting most of the segments were assigned to a single class representing a mixture of different views. More conservative regularisations with T=8 yielded a more representative number of different 2D class averages that still showed clear β-strand separation.
We then selected those segments from the PHF and SF data sets that were assigned to 2D class averages with β-strand separation for subsequent 3D classification runs. For these calculations, we used a single class (K=1); a T-value of 20; and we used the previously obtained sub-nanometer PHF and SF reconstructions, low-pass filtered to 15 Å, as initial models. Imposing a helical rise of 4.7 Å and a helical twist of −1° (as estimated from the cross-over distances of filaments in the micrographs) led to 3D reconstructions of both PHFs and SFs in which β-strands were clearly resolved. At this point, we tested a range of different helical parameters. Optimizations of the helical twist all converged onto values of approximately −1° per 4.7 Å rise. Helical rises of multiples of 4.7 Å all led to β-strand separation, but in agreement with the observed absence of layer lines between 50 and 4.7 Å (Extended Data Figure 3d) we were unable to detect any repeating patterns among successive rungs of β-strands. Therefore, we decided to impose a helical rise of 4.7 Å, which results in a reconstruction in which all rungs of β-strands are identical. Finally, as we realised that PHFs consist of two identical protofilaments related by an approximate 21 screw, we imposed additional helical symmetry on the PHFs by using a rise of 4.7/2Å and a twist of (360°-1°)/2. This additional symmetry was absent in SFs, where the two protofilaments pack in an asymmetrical manner.
3D classifications with local optimisation of the helical twist and rise were used for all four data sets to further select segments for high-resolution refinement. Although these classifications resulted in relatively small subsets of the initial data sets (Extended Data Table 1), reconstructions from much larger subsets gave similar reconstructions, albeit to lower resolutions, that were still in full agreement with our atomic interpretation (see below). This indicates that image classification did not select for a specific structure from a conformationally heterogeneous data set, but instead was successful in distinguishing the better segments from data sets with images of varying quality, which is in line with observations in single-particle analysis. Image classification also did not separate variable twists of PHFs and SFs. Instead, RELION combines segments from filaments with variable twists into a single 3D reconstruction and reduces the corresponding blurring effects by only using the central part of an intermediate asymmetrical reconstruction for real-space helical symmetrisation21. For both PHFs and SFs, we used a 10% value for the corresponding helical_z_percentage parameter.
High-resolution refinements were performed using RELION’s 3D auto-refinement, during which both the helical twist and rise were optimised21. As initial models, we used 7 Å low-pass filtered maps of the PHF and SF from the 3D classification runs with K=1. In some refinements, the two half-maps converged onto different translations along the helical axis. In such cases, it was necessary to halt the run, align the two half-maps outside RELION, before resuming the refinement. Final, overall resolution estimates were calculated from Fourier shell correlations at 0.143 between the two independently refined half-maps, using phase-randomisation to correct for convolution effects of a generous, soft-edged solvent mask53. Local resolution estimates were obtained using the same phase-randomisation procedure, but with a soft spherical mask that was moved over the entire map. Final PHF and SF reconstructions were sharpened using standard post-processing procedures in RELION, resulting in B-factors of approximately −100 Å2 on all maps (Extended Data Table 1), and helical symmetry was imposed on the post-processed maps using the relion_helix_toolbox program21. The PHF reconstructions from the FL and PT data sets were very similar, with overall resolution estimates of 3.4 and 3.5 Å (cf. Figure 2 and Extended Data Figure 6). The SF map from the FL data set had an overall resolution of 3.4 Å, but due to a lower abundance of SFs after pronase treatment, the corresponding map only reached 5.1 Å and β-strands were thus not separated.
Model Building and Refinement
The PHF and SF maps from the PT and FL data sets have complementary regions for model building. The β-helix motif at the tips of the C-shaped protofilaments is better ordered in the PHFs than in SFs, whereas the amino- and carboxy-terminal ends of the protofilament cores are best ordered in the SF map from the FL data set. Moreover, density for the cross-β structures near the termini of the PHFs is better defined in the PT reconstruction than in the FL reconstruction. Combined, the different maps allow for unambiguous main-chain tracing of the atomic models (Figure 2; Extended Data Figures 4 and 6). Initial model building started by fitting the L-type β-helix from the magic-angle spinning NMR-derived structure of filaments of the prion-forming domain HET-s218-289 (PDB-entry 2RNM) in the tip of the protofilaments of the PT PHF reconstruction. The sequence of only one of the four microtubule-binding repeats of Tau had a sequence that was compatible with the observed density in both PHFs and SFs (Extended Data Figure 4). Using the β-helix region as a starting point, we then built the remainder of the amino- and carboxy-terminal regions by manually adding amino acids and targeted real-space refinement in COOT54.
Fourier-space refinement of the complete atomic model against the PHF and SF maps was performed in REFMAC55. A stack of three consecutive monomers from each of the protofilaments was refined in order to preserve nearest-neighbour interactions for the middle chain. Local symmetry restraints were imposed in REFMAC to keep all β-strand rungs identical. Since most of the structure adopts a β-strand conformation, hydrogen-bond restraints were imposed to preserve a parallel, in-register hydrogen-bonding pattern in earlier stages of the model building process. In addition, ϕ/ψ angle restraints were imposed for the polyglycine II region, 333GGG335, and the three-residue β-arc, 347KDR349. The dihedral angles for polyglycine II were taken from Crick’s proposed fold27 (ϕ = −80, ψ =150) and those for the three-residue β-arc from the crystal structure of PDB-entry 1QRE56. Side-chain clashes were detected using MOLPROBITY57 and corrected by iterative cycles of real-space refinement in COOT and Fourier-space refinement in REFMAC. Refinements of atomic models were performed for the FL and PT PHFs and for the FL SF. The refined model from the FL SF was rigid-body fitted into the PT SF map. For each refined structure, separate model refinements were performed against a single half-map, and the resulting model was compared to the other half-map to confirm the absence of overfitting. The final models were stable in refinements without additional restraints. Statistics for the final models are shown in Extended Data Table 1.
Ethical Review Board and informed consent
The Indiana Alzheimer Disease Center studies are reviewed and approved by the Indiana University Institutional Review Board. Informed consent was obtained from the patient’s next of kin.
Extended Data
Extended Data Figure 1. Immunolabeling of the brain sample.
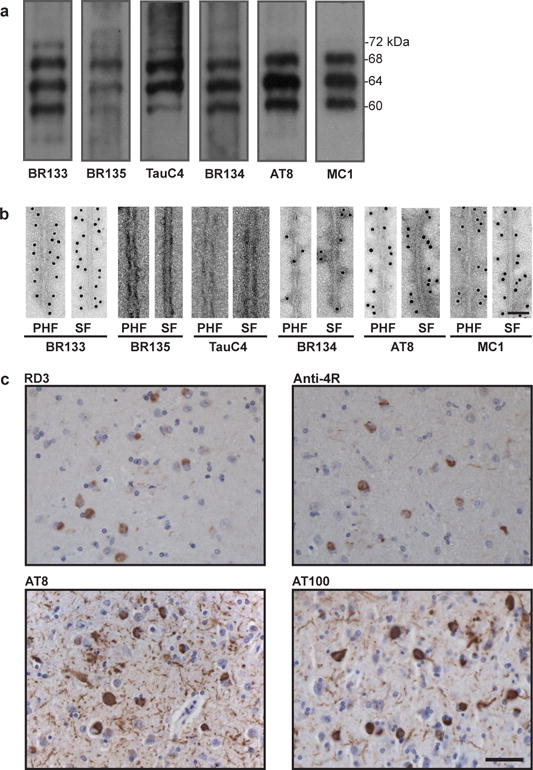
a,b. Immunolabeling of the sarkosyl-insoluble fraction from the patient’s temporal cortex. Immunoblots (a) using anti-Tau antibodies BR133 (amino-terminus), BR135 (R3), TauC4 (R4), BR134 (carboxy-terminus), AT8 (pS202/pT205) and MC1. Immunogold negative-stain electron microscopy (b) of PHFs and SFs with BR133, BR135, TauC4, BR134, AT8 and MC1. Scale bar, 500 Å. c. Light microscopy of sections from the temporal cortex showing staining of neurofibrillary tangles, neuropil threads and plaque neurites using RD3 (3R), Anti-4R (4R), AT8 and AT100 (pT212/pS214/pT217). Nuclei are counterstained blue. Scale bar, 50 μm.
Extended Data Figure 2. PHFs and SFs at various stages in the purification.
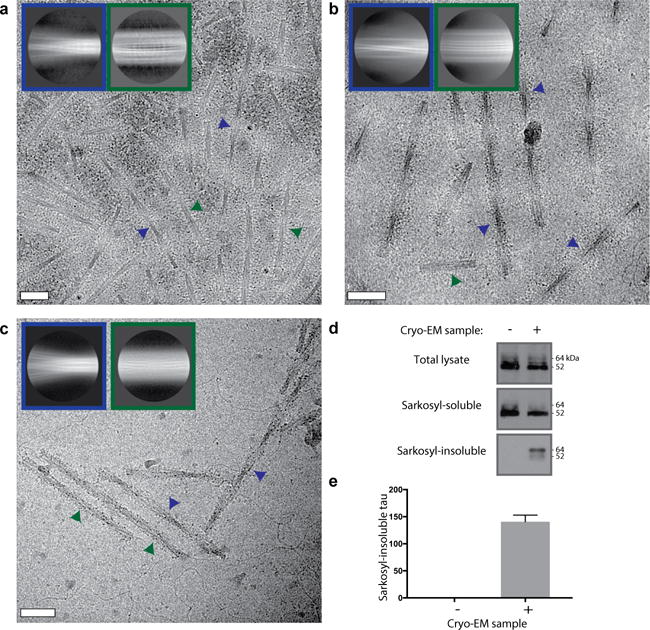
a–c. Cryo-EM micrographs and reference-free 2D class averages for PHFs (blue insets) and SFs (green insets) for the Tau sample after the sucrose step (a), gel filtration (b), and pronase treatment (c). Examples of PHFs and SFs in the micrographs are indicated with blue and green arrows, respectively. Scale bars, 500 Å. d. Western blots with antibody HT7 (Thermo; catalogue nr. MN1000) of the total lysate, sarkosyl-soluble and sarkosyl-insoluble fractions of HEK 293T cells expressing wild-type 0N4R human tau and treated with (+) or without (−) the sarkosyl-insoluble fraction from the patient’s temporal cortex following gel filtration show that the cryo-EM sample is capable of seeding aggregation of human Tau. e. Densitometric analysis (mean ± standard error of the mean, n=3) of HT7 blots of sarkosyl-insoluble fractions from cells.
Extended Data Figure 3. Cryo-EM map and model comparisons.
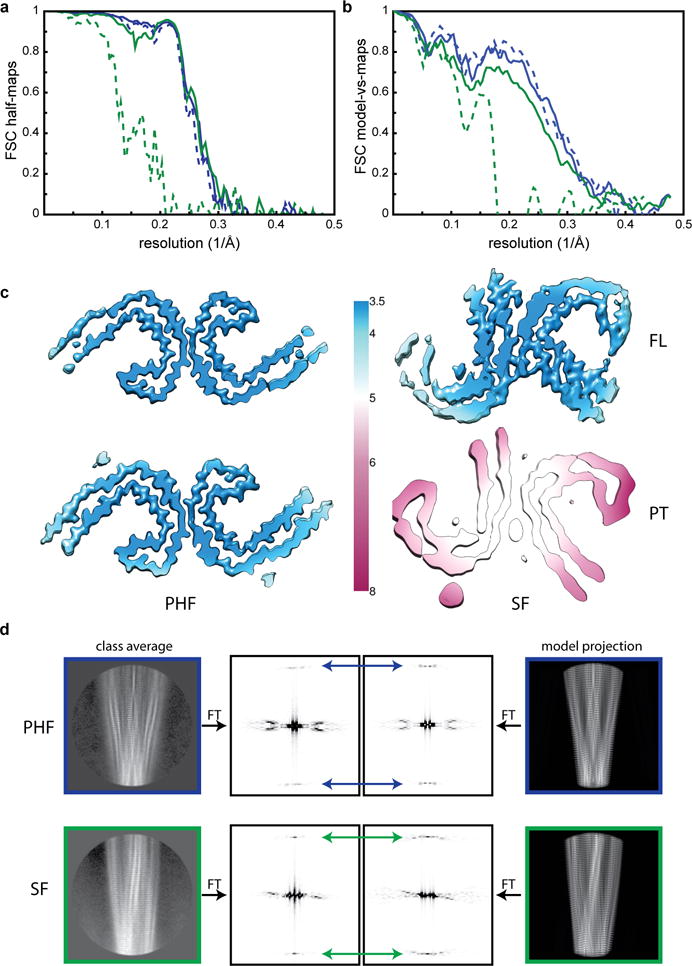
a. Fourier Shell Correlation (FSC) curves between two independently refined half-maps for the FL PHFs (blue, solid); FL SF (green, solid); PT PHF (blue, dashed) and PT SFs (green, dashed). b. FSC curves between the cryo-EM reconstructions and the refined atomic models, using the same colour coding as in a. c. Local resolution estimates for the four cryo-EM reconstructions. d. Comparison of power spectra (the squared amplitudes of the Fourier Transform, FT) of reference-free 2D class averages with those of corresponding projections of the atomic models. In PHFs the approximate 21 screw symmetry between subunits on the two protofilaments leads to off-meridional n=1 Bessel function peaks on the 1/(4.7 Å) layer line (blue arrows). For SFs, where the asymmetric unit consists of two subunits at the same level, one from each protofilament, there is a meridional n=0 Bessel function peak on the 1/(4.7 Å) layer line (green arrows).
Extended Data Table 1.
Cryo-EM structure determination and model statistics for full-length (FL) and pronase-treated (PT) filaments
| Data Collection | FL PHF | PT PHF | FL SF | PT SF |
|---|---|---|---|---|
| Magnification | ×105,000 | ×105,000 | ×105,000 | ×105,000 |
| Defocus range (μm) | −1.0 to −3.0 | −1.0 to −3.0 | −1.0 to −3.0 | −1.0 to −3.0 |
| Voltage (kV) | 300 | 300 | 300 | 300 |
| Microscope | Krios | Krios | Krios | Krios |
| Camera | K2 Summit | K2 Summit | K2 Summit | K2 Summit |
| Frame exposure time (s) | 0.2 | 0.8 | 0.2 | 0.8 |
| # movie frames | 50 | 20 | 50 | 20 |
| Total electron dose (e−/Å−2) | 60 | 55 | 60 | 55 |
| Pixel size (Å) | 1.04 | 1.15 | 1.04 | 1.15 |
|
| ||||
| Reconstruction | FL PHF | PT PHF | FL SF | PT SF |
|
| ||||
| Box size (pixel) | 270 | 270 | 270 | 270 |
| Inter-box distance (Å) | 28 | 28 | 28 | 28 |
| # segments extracted | 214,757 | 66,585 | 84,701 | 9,273 |
| # segments after Class2D | 214,262 | 61,321 | 84,089 | 8,891 |
| # segments after Class3D | 23,086 | 20,778 | 24,689 | 8,627 |
| Resolution (Å) | 3.4 | 3.5 | 3.4 | 4.9 |
| Map sharpening B-factor (Å2) | −105 | −106 | −114 | −79 |
| Helical rise (Å) | 2.36 | 2.36 | 4.74 | 4.78 |
| Helical twist (°) | 179.4 | 179.4 | −1.05 | −1.01 |
|
| ||||
| Atomic model | FL PHF | PT PHF | FL SF | PT SF |
|
| ||||
| # unique non-hydrogen atoms | 555 | 555 | 555 | – |
| R.m.s.d. bonds (Å) | 0.01 | 0.01 | 0.01 | – |
| R.m.s.d. angles (°) | 1.04 | 0.82 | 0.81 | – |
| Molprobity clashscore, all atoms | 5.13 | 7.42 | 5.78 | – |
| Molprobity score | 2.03 | 1.88 | 1.91 | – |
| Favored rotamers (%) | 98.44 | 96.88 | 96.85 | – |
| Ramachandran outliers (%) | 0.00 | 0.00 | 0.00 | – |
| Ramachandran favoured (%) | 78.4 | 92.49 | 88.11 | – |
| Cβ deviations > 0.25 Å (%) | 0.00 | 0.00 | 0.00 | – |
| Bad bonds (%) | 0.00 | 0.00 | 0.00 | – |
| Bad angles (%) | 0.00 | 0.00 | 0.00 | – |
| Average FSC model-vs-map | 0.80 | 0.80 | 0.75 | – |
Supplementary Material
The R3:R4 model provides the best fit to cryo-EM densities in the β-helix region (top row) and the cross-β region near the termini (second row) of the PHF protofilaments. The same model also provides the best fit to densities at the cross-β regions near the termini of the two protofilaments (third and fourth row) in SFs. Major discrepancies between the cryo-EM density and alternative atomic models (consisting of R2:R3, R1:R3 and R1:R2 cores) are highlighted in red.
a. The PHF backbone atoms are shown in blue, the SF backbone atoms in green. The r.m.s.d. between the backbone atoms is 1.2 Å. b. As in a, but showing all atoms.
Sharpened maps are shown in blue for PHFs (a) and green for SFs (b). Additional densities in contact with lysines 317 and 321 are indicated with red arrows. Unsharpened density, low-pass filtered to 4.5 Å resolution in case of the PHFs, is shown in grey. The unsharpened density that is highlighted with an orange background is reminiscent of a less-ordered β-sheet and could accommodate an additional 16 amino acids, which would correspond to a mixture of residues 259–274 (R1) from 3R Tau and residues 290–305 (R2) from 4R Tau.
a. Relative abundance of identified peptides from the pronase-treated filaments by mass-spectrometry are shown in green. The peptide used to produce Anti-R4 is shown in blue (with a D instead of an N at position 279). The epitope of MN423 is shown in orange. b. Immunogold negative-stain electron microscopy of PHFs and SFs with Anti 4R-tau (blue) and MN423 (orange) with (+) and without (-) pronase treatment. Scale bar, 100 nm.
a. Overlay of the backbones of the β-helix in Tau (cyan) and the β-solenoid of Het-s (orange and purple). The r.m.s.d. between the backbone atoms is 1.3 Å. b-c. All-atom overlays of the Tau and Het-s β-helices.
Schematic views of hypothetical core structures of R1:R3 from 3R Tau (a), R2:R3 from 4R Tau (b); and R1:R2 from 4R Tau (c). Note that in these conformations, the β-helices would only be 18 instead of 19 residues long, because of an additional residue (D348) in R4. Still, the β-helices would have mainly hydrophobic residues in the core, polar residues on the outside, and the pivotal glycine closing the motif. Each hypothetical core structure also contains the first 10 amino acids of the next repeat (R4 for R1:3 and R2:R3; R3 for R1:R2). It could also be that there are cores of more than two repeats.
Acknowledgments
These findings mark the culmination of a conversation at the MRC Laboratory of Molecular Biology 34 years ago between Aaron Klug, the late Martin Roth and R.A.C. about the structural analysis of Alzheimer filaments. We are grateful to the patient’s family for donating brain tissue; to Francine Epperson, Rose Marie Richardson and Urs Kuederli for human brain collection and analysis; to Peter Davies, Masato Hasegawa and Michal Novak for antibodies MC-1, TauC4 and MN423, respectively; to Hong Zhou for use of the Titan Krios at the University of California at Los Angeles; to Shaoxia Chen, Christos Savva and Giuseppe Cannone for support with electron microscopy at the MRC Laboratory of Molecular Biology; to Toby Darling and Jake Grimmett for help with computing; and to Mark Skehel for support with mass spectrometry. M.G. is an Honorary Professor in the Department of Clinical Neurosciences of the University of Cambridge. This work was supported by the UK Medical Research Council (MC_UP_A025_1012 to G.M., MC_U105184291 to M.G. and MC_UP_A025_1013 to S.H.W.S.), the European Union (Marie Curie International Outgoing Fellowship to A.W.P.F., Joint Programme-Neurodegeneration Research to M.G. and B.F., and Horizon 2020 IMPRiND to M.G. and A.W.P.F.), the US National Institutes of Health (grant P30-AG010133 to B.G.) and the Department of Pathology and Laboratory Medicine, Indiana University School of Medicine (to B.G.).
Footnotes
The authors declare no competing financial interests.
Author contributions
B.G. performed neuropathology; H.J.G. performed genetic analysis; B.F. conducted filament extraction and immunolabeling; A.W.P.F. performed cryo-EM; S.H. and S.H.W.S. provided cryo-EM software; A.W.P.F. and S.H.W.S. analysed cryo-EM data; A.W.P.F., A.G.M and G.M. built the atomic model; R.A.C. contributed to the inception of the study. M.G. and S.H.W.S. supervised the project. All authors contributed to writing the manuscript.
Data availability
Cryo-EM maps have been deposited to the Electron Microscopy Data Bank (accession numbers 3741 for FL PHF; 3742 for PT PHF; 3743 for FL SF; and 3744 for PT SF). Refined atomic models have been deposited to the Protein Data Bank (accession numbers 5O3L for FL PHF; 5O3O for PT PHF; and 5O3T for FL SF).
References
- 1.Wilcock GK, Esiri MM. Plaques, tangles and dementia. A quantitative study. J Neurol Sci. 1982;56:343–356. doi: 10.1016/0022-510x(82)90155-1. [DOI] [PubMed] [Google Scholar]
- 2.Ghetti B, et al. Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41:24–46. doi: 10.1111/nan.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kidd M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature. 1963;197:192–193. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 4.Terry RD. The fine structure of neurofibrillary tangles in Alzheimer’s disease. J Neuropathol Exp Neurol. 1963;22:629–642. doi: 10.1097/00005072-196310000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Yagishita S, Itoh Y, Nan W, Amano N. Reappraisal of the fine structure of Alzheimer’s neurofibrillary tangles. Acta Neuropathol. 1981;54:239–246. doi: 10.1007/BF00687747. [DOI] [PubMed] [Google Scholar]
- 6.Crowther RA. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci U S A. 1991;88:2288–2292. doi: 10.1073/pnas.88.6.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wischik CM, et al. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85:4884–4888. doi: 10.1073/pnas.85.13.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braak H, Del Tredici K. Potential pathways of abnormal Tau and α-synuclein dissemination in sporadic Alzheimer’s and Parkinson’s diseases. Cold Spring Harb Perspect Biol. 2016;8:a023630. doi: 10.1101/cshperspect.a023630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson SJ, et al. Short fibrils constitute the major species of seed-competent Tau in the brains of mice transgenic for human P301S Tau. J Neurosci. 2016;36:762–772. doi: 10.1523/JNEUROSCI.3542-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 11.Goedert M, Masuda-Suzukake M, Falcon B. Like prions: the propagation of aggregated tau and α-synuclein in neurodegeneration. Brain. 2017;140:266–278. doi: 10.1093/brain/aww230. [DOI] [PubMed] [Google Scholar]
- 12.Crowther RA, Goedert M. Abnormal tau-containing filaments in neurodegenerative diseases. J Struct Biol. 2000;130:271–279. doi: 10.1006/jsbi.2000.4270. [DOI] [PubMed] [Google Scholar]
- 13.Guo JL, et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med. 2016;213:2635–2654. doi: 10.1084/jem.20160833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt M, et al. Peptide dimer structure in an Aβ(1-42) fibril visualized with cryo-EM. Proc Natl Acad Sci U S A. 2015;112:11858–11863. doi: 10.1073/pnas.1503455112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colvin MT, et al. Atomic resolution structure of monomorphic Aβ42 amyloid fibrils. J Am Chem Soc. 2016;138:9663–9674. doi: 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu JX, et al. Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wälti MA, et al. Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. Proc Natl Acad Sci U S A. 2016;113:E4976–4984. doi: 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuttle MD, et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat Struct Mol Biol. 2016;23:409–415. doi: 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- 20.McEwan WA, et al. Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc Natl Acad Sci U S A. 2017;114:574–579. doi: 10.1073/pnas.1607215114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He S, Scheres SHW. Helical reconstruction in RELION. J Struct Biol. 2017 doi: 10.1016/j.jsb.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fitzpatrick AWP, et al. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc Natl Acad Sci U S A. 2013;110:5468–5473. doi: 10.1073/pnas.1219476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jakes R, Novak M, Davison M, Wischik CM. Identification of 3- and 4-repeat tau isoforms within the PHF in Alzheimer’s disease. EMBO J. 1991;10:2725–2729. doi: 10.1002/j.1460-2075.1991.tb07820.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wischik CM, et al. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85:4506–4510. doi: 10.1073/pnas.85.12.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taniguchi-Watanabe S, et al. Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol. 2016;131:267–280. doi: 10.1007/s00401-015-1503-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dan A, et al. Extensive deamidation at asparagine residue 279 accounts for weak immunoreactivity of tau with RD4 antibody in Alzheimer’s disease brain. Acta Neuropathol Commun. 2013;1:54. doi: 10.1186/2051-5960-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crick FH, Rich A. Structure of polyglycine II. Nature. 1955;176:780–781. doi: 10.1038/176780a0. [DOI] [PubMed] [Google Scholar]
- 28.Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 29.Bibow S, et al. The dynamic structure of filamentous tau. Angew Chem Int Ed Engl. 2011;50:11520–11524. doi: 10.1002/anie.201105493. [DOI] [PubMed] [Google Scholar]
- 30.Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem. 1996;271:32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- 31.Poorkaj P, et al. An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol. 2002;52:511–516. doi: 10.1002/ana.10340. [DOI] [PubMed] [Google Scholar]
- 32.Hayashi S, et al. Late-onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol. 2002;51:525–530. doi: 10.1002/ana.10163. [DOI] [PubMed] [Google Scholar]
- 33.Clavaguera F, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110:9535–9540. doi: 10.1073/pnas.1301175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sunde M, et al. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 35.Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc Natl Acad Sci U S A. 2004;101:8342–8347. doi: 10.1073/pnas.0402254101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kadavath H, et al. Folding of the Tau protein on microtubules. Angew Chem Int Ed Engl. 2015;54:10347–10351. doi: 10.1002/anie.201501714. [DOI] [PubMed] [Google Scholar]
- 37.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 38.von Bergen M, et al. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci U S A. 2000;97:5129–5134. doi: 10.1073/pnas.97.10.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie C, et al. Identification of key amino acids responsible for the distinct aggregation properties of microtubule‐associated protein 2 and tau. J Neurochem. 2015;135:19–26. doi: 10.1111/jnc.13228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sawaya MR, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 41.Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33:9511–9522. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- 42.Kaufman SK, et al. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron. 2016;92:796–812. doi: 10.1016/j.neuron.2016.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kajava AV, Steven AC. Beta-rolls, beta-helices, and other beta-solenoid proteins. Adv Protein Chem. 2006;73:55–96. doi: 10.1016/S0065-3233(06)73003-0. [DOI] [PubMed] [Google Scholar]
- 44.Wasmer C, et al. Amyloid fibrils of the HET-s(218-289) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 45.Riek R, Eisenberg DS. The activities of amyloids from a structural perspective. Nature. 2016;539:227–235. doi: 10.1038/nature20416. [DOI] [PubMed] [Google Scholar]
- 46.Herrmann US, et al. Structure-based drug design identifies polythiophenes as antiprion compounds. Sci Transl Med. 2015;7:299ra123. doi: 10.1126/scitranslmed.aab1923. [DOI] [PubMed] [Google Scholar]
- 47.Åslund A, et al. Novel pentameric thiophene derivatives for in vitro and in vivo optical imaging of a plethora of protein aggregates in cerebral amyloidoses. ACS Chem Biol. 2009;4:673–684. doi: 10.1021/cb900112v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kolb HC, Andrés JI. Tau positron emission tomography imaging. Cold Spring Harb Perspect Biol. 2017;9:a023721. doi: 10.1101/cshperspect.a023721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spina S, et al. The tauopathy associated with mutation +3 in intron 10 of Tau: characterization of the MSTD family. Brain. 2008;131:72–89. doi: 10.1093/brain/awm280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Falcon B, et al. Conformation determines the seeding potencies of native and recombinant Tau aggregates. J Biol Chem. 2015;290:1049–1065. doi: 10.1074/jbc.M114.589309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang K. Gctf: Real-time CTF determination and correction. J Struct Biol. 2016;193:1–12. doi: 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen S, et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy. 2013;135:24–35. doi: 10.1016/j.ultramic.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 56.Iverson TM, Alber BE, Kisker C, Ferry JG, Rees DC. A closer look at the active site of gamma-class carbonic anhydrases: high-resolution crystallographic studies of the carbonic anhydrase from Methanosarcina thermophila. Biochemistry. 2000;39:9222–9231. doi: 10.1021/bi000204s. [DOI] [PubMed] [Google Scholar]
- 57.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The R3:R4 model provides the best fit to cryo-EM densities in the β-helix region (top row) and the cross-β region near the termini (second row) of the PHF protofilaments. The same model also provides the best fit to densities at the cross-β regions near the termini of the two protofilaments (third and fourth row) in SFs. Major discrepancies between the cryo-EM density and alternative atomic models (consisting of R2:R3, R1:R3 and R1:R2 cores) are highlighted in red.
a. The PHF backbone atoms are shown in blue, the SF backbone atoms in green. The r.m.s.d. between the backbone atoms is 1.2 Å. b. As in a, but showing all atoms.
Sharpened maps are shown in blue for PHFs (a) and green for SFs (b). Additional densities in contact with lysines 317 and 321 are indicated with red arrows. Unsharpened density, low-pass filtered to 4.5 Å resolution in case of the PHFs, is shown in grey. The unsharpened density that is highlighted with an orange background is reminiscent of a less-ordered β-sheet and could accommodate an additional 16 amino acids, which would correspond to a mixture of residues 259–274 (R1) from 3R Tau and residues 290–305 (R2) from 4R Tau.
a. Relative abundance of identified peptides from the pronase-treated filaments by mass-spectrometry are shown in green. The peptide used to produce Anti-R4 is shown in blue (with a D instead of an N at position 279). The epitope of MN423 is shown in orange. b. Immunogold negative-stain electron microscopy of PHFs and SFs with Anti 4R-tau (blue) and MN423 (orange) with (+) and without (-) pronase treatment. Scale bar, 100 nm.
a. Overlay of the backbones of the β-helix in Tau (cyan) and the β-solenoid of Het-s (orange and purple). The r.m.s.d. between the backbone atoms is 1.3 Å. b-c. All-atom overlays of the Tau and Het-s β-helices.
Schematic views of hypothetical core structures of R1:R3 from 3R Tau (a), R2:R3 from 4R Tau (b); and R1:R2 from 4R Tau (c). Note that in these conformations, the β-helices would only be 18 instead of 19 residues long, because of an additional residue (D348) in R4. Still, the β-helices would have mainly hydrophobic residues in the core, polar residues on the outside, and the pivotal glycine closing the motif. Each hypothetical core structure also contains the first 10 amino acids of the next repeat (R4 for R1:3 and R2:R3; R3 for R1:R2). It could also be that there are cores of more than two repeats.