

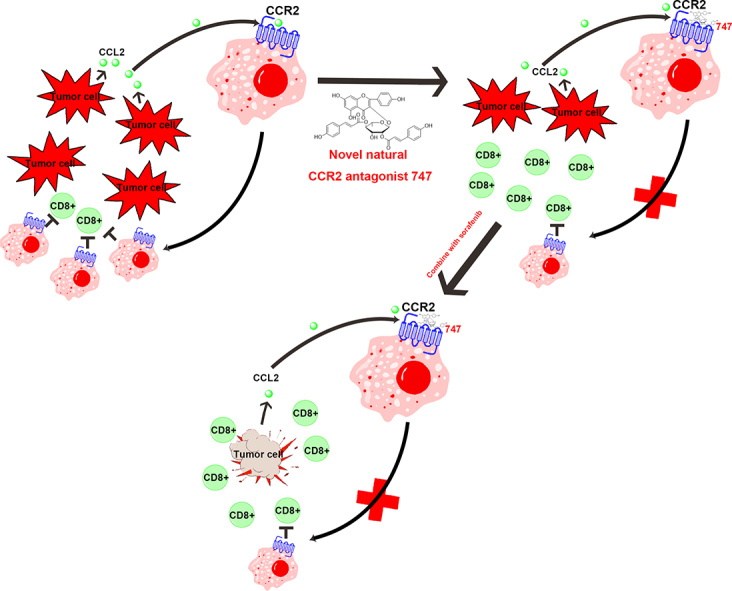
Abstract
Hepatocellular carcinoma (HCC) is a common malignant tumor in the digestive tract with limited therapeutic choices. Although sorafenib, an orally administered multikinase inhibitor, has produced survival benefits for patients with advanced HCC, favorable clinical outcomes are limited due to individual differences and resistance. The application of immunotherapy, a promising approach for HCC is urgently needed. Macrophage infiltration, mediated by the CCL2/CCR2 axis, is a potential immunotherapeutic target. Here, we report that a natural product from Abies georgei, named 747 and related in structure to kaempferol, exhibits sensitivity and selectivity as a CCR2 antagonist. The specificity of 747 on CCR2 was demonstrated via calcium flux, the binding domain of CCR2 was identified in an extracellular loop by chimera binding assay, and in vivo antagonistic activity of 747 was confirmed through a thioglycollate-induced peritonitis model. In animals, 747 elevated the number of CD8 + T cells in tumors via blocking tumor-infiltrating macrophage-mediated immunosuppression and inhibited orthotopic and subcutaneous tumor growth in a CD8 + T cell-dependent manner. Further, 747 enhanced the therapeutic efficacy of low-dose sorafenib without obvious toxicity, through elevating the numbers of intra-tumoral CD8 + T cells and increasing death of tumor cells. Thus, we have discovered a natural CCR2 antagonist and have provided a new perspective on development of this antagonist for treatment of HCC. In mouse models of HCC, 747 enhanced the tumor immunosuppressive microenvironment and potentiated the therapeutic effect of sorafenib, indicating that the combination of an immunomodulator with a chemotherapeutic drug could be a new approach for treating HCC.
Keywords: Preclinical models, Combination therapy, Experimental therapeutics, Kaempferol, Immunotherapy
Graphical Abstract
Highlights
-
•
We identified a natural product, 747, as an antagonist of CCR2.
-
•
747 exhibited anticancer properties and potentiated the efficacy of sorafenib in mouse models of HCC.
-
•
A combination of immunotherapy and chemotherapy could be a new approach for treating HCC.
Hepatocellular carcinoma (HCC) is a common malignant tumor that arises in people with a chronic liver disorder and inflammation. Macrophage infiltration, controlled by the CCL2/CCR2 axis, is evident in various liver diseases, including hepatitis, cirrhosis, and tumor progression, making it a therapeutic immune target for liver cancer. In the present investigation, we identified a CCR2 antagonist, 747, from Abies georgei. In a mouse model of HCC, 747 alone exhibited anticancer properties and potentiated the antitumor efficacy of a low dose of sorafenib. Our findings indicate that a combination of immunotherapy and chemotherapy could be a new approach for treating HCC.
1. Introduction
Hepatocellular carcinoma (HCC), the second leading cause of cancer deaths worldwide (Torre et al., 2015), arises in people with chronic liver disorders and inflammation. Most patients are not suitable for hepatic resection or transplantation. To treat these patients, chemotherapeutic agents, such as cisplatin, oxaliplatin, gemcitabine, and 5-fluorouracil, have been used, but they have low response rates. Sorafenib is the only agent approved for treating advanced HCC, but it has a limited effect on clinical outcome (Chen et al., 2015). Therefore, it is urgent to develop new approaches to treat HCC.
Immunotherapy has been regarded as the best hope for cancer therapy, but the clinical benefits vary widely (Prieto et al., 2015, Koyama et al., 2016). Immune checkpoints, such as CTLA-4, PD-1 and TIM-3, have been the most widely studied targets for immunotherapy. For application to HCC, a recent phase I/II trial of a PD-1 antibody, CT-011, was halted because of slow accrual (ClinicalTrials.gov, Identifier: NCT00966251). A more appropriate immunotherapy for HCC needs to be found (Prieto et al., 2015, Chen et al., 2015).
Infiltration of various immune cells leads to an immunosuppressive microenvironment that is responsible for HCC initiation and progression; macrophages are a primary component of these cells (Sun and Karin, 2012, Li et al., 2015, Baeck et al., 2012). CCR2 is a feature of inflammatory monocytes, which are circulating precursors of tissue macrophages. In acute or chronic inflammation, CCR2 controls trafficking of bone marrow monocytes into the bloodstream and their migration to inflammatory sites (Kurihara et al., 1997). The CCL2/CCR2 axis and corresponding macrophage infiltration are involved in liver pathology, including acute and chronic hepatitis, cirrhosis, tumor progression, and metastasis (Huang et al., 2015, Li et al., 2015, Marra and Tacke, 2014), making it a potential immunotherapeutic target for liver cancer.
CCR2 antagonists have been developed and evaluated in pre-clinical and clinical trials mainly due to the need for eliminating monocytes/macrophages, which would ameliorate inflammation associated with diseases. Although various agents have been discovered, and several have entered clinical trials, none have proved effective due to the selectivity of other chemokine receptors, their lack of potency in binding to rodent receptors, and poor PK/PD profiles (Struthers and Pasternak, 2010, Zimmermann et al., 2014).
In the present study, we identified a natural product, 747, from Abies georgei (Fig. 1A), as a CCR2 antagonist and, with murine HCC models, evaluated it for activity in blocking CCL2/CCR2-mediated recruitment of monocytes/macrophages and for therapy of liver cancer. Further, we determined the effect of 747 combined with low-dose sorafenib for treatment of HCC in mice.
Fig. 1.

747 is a promising CCR2 antagonist, as determined by tests in vitro and in mice.
(A) The chemical structure of 747 is kaempferol 3-(2,4-di-E-p-coumaroylrhamnoside. (B) Inhibitory effect of 747 on chemotaxis of hCCL2-induced THP-1 cells and mCCL2-induced murine peritoneal macrophages. (C) Inhibitory effect of 747 on binding of 125I-MCP-1 in CHO-K1-hCCR2/mCCR2 cells and binding of 125I-MIP1α in CHO-K1-hCCR5/mCCR2 cells. (E) Inhibitory effect of 747 on CCL2-induced calcium flux in 293FT-Gcamp6-CCR2 cells and RANTES-induced calcium flux in 293FT-Gcamp6-CCR5 cells. (F) 747 inhibition of thioglycollate-induced infiltration of peritoneal cells. *p < 0.05.
2. Materials and Methods
2.1. Chemicals and Reagents
All chemicals were of analytical grade. The CCR2 antagonist (747) with a purity of > 97% was isolated from Abies georgei in the Natural Products Laboratory at the Second Military Medical University (Shanghai, PR China). Sorafenib was obtained from ChemPartner (Shanghai, China), and D-luciferin was acquired from Xenogen Corp (Alameda, CA, USA). For daily injection, compounds were formulated as solutions in 0.5% hydroxypropyl methylcellulose with 0.2% Tween. 747, at 50 or 100 mg/kg of body weight, was administered intraperitoneally. Sorafenib, at 10 or 30 mg/kg of body weight, was administered intragastrically. Macrophage depletion was accomplished with clodronate liposomes (FormuMax Scientific, Inc., Palo Alto, CA), and CD8 T cell neutralization was achieved with anti-mouse CD8a antibody (eBioscience, San Diego, CA, USA).
2.2. Homology Modeling
The structure of CCR2 was modeled according to a previously reported method (Kim et al., 2011). The crystal structure of C-X-C chemokine receptor-4 (CXCR4, PDBID: 3ODU) was used as a template. Homology modeling was accomplished with the program, MODELLER (Sali and Blundell, 1993) in the DS software package. During the calculation, the ligand IT1t in the crystal structure of CXCR4 was copied to maintain a cavity in the space surrounded by the seven transmembrane helices. Ten models were generated and sorted by PDF total energy. Three models with the lowest energy were selected and refined by energy minimization with a fixed backbone constraint. The CHARMM force field and conjugate gradient algorithms with the maximum of 500 iterations were applied. The models were evaluated by PROCHECK (Morris et al., 1992). Finally, one was selected for virtual screening.
2.3. Virtual Screening
Hydrogen atoms and charges were added to the modeled structure of CCR2 during a brief relaxation performed using the ‘Protein Preparation Wizard’ workflow in Maestro 9.0. After optimizing the hydrogen bond network, the crystal structure was minimized using OPLS 2005 force field with the maximum RMSD value of 0.3 Å. The grid-enclosing box was centered on the cavity generated with the ligand of IT1t and defined to enclose residues located within 14 Å from the ligand, and a scaling factor of 1.0 was set to van der Waals radii with the partial atomic charges of 0.25 to soften the nonpolar parts of the receptor. The three-dimensional structures of compounds in the natural product database, containing about 4000 molecules, were generated with the Ligprep module. In the virtual screening process, standard precision (SP) and extra precision (XP) approaches were adopted successively, and 1000 compounds were reserved after being screened with the SP mode. The top 200 compounds were retrieved and ranked by GlideScore with the XP mode, and these hits were visually inspected for their binding modes. Among the selected compounds, 747 exhibited the most functional (chemotaxis) inhibition (Fig. S1).
2.4. Ca2 + Flux
Gcamp6 (Addgene), a new genetically encoded Ca2 + indicator superior to synthetic indicator dyes (Nakai et al., 2001), was stably expressed in 293FT cells. 293FT-Gcamp6 cells were transiently transfected with relevant chemokine receptors using Lipofectamine 2000 transfection reagent (Invitrogen). Cells were harvested 48 h after transfection, washed three times, and incubated in 96-well black plates with various concentrations of 747 in Ringer's buffer at 37 °C for 3 h (Corning, cat. No. 3603) with 5 × 105 cells per well. Then 2 mM CaCl2 and corresponding ligands were added into the wells to induce Ca2 + flux. The plates were subsequently read and recorded for fluorescence intensity with excitation at 488 nm and emission at 525 nm (EnSpire® Multimode Plate Readers, PerkinElmer).
2.5. Construction of Chemokine Receptors and Cell Transfection
The complementary DNAs (cDNAs) encoding the C-C chemokine receptors and CXC-receptors (R&D, Minneapolis, MN, USA) were cloned into the expression vector pcDNA3.1/V5-His-A (Invitrogen) at the BamHI and XholI sites. Single mutants were constructed by PCR-based site-directed mutagenesis. CHO-K1 cells were seeded onto 96-well, poly-d-lysine-treated cell culture plates (PerkinElmer) at a density of 2.7 × 104 cells per well. After overnight culture, the cells were transiently transfected with plasmid DNA using Lipofectamine 2000 transfection reagent (Invitrogen).
2.6. Whole-cell Binding Assay
At 24 h after transfection, cells were harvested, washed twice, and incubated with blocking buffer (F12 supplemented with 33 mM HEPES (pH 7.4) and 0.1% bovine serum albumin (BSA)) for 2 h at 37 °C. For competition binding, the cells were incubated in binding buffer with a constant concentration of 125I-MCP1 or 125I-MIP1α (40 pM) and different concentrations of 747 at room temperature for 3 h. Cells were washed three times with ice-cold PBS and lysed by 50 μL lysis buffer (PBS supplemented with 20 mM Tris-HCl (pH 7.4) and 1% Triton X-100). The plates were counted for radioactivity in a scintillation counter (MicroBeta2 Plate Counter, PerkinElmer) using a scintillation cocktail (OptiPhase SuperMix, PerkinElmer). Specific binding was determined by subtracting non-specific binding observed in the presence of 100 nM unlabeled MCP1 or MIP1α.
2.7. Thioglycollate Model of Sterile Peritonitis
Mice were intraperitoneally dosed with 747 or vehicle 3 h before an intraperitoneal injection with 1 mL of 4% thioglycollate and with 747 or vehicle once daily thereafter. At 3 days after thioglycollate challenge, peritoneal cells were harvested by lavage with 5 mL PBS. After lysis of red blood cells, peritoneal macrophages were counted (Bio-Rad, TC10) and subjected to flow cytometry with antibodies PE-CD11b (eBioscience), BV421-F4/80 (Biolegend), and APC-mCCR2 (R&D).
2.8. Biochemical Parameters
Serum levels of blood urea nitrogen (BUN) and creatinine (Cr) and serum activities of aspartate aminotransferase (AST), alanine aminotransferase (ALT), were qualified by using standard auto-analyzer methods on chemray 240 (Rayto).
2.9. Quantitative Real-time PCR
Total RNA was extracted with the TRIzol reagent (Invitrogen) and reverse-transcribed into cDNA. Quantitative real-time PCR was performed on an ABI7900HI (Applied Biosystems). Target gene expression was normalized to GAPDH and L32 as the control genes.
2.10. Cell Cultures
Mouse liver cancer cells (Hepa1-6), human monocyte cells (THP-1), and human HCC cells (HepG2) were purchased from ATCC (Manassas, VA); mouse liver cancer cells (LPC-H12), human immortalized non-tumorigenic liver cells (7702), and HCC cells (BEL-7404 and SMMC-7721) were obtained from the Cell Bank of the Shanghai Institutes for Biological Sciences (Chinese Academy of Sciences, Shanghai, China). The portal vein tumor thrombus 1 (PVTT-1) cell line was a gift from Dr. Dong Xie (Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, China).All cells maintained in DMEM or 1640 media () with 25 μM glutamine, 10%FBS, 1% penicillin-streptomycin under routine atmosphere at 37 °C.
2.11. Immunohistochemistry
Immunohistochemistry was performed with primary antibodies against CD8 (Novus) and cleaved caspase-3 (Cell Signaling Technology). In brief, paraffin sections of tumors were deparaffinized and rehydrated, and, after antigen retrieval, stained with primary antibodies overnight at 4 °C and applied to HRP-conjugated secondary antibodies at 37 °C for 1 h. Proteins were visualized after diaminobenzidine staining (Glostrup, Denmark).
2.12. Flow Cytometry
Fresh mouse tumor tissues were dissociated into single-cell suspensions with mouse tumor dissociation kits (Miltenyi Biotec) following the manufacturer's instructions. After lysis of red blood cells, the cells were centrifuged, suspended in FACS buffer, and incubated with anti-mouse CD16/32 for 15 min to avoid non-specific binding. Then cells were stained for 45 min at 4 °C in the dark. The cells were washed once with PBS, suspended in 200 μL PBS, and analyzed by BD FACS Aria II (BD Biosciences, San Jose, CA). For detection of tumor-infiltrated cells, cell staining included the following antibodies: PE-CD11b, APC-CD4, and PE-cy7-CD8 from eBioscience; PE-cy7-Gr1, BV421-F4/80, and APC-human CCR2 from Biolegend (San Diego, CA, USA); and APC-conjugated mouse CCR2 from R&D (Minneapolis, MN, USA). A minimum of 10,000 events were acquired for each sample.
2.13. Animal Studies
BALB/c athymic nude mice and C57BL/6 mice (male, 4–6 weeks old) were obtained from Shanghai Slac Laboratory Animal Co. and maintained in a pathogen-free vivarium under standard conditions. The strain of the CCR2 knockout mice is B6.129S4-Ccr2tm1Ifc/J, purchased from the Jackson Laboratory. The animal use and experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute for Nutritional Sciences. SIBS, CAS.
2.14. Subcutaneous HCC Model
Hepa1-6 or LPC-H12 cells (1.5 × 106 in 150 μL of medium) were injected into the right flanks of recipient mice. Tumor-bearing (about 0.5 cm in diameter) mice were randomized into control and treatment groups. Tumor volume were determined with the formula 1/2a × b2 (where a is the long diameter, and b is the short diameter).
2.15. Luciferase-expressing Orthotopic HCC Model
Hepa1-6 cells stably expressing firefly luciferase (1.0 × 106 in 100 μL of medium) were orthotopically injected into the livers of recipient mice under anesthesia with tribromoethanol (240 mg/kg, Sigma). The growth of tumors was monitored by bioluminescent imaging with the Xenogen IVIS imaging system (Perkin-Elmer, Fremont, CA, USA). Ten days after being implanted with tumor cells, tumor-bearing mice were evaluated by imaging and randomized into control and treatment groups. Mice were imaged once every 6 days during the course of the treatment.
2.16. Statistical Analyses
All values were recorded as the means ± SEM from at least 2 or 3 independent experiments. The data were analyzed using GraphPad Prism version 5.01 (GraphPad Software, Inc.). Statistical significance was determined through two-tailed Student's t-tests. P values < 0.05 were considered statistically significant.
3. Results
3.1. Characterization of 747 as a CCR2 Antagonist
After homology modeling and virtual screening, 43 natural products were chosen. Among the selected compounds, 747 exhibited the strongest inhibition of hCCL2-THP1 chemotaxis (Fig. S1). In a concentration-dependent manner, it inhibited migration of hCCL2/THP-1 cells and mCCL2/thioglycollate-stimulated mouse primary peritoneal macrophages (Fig. 1B). Whole-cell radio ligand binding assays were performed to evaluate the capacity of 747 to compete with 125I-MCP1 and 125I-MIP1α binding to CCR2 and CCR5 (mouse and human, due to high sequence homology). 747 inhibited the binding of 125I-MCP1 to hCCR2 and mCCR2 with IC50 values of 82 nM and 414 nM, respectively (Fig. 1C). However, 747 inhibited the binding of 125I-MIP1α to hCCR5 with an IC50 value of 16 μM and showed no inhibition of 125I-MIP1α binding to mCCR5 (Fig. 1D).
Calcium flux is a feature of activation of G-protein-coupled receptors (GPCRs). The 293FT-Gcamp6 system transfected with chemokine receptors was employed to measure calcium mobilization. 747 inhibited CCL2-induced calcium flux with an IC50 of 30 nM, but showed no substantial inhibition of RANTES-induced calcium flux (Fig. 1E). For CCR2-dependent macrophage recruitment in mice by thioglycollate, 747, at doses of 50, 100, 150 mg/kg, suppressed infiltration by peritoneal macrophages by 75.7%, 87.0%, and 92.8%, respectively (Fig. 1F). In CCR2−/− mice, 747 failed to reduce macrophage infiltration (data not shown). A panel of C-C chemokine and C-X-C chemokine receptors was evaluated in Ca2+ flux assays. Relative to CCR2, 747 exhibited no appreciable inhibition of any tested receptors (Table 1).
Table 1.
Chemokine receptor selectivity profile of 747.
| Receptor | Cell type | Assay format | IC50 (μM) |
|---|---|---|---|
| CCR1 | 293FT-Gcamp6-CCR1 transf. | Calcium flux | > 100 |
| CCR2 | 293FT-Gcamp6-CCR2 transf. | Calcium flux | ~ 0.03 |
| CCR3 | 293FT-Gcamp6-CCR3 transf. | Calcium flux | > 10 |
| CCR4 | 293FT-Gcamp6-CCR4 transf. | Calcium flux | > 10 |
| CCR5 | 293FT-Gcamp6-CCR5 transf. | Calcium flux | ~ 10 |
| CCR6 | 293FT-Gcamp6-CCR6 transf. | Calcium flux | > 10 |
| CCR7 | 293FT-Gcamp6-CCR7 transf. | Calcium flux | > 1 |
| CCR8 | 293FT-Gcamp6-CCR8 transf. | Calcium flux | > 1 |
| CCR9 | 293FT-Gcamp6-CCR9 transf. | Calcium flux | > 10 |
| CXCR1 | 293FT-Gcamp6-CXCR1 transf. | Calcium flux | > 1 |
| CXCR2 | 293FT-Gcamp6-CXCR2 transf. | Calcium flux | 1–10 |
| CXCR3 | 293FT-Gcamp6-CXCR3 transf. | Calcium flux | ~ 10 |
| CXCR4 | 293FT-Gcamp6-CXCR4 transf. | Calcium flux | 1–10 |
| CXCR5 | 293FT-Gcamp6-CXCR5 transf. | Calcium flux | > 10 |
| CXCR6 | 293FT-Gcamp6-CXCR6 transf. | Calcium flux | > 100 |
| CXCR7 | 293FT-Gcamp6-CXCR7 transf. | Calcium flux | > 100 |
CCR, chemokine (C-C motif) receptor; CXCR, chemokine (C-X-C motif) receptor.
3.2. Characterization of the Binding Mode of CCR2-747 by Use of CCR2-CCR5 Chimeras
As described above, 747 exhibited distinct binding affinities for CCR2 and CCR5, despite their high similarity. A CCR2-CCR5 chimeric approach (Zweemer et al., 2013) was utilized to clarify the role of the regions of the receptor in binding 747 (Fig. 2A and B). After the CCR5 extracellular loops were replaced with CCR2 extracellular loops, the binding affinity of 747-125IMCP1/CCR5 was increased, with the IC50 of 15 μM changing to 639.9 nM. 747-125IMCP1/CCR2 binding affinity was not appreciably altered after the CCR2 C-terminal was replaced by the CCR5 C-terminal (Fig. 2C). Consistently, when the extracellular loop was moved from CCR5 to CCR2, 747-125IMIP1α/CCR5 binding affinity was increased, with an IC50 of 18 μM changing to 1 μM. Substituting the CCR5 C-terminal with the CCR2 C-terminal resulted in a change of binding affinity from 18 μM to 14 μM (Fig. 2D). To access the binding of 747 with CCR2, a CCR2 homology model and a docked 747 model were utilized. In the structure of 747, three phenol moieties bind to the rhamnoside group, which makes it flexible, generates various conformers, and provides hydroxyls that can form hydrogen bonds with protein. From the docking results (Fig. 2E), we found that one conformer of compound 747 could bind to an extracellular ligand pocket with a preferable mode according to the scoring and coinciding with experimental results (Singh and Sobhia, 2013, Kim et al., 2011). Moreover, we determined the effect of residues of mutants from the docking results in binding 747. The binding affinity of 747 for an N199A mutant receptor was 2-fold lower compared with the wild type (Fig. 2F).
Fig. 2.

Mode of binding of 747 with CCR2.
(A) Sequence alignment of CCR2 and CCR5, the transmembrane regions, intracellular and extracellular loops are marked. From the docking results, 747 bonds with CCR2 residues are marked in red. (B) Schematic diagram of CCR2, CCR5, and the chimeric receptors CCR2-CCR5 (C-term), CCR5-CCR2 (C-term), and CCR5-CCR2 (Extracellular loops). (C) Inhibitory effect of 747 on binding of 125I-MCP-1 for CCR2, CCR5, and chimeric receptors expressed in CHO-K1 cells. (D) Inhibitory effect of 747 on binding of 125I-MIP-1α for CCR2, CCR5, and chimeric receptors expressed in CHO-K1 cells. (E) Induced-fit docking of 747 in a homology model of CCR2 based on the C-X-C chemokine receptor-4 crystal structure. (F) Inhibitory effect of 747 on binding of 125I-MIP-1α for CCR2 wild type and mutant receptors expressed in CHO-K1 cells. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
In an orthotopic model of murine liver cancer, 747 suppressed liver tumor growth, decreased infiltration of tumor-associated macrophages, and increased CD8 T cells.
The CCL2/CCR2 axis and corresponding macrophage infiltration are involved in liver pathology, including acute and chronic hepatitis, cirrhosis, tumor progression, and metastasis (Huang et al., 2015, Li et al., 2015, Marra and Tacke, 2014), making it a therapeutic target for liver cancer. With IC50 values > 10 μM, 747 had little cytotoxicity to Hepa1-6, LPC, THP-1, or other human liver cancer cells (Fig. S2). To evaluate the therapeutic effect of 747 as a CCR2 antagonist, an orthotopic model of murine liver cancer (Hepa1-6) was established (Fig. 3A). Liver tumor growth was inhibited after administration of 747 at doses of 50 or 100 mg/kg (Fig. 3B and C). The average tumor volumes of the groups treated with 747 were smaller than those for mice treated with the vehicle and were comparable to those for mice treated with sorafenib (30 mg/kg), the only first-line drug for treating advanced HCC (Wilhelm et al., 2004). Flow-cytometric analysis of tumor tissues after treatment with 747 showed a 43% decrease of TAMs and a 2-fold increase of CD8 T cells (Fig. 3D and E). In nude mice (T-cell deficient) bearing subcutaneous Hepa1-6 tumors, 747 had little effect on tumor growth (Fig. S4A and B). Further, 747 caused little toxicity, as determined by effects on weights of these mice and the serum levels of ALT, AST, blood urea nitrogen, and creatinine (Fig. 3F, Figs. S3 and S4C).
Fig. 3.

In an orthotopic liver cancer model, 747 suppresses liver cancer growth, reduces TAMs, and increases CD8 T cells.
(A) The schedule for liver cancer treatment and imaging. (B) Representative bioluminescence images of mice treated with 747 intraperitoneally (low: 50 mg/kg, high: 100 mg/kg), sorafenib intragastrically (30 mg/kg), or the vehicle (n = 6–7 mice/group). Representative photographs of liver tumors after treatment; the black dotted lines indicate the tumor regions. (C) Relative luminescence of liver tumors depicted in (B). (D) Representative flow cytometry results showing the proportion of CD4 T cells, CD8 T cells, and TAMs (CD11b + F4/80 +) in Hepa1-6 orthotopic tumor tissues of C57BL/6 mice. (E) Quantification of immune cells by flow-cytometric analysis in tumor tissues. Data are shown as means ± SEM. *p < 0.05. (F) Body weight changes of mice during treatment. Data are shown as means ± SEM.
3.3. 747 Possesses Anti-liver Cancer Activity Via Reduction of TAMs, Leading to Expansion of CD8 T Cells
These findings indicated that TAMs and CD8 T cells were responsible for the anti-tumor effects of 747. To gain insight into the relationships among 747, TAMs, and CD8 T cells, clodronate liposomes and anti-mouse CD8a antibody were used to deplete macrophages and CD8 T cells, respectively, in C57/BL6 mice bearing Hepa1-6 subcutaneous tumors (Fig. 4A). For the group with TAMs depleted by clodronate liposomes, tumor volumes were lower, similar to the 747 group. The therapeutic effect was not enhanced by treatment with the combination (Fig. 4B and C). Clodronate liposomes, 747, or the combination enhanced the proportion of CD8 T cells, resulting in reduced TAMs in the tumor microenvironment (Fig. 4D and E). These anti-tumor effects were abolished by depletion of CD8 T cells by anti-mouse CD8a antibody. The tumor volumes of the group depleted of CD8 T cells were larger than those for the group dosed with the vehicle (Fig. 4B and C). Consistent with the inhibition of tumor growth, flow-cytometric analysis of tumor tissue revealed that the anti-tumor activity of 747 was dependent on expansion of CD8 T cells mediated by reduction of macrophages (Fig. 4D and E). Other immune cells, such as CD4 T cells (Fig. 4E), myeloid-derived suppressor cells (MDSCs) (Fig. S5), B cells, and NK T cells (data not shown) were not changed appreciably. Thus, 747 blocks infiltration by TAMs, leading to elevated numbers of CD8 T cells, which kill tumor cells. Additionally, for mice dosed with 747, CD8 T cells are a downstream target of TAMs.
Fig. 4.

Depletion of CD8 T cells eliminates the anti-tumor effect of 747.
(A) The schedule of 747 treatment and depletion of macrophages and CD8 T cells in Hepa1-6 subcutaneous tumors (n = 5 mice/group). (B) Representative photographs of subcutaneous liver tumors after treatment with either 747. macrophage/CD8 T cell depletion, or both. (C) The tumors were measured and the proportion of TAMs, CD4 T cells, and CD8 T cells were quantified by FACS (D, E). Data are presented as means ± SEM. *p < 0.05.
3.4. Blockage of TAMs With 747 Enhances the Anti-tumor Efficacy of Sorafenib
For sorafenib, a multi-kinase inhibitor and the only systemic treatment for advanced HCC, about 30% of patients benefit from treatment, with the modest efficacy due to individual differences, intrinsic and acquired resistance, tumor heterogeneity, and an immunosuppressive microenvironment (Llovet et al., 2008, Cheng et al., 2009, Chen et al., 2015). Toxicity at high dosages is another concern. As established by clinical results, immunotherapy is a promising approach to treat cancer (Prieto et al., 2015). Since our previous results indicated that treatment with 747 increases intra-tumoral CD8 T cells through blocking TAMs, a combination of 747 and low dose (10 mg/kg) sorafenib were tested in mice bearing subcutaneous Hepa1-6 liver cancers (Fig. 5A). 747 treatment alone showed an appreciable therapeutic effect, the low dose of sorafenib showed no efficacy, and the combination exhibited the most efficacy (Fig. 5B). Measurements of body weights indicated no appreciable toxicity among the groups during treatment (Fig. 5C). Consistent with these results, 747 treatment blocked infiltration of TAMs and enhanced intratumoral CD8 T cells to kill tumor cells. The combination with sorafenib had no effect on the reduction of TAMs or the increase of CD8 T cells (Fig. 5D and E). To exclude a cell line-dependent effect, LPC-H12 (another murine HCC cell line) model of subcutaneous liver cancer was utilized, with similar results (Fig. S6).
Fig. 5.

747 potentiates the therapeutic effect of sorafenib.
(A) The schedule for 747 treatment and tumor measurement. (B) Representative photographs of subcutaneous liver tumors after treatment with 747, sorafenib, or the combination, and measured by tumor growth and weight. (C) Body weight changes of mice during treatment. (D, E) Proportions of TAMs, CD4 T cells, and CD8 T cells were quantified by FACS. Data are presented as means ± SEM. *p < 0.05.
747 potentiates the therapeutic effects of sorafenib by increasing tumor necrosis and apoptosis and enhancing cytotoxicity of intra-tumoral CD8 T cells.
To determine if the efficacy observed with the combination of 747 and sorafenib was a result of increased tumor cell death, tumors in the various groups were evaluated for the extent of necrosis (Fig. 6A). The staining for cleaved caspase-3 indicated that apoptosis was increased in the combination group compared to the other groups (Fig. 6B). 747 decreased TAM recruitment (Fig. 6C) and changed the macrophage phenotype. IHC of Arg-1 showed that the M2 macrophages in tumors were decreased after 747 treatment. Study of bone marrow-derived macrophages (BMDMs) showed that 747 enhanced the expression of M1-specific genes and depressed expression of M2-specific genes (Fig. 6E). Since M1 macrophages have antitumor effects and are involved in activation of T cells, we determined if 747 treatment increased CD8 T cells through blocking macrophage infiltration. We sorted CD8 T cells from tumors to examine their activation status, and found that the cytotoxic marker genes were increased (Fig. 6F). With BMDM-CD8 T cell co-cultures, pretreatment of BMDMs with 747 caused CD8 T cells to secrete TNF-α, an inducer of apoptosis, in a dose-dependent manner. These findings indicate that, in tumors, 747 blocks TAM infiltration and increases CD8 T cells. Concurrently, it changes TAM polarization towards the M1 phenotype and enhances CD8 T cell cytotoxicity, which explains, in part, its effect in combination therapy.
Fig. 6.

Treatment with the combination of 747 and sorafenib enhances tumor cell death and improves the cytotoxicity of CD8 T cells.
(A) Representative hematoxylin and eosin staining of subcutaneous tumors after treatment with 747, sorafenib, or the combination (Scale Bar: 50 μm). (B) Representative immunohistochemistry of cleaved caspase-3 in tumor sections (Scale Bar: 50 μm). Representative immunohistochemistry of F4/80 (macrophage cells) in the tumor sections (C) and Arg-1 (M2 marker) in the tumor sections (D) (Scale Bar: 50 μm). (E) BMDM treated with vehicle, 747 (5 μM) for 24 h. qRT-PCR results indicating the expression of M1 and M2 macrophage markers in BMDM. (F) CD8 + T cells were sorted from tumor at the end point. qRT-PCR showing the expression of cytotoxic marker in CD8 + T cells.(G) BMDM treated with vehicle,747 (5 μM) for 24 h and subsequently co-cultured with mice primary CD8 + T cells for 72 h. Cytometric Bead Array kits were employed to measure cytokine expression in the culture supernatants.
4. Discussion
Due to its involvement in various biological and pathological functions, CCR2 is an attractive target for drug discovery. However, development of CCR2 antagonists has been disappointing because of receptor redundancy, species differences, selectivity of other chemokine receptors, and limited clinical efficacy (Zimmermann et al., 2014). Since none of the present CCR2 antagonists has passed clinical trials, new concepts and strategies are needed. Natural products and their pharmacophores have been considered in the drug development process, and, from 1981 to 2014, they have accounted for nearly half of all newly approved drugs (Newman and Cragg, 2016). In the present study, a CCR2 antagonist, 747, was selected from a natural product library. 747 binds to human and mouse CCR2 with high affinity and selectivity relative to other chemokine receptors, particularly CCR5, which shares 72% homology (Zhao, 2010). Further, 747 binds to the major pocket of CCR2 through extracellular loops, whereas, among the mutants, only N199A mutant receptors decrease the binding affinity of 747 with CCR2. As determined from docking results, 747 forms hydrogen bonds with residues H121, F194, N199, N266, and T267, which partially explains why a single mutation may not show an appreciable effect on binding. Thus, 747, a CCR2 antagonist with a structure different from current antagonists, has antitumor potential and provides new insight for discovery of CCR2 antagonists.
For HCC, the second leading cause of cancer death worldwide, there is a lack of effective drugs. The only approved drug, sorafenib, has modest survival benefits (Llovet et al., 2008, Abou-Alfa et al., 2006, Cheng et al., 2009). Immunotherapy has been considered as an effective approach for cancer treatment, but the clinical benefits vary due to individual responses, tumor type (especially solid tumors), different immune targets, and relevant drugs (Prieto et al., 2015, Koyama et al., 2016). Additionally, immunomodulators alone generally possess modest antitumor activity as a consequence of their indirect inhibition of tumors (Prieto et al., 2015, Beatty and Gladney, 2015, Khalil et al., 2016, Restifo et al., 2016). An approach for immunotherapy, targeting of immune checkpoints, including CTLA-4, PD-1, TIM-3, LAG-3, and BTLA, is being considered. A clinical trial of the anti-PD-1 monoclonal antibody, CT-011, was terminated for patients with HCC because of slow accrual (ClinicalTrials.gov Identifier: NCT00966251) (Prieto et al., 2015, Chen et al., 2015). Although pembrolizumab and nivolumab, both PD-1 antibodies, entered phase III trials and showed a favorable outcomes, more exploration is needed to develop therapeutic strategies against HCC (Kudo, 2017).
We previously demonstrated that the CCL2/CCR2 axis mediates recruitment of TAMs and is involved in liver cancer. Thus, blocking of CCR2 could inhibit progression of liver cancers (Li et al., 2017). The present research shows that 747 alone reduces growth of liver tumors by relieving TAMs-mediated immunosuppression. Compared to mice dosed with clodronate liposomes, 747 elevated CD8 T cells with commensurate macrophage depletion, resulting in tumor inhibition. 747 also reduced TAMs in the tumor microenvironment and shifted macrophages towards the M1 phenotype (data not shown), which explains, in part, why the enhancement of CD8 T cells leads to better efficacy. Unlike some other immunomodulators, 747 alone exhibited anticancer properties, potentiated the antitumor efficacy of a low dose of sorafenib, and reduced its toxicity as reflected in body weight changes. Furthermore, the combination possessed the strongest inhibition of tumors via increased CD8 T cell counts and improved distribution within the tumor. Thus, we have presented proof-of-principle evidence that 747 is an immunomodulator that, combined with sorafenib, potentiates therapeutic efficacy by reducing numbers of TAMs, shifting polarization of TAMs, enhancing numbers CD8 T cells, improving CD8 T cell distribution in the tumor microenvironment, and increasing necrosis and apoptosis of tumor cells.
In summary, we identified 747, a natural CCR2 antagonist that meets demands of CCR2 antagonist design and development. 747 exhibited anti-liver cancer effects through elevating CD8 T cells via blocking CCR2-mediated recruitment of TAMs. Further, 747, combined with low-dose sorafenib, displayed anti-tumor effects through improving the quantity and distribution of CD8 T cells within tumors, without obvious toxicity. This combination of immunotherapy and chemotherapy provides a new approach for therapy of HCC (Marabelle et al., 2014).
5. Significance
Hepatocellular carcinoma (HCC) is a common malignant tumor of the digestive tract with limited therapeutic choices. Although sorafenib, an orally administered multikinase inhibitor, has produced survival benefits for patients with advanced HCC, favorable clinical outcomes are limited due to individual differences and resistance. Immunotherapy is a promising approach, and its application to HCC is urgently needed. Infiltration of macrophages, mediated by the CCL2/CCR2 axis, is a potential immunotherapeutic target for HCC. Here, we report that a natural product from Abies georgei, named 747 and related in structure to kaempferol, exhibits sensitivity and selectivity as a CCR2 antagonist. The specificity of 747 on CCR2 was demonstrated via calcium flux, the binding domain of CCR2 was identified in an extracellular loop by chimeras binding assay, and in vivo antagonistic activity of 747 was confirmed with a TG-induced peritonitis model. In vivo, 747 elevated the number of CD8 + T cells in tumors via blocking tumor-infiltrating macrophage-mediated immunosuppression and inhibited orthotopic and subcutaneous tumor growth in a CD8 + T cell-dependent manner. Further, 747, without obvious toxicity, enhanced the therapeutic efficacy of low-dose sorafenib through elevating intra-tumoral CD8 + T cells and increasing death of tumor cells. Thus, we have discovered a natural CCR2 antagonist, 747, and have provided a new perspective on development of CCR2 antagonists for treatment of related diseases. In mouse models of HCC, 747 improves the tumor immunosuppressive microenvironment and potentiates the therapeutic effect of sorafenib, indicating that the combination of immunomodulators and chemotherapeutic drugs could be a new approach for treating HCC.
Author Contributions
W.Y. and H.W. conceived and designed experiments. W.Y. and H.W. analyzed data and wrote the manuscript. H.W. and W.Z. supervised the project. W.Y., Q.B., X.L., H. L, S. Z, Y. Y, F.W., X.D., and J.L. performed the in vitro and in vivo studies. All authors reviewed and approved the manuscript.
Competing Interests
The authors have no financial conflicts of interest.
Acknowledgements
This work was supported by grants from the National Nature Science Foundation (81630086, 91529305, 81230090, 81520108030, and 31401611), supported by Professor of Chang Jiang Scholars Program, the Strategic Priority Research Program (XDA12020319) of the Chinese Academy of Sciences, and the Science and Technology Commission of Shanghai Municipality (16391903700 and 14391901800). The authors thank Dr. Donald L. Hill for editorial assistance.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.ebiom.2017.07.014.
Appendix A. Supplementary data
Supplementary figures
References
- Abou-Alfa G.K., Schwartz L., Ricci S., Amadori D., Santoro A., Figer A., De Greve J., Douillard J.Y., Lathia C., Schwartz B., Taylor I., Moscovici M., Saltz L.B. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2006;24:4293–4300. doi: 10.1200/JCO.2005.01.3441. [DOI] [PubMed] [Google Scholar]
- Baeck C., Wehr A., Karlmark K.R., Heymann F., Vucur M., Gassler N., Huss S., Klussmann S., Eulberg D., Luedde T., Trautwein C., Tacke F. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut. 2012;61:416–426. doi: 10.1136/gutjnl-2011-300304. [DOI] [PubMed] [Google Scholar]
- Beatty G.L., Gladney W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015;21:687–692. doi: 10.1158/1078-0432.CCR-14-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Jin R., Zhao J., Liu J., Ying H., Yan H., Zhou S., Liang Y., Huang D., Liang X., Yu H., Lin H., Cai X. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015;367:1–11. doi: 10.1016/j.canlet.2015.06.019. [DOI] [PubMed] [Google Scholar]
- Cheng A.L., Kang Y.K., Chen Z., Tsao C.J., Qin S., Kim J.S., Luo R., Feng J., Ye S., Yang T.S., Xu J., Sun Y., Liang H., Liu J., Wang J., Tak W.Y., Pan H., Burock K., Zou J., Voliotis D., Guan Z. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- Huang W., Chen Z., Zhang L., Tian D., Wang D., Fan D., Wu K., Xia L. Interleukin-8 induces expression of FOXC1 to promote transactivation of CXCR1 and CCL2 in hepatocellular carcinoma cell lines and formation of metastases in mice. Gastroenterology. 2015;149:1053–1067.e14. doi: 10.1053/j.gastro.2015.05.058. [DOI] [PubMed] [Google Scholar]
- Khalil D.N., Smith E.L., Brentjens R.J., Wolchok J.D. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016;13:273–290. doi: 10.1038/nrclinonc.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.H., Lim J.W., Lee S.W., Kim K., No K.T. Ligand supported homology modeling and docking evaluation of CCR2: docked pose selection by consensus scoring. J. Mol. Model. 2011;17:2707–2716. doi: 10.1007/s00894-010-0943-x. [DOI] [PubMed] [Google Scholar]
- Koyama S., Akbay E.A., Li Y.Y., Herter-Sprie G.S., Buczkowski K.A., Richards W.G., Gandhi L., Redig A.J., Rodig S.J., Asahina H., Jones R.E., Kulkarni M.M., Kuraguchi M., Palakurthi S., Fecci P.E., Johnson B.E., Janne P.A., Engelman J.A., Gangadharan S.P., Costa D.B., Freeman G.J., Bueno R., Hodi F.S., Dranoff G., Wong K.-K., Hammerman P.S. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016;7 doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo M. Immune checkpoint inhibition in hepatocellular carcinoma: basics and ongoing clinical trials. Oncology. 2017;92(Suppl. 1):50–62. doi: 10.1159/000451016. [DOI] [PubMed] [Google Scholar]
- Kurihara T., Warr G., Loy J., Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J. Exp. Med. 1997;186:1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Yao W., Yuan Y., Chen P., Li B., Li J., Chu R., Song H., Xie D., Jiang X., Wang H. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;Jan;66(1):157–167. doi: 10.1136/gutjnl-2015-310514. [DOI] [PubMed] [Google Scholar]
- Llovet J.M., Ricci S., Mazzaferro V., Hilgard P., Gane E., Blanc J.F., De Oliveira A.C., Santoro A., Raoul J.L., Forner A., Schwartz M., Porta C., Zeuzem S., Bolondi L., Greten T.F., Galle P.R., Seitz J.F., Borbath I., Haussinger D., Giannaris T., Shan M., Moscovici M., Voliotis D., Bruix J. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- Marabelle A., Kohrt H., Caux C., Levy R. Intratumoral immunization: a new paradigm for cancer therapy. Clin. Cancer Res. 2014;20:1747–1756. doi: 10.1158/1078-0432.CCR-13-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra F., Tacke F. Roles for chemokines in liver disease. Gastroenterology. 2014;147:577–594.e1. doi: 10.1053/j.gastro.2014.06.043. [DOI] [PubMed] [Google Scholar]
- Morris A.L., Macarthur M.W., Hutchinson E.G., Thornton J.M. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]
- Nakai J., Ohkura M., Imoto K. A high signal-to-noise Ca(2 +) probe composed of a single green fluorescent protein. Nat. Biotechnol. 2001;19:137–141. doi: 10.1038/84397. [DOI] [PubMed] [Google Scholar]
- Newman D.J., Cragg G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- Prieto J., Melero I., Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2015;12:681–700. doi: 10.1038/nrgastro.2015.173. [DOI] [PubMed] [Google Scholar]
- Restifo N.P., Smyth M.J., Snyder A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer. 2016;16:121–126. doi: 10.1038/nrc.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Singh R., Sobhia M.E. Structure prediction and molecular dynamics simulations of a G-protein coupled receptor: human CCR2 receptor. J. Biomol. Struct. Dyn. 2013;31:694–715. doi: 10.1080/07391102.2012.707460. [DOI] [PubMed] [Google Scholar]
- Struthers M., Pasternak A. CCR2 antagonists. Curr. Top. Med. Chem. 2010;10:1278–1298. doi: 10.2174/156802610791561255. [DOI] [PubMed] [Google Scholar]
- Sun B., Karin M. Obesity, inflammation, and liver cancer. J. Hepatol. 2012;56:704–713. doi: 10.1016/j.jhep.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre L.A., Bray F., Siegel R.L., Ferlay J., Lortet-Tieulent J., Jemal A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- Wilhelm S.M., Carter C., Tang L., Wilkie D., Mcnabola A., Rong H., Chen C., Zhang X., Vincent P., Mchugh M., Cao Y., Shujath J., Gawlak S., Eveleigh D., Rowley B., Liu L., Adnane L., Lynch M., Auclair D., Taylor I., Gedrich R., Voznesensky A., Riedl B., Post L.E., Bollag G., Trail P.A. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- Zhao Q. Dual targeting of CCR2 and CCR5: therapeutic potential for immunologic and cardiovascular diseases. J. Leukoc. Biol. 2010;88:41–55. doi: 10.1189/jlb.1009671. [DOI] [PubMed] [Google Scholar]
- Zimmermann H.W., Sterzer V., Sahin H. CCR1 and CCR2 antagonists. Curr. Top. Med. Chem. 2014;14:1539–1552. doi: 10.2174/1568026614666140827144115. [DOI] [PubMed] [Google Scholar]
- Zweemer A.J., Nederpelt I., Vrieling H., Hafith S., Doornbos M.L., De Vries H., Abt J., Gross R., Stamos D., Saunders J., Smit M.J., Ijzerman A.P., Heitman L.H. Multiple binding sites for small-molecule antagonists at the CC chemokine receptor 2. Mol. Pharmacol. 2013;84:551–561. doi: 10.1124/mol.113.086850. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures