

Abstract
Macrophages exhibit diverse phenotypes and functions; they are also a major cell type infiltrating chronically rejected allografts. The exact phenotypes and roles of macrophages in chronic graft loss remain poorly defined. In the present study, we used a mouse heart transplant model to examine macrophages in chronic allograft rejection. We found that treatment of C57BL/6 mice with CTLA4 immunoglobulin fusion protein (CTLA4-Ig) prevented acute rejection of a Balb/c heart allograft but allowed chronic rejection to develop over time, characterized by prominent neointima formation in the graft. There was extensive macrophage infiltration in the chronically rejected allografts, and the graft-infiltrating macrophages expressed markers associated with M2 cells but not M1 cells. In an in vitro system in which macrophages were polarized into either M1 or M2 cells, we screened phenotypic differences between M1 and M2 cells and identified purinergic receptor P2×7 (P2×7r), an adenosine triphosphate (ATP)–gated ion channel protein that was preferentially expressed by M2 cells. We further showed that blocking the P2x7r using oxidized ATP (oATP) inhibited M2 induction in a dose-dependent fashion in vitro. Moreover, treatment of C57BL/6 recipients with the P2x7r antagonist oATP, in addition to CTLA4-Ig treatment, inhibited graft-infiltrating M2 cells, prevented transplant vasculopathy, and induced long-term heart allografts survival. These findings highlight the importance of the P2x7r–M2 axis in chronic rejection and establish P2x7r as a potential therapeutic target in suppression of chronic rejection.
Introduction
The short-term transplant survival has been excellent and continues to improve in the clinic, and the 1-year graft survival is >80% for most organs transplanted (1). This is mainly due to improved immunosuppression that effectively controls acute rejection. Nevertheless, this remarkable success in the short term has not translated into long-term benefit. In fact, long-term transplant survival in the clinic remains largely unchanged over the past few decades, and most transplants are continuously lost to rejection over time. Histologically, late graft loss is often characterized by concentric and progressive arteriosclerosis, which is accompanied by interstitial fibrosis in the grafts, and this pathology is commonly referred to as chronic rejection (2). In fact, chronic rejection has become a major hurdle to transplant success in the clinic. To date, there are limited means to intervene therapeutically in this type of graft rejection.
The current belief is that development of chronic rejection is a multifactorial process involving both immune and nonimmune system mechanisms (3). In animal models, chronic rejection frequently occurs in immunosuppressed hosts in which acute cellular rejection is inhibited (4). The chronically rejected allografts are infiltrated by innate inflammatory cells, especially macrophages, and T cells are often few and far between in the graft (5). In the clinical setting, it has been shown that under broad immunosuppression protocols, the intensity of macrophage infiltration in the graft is correlated with increased incidence of chronic rejection and poor graft outcomes (6–8). These data suggest that activation of the innate immune cells, especially macrophages in the graft, may produce a protracted response of inflammation and repair that eventually results in the loss of allografts. This hypothesis would place local graft inflammation as the common pathway to chronic rejection and macrophages as key drivers of tissue inflammation (9). A major challenge to this hypothesis is that macrophages are an extremely plastic and dynamic cell type (10). Depending on the organ types, the local cytokine milieu, and the activation of other immune cells, macrophages can differentiate into phenotypically and functionally distinct subsets that exert diverse impact on the nature and outcomes of immune responses (11, 12). Activation of Toll-like receptors and/or Th1-type immunity, for example, often polarizes macrophages to M1 cells (also called classically activated macrophages), which are potent producers of nitric oxide and proinflammatory cytokines that mediate acute inflammation and cellular toxicity. In contrast, presence of IL-4 and IL-13 or activation of Th2 cells usually skews macrophages to M2 cells or alternatively activated macrophages that are involved in immune regulation and wound healing as well as tissue repair and regeneration (13). In fact, M1 and M2 cells most likely represent the extreme ends of a wide spectrum in which multiple other subsets exist, especially under in vivo settings (14). Nonetheless, the exact identity and function of macrophages in chronic rejection, the conditions under which they develop and survive, and their impact on transplant outcomes remain incompletely defined.
In the present study, we examined features of graft-infiltrating macrophages in chronically rejected heart allografts in the mouse and found that macrophages infiltrating the heart allografts adopted an M2 phenotype; they preferentially expressed the purinergic receptor P2X7 (P2x7r), an adenosine triphosphate (ATP)–gated ion channel protein on the cell surface (15). Importantly, blocking the P2x7r inhibited M2 polarization in vitro and suppressed the development of chronic allograft rejection in vivo, demonstrating the importance of the P2x7r–M2 axis in chronic rejection.
Materials and Methods
Animals
Male C57BL/6 (H-2b) and BALB/c (H-2d) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All mice were maintained under specific pathogen-free conditions. Animal use and care conformed to the guidelines established by the American Association for Accreditation of Laboratory Animal Care and approved by the institutional animal care committee at Houston Methodist Hospital in Houston, Texas.
Antibodies and reagents
The fluorochrome- or biotin-tagged antibodies were purchased from BD Pharmingen (San Diego, CA), eBiosciences (San Diego, CA), BioLegend (San Diego, CA) and R&D Systems (Minneapolis, MN): CD16/32 (clone 93), CD11b (M1/70), CD206 (C068C2), CD45 (30-F11), Ly6G (1A8), F4/80 (BM8), CD80 (16-10A1), CD86 (GL-1), Dectin-1 (CLEC7A). A Zombie Aqua viability kit was purchased from BioLegend. Polyclonal anti-P2x7r antibody was purchased from Abcam (Cambridge, MA). Human recombinant CTLA4 immunoglobulin (CTLA4-Ig) was purchased from BioXCell (West Lebanon, NH). Lipopolysaccharide (LPS) and oxidized ATP (oATP) were obtained from Sigma-Aldrich (St. Louis, MO). Mouse recombinant macrophage colony-stimulating factor (rM-CSF), recombinant interferon γ (IFN-γ), rIL-4 and rIL-13 were purchased from Peprotech (Rocky Hill, NJ).
Flow cytometry
Graft-infiltrating cells were collected, as described previously (16). Cells were preincubated with Zombie Aqua viability kit for 10 min, followed by anti-CD16/32 for 10 min, and then stained with fluorochrome-labeled anti-CD45, anti-Ly6G, anti-CD11b, anti-F4/80, anti-CD80, anti-CD86, anti-Dectin-1, anti-CD206 and anti-P2x7r antibodies for 30 min at 4°C. After staining, samples were washed and then analyzed using a BD LSRFortessa flow cytometer (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo software (FlowJo LLC, Ashland, OR).
Polarization of macrophages in vitro
For in vitro polarization of macrophages, macrophages were first induced from bone marrow cells, as described previously (17). Briefly, bone marrow cells were isolated from the femur and tibia of C57BL/6 mice and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin in the presence of 10 ng/mL murine rM-CSF. The medium was replenished on days 3 and 6, and cells cultured under such conditions were shown to differentiate into macrophages 6 days later. In some experiments, mouse peritoneal macrophages were obtained from the peritoneal cavity of C57BL/6 mice. To polarize macrophages into the M1 or M2 subset, bone marrow–derived macrophages were incubated with 100 ng/mL LPS and 20 ng/mL IFN-γ (for M1 induction) or incubated with 20 ng/mL IL-4 and 20 ng/mL IL-13 (for M2 induction) for 24 h. Cells were harvested and then assessed for differentiation into M1 and M2 cells and expression of M1- or M2-associated markers (18). In some experiments, the polarized M1 and M2 cells were used for phenotypic and functional assays.
Quantitative reverse transcriptase polymerase chain reaction
Total RNA was exacted using the TRIzol reagent (Qiagen, Valencia, CA), and mRNA was reverse transcribed into cDNA with the cDNA synthesis kit (Promega, Fitchburg, WI). The reaction protocol included a 10-min incubation time at 95°C. The amplification cycles consisted of 95°C for 10 s, 60°C for 20 s and 72°C for 20 s for each cycle for a total of 40 cycles, followed by a final elongation at 72°C for 5 min. Primers used are listed in Table 1. Quantitative polymerase chain reaction (PCR) was performed in a StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA) using SYBR Green master mix (TAKARA, Tokyo, Japan). HPRT was used as a housekeeping gene for sample standardization. Fold changes in target genes were calculated using the ΔCt value, as reported previously (19).
Table 1.
Primer sequences used for quantitative reverse transcriptase polymerase chain reaction
| Forward primer | Reverse primer | |
|---|---|---|
| iNOS | 5′-gtt ctc agc cca aca ata caa ga-3′ | 5′-gtg gac ggg tcg atg tca c-3′ |
| TNF-α | 5′-gcc tct tct cat tcc tgc ttg-3′ | 5′-ggg tct ggg cca tag aac tg-3′ |
| IL-6 | 5′-tgc aag aga ctt cca tcc agt-3′ | 5′-taa gcc tcc gac ttg tga agt-3′ |
| ARG-1 | 5′-ctc caa gcc aaa gtc ctt aga g-3′ | 5′-agg agc tgt cat tag gga cat c-3′ |
| CD206 | 5′-ttg gac gga tag atg gag gg-3′ | 5′-cca ggc agt tga gga ggt tc-3′ |
| Fizz1 | 5′-cca atc cag cta act atc cct cc-3′ | 5′-cca gtc aac gag taa gca cag-3′ |
| HPRT | 5′-agt aca gcc cca aaa tgg tta-3′ | 5′-ctt agg ctt tgt att tgg ctt t-3′ |
Arg-1, arginase 1; iNOS, inducible nitric oxide synthase; TNF-α, tumor necrosis factor α.
Immunoblotting assay
Total cellular extracts from macrophages were boiled, electrophoresed in sodium dodecyl sulfate polyacrylamide gels (Bio-Rad Laboratories, Hercules, CA), and then transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA). The membrane was blocked in phosphate-buffered saline (PBS) containing 5% bovine serum albumin. The membrane was washed, then incubated overnight with anti–mouse inducible nitric oxide synthase (iNOS; eBioscience, San Diego, CA), anti–mouse arginase 1 (Arg-1; R&D Systems), anti–mouse CD206 (Abcam, Cambridge, MA), or anti–mouse β-actin (R&D Systems) antibodies in Tris-buffered saline containing 0.5% Tween 20. After washing, the membrane was incubated for 1 h with appropriate horseradish peroxidase–coupled secondary antibodies. The specific bands were visualized using the enhanced chemiluminescence reagents (Thermo Scientific, Waltham, MA). For all experiments, β-actin was used as a loading control (19).
Heart transplantation
Heterotopic heart transplantation in the mouse was performed, as described previously (20). Briefly, heart grafts were harvested from donor mice and transplanted into the abdominal cavity of recipient mice by anastomosing the aorta and pulmonary artery of the graft end-to-side to the recipient’s aorta and vena cava, respectively. Graft survival was monitored by daily transabdominal palpation, and graft rejection was defined as cessation of palpable heartbeats, verified by laparotomy.
Treatment of recipient mice with CTLA4-Ig consisted of one dose of CTLA4-Ig (0.25 mg intraperitoneally) given 1 day after transplantation. Treatment with oATP consisted of 0.25 mg on days 0, 1, 3, 5, and 7 after transplantation. Groups of recipient mice were also treated with a combination of CTLA4-Ig and oATP in which one dose of CTLA4-Ig (0.25 mg) was given 1 day after heart grafting, and oATP (0.25 mg) was given on days 14, 16, 18, 20, and 22 after transplantation. Day 0 was defined as the day of heart transplantation.
Isolation of graft-infiltrating cells
Recipient mice bearing the heart allografts were sacrificed, and grafts were removed after perfusing the grafts with PBS plus 0.5% heparin to flush cells from the circulation. The heart grafts were chopped into pieces and digested at 37°C for 30 min in DMEM containing 300 U/mL type II collagenase (Worthington, Lakewood, NJ) and 40 U/mL DNase I (Roche, Indianapolis, IN) before pressing through a 40-μm filter. The collected cells were washed twice in PBS; stained for surface markers CD45, Ly6G, CD11b, and CD206; and then analyzed by fluorescence-activated cell sorting.
Tissue histology and immunofluorescence
The heart grafts and, in some cases, the native hearts were harvested, formalin fixed and paraffin embedded. Tissue blocks were sectioned at 2 μm, and slides were baked at 60°C for 1 h, deparaffinized and rehydrated, followed by staining with hematoxylin and eosin or Masson’s trichrome stain and evaluated by light microscopy. For immunofluorescence staining, tissue slides were prepared by microwaving paraffin-fixed sections in 0.1 M citrate buffer (pH 6.0), followed by staining for CD11b, F4/80, CD206 or iNOS and visualized with a fluorescence microscope (Nikon Eclipse 80i; Nikon, Tokyo, Japan). To quantify levels of target cell types, positively stained cells were counted in a minimum of five random microscopic fields, and the proportion of CD11b and CD206 double-positive cells in CD11b-positive (CD11b+) cells were calculated and presented.
Statistics
Statistical differences between groups were analyzed using the one-way analysis of variance. Graft survival was plotted using the Kaplan–Meier method, and the log-rank test was used to assess significance. All statistical analyses were performed using GraphPad Prism 6 (GraphPad Software, La Jolla, CA), and p < 0.05 was considered significant.
Results
CTLA4-Ig treatment inhibited acute rejection but allowed chronic rejection to develop in the graft
We transplanted Balb/c heart allografts into B6 recipients, and groups of recipient mice were treated with CTLA4-Ig; graft survival and graft histology were examined in a timely fashion. As shown in Figure 1, CTLA4-Ig treatment significantly prolonged heart allograft survival (mean survival time [MST] 31.5 days, n = 6) compared with untreated recipients (MST 8 days, n = 10; p < 0.05) (Figure 1A). Histology assessments of Balb/c heart allografts from CTLA4-Ig–treated recipients at 30 days after transplantation revealed extensive neointimal hyperplasia and lumen occlusion as wells mononuclear cell infiltration, which affected most vessels examined in the grafts (Figure 1B). Moreover, Masson’s trichrome staining of the heart allografts from the CTLA4-Ig–treated mice showed prominent vascular fibrosis in the grafts (Figure 1C). These features are consistent with the development of chronic allograft rejection (2). None of these changes were observed in heart grafts transplanted into syngeneic C57BL/6 recipients 30 days later (Figures 1B and C).
Figure 1. CTLA4-Ig treatment prevents acute heart allograft rejection but induces chronic allograft rejection (transplant vasculopathy).
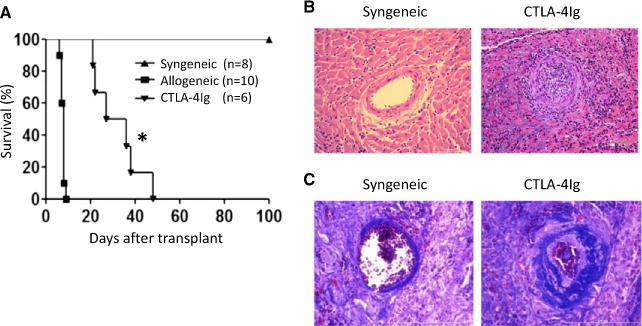
(A) Survival curves of heart grafts transplanted into syngeneic and allogeneic C57BL/6 hosts with or without CTLA4-Ig treatment. The MST of Balb/c heart allografts in CTLA4-Ig-treated mice was ≈31 days, and the MST of the control group was 8 days. The syngeneic heart grafts survived indefinitely. (B) Hematoxylin and eosin staining of syngeneic and CTLA4-Ig-treated grafts at 30 days after transplantation showing tissue and vascular structure in the grafts as well as cellular infiltration. (C) Masson’s trichrome staining of heart transplants at 30 days after transplantation showing tissue and vascular fibrosis. Representative pictures of one of three grafts examined in each group are shown (*p < 0.05). CTLA4-Ig, CTLA4 immunoglobulin; MST, mean survival time.
Graft-infiltrating macrophages preferentially expressed M2-associated markers in chronically rejected heart allografts
CTLA4-Ig is known to inhibit T cell activation (21). In this study, we focused on activities of macrophages in chronic rejection. As shown in Figure 2A, immunofluorescence staining of heart grafts showed that F4/80+ and CD11b+ macrophages infiltrated the heart allografts in CTLA4-Ig treated hosts, and this infiltration was especially prominent in or around chronic rejection lesions, whereas macrophages were not readily observed in syngeneic heart grafts (Figure 2A, top panel). To further examine the phenotypes of such infiltrating cells, we stained the heart allografts for cell surface or intracellular markers that usually mark polarized M1 and M2 cells (22) and analyzed them by fluorescence microscopy. As shown in Figure 2A (bottom panel), a substantial number of graft-infiltrating macrophages coexpressed CD206 (i.e. CD11b+CD206+), whereas expression of iNOS by infiltrating macrophages was minimal in CTLA4-Ig–treated mice. As presented by cell counts per microscopic view, there were roughly threefold more CD206+ macrophages than iNOS+ macrophages in the grafts (Figure 2B), suggesting that the graft-infiltrating macrophages may have adopted an M2 phenotype in chronic rejection. To further address this issue, we isolated graft-infiltrating cells from syngeneic and chronically rejected heart grafts (30 days after transplantation) and examined the phenotypes of such cells by flow cytometry. As shown in Figure 2B, among the CD45+ cells recovered from the grafts, ≈30% were CD11b+Ly6G− macrophages from the syngeneic grafts and ≈60% were CD11b+Ly6G− cells from CTLA4-Ig-treated heart allografts (Figure 2D). The absolute number of CD11b+cells in an allograft was ≈1 × 106, whereas those in a syngeneic graft were ≈2 × 105 (Figure 2E). Furthermore, among the CD11b+ Ly6G− cells, the relative percentage as well as the absolute number of CD11b+CD206+ macrophages (M2 cells) were significantly higher in the CTLA4-Ig-treated group compared with the syngeneic controls (Figures 2F–H), further demonstrating the selective enrichment of M2 cells in chronically rejected allografts.
Figure 2. Assessment of graft-infiltrating macrophages in heart allograft at 30 days after transplantation.
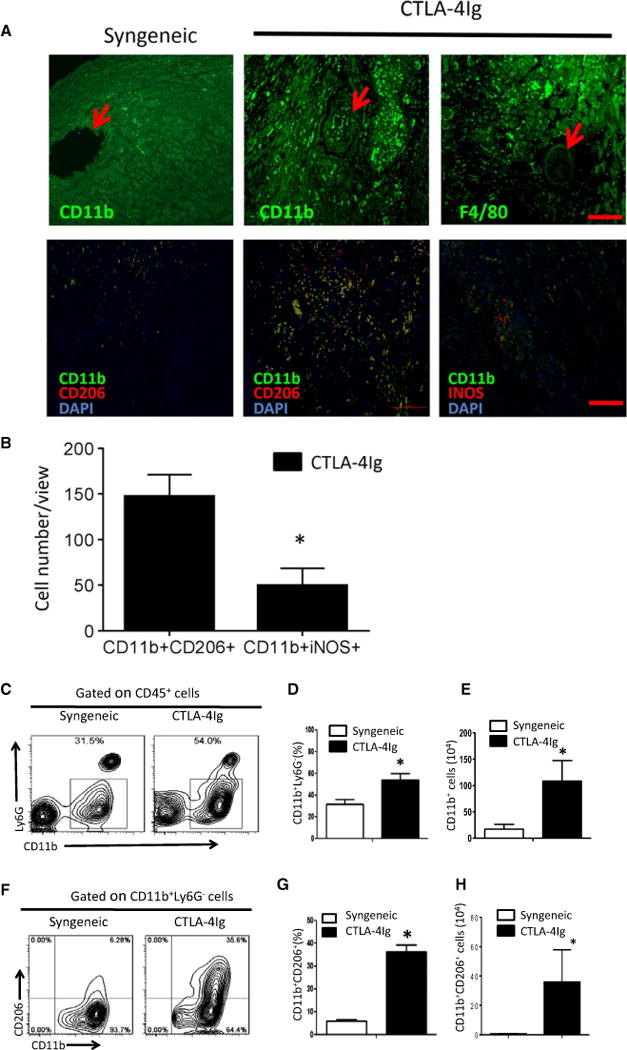
(A) Immunofluorescence staining of graft-infiltrating macrophages in CTLA4-Ig–treated mice was shown. Syngeneic heart grafts were used as controls. The arrows indicate arteries in the grafts (upper panel). Expression of CD206 or iNOS (red) by infiltrating macrophages was determined by double staining for DAPI (blue), CD11b (green) and CD206 (red) or iNOS (red). The merged fluorescence (yellow) identifies cells staining for both CD11b and CD206 or CD11b and CD206. Scale bar = 100 μm. (B) Cell counts from immunofluorescence staining in (A) showing CD11b+CD206+ (M2) and CD11b+iNOS+ (M1) cells per microscopic view. Data shown are mean cell number ± SE of five random views per sample for a total of three samples. (C) FACS plot showing gating strategy of graft infiltrating cells among CD45+ cells obtained from syngeneic and CTLA4-Ig–treated allografts. (D) Relative percentage of CD11b+ macrophages retrieved from each heart graft. Data shown are mean ± SE of four transplants in each group. (E) The absolute number of CD11b+ cells from each heart transplant. Data shown are mean ± SE of four transplants in each group. (F) FACS plot showing gating strategy for CD206+ cells among the total CD11b+ population. (G) Relative percentage of CD206+ cells among the total CD11b+ cells from syngeneic and CTLA4-Ig–treated allografts. Data shown are mean ± SE of four transplants in each group. (H) The absolute number of CD11b+CD206+ cells in each graft. Data shown are mean ± SE of four transplants in each group (*p < 0.05). CTLA4-Ig, CTLA4 immunoglobulin; DAPI, 4′,6-diamidino-2-phenylindole; FACS, fluorescence-activated cell sorting; iNOS, inducible nitric oxide synthase; SE, standard error.
Macrophage polarization in vitro and the identification of P2x7r
To better appreciate unique features of M2 cells in rejecting allografts and whether such cells could be therapeutically targeted, we set up an in vitro model in which macrophages were first derived from C57BL/6 bone marrow cells and then polarized into either M1 or M2 cells in vitro. Specifically, bone marrow cells were cultured in the presence of M-CSF for 6 days, followed by stimulation with LPS plus IFN-γ to polarize them into M1 cells or with IL-4 plus IL-13 to polarize them into M2 cells. Bone marrow–derived macrophages without polarizing cytokines were used as controls. As shown in Figure 3(A), stimulation of bone marrow–derived macrophages with IL-4 and IL-13 resulted in marked expression of Arg-1, CD206 and Fizz1 (M2 markers), as assessed by real-time PCR, whereas expression of iNOS, tumor necrosis factor α (TNF-α) and IL-6 (M1 markers) was not detectable. Conversely, stimulation with LPS and IFN-γ markedly upregulated the expression of iNOS, TNF-α and IL-6 transcripts but not Arg-1, CD206 and Fizz1 transcripts, confirming the successful polarization of M1 and M2 cells. This polarization was further confirmed using immunoblotting assays with antibodies specific for CD206, Arg-1 and iNOS (Figure 3B).
Figure 3. Polarization of bone marrow–derived macrophages into M1 and M2 cells in vitro.
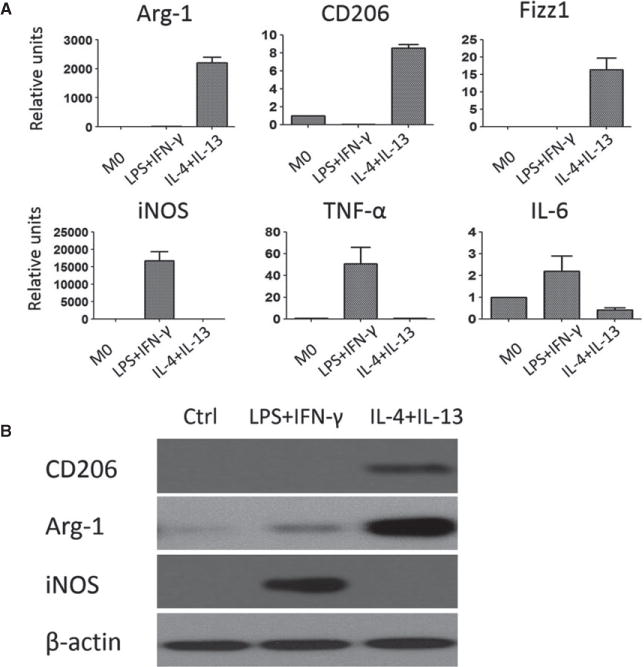
(A) Mouse bone marrow–derived macrophages were polarized into M1 cells with LPS and IFN-γ or to M2 cells with IL-4 and IL-13. Expression of M1- and M2-associated molecules were assessed by quantitative reverse transcriptase polymerase chain reaction 24 h later. Bone marrow–derived macrophages without polarizing cytokines were included as controls and referred to as M0 cells. The relative levels for Arg-1, CD206, Fizz1, iNOS, TNF-α, and IL-6 gene transcripts were normalized against HPRT and shown as mean ± SE of four independent experiments. (B) Western blot showing expression of CD206, Arg-1 and iNOS proteins in polarized M1 and M2 cells. The blot shown is representative of four independent experiments. Arg-1, arginase 1; IFN-γ, interferon γ; iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; SE, standard error; TNF-α, tumor necrosis factor α.
By using the polarized M1 and M2 cells as a model, we screened a panel of cell surface molecules that potentially differentiate M1 and M2 cells, which may serve as potential targets for therapeutic interventions. Flow cytometry showed that M1 cells expressed higher levels of CD80 and CD86 than did M2 cells, whereas M2 cells preferentially expressed CD206 and Dectin-1 on the cell surface (Figure 4A). Of particular interest was P2x7r, an ion channel protein that uses extracellular ATP as ligands (23), which was expressed at much higher levels in M2 cells than in M1 cells. This preferential expression of P2x7r by M2 cells was further demonstrated by both flow cytometry and western blotting assays using P2x7r-specific antibody (Figures 4B and C).
Figure 4. Analysis of cell surface markers expressed by polarized M1 and M2 cells.
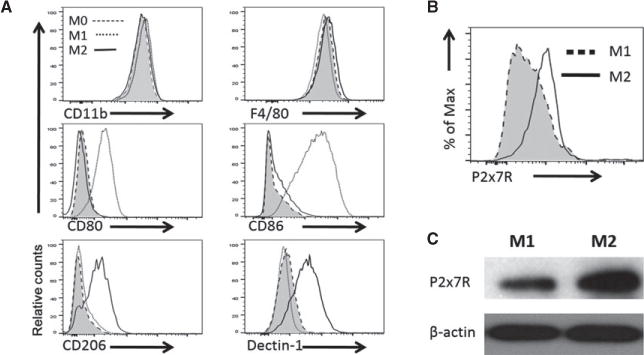
(A) Mouse bone marrow–derived macrophages were polarized into M1 cells (cultured in LPS and IFN-γ) or M2 cells (cultured in IL4- and IL-13), and macrophages cultured without polarizing cytokines were referred as M0 cells. The polarized macrophages were analyzed for expression of CD11b, F4/80, CD80, CD86, CD206, and Dectin-1 expression by FACS. (B) The FACS plot showing the expression of P2x7r by polarized M1 and M2 cells. (C) Western blot showing P2x7r protein expression in polarized M1 and M2 cells. Representative data of one of three experiments are shown. FACS, fluorescence-activated cell sorting; P2x7r, purinergic receptor P2X7.
The P2x7r antagonist oATP inhibited the induction of M2 cells in vitro
Because extracellular ATP accumulates at sites of tissue injury and serves as a danger molecule (23), expression of P2x7r by macrophage suggests that P2x7r engagement may govern macrophage functions. To test this possibility, we examined whether blocking the P2x7r using oATP, a receptor-specific antagonist (24), would inhibit macrophage polarization to M1 or M2 cells. We cultured bone marrow–derived macrophages under either M1- or M2-polarizing conditions. In those cultures, different concentrations of oATP were added to block P2x7r, and induction of M1 and M2 associated molecules was determined 24 h later by real-time reverse transcriptase PCR. As shown in Figure 5A, oATP strongly inhibited the induction of M2 cells, as shown by reduced expression of Arg-1, CD206 and Fizz1, whereas induction of M1 cells was not affected by oATP. In addition, western blotting assay also showed that oATP inhibited Arg-1 and CD206 expression in a dose-dependent manner under M2-polarizing conditions, and at a concentration of 200 μM, oATP strongly inhibited the expression of Arg-1 and CD206 by M2 cells (Figure 5B). We did not observe cell death following oATP treatment within this time period (24 h), as assessed by annexin V staining and flow cytometry (Figure 5C).
Figure 5. The purinergic receptor P2X7 antagonist oATP inhibits macrophage polarization to M2 cells in vitro.
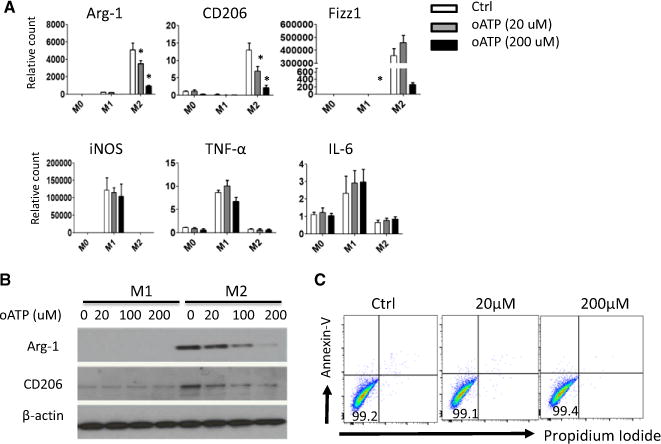
(A) Mouse bone marrow–derived macrophages were polarized toward M1 or M2 macrophages in the presence of different concentrations of oATP for 24 h. Cells were harvested; induction of Arg-1, CD206, Fizz1, iNOS, TNF-α, and IL-6 mRNA was analyzed by quantitative reverse transcriptase polymerase chain reaction; and relative levels normalized against HPRT were shown. (B) Western blot analysis of CD206 and Arg-1 protein expression by M2 cells with or without oATP. Results shown are from one of three independent experiments. (C) FACS plot showing viability of M2 cells cultured with oATP in vitro. Live bone marrow–derived macrophages were cultured under M2 conditions with or without oATP for 24 h, stained with PI and annexin V, and assessed by FACS (*p < 0.05). Arg-1, arginase 1; Ctrl, control; FACS, fluorescence-activated cell sorting; iNOS, inducible nitric oxide synthase; oATP, oxidized adenosine triphosphate; TNF-α, tumor necrosis factor α.
P2x7r antagonist oATP inhibited chronic allograft rejection in vivo in combination with CTLA4-Ig
To further determine the role of M2 cells and the effect of P2x7r blockade in suppression of chronic allograft rejection, we again transplanted BALB/c heart allografts into C57BL/6 recipients, and groups of transplant recipients were treated with CTLA4-Ig alone, oATP alone or a combination of CTLA4-Ig and oATP, and graft survival was determined. Tissue histology was performed on a cohort of heart allografts from the treated groups for signs of chronic rejection. As shown in Figure 6(A), treatment of B6 recipients with a combination of CTLA4-Ig and oATP induced long-term heart allograft survival, and seven of eight grafts survived long term (>90 days). Moreover, histological assessment on day 30 after transplantation revealed no obvious signs of vascular damage and vascular occlusion in the grafts (Figure 6B). In contrast, CTLA4-Ig treatment alone prolonged heart allograft survival (MST 31 days), but all grafts developed histological signs of chronic rejection, which was characterized by occlusion of intragraft vessels (Figure 6B). The MST of heart allografts in oATP-treated mice was ≈18 days (Figure 6A), and tissue histology was consistent with features of T cell–mediated acute rejection (data not shown). In addition, analysis by flow cytometry showed that in recipients treated with both CTLA4-Ig and oATP, there was a marked reduction of CD11b+CD206+ M2 cells in both relative percentage (Figure 6C) and absolute cell number (Figure 6E) in the heart allografts compared with mice treated with CTLA4-Ig alone. These data suggest that suppression of M2 cells is critically involved in preventing chronic rejection.
Figure 6. Treatment with CTLA4-Ig and oATP inhibited chronic rejection and induced long-term heart allograft survival.
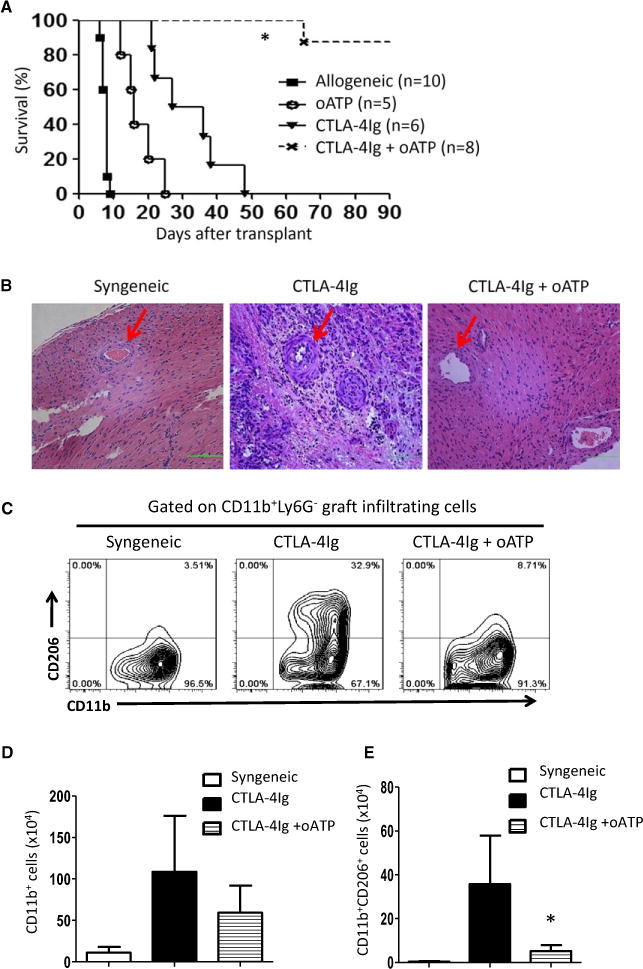
(A) Survival curves for Balb/c heart allografts in B6 recipients treated with CTLA4-Ig, oATP or a combination of CTLA4-Ig and oATP. (B) Sections of hematoxylin and eosin staining of heart transplants from recipients treated with CTLA4-Ig alone or combined CTLA4-Ig and oATP showing vascular changes and tissue damage. Arrows indicate small arteries in the graft. Syngeneic heart transplants were used as controls for comparison. (C) Representative fluorescence-activated cell sorting plots showing CD206+ cells among CD11b+ graft-infiltrating cells in recipient mice treated with CTLA4-Ig or combined CTLA4-Ig and oATP 30 days after transplantation. (D) The number of CD11b+ cells retrieved from heart allografts. Data shown are mean ± SE of three transplants in each group. (E) The number of CD11b+CD206+ cells from heart allografts is shown. Data shown are mean ± SE of three transplants in each group. (*p < 0.05). CTLA4-Ig, CTLA4 immunoglobulin fusion protein; oATP, oxidized adenosine triphosphate; SE, standard error.
Discussion
In the present study, we made several interesting and important findings. First, macrophages are very dynamic during graft rejection, as suggested previously (5). They adopted features of M2 cells in the grafts when acute cellular rejection was inhibited and contributed significantly to chronic rejection. Second, M2 cells preferentially express P2x7r and are functionally involved in M2 induction. Because the ligand for P2x7r is extracellular ATP, which is often enriched at sites of tissue injury and repair (25), ATP may provide a danger signal in driving activation of infiltrating macrophages through engagement of P2x7r. Our data also highlight the importance of cell surface channel proteins, in addition to cytokine receptors and Toll-like receptors, in mediating inflammatory responses (25). Finally, we provided evidence that the P2x7r antagonist oATP inhibits M2 induction; in the heart transplant model, oATP inhibits chronic allograft rejection, which is associated with a marked reduction of intragraft M2 cells, demonstrating a critical role for the P2x7r–M2 axis in the pathogenesis of chronic rejection.
What causes the predominance of M2 cells over M1 cells in the chronically rejected heart allografts remains unknown, and whether M2 cells are induced in situ in the heart grafts or migrate to the grafts from other places deserves further clarification. Nonetheless, the local milieus of acute and chronic rejection are very different. In acute rejection, activation of T cells is robust and followed by swift release of Th1 cytokines, during which graft-infiltrating macrophages are favored to an M1 phenotype (22). When the acute T cell response—especially the Th1 type of immunity—is inhibited, as in the case of CTLA4-Ig treatment (26), a different type of immune response involving different cell types often ensues and usually manifests as chronic rejection. In fact, the innate immune cells are more closely involved in graft injury than adaptive immune cells in transplant recipients under broad immune suppression in which acute T cell activation is profoundly inhibited (27). There are reports that CTLA4-Ig inhibits Th1 responses but spares Th2 responses (26), a condition that is compatible with M2 induction because Th2 cells are potent producers of IL-4, IL-5, and IL-13 (28). Nevertheless, the T cell–derived cytokines, including Th2 cytokines, are mostly prominent in the initial phase of rejection, and the T cell activities are diminished 30 or 40 days later when M2 cells and chronic rejection become prominent. Consequently, other mechanisms are likely present and involved in sustaining local inflammation and M2 induction. It was recently demonstrated that group 2 innate lymphoid cells, which are widely distributed in all tissues and organs, produce copious amounts of Th2-type cytokines (29). These cells are potential candidates in mediating chronic inflammation, including chronic allograft rejection, but how these cells become activated in transplant settings remains to be defined.
Our finding that P2x7r signaling can regulate M2 differentiation provides another possible mechanism in chronic rejection. It is well known that P2x7r uses ATP as a ligand; ATP is released in the presence of cell death and tissue injury and serves as a danger signal for inflammatory cells (23). Consequently, graft injury and repair may lead to ATP release and, on engaging P2x7r, induces the generation of M2 cells to propagate tissue inflammation. It is also possible that M2 cells are derived from M1 in the grafts when the M1 milieu subsides over time. Recent studies indicated that M1 macrophages can convert to M2 macrophages in situ. A switch from M1 to M2 cells occurs, for example, in the central nervous system, in which the M1 type of resident microglia as well as infiltrating macrophages change to M2 cells following demyelination (30). Whether this type of switch also occurs in transplants remains to be determined.
In addition to macrophages, other cell types express P2x7r, including T cells; therefore, P2x7r signaling potentially affects diverse responses and cell types (15). In fact, P2x7r antagonists can inhibit T cell activation in vitro, and in a minor antigen mismatched heart transplant model, they can induce heart allograft survival in vivo (31). The effects on macrophages were not examined, but graft histology revealed marked reduction of infiltrating macrophages in heart allografts surviving long term (31), a finding that is consistent with our observations. Furthermore, we showed that the P2x7r antagonist oATP appears to inhibit M2 cells because in vitro polarization of macrophages to M2 cells was strongly suppressed by oATP. Moreover, in the heart transplant model in which transplant recipients were treated with oATP at 14 days after CTLA4-Ig—a time point at which the T cell response was substantially diminished—long-term graft survival was readily achieved. Our data do not rule out the possibility that oATP may also inhibit other cell types besides M2 cells in prolonged transplant survival; however, oATP alone showed marginal effects in blocking acute rejection, and treatment with oATP first for 7 days followed by CTLA4-Ig also failed to produce long-term allograft survival (unpublished data). Consequently, the target cells affected by oATP and CTLA4-Ig are very different, and in our studies, both graft histology and flow cytometry suggest preferential suppression of M2 cells by the P2x7r antagonist oATP. Analysis of this issue awaits the generation of P2x7r floxed mice in future studies.
In other models, P2x7r engagement can activate inflammasomes in macrophages, resulting in the production of IL-1β, which is a feature of M1 cells. Most data suggest that when triggered by extracellular ATP, P2x7r forms a nonselective cation channel, leading to the rapid influx of calcium as well as the efflux of sodium and potassium, resulting in activation of the inflammasome (32). A recent study, however, demonstrated that the P2x7r pathway can also uncouple inflammasome activation from IL-1β production if the inflammasome is trapped in actin filaments, and thus M1 cell induction is inhibited and M2 cells are favored (33). Furthermore, it has been reported that P2x7r variants exist, some of which are gain-of-function variants, whereas others are loss-of-function variants (34). Whether P2x7r variants are involved in differential induction of M1 and M2 cells warrants further investigation. Regardless, the outcome of macrophage activation and the effects of P2x7r signaling appear to be context or model dependent.
Our data support the importance of innate immune cells, especially macrophage subsets in chronic allograft rejection (6), and suggest that the local graft environment may affect the character of innate immune cells. ATP is a key component of tissue injury and repair, and it promotes M2 induction in the local inflammatory milieu. This is consistent with other reports highlighting the role of macrophages, especially M2 cells in chronic graft loss (7); however, how M2 cells lead to vessel occlusion, which involves proliferation of other cell types both inside and outside of the blood vessels, is still poorly understood. M2 cells can produce growth factors such as TGF-β and platelet-derived growth factor, which can induce smooth muscle cell proliferation, activation of myofibroblasts and extracellular matrix deposition (35). Those are key elements of neointima during chronic rejection (5). It should be noted that in heart allografts surviving long term, we noticed a substantial number of CD11b+ myeloid cells that do not express features of M1 and M2 cells (Figure 6); whether they belong to myeloid suppressor cells that promote graft survival, as recently reported (36), remains to be determined in future studies.
In conclusion, we provided evidence that M2 cells contribute significantly to chronic rejection and that P2x7r blockade inhibits M2 cells and prevents neointima formation and graft loss to chronic rejection. Our data suggest that the P2x7r–M2 pathway may be a potential therapeutic target in treatment or prevention of M2-induced graft inflammation and chronic graft rejection.
Acknowledgments
This project was supported by the National Institutes of Health R01AI080779 (X.C.L.), the National Science Foundation of China scholarship council (C.W.) and the International Collaboration Center in Transplantation (2015B050501002). We thank Laurie Minze and Lindsay Mumma for operational support during these studies.
Abbreviations
- Arg-1
arginase 1
- ATP
adenosine triphosphate
- CD11b+
CD11b-positive
- CTLA4-Ig
CTLA4 immunoglobulin fusion protein
- Ctrl
control
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco’s modified Eagle’s medium
- FACS
fluorescence-activated cell sorting
- IFN-γ
interferon γ
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- MST
mean survival time
- oATP
oxidized adenosine triphosphate
- P2x7r
purinergic receptor P2X7
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- SE
standard error
- TNF-α
tumor necrosis factor α
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
References
- 1.Stehlik J, Mehra MR, Sweet SC, et al. The International Society for Heart and Lung Transplantation Registries in the Era of Big Data With Global Reach. J Heart Lung Transplant. 2015;34:1225–1232. doi: 10.1016/j.healun.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Loupy A, Toquet C, Rouvier P, et al. Late Failing Heart Allografts: Pathology of Cardiac Allograft Vasculopathy and Association With Antibody-Mediated Rejection. Am J Transplant. 2016;16:111–120. doi: 10.1111/ajt.13529. [DOI] [PubMed] [Google Scholar]
- 3.Tullius SG, Tilney NL. Both alloantigen-dependent and -independent factors influence chronic allograft rejection. Transplantation. 1995;59:313–318. [PubMed] [Google Scholar]
- 4.Meier-Kriesche HU, Schold JD, Kaplan B. Long-term renal allogrft survial: Have we made significant progress or is it time to rethink our analytic and therapeutic strategies? Am J Transplant. 2004;4:1289–1295. doi: 10.1111/j.1600-6143.2004.00515.x. [DOI] [PubMed] [Google Scholar]
- 5.Kaul AM, Goparaju S, Dvorina N, et al. Acute and chronic rejection: compartmentalization and kinetics of counterbalancing signals in cardiac transplants. Am J Transplant. 2015;15:333–345. doi: 10.1111/ajt.13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mannon RB. Macrophages: Contributors to allograft dysfunction, repair, or innocent bystanders? Curr Opin Organ Transplant. 2012;17:20–25. doi: 10.1097/MOT.0b013e32834ee5b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu GW, Kobashigawa JA, Fishbein MC, et al. Asymptomatic antibody-mediated rejection after heart transplantation predicts poor outcomes. J Heart Lung Transplant. 2009;28:417–422. doi: 10.1016/j.healun.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toki D, Zhang W, Hor KL, et al. The role of macrophages in the development of human renal allograft fibrosis in the first year after transplantation. Am J Transplant. 2014;14:2126–2136. doi: 10.1111/ajt.12803. [DOI] [PubMed] [Google Scholar]
- 9.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dal-Secco D, Wang J, Zeng Z, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med. 2015;212:447–456. doi: 10.1084/jem.20141539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ochando J, Kwan WH, Ginhoux F, Hutchinson JA, Hashimoto D, Collin M. The mononuclear phagocyte system in organ transplantation. Am J Transplant. 2016;16:1053–1069. doi: 10.1111/ajt.13627. [DOI] [PubMed] [Google Scholar]
- 12.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mills CD. M1 and M2 macrophages: Oracles of health and disease. Crit Rev Immunol. 2012;32:463–488. doi: 10.1615/critrevimmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- 14.McNelis JC, Olefsky JM. Macrophages, immunity, and metabolic disease. Immunity. 2014;41:36–48. doi: 10.1016/j.immuni.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 15.Rissiek B, Haag F, Boyer O, Koch-Nolte F, Adriouch S. P2X7 on mouse T cells: One channel, many functions. Front Immunol. 2015;6:204. doi: 10.3389/fimmu.2015.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Conde P, Rodriguez M, van der Touw W, et al. DC-SIGN(+) Macrophages Control the Induction of Transplantation Tolerance. Immunity. 2015;42:1143–1158. doi: 10.1016/j.immuni.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jablonski KA, Amici SA, Webb LM, et al. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE. 2015;10:e0145342. doi: 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zajac E, Schweighofer B, Kupriyanova TA, et al. Angiogenic capacity of M1- and M2-polarized macrophages is determined by the levels of TIMP-1 complexed with their secreted proMMP-9. Blood. 2013;122:4054–4067. doi: 10.1182/blood-2013-05-501494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao X, Shi X, Fan Y, et al. GITR subverts Foxp3(+) Tregs to boost Th9 immunity through regulation of histone acetylation. Nat Commun. 2015;6:8266. doi: 10.1038/ncomms9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li XC, Li Y, Dodge I, et al. Induction of allograft tolerance in the absence of Fas-mediated apoptosis. J Immunol. 1999;163:2500–2507. [PubMed] [Google Scholar]
- 21.Li XC, Rothstein DM, Sayegh MH. Costimulatory pathways in transplantation: Challenges and new developments. Immunol Rev. 2009;229:271–293. doi: 10.1111/j.1600-065X.2009.00781.x. [DOI] [PubMed] [Google Scholar]
- 22.Mills CD. Anatomy of a discovery: M1 and M2 macrophages. Front Immunol. 2015;6:212. doi: 10.3389/fimmu.2015.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solini A, Usuelli V, Fiorina P. The dark side of extracellular ATP in kidney diseases. J Am Soc Nephrol. 2015;26:1007–1016. doi: 10.1681/ASN.2014070721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sullivan JA, Jankowska-Gan E, Shi L, et al. Differential requirement for P2X7R function in IL-17 dependent vs. IL-17 independent cellular immune responses. Am J Transplant. 2014;14:1512–1522. doi: 10.1111/ajt.12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arulkumaran N, Unwin RJ, Tam FW. A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin Investig Drugs. 2011;20:897–915. doi: 10.1517/13543784.2011.578068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sayegh MH, Akalin E, Hancock WW, Russell ME, Carpenter CB. CD28-B7 blockade after alloantigenic challenge in vivo inhibits Th1 cytokines but spares Th2. J Exp Med. 1995;181:1869–1874. doi: 10.1084/jem.181.5.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li XC. The significance of non-T cell pathways in graft rejection: Implications for transplant tolerance. Transplantation. 2010;90:1043–1047. doi: 10.1097/TP.0b013e3181efcfe9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strom TB, Roy-Chaudhury P, Manfro R, et al. The Th1/Th2 paradigm and the allograft response. Curr Opin Immunol. 1996;8:688–693. doi: 10.1016/s0952-7915(96)80087-2. [DOI] [PubMed] [Google Scholar]
- 29.Huntington ND, Carpentier S, Vivier E, Belz GT. Innate lymphoid cells: Parallel checkpoints and coordinate interactions with T cells. Curr Opin Immunol. 2016;38:86–93. doi: 10.1016/j.coi.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 30.Miron VE, Boyd A, Zhao JW, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vergani A, Tezza S, D’Addio F, et al. Long-term heart transplant survival by targeting the ionotropic purinergic receptor P2X7. Circulation. 2013;127:463–475. doi: 10.1161/CIRCULATIONAHA.112.123653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qu Y, Franchi L, Nunez G, Dubyak GR. Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol. 2007;179:1913–1925. doi: 10.4049/jimmunol.179.3.1913. [DOI] [PubMed] [Google Scholar]
- 33.Pelegrin P, Surprenant A. Dynamics of macrophage polarization reveal new mechanism to inhibit IL-1beta release through pyrophosphates. EMBO J. 2009;28:2114–2127. doi: 10.1038/emboj.2009.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Englezou PC, Rothwell SW, Ainscough JS, et al. P2X7R activation drives distinct IL-1 responses in dendritic cells compared to macrophages. Cytokine. 2015;74:293–304. doi: 10.1016/j.cyto.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nykanen AI, Krebs R, Tikkanen JM, et al. Combined vascular endothelial growth factor and platelet-derived growth factor inhibition in rat cardiac allografts: beneficial effects on inflammation and smooth muscle cell proliferation. Transplantation. 2005;79:182–189. doi: 10.1097/01.tp.0000147199.60464.f9. [DOI] [PubMed] [Google Scholar]
- 36.Garcia MR, Ledgerwood L, Yang Y, et al. Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. J Clin Invest. 2010;120:2486–2496. doi: 10.1172/JCI41628. [DOI] [PMC free article] [PubMed] [Google Scholar]